

Abstract
Adoptive cell therapy (ACT) consisting of genetically engineered T cells expressing tumor antigen-specific T-cell receptors displays robust initial antitumor activity, followed by loss of T-cell activity/persistence and frequent disease relapse. We characterized baseline and longitudinal T-cell phenotype variations resulting from different manufacturing and administration protocols in patients who received ACT. Patients with melanoma who enrolled in the F5-MART-1 clinical trial (NCT00910650) received infusions of MART-1 T-cell receptors transgenic T cells with MART-1 peptide-pulsed dendritic cell vaccination. Patients were divided into cohorts based on several manufacturing changes in the generation and administration of the transgenic T cells: decreasing ex vivo stimulation/expansion time, increased cell dose, and receiving fresh instead of cryopreserved cells. T-cell phenotypes were analyzed by flow cytometry at baseline and longitudinally in peripheral blood. Transgenic T cells with shorter ex vivo culture/expansion periods displayed significantly increased expression of markers associated with less differentiated naive/memory populations, as well as significantly decreased expression of the inhibitory receptor programmed death 1 (PD1). Patients receiving fresh infusions of transgenic cells demonstrated expansion of central memory T cells and delayed acquisition of PD1 expression compared with patients who received cryopreserved products. Freshly infused transgenic T cells showed persistence and expansion of naive and memory T-cell populations and delayed acquisition of PD1 expression, which correlated with this cohort’s superior persistence of transgenic cells and response to dendritic cell vaccines. These results may be useful in designing future ACT protocols.
Key Words: melanoma, adoptive cell therapy, T-cell phenotype, memory T cells, PD1
Immunotherapy can be an effective means of treating or curing a variety of advanced malignancies. These approaches generally rely on CD8+ cytotoxic T lymphocytes (CTL), which recognize tumor antigens via their T-cell receptor (TCR), and then induce tumor cell death via effector molecules such as granzyme B, engaging tumor necrosis factor family of death receptors, and interferons. Adoptive cell therapy (ACT) involves the ex vivo generation of CD8+ T cells which target specific tumor antigens.1 These T cells can be generated ex vivo by expansion of tumor-infiltrating lymphocytes (TILs) from a given tumor,2 expansion of endogenous, low-frequency CD8+ T cells which target a tumor antigen,3,4 or, most recently, by genetic transfer of a TCR with specificity to a given tumor antigen.5 Such products are then expanded ex vivo and reinfused into the patient. Several groups have previously demonstrated that ACT typically results in frequent initial tumor responses, but with infrequent long-term responses. Prior landmark ACT clinical trials by Rosenberg and colleagues at the National Cancer Institute, Surgery Branch demonstrated complete responses in a minority of patients treated with genetically engineered T cells directed against melanoma-specific antigens MART-1 and gp100.6,7 Additional antigens and their associated malignancies have been targeted in this manner as well, most notably with anti-NY-ESO-1 TCR-engineered T cells used to treat NY-ESO-1+ melanomas and synovial sarcomas.8,9 However, despite robust initial tumor responses in the majority of patients treated in these trials, complete responses without recurrence were only seen in a minority of patients. This widely reproduced phenomenon underscores the need to better understand the behavior of the genetically modified T cells utilized in ACT to enhance the duration of their initially strong antitumor effects.
Our group’s prior experience with ACT, the University of California, Los Angeles/CalTech F5-MART-1 clinical trial, utilized autologous T cells which had undergone retroviral transduction to express a TCR against MART-1 and subsequently expanded ex vivo, in conjunction with autologous MART-1 peptide-pulsed dendritic cells (DCs) to treat patients with locally advanced or metastatic melanoma.10 Nine of the 13 treated patients (69%) showed evidence of transient tumor regression by computed tomography or positron emission tomography/computer tomography. Peripheral blood reconstitution with MART-1-specific T cells peaked within 2 weeks of ACT, indicating dramatic in vivo expansion. However, none of the initial tumor responses were durable beyond 6 months. We had previously analyzed TCR transgenic T cells administered and recovered from 3 of the patients in this series using new generation microfluidics-based miniaturized assays. These assays allow us to simultaneously study multiple functional cytokine responses of T cells based on defined antigen specificities.11,12 These studies showed that the initial polyfunctionality and cytotoxicity resulting in high antitumor activity of the TCR transgenic T cells is gradually lost in vivo, which was temporally associated with the clinical course of initial tumor response followed by progression.
The need for improving ACT’s efficacy has led to the characterization of factors responsible for the success or failure of these therapies. Prior work by Restifo and colleagues has demonstrated the role of less differentiated T cells in successful ACT, primarily the naive T cell (TN) and central memory T cell (TCM) compartments. Mouse models have revealed that ACT with these less differentiated T-cell subsets demonstrate superior in vivo expansion, persistence, and antitumor activity when compared with the more terminally differentiated effector memory (TEM) and effector (TEFF) T-cell compartments.13,14 Furthermore, they have shown that TEFF cells derived from naive T cells resist terminal differentiation and have greater proliferative potential than TEFF derived from memory T-cell populations.15,16 In addition, the loss of antitumor activity in mouse models of ACT has been associated with the acquisition of inhibitory receptors such as programmed death 1 (PD1) on both tumor-infiltrating lymphocytes (TILs) and circulating CTLs, and the addition of agents blocking these inhibitory receptors potentiates the activity of ACT in these preclinical models.17
Our F5-MART-1 trial utilized several manufacturing changes in the generation and administration of the modified T cells, namely decreasing ex vivo stimulation/expansion time from 7 to 6 days, increasing the number of cells infused, and the use of fresh instead of cryopreserved cells. Of note, our patients who received fresh TCR transgenic T cells were noted to experience a more prolonged persistence of circulating transgenic T cells, as well as reexpansion of the transgenic cells in response to the MART-1 peptide DC vaccinations.10 We therefore chose to longitudinally study the transgenic T cells in our patients to understand the phenotypic effect of the differences in manufacturing techniques employed. We report that shorter ex vivo culture/expansion periods are associated with increases in expression of T-cell markers of naive/memory phenotypes, and that this phenotype persists longer in freshly infused cells compared with cryopreserved cells. In addition, while shorter culture time was associated with decreased initial expression of the inhibitory TCR programmed death 1 (PD1), freshly infused cells demonstrated delayed acquisition of PD1 when compared with the cryopreserved cells. These differences may be responsible for these patients displaying increased persistence of their transgenic T-cell products, as well as their responsiveness to DC vaccinations. These findings may help inform the optimization of future ACT protocols, and lead to improved treatment outcomes.
METHODS
Clinical Trial, Patients, and Manufacturing Cohorts of MART-1 Engineered T Cells
Patients positive for HLA-A*0201 and with a MART-1+ metastatic melanoma were enrolled in the F5-MART-1 clinical trial (NCT00910650) from April 2009 to September 2011. A written consent form for the University of California Los Angeles (UCLA) Institutional Review Board (#08-02-020 and #10-001212) was obtained from each patient, under investigational new drug 13859 from the US Food and Drug Administration.10,12 Detailed descriptions of the clinical trial design and manufacturing of the MART-1 TCR transgenic T cells are previously described.10 We divided the patients into 4 cohorts for immune monitoring analyses following sequential amendments to the TCR transgenic T-cell manufacturing. Briefly, cohort 1 (patients F5-1 through F5-4) was treated under the original protocol, in which nonmobilized peripheral blood mononuclear cells (PBMCs) were stimulated in culture with IL-2/OKT3 and were transduced with our clinical grade retrovirus vector expressing the MART-1 F5 TCR on 2 consecutive days, and were then continually expanded for 96 hours (7 d total culture time), after which they were cryopreserved as soon as the lot release criteria were cleared, and subsequently receiving up to 1×109 previously cryopreserved TCR transgenic lymphocytes. Cells were infused into patients following a conditioning regimen with cyclophosphamide and fludarabine, along with up to 14 doses of IL-2, as previously described.10 Cohort 2 (patients F5-5 through F5-9) limited the period of ex vivo expansion to 72 hours following transduction (6 d total culture time) before cryopreservation and subsequent administration of 1×109 TCR transgenic lymphocytes. Cohort 3 (patients F5-10 and F5-11) increased the total number of cryopreserved TCR transgenic lymphocytes given to up to a maximum of 1×1010, while total culture time remained 6 days total. Cohort 4 (patients F5-12 through F5-14) received fresh infusions of up to 1×1010 TCR transgenic lymphocytes, rather than cryopreserved. In addition, these patients received 1 day less of fludarabine conditioning, and were given only a maximum of 9 doses of IL-2 instead of 14. Culture time remained 6 days total for this cohort. Patient F5-15 also received freshly infused cells, but with a maximum of only 1×109 cells. This patient also received more frequent administration of IL-2, but at lower dosage.
Flow Cytometry Surface Staining
PBMCs were centrifuged (400g for 5 min), resuspended in 100 μL of adult bovine serum (Omega Scientific, Tarzana, CA) and stained with preconjugated antibodies for flow cytometry,18 and acquired using 2 LSR II Flow Cytometers, one with 3 lasers (blue, red, and violet) and the other with 4 lasers (blue, red, violet, and ultraviolet; BD Biosciences, San Jose, CA). A minimum of 500,000 events were captured for each experiment. Antibodies against CD3, CD8, CD4, CD25, HLA-DR, CD45RO, CCR7, CCR5, PD1, CD45RA, CD27, CD28 and CD62L, as well as 7-Aminoactinomycin D, were purchased from BD Biosciences, Beckman Coulter (Brea, CA), Biolegend (San Diego, CA), and Thermo Fisher Scientific (Waltham, MA). MART-1 HLA-A*0201 Tetramers and negative controls were purchased from Beckman Coulter. Detailed description of the antibodies and staining is described in previously published articles.10,12 For CD8+ T-cell phenotype characterization, TN were classified as CD45RA+/CCR7+/CCR5−/PD1−, CD45RA+/CCR7+/CCR5−/PD1+, CD45RA+/CCR7+/CCR5+/PD1−, and CD27+/CD28+/CD62L+; TCM as CD45RO+/CD25−/HLA-DR−/CD127+, CD45RA−/CCR7+/CCR5−/PD1−, and CD27+/CD28−/CD62L+; TEM as CD45RA−/CCR7−/CCR5−/PD1−, CD45RA−/CCR7−/CCR5−/PD1+, CD45RA−/CCR7−/CCR5+/PD1−, and CD45RA−/CCR7−/CCR5+/PD1+; effector memory RA (TEMRA) as CD45RA+/CCR7−/CCR5+/PD1−, CD45RA+/CCR7−/CCR5+/PD1−, CD45RA+/CCR7−/CCR5+/PD1−, CD45RA+/CCR7−/CCR5+/PD1−; and TEFF as CD45RO+/CD25+/HLA-DR+/CD127−, CD45RO+/CD25+/HLA-DR−/CD127−, and CD45RO+/CD25-/HLA-DR−/CD127−. For CD4 phenotype characterization, suppressor T regulatory cells (Treg) were defined as CD4+/CD25+/CD127−.
Flow Cytometry Analysis
All flow data analyses were done with either FlowJo (Tree Star Inc., Asland, OR) or Cytobank (www.cytobank.com).19 Biexponential and arcsinh displays were used in the analyses.
Statistical Analysis
Graphing, heatmaps, and descriptive statistical analyses were carried out with GraphPad Prism version 7.0 (GraphPad, San Diego, CA). For the comparison of the characteristics of the 7 day versus 6 day culture cohorts’ infusion products, unpaired Student t test was used. Log-transformation was performed if normality assumption was violated according to the Shapiro-Wilk test. P-values of <0.05 were considered statistically significant.
RESULTS
Patient Characteristics and Outcomes
As previously described,10 there were multiple protocol amendments during this trial, which significantly altered the manufacturing of the infused cell products as described previously. The 4 manufacturing cohorts and their associated differences are summarized and subdivided on Table 1, along with patient characteristics and outcomes. There was transient evidence of initial tumor response to ACT in 9 of 13 patients as determined by day 30 computed tomography or positron emission tomography/computer tomography scans. In patients who survived to the end of the study, 8 demonstrated stable disease, while 4 showed progressive disease. One subject, F5-5, was ultimately ineligible for the trial due to the discovery of brain metastases shortly after the subject was enrolled, and did not receive his transgenic T-cell infusion product. Another subject, F5-15, was enrolled after an additional amendment to our protocol changing the IL-2 administration from high dose intravenously to low dose subcutaneously bid for up to 14 days, therefore this patient received more frequent dosing of IL-2, but at lower dosing. Patient F5-15 also had reduced number of infused cells (the original 1×109 cells used in the earlier cohorts). All patients ultimately died of their underlying metastatic melanoma. Progression-free survival ranged from 0 to 7 months, while overall survival ranged from 1 to 86 months (Table 1).
TABLE 1.
Patient Demographics, Outcomes, and Distribution by Manufacturing Cohort

Patient F5-10 suffered bone marrow failure secondary to disease progression, and we were unable to obtain any longitudinal samples beyond the first 15 days. We were also unable to obtain any samples between day +30 and day +100 in patient F5-11 due to significant adverse events (SAEs) during this period. Patients F5-12 and F5-13 experienced SAEs related to IL-2 administration, and samples before day +15 were unable to be obtained. We were also unable to obtain samples longitudinally for patients F5-14 and F5-15 due to SAEs within the first month, and they passed away in the first month following T cell infusion.
Baseline Phenotypic Differences Between Manufacturing Cohorts of MART-1 Transgenic T Cells
We first examined the phenotypic differences between cohort 1 and cohorts 2, 3, and 4 to explore the effects of shorter ex vivo culture and expansion time (7 vs. 6 days). We found no significant differences in the CD4+/CD8+ ratio between 7-day and 6-day culture/expansion times (1.78 vs. 2.70, Fig. 1B). Eleven surface markers were used to phenotypically classify the MART-1 transgenic T-cell products as described previously. The transgenic CD4+ T-cell subset (Fig. 1D) demonstrated significantly higher CCR7 expression in the 6-day culture time cohorts than in the 7-day culture cohort (25.3% vs. 1.38%, P<0.05), as well as higher expression of CD62L (66.2% vs. 30.3%, P<0.05). In addition, there was increased CD25 expression noted in the 6-day culture time cohorts compared with the 7-day culture cohort (84.0% vs. 43.3%, P<0.01). In the transgenic CD8+ subset (Fig. 1E), CCR7 elevation in shorter culture time nearly achieved significance (23.1% vs. 1.4%, P=0.066). However, CD62L expression was significantly higher in the shorter culture time cohorts compared with cohort 1 (70.4% vs. 34.0%, P<0.01). Furthermore, the inhibitory receptor PD1 displayed significantly lower expression in the cell products which had been cultured for 6 days compared with 7 days (7.0% vs. 27.3%, P<0.05).
FIGURE 1.
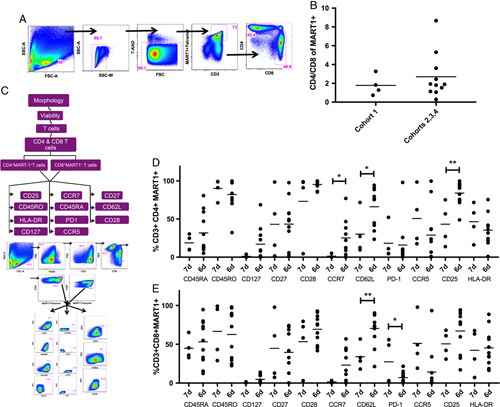
Immune phenotypic analysis of MART-1 transgenic T cells in the final product. A, Gating strategy used to extract the percentage of transgenic CD4 and CD8 T cells. B, Ratio of CD4 versus CD8 cells. C, Gating strategy to extract the immune markers. D and E, Comparison of immune marker expression in MART1+ CD4+ (D) and CD8+ (E) T cells, comparing the 7-day culture/expansion cohort—cohort 1 (n=4), to the 6-day culture/expansion cohorts—cohorts 2, 3, and 4 (n=11). Dots represent individual data points, lines indicate means. *P<0.05; **P<0.01.
Persistence of Naive/Memory T Cells in Freshly Infused Transgenic Product
We compared the changes in transgenic T-cell phenotype between the different manufacturing/administration protocols over time for patients in whom we were able to obtain longitudinal PBMC samples (patients F5-1 through F5-4 in cohort 1, patients F5-6 through F5-9 in cohort 2, patients F510 and F5-11 in cohort 3, and patients F5-12 and F5-13 in cohort 4). The most striking trends in both the CD4+ and CD8+ populations displayed were a persistence of the naive/memory T-cell markers CD127 and CCR7 in cohorts 2, 3, and 4, as well as a significant delay in the acquisition of PD1 in cohort 4 (Fig. 2). While there was negligible expression of CD127 at baseline and longitudinally in the CD8 transgenic cells in cohort 1 (<1% positive average, Fig. 2A), this memory T-cell marker expanded beyond its baselines in cohort 2 (from 5.99% to 47% at end of study), cohort 3 (from 7.5% to 21.3%) and cohort 4 (from 5.4% to 70.5%). The marker of memory/naivety CCR7 remained at <1% of transgenic CD8+ T cells for cohort 1. Despite starting off at higher levels in cohorts 2 and 3 (27.7% and 36.7%, respectively), CCR7 average expression levels gradually diminished over time in these cohorts. Cohort 4, however, displayed a longitudinal expansion of CCR7, reaching a peak of 62.1% average positivity at day +45, and a persistence of this expression up to day +100 (31%). When transgenic CD8+ T cells were stratified by naive/central memory/effector memory/effector memory RA/effector phenotypes (Fig. 2B), we observed a contraction of TN and TCM compartments in cohort 1 (from 8.2% and 6.5% to 4.2% and 2.6% of total, respectively), compared with a starting higher average proportion TN and TCM subpopulations of T cells within cohort 2 (TN 23.3%, TCM 5.9%) and cohort 3 (TN 18.1%, TCM 3.8%), as well as overall persistence of these compartments by the end of study (cohort 2—TN 17.7%, TCM 5.9%; cohort 3—TN 19.6%, TCM 8.7%). Furthermore, while cohort 4 displayed a higher average amount of TN (15.0%), which contracted to 5.5% of total by the end of the study, there was an overall expansion of TCM cells in this cohort (from 3% to 34.9%) by day +100.
FIGURE 2.
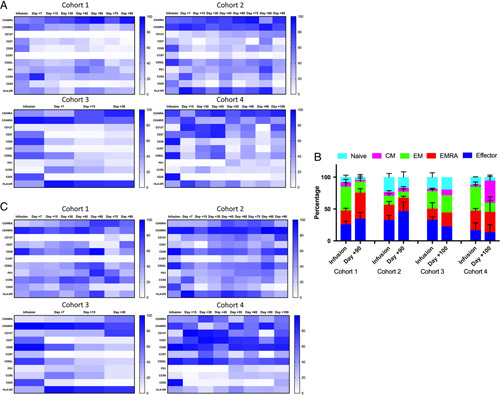
Longitudinal changes in phenotypic markers on MART-1 transgenic T cells. A and C, Heatmap representing changes in average percentages of transgenic CD8 T cells (A) and CD4 T cells (C) between the 4 manufacturing cohorts over time. Average percent positivity of a given marker is expressed in color scale from white (0%) to blue (100%), color scales are expressed to the right. Figures represent pooled data within each cohort. B, Phenotypic changes of CD8+MART-1+ T cells. The average percentage of each T-cell phenotype (naive, central memory, effector memory, effector memory RA, and effector) is represented by a different color. Gating strategies are the same as in Figure 1C. Error bars represent SEM. Cohort 1: n=4, cohort 2: n=4, cohort 3: n=2, cohort 4: n=2.
Several trends were also apparent within the CD4+ population of transgenic T cells (Fig. 2C): CD127 expression was negligible in cohort 1, achieving a maximum expression of 15.8% on day +75, which was not sustained. Conversely, cohort 2 achieved continual expansion of CD127 to a maximum of 76.1% by day +90, while cohort 4 achieved a rapid expansion of CD127 to an average of 97.3% positive, with persistence of 67.5% positive by day +100 (there were no detectable CD4+ transgenic T cells after day +30 in cohort 3). CCR7 also remained negligible in cohort 1 throughout the study. Cohort 2 displayed a late expansion of this memory marker to 49.3% at day +80, while cohort 4 displayed an expansion of this marker to 64.8% at day +30, with a persistence of 32.4% positive by day +100.
Delayed PD1 Acquisition in Freshly Infused Transgenic Product Compared With Cryopreserved Product
The T cell inhibitory marker PD1 also displayed different longitudinal dynamics in cohort 4, which received fresh infusions of transgenic cells, as compared with the cryopreserved cohorts. Although cohorts 2, 3, and 4 all displayed significantly lower PD1 expression than cohort 1 at baseline, cohorts 2 and 3 displayed progressive acquisition of PD1 within a week of infusion. Cohort 1, which displayed an average PD1 expression of 27.3% and 18.2% in the transgenic CD8+ and CD4+ compartments, respectively, increased steadily to peaks of 57.7% and 84% within the transgenic CD8+ and CD4+ compartments, respectively (Figs. 2A, C). Cohorts 2 and 3, which initially had only 7.3% and 7.8% average PD1 expression in the CD8+ transgenic cells at infusion, displayed a rapid and steady increase in the acquisition of this marker within the first week, rising to maximums of 75.6% and 27.5%, respectively. This trend was mirrored within the CD4+ transgenic T-cell population as well, with rapid rises from baselines of 12.0% and 9.2% to maximums of 56.4% and 25.0% within cohorts 2 and 3, respectively. Cohort 4 displayed low PD1 expression at baseline within the CD8+ and CD4+ transgenic populations (1.0% and 2.0%, respectively). However, PD1 expression remained nearly undetectable (<1%) in both the CD4+ and CD8+ transgenic cell population in this cohort until day +55, reaching maximums of only 28.4% and 28.7%, respectively.
We also examined the total proportion of CD8+PD1+ circulating lymphocytes in our patients, to examine the overall trends of this population, as well as proportion of transgenic CD8+PD1+ cells which made up the total of this phenotype (Fig. 3). Overall, the proportion of transgenic CD8+PD1+ T cells which made up the total quantity of CD8+PD1+ T cells remained the same or was slightly augmented within the first 30 to 45 days postinfusion, after which it remained roughly proportional in cohort 2, while modestly contracting in cohorts 1 and 3. Cohort 4, by comparison, maintained the lowest overall population of CD8+PD1+ T cells, only minimally expanding both the total and transgenic populations after day +55.
FIGURE 3.
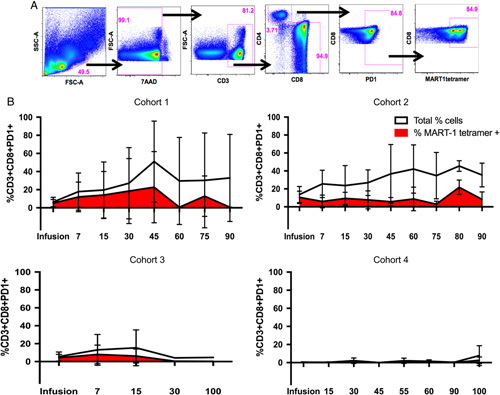
Baseline and longitudinal proportions of total and transgenic CD8+PD1+ T cells in patients. A, Gating strategy to obtain the percentage of CD8+PD1+ T cells, and percentage of MART-1 tetramer expressing cells within that population. B, Total frequency of PD1+CD8+ expressing T cells (white), and the proportion of those cells expressing the MART-1 TCR (red) in the final infusion product and on circulating peripheral lymphocytes of the patients over time (measured in days). Error bars represent SDs. Cohort 1: n=4, cohort 2: n=4, cohort 3: n=2, cohort 4: n=2.
Presence of Suppressor Tregs Within Infusion Products, Followed by Rapid Clearance
In order to understand the progressive loss of transgenic cell number and activity over time, we explored the presence of suppressor Tregs within the infusion product and over time (Fig. 4). Significant proportions of both transgenic and nontransgenic T cells with a Treg phenotype were present within the infusion product in all cohorts, with no significant differences between manufacturing cohorts. However, all transgenic Tregs were cleared from circulation within the first 15 days. This was accompanied by an overall decrease in total Tregs as well. Cohort 4 did not have sufficient samples collected for day +15 analysis of Tregs, but all total and transgenic Tregs were cleared from this cohort by day +30.
FIGURE 4.
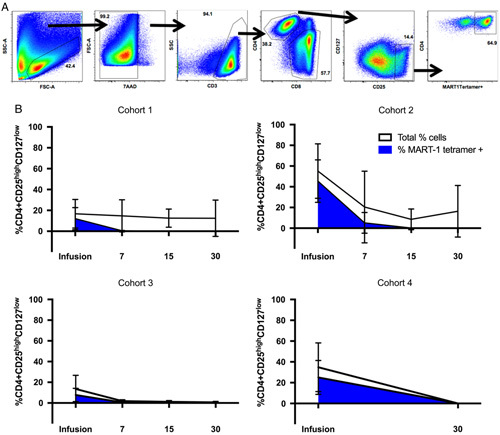
Baseline and longitudinal proportion of total and transgenic suppressor Tregs in patients. A, Gating strategy to obtain the percentage of Treg cells, and percentage of MART-1 tetramer expressing cells within that population. B, Total frequency of Tregs T cells, defined by CD4+CD25highCD127Low (white), and the proportion of those cells expressing the MART-1 TCR (blue) in the final infusion product and on circulating peripheral lymphocytes of the patients over time (measured in days). Error bars represent SDs. Cohort 1: n=4, cohort 2: n=4, cohort 3: n=2, cohort 4: n=2.
MART-1 Transgenic CD8 T Cells Track to Tumors, and these TILs Acquire Expression of PD1
We next characterized the ability of transgenic T cells to track to the tumor sites, as well as their phenotypic changes within the tumor microenvironment, by examining on-treatment tumor biopsies in the 2 patients from whom we were able to obtain such biopsies (F5-1 and F5-2) at day +24 and day +30, respectively. We then compared the infusion T cells, day +30 peripheral blood CD8+ T cells, and tumor biopsy TILs (Fig. 5). Patient F5-1, who displayed 82.3% expression of MART-1 tetramer in the infusion product, displayed persistence of 38.8% in peripheral blood CTLs at day +30, while TILs isolated from the tumor biopsy displayed 10.7% MART-1 tetramer positivity (Fig. 5A). Patient F5-2 similarly displayed a robust expression of MART-1 tetramer in the infusion product (63.1%), which both persisted in the peripheral blood (43.5% positive at day +30), and were detectable in the tumor at day +30 as well (9.85% positive in TILs). We further characterized the degree of PD1 acquisition within both the peripheral blood transgenic CTLs and TILs. Patient F5-1, who displayed 22.6% PD1+ CD8+MART-1+ cells in the infusion product (Fig. 5C), showed elevated PD1 expression within the transgenic CD8+ population within both the peripheral blood and TIL compartments (83.4% and 64.4%, respectively). While patient F5-2 showed a transient decrease in overall PD1 expression in the peripheral blood by day +30 (from 19.5% to 1.3%), the CD8+MART-1+ TILs demonstrated increased expression of PD1 (38.2%).
FIGURE 5.
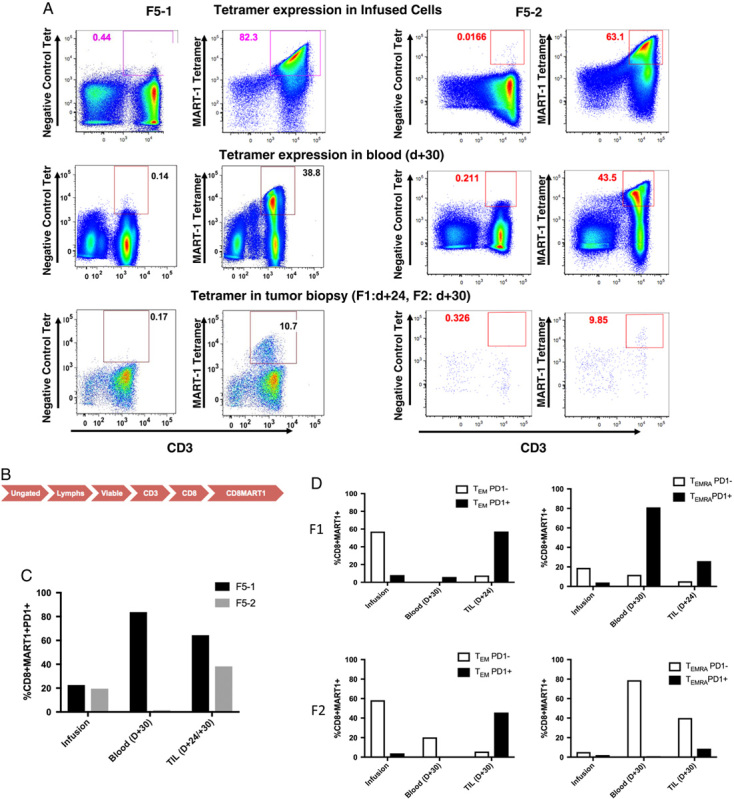
Transgenic CD8 T cells track to the site of tumor and acquire expression of PD1. A, Percent expression of MART-1+ T cells in infused, blood and biopsy samples from patients F1 (left) and F2 (right). Scramble peptide (negative) was used for gating purposes. B, Flow of gating strategy for the plots in (C) and (D). Following this gating, additional phenotypic markers were examined per the gating strategy in Figure 1C. C, Percent expression of PD1 on CD8+MART-1+ T cells in patients F5-1 and F5-2 in the infusion product, peripheral blood, and TILs. D, Percentage of PD1− and PD1+ CD8+MART-1+ T cells with the effector memory (TEM) and effector memory RA (TEMRA) compartments in the infusion product, peripheral blood, and tumor-infiltrating lymphocytes.
In order to better characterize this acquisition of PD1 expression within the TILs of these patients, we examined PD1 acquisition within the TEM and TEMRA compartments using the canonical Boolean gate approach.20 As before, TEM was defined as CD45RA−/CCR7−/CCR5+/−/PD1+/−, while TEMRA was defined as CD45RA+/CCR7−/CCR5+/−/PD1+/−. In patient F5-1, PD1− transgenic CD8 T cells represented 57.2% of the TEM compartment in the infusion product (Fig. 5D). These PD1− cells had disappeared completely from the peripheral blood by day +30, and remained only minimally present within the TILs (7.6%). However, the PD1+ transgenic CD8+ T-cell population increased from 8.1% of the TEM compartment in the infusion product to 57.3% of this compartment within the TILs. This trend was also seen in the TEMRA compartment as well: PD1− transgenic TEMRA cells decreased from 19% in the infusion product to 11.9% in the peripheral blood and 5.3% in the TILs, while PD1+ transgenic TEMRA increased in both the peripheral blood and within the TILs (81% and 25.9%, respectively). Patient F5-2 also displayed acquisition of PD1 expression within the TEM compartment, with PD1− transgenic TEM cells falling from 58.3% in the infusion product to 20.3% and 5.7% in the peripheral blood and TILs at day +30, respectively (Fig. 5D). Conversely, PD1 expression in the TEM compartment rose from 3.9% in the infusion product to 45.7% in the TILs (while disappearing from the peripheral blood). Within the TEMRA compartment, patient F5-2 demonstrated an expansion of PD1− transgenic cells in the peripheral blood, while still showing acquisition of PD1 expression within the transgenic TILs (from 2.1% to 8.6% of total).
DISCUSSION
Given the ability of ACT to generate robust initial antitumor responses, there is interest in exploring ways of enhancing its therapeutic duration. In addition, as manufacturing protocols of cellular products can vary between institutions, characterization of phenotypic differences resulting from variations in manufacturing practices is an important topic with therapeutic implications. Previous preclinical models of ACT have demonstrated that prolonged ex vivo expansion resulted in transgenic T cells which were more terminally differentiated, had decreased proliferative capacity, and lower antitumor activity in vivo when compared with products with shorter ex vivo expansion periods.13,21,22 Therefore, our F5-MART-1 trial had utilized a 7-day, and later a 6-day culture period, both of which were shorter than the previously utilized 2 to 4 week expansion periods in other ACT trials.6,7 We also infused fresh rather than cryopreserved cells in the later patients enrolled in the trial, in an effort to improve clinical outcomes by avoiding the physiologic stressors of freezing/thawing. These patients’ responses differed in that they demonstrated longer persistence of transgenic T-cell products, and were the only cases to display evidence of boosting effects in response to the DC vaccines, as previously reported.10 Two patients (F5-12 and F5-14) who received fresh cell infusions were the only patients noted to have severe cytokine storm syndrome, which was not seen in any patients who received cyropreserved cells, as previously reported.10 Furthermore, another patient who received fresh cells (F5-13) displayed recall whole body rash which accompanied the reexpansion of TCR transgenic cells in response to DC vaccination. These results further imply a superior functionality of the fresh transgenic cells, although the small sample size prevents us from drawing firm conclusions.
Given the prolonged exposure to IL-2 in our culture/expansion protocols, we were concerned that differences in suppressor Treg populations, as well as CD4/CD8 ratios, would contribute to loss of antitumor functionality. Using the conventional CD25+/CD127− surface markers23–26 to characterize Tregs (FoxP3 was not used due to its potential presence on effector cells),27,28 we found that while suppressor Tregs were present within all infusion products, they were rapidly cleared from circulation. Furthermore, culture time had no effect on the CD4/CD8 ratio in the infusion product, implying that the final product maintains a ratio similar to that of healthy donors (∼2) despite several days of culture/expansion with IL-2. However, our patients in shorter culture time demonstrated significantly increased surface markers associated with less differentiated naive/memory T-cell subtypes (CCR7, CD62L, and CD127), as well as decreased expression of the inhibitory receptor PD1. Shorter culture times were also associated with increased relative amounts of naive/memory T cells, which persisted over time. While these trends have previously been observed in preclinical models of ACT and in chimeric antigen receptor T cells,13,21,22,29 it is remarkable that these differences were apparent in our study of TCR-engineered clinical ACT products despite only one day less spent in culture.
Despite evidence from our group and others that cryopreserved T cells still maintain transient antitumor activity and specificity,10,12,30 all cryopreserved cohorts exhibited a relatively rapid and steady increase of PD1 expression in transgenic T cells. This was seen both within the peripheral circulation as well as the tumor microenvironment, in the cases examined. Only the patients who received freshly infused cell products experienced significant delays in acquisition of PD1 expression, as well as an expansion of the central memory compartment over time. These 2 factors are in keeping with increased persistence of transgenic CD4 and CD8 cells noted in this cohort. While the reduced IL-2 dosing utilized in cohort 4 may have play a role in the delayed acquisition of PD1 and persistence/expansion of memory/naive T cells in this cohort, it would not have affected the baseline phenotypic differences noted. Furthermore, the rapid clearance of Tregs from circulation (both in transgene positive and negative cells) implies that the IL-2 administration differences may not have yielded a significant overall phenotypic effect to the T cells. However, there may be other confounding host immunomodulatory factors which contributed to the phenotypic differences noted in cohort 4, due to the reduced fludarabine conditioning regimen which these patients received.
Previous studies have demonstrated the importance of TN and TCM populations in maintaining proliferative capacity and antitumor activity of the other T-cell populations.14–16,21 However, increased central memory T cells and delayed/decreased PD1 expression may also explain the boosting response to DC vaccination in the cohort receiving fresh infused cells. DCs have been shown to maximize the response of TCM cells to antigenic challenge.31 Therefore, a larger reserve of TCM cells could lead to enhanced response to the DCs themselves. Furthermore, although terminally differentiated TEFF can function as suppressor cells by killing DCs in a perforin/granzyme B-dependent manner,32,33 TCM cells themselves have been shown to protect DCs from this TEFF-mediated killing.34 Finally, the expression of PD1 on T cells can inhibit their interactions with DCs themselves via PD-L1 and PD-L2 expression on the DCs, and PD1 blockade can restore such interactions.35–39 Taken together, these findings may collectively explain why fresh infused transgenic cells subjected to shorter expansion times displayed superior DC vaccine responses.
Unfortunately, all patients, including those who received fresh transgenic cells, ultimately succumbed to progressive disease. Given that all patient cohorts eventually developed PD1 expression, this is a likely contributor to the eventual decrease in T cell functionality despite any persistence of the cells. PD1 is well known to be progressively upregulated after TCR engagement, leading to loss of T cell functionality in multiple malignancies.40,41 With the success of preclinical models in enhancing transgenic T-cell cancer therapies via addition of PD1 blockade,17,42 this may represent an attractive means of perpetuating the antitumor activity of these therapies. When combined with other factors that further optimize T-cell persistence and activity, such as increased naive/memory T cells and DC vaccine boosts, these measures may yield optimal ACT responses. Furthermore, given the apparent advantages of fresh transgenic cells over cryopreserved cells, as well the progressive loss of transgenic cells in all cases of ACT, there may be utility in modalities which provide a continuous supply of transgenic T cells to the patient. Previous preclinical models have demonstrated that TCR-transduced CD34+ peripheral blood stem cells can endogenously differentiate into fully functional T cells expressing the TCR of interest.43–45 Such models may yield improved clinical outcomes by circumventing many of the short and long-term phenotypic disadvantages of current ACT manufacturing practices which have been demonstrated.
Our study has several limitations. The relatively low number of patients per cohort, combined with the possible statistical effect of multiple parameter testing, limited our ability to calculate significant population statistics for comparison. Analysis of larger data sets, as well as utilizing more rigorously controlled in vitro and in vivo models for longer periods, may yield additional phenotypic and functional variations between different manufacturing and administration protocols. In addition, we were only able to obtain biopsies to examine postinfusion TILs in 2 patients, and these represented only a small fraction of live lymphocytes (∼1400 for patient F5-1, and ∼130 for patient F5-2), likely due to the variability of T-cell infiltration between individual tumors. However, the relatively high proportion of transgenic T cells present within the tumors, combined with the differences when compared with the negative control tetramer, support the inference that transgenic T cells were able to track to the tumors analyzed. Furthermore, limitations in the surface marker panels utilized prevented us from describing the changes in individual surface markers over time stratified by different transgenic T-cell phenotypes, as well as characterizing additional subsets of T cells, such as stem cell memory T cells (TSCM), which are also of great importance in maintaining successful ACT immunotherapy. The use of newer technologies, such as mass cytometry by time-of-flight, which merges single-cell flow cytometry with mass spectrometry, enables the monitoring of many more simultaneous factors than conventional flow cytometry.46,47 Use of this technology will allow for more sophisticated monitoring of many more T-cell phenotypes in our future ACT studies.
In conclusion, our study demonstrates that shorter ex vivo expansion times of transgenic T cells for ACT are associated with increased markers of naive and memory populations of T cells, as well as decreased PD1 expression. Furthermore, fresh cell product infusions were associated with persistence/expansion of memory compartments of transgenic T cells, as well as delayed acquisition of PD1 expression. These dual characteristics may explain our previous experiences in which patients receiving fresh cell products displayed superior persistence of transgenic T cells, as well as superior augmentation of this persistence in response to DC vaccination. These results may be useful in design and optimization of future ACT manufacturing protocols, as well as augmenting their effectiveness via addition of immunomodulatory agents blocking inhibitory receptors.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the NHLBI National Gene Vector Biorepository at Indiana University (PI Cornetta, P40HL116212) for assistance in the generation and maintenance of the F5 Vector. Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by National Institutes of Health awards P30 CA016042 and 5P30 AI028697, and by the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA.
Conflicts of Interest/Financial Disclosures
Supported in part by NIH grants R01 CA129816 (to J.S.E.), P01 CA132681 (to D.B.), R35 CA197633 and P01 CA168585 (to A.R.), as well as the Parker Institute for Cancer Immunotherapy, the Ressler Family Fund, the Grimaldi Family Fund, the Samuels Family Fund and the Garcia-Corsini Family Fund (to A.R.). T.S.N. is supported by the NIH/NICHD grant K12-HD000850 (Pediatric Scientist Development Program).
All authors have declared that there are no financial conflicts of interest with regard to this work.
Footnotes
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of the NIH, the NCI, the NICHD, the Parker Institute for Cancer Immunotherapy, the Grimaldi Family Fund, the Samuels Family Fund, the Garcia-Corsini Family Fund, or the Ressler Family Foundation.
REFERENCES
- 1.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunder NN, Wallen H, Cao J, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pollack SM, Jones RL, Farrar EA, et al. Tetramer guided, cell sorter assisted production of clinical grade autologous NY-ESO-1 specific CD8(+) T cells. J Immunother Cancer. 2014;2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.June CH, Maus MV, Plesa G, et al. Engineered T cells for cancer therapy. Cancer Immunol Immunother. 2014;63:969–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robbins PF, Kassim SH, Tran TL, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chodon T, Comin-Anduix B, Chmielowski B, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res. 2014;20:2457–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma C, Fan R, Ahmad H, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17:738–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma C, Cheung AF, Chodon T, et al. Multifunctional T-cell analyses to study response and progression in adoptive cell transfer immunotherapy. Cancer Discov. 2013;3:418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klebanoff CA, Gattinoni L, Torabi-Parizi P, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102:9571–9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klebanoff CA, Gattinoni L, Palmer DC, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res. 2011;17:5343–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinrichs CS, Borman ZA, Gattinoni L, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117:808–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother. 2012;35:651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moon EK, Ranganathan R, Eruslanov E, et al. Blockade of programmed death 1 augments the ability of human T cells engineered to target NY-ESO-1 to control tumor growth after adoptive transfer. Clin Cancer Res. 2016;22:436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibarrondo FJ, Yang OO, Chodon T, et al. Natural killer T cells in advanced melanoma patients treated with tremelimumab. PLoS One. 2013;8:e76829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen TJ, Kotecha N. Cytobank: providing an analytics platform for community cytometry data analysis and collaboration. Curr Top Microbiol Immunol. 2014;377:127–157. [DOI] [PubMed] [Google Scholar]
- 20.Sallusto F, Lenig D, Forster R, et al. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. [DOI] [PubMed] [Google Scholar]
- 21.Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seddiki N, Santner-Nanan B, Martinson J, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203:1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartigan-O'Connor DJ, Poon C, Sinclair E, et al. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J Immunol Methods. 2007;319:41–52. [DOI] [PubMed] [Google Scholar]
- 26.Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the Human Immunology Project. Nat Rev Immunol. 2012;12:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuczma M, Pawlikowska I, Kopij M, et al. TCR repertoire and Foxp3 expression define functionally distinct subsets of CD4+ regulatory T cells. J Immunol. 2009;183:3118–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roncarolo MG, Gregori S. Is FOXP3 a bona fide marker for human regulatory T cells? Eur J Immunol. 2008;38:925–927. [DOI] [PubMed] [Google Scholar]
- 29.Neeson P, Shin A, Tainton KM, et al. Ex vivo culture of chimeric antigen receptor T cells generates functional CD8+ T cells with effector and central memory-like phenotype. Gene Ther. 2010;17:1105–1116. [DOI] [PubMed] [Google Scholar]
- 30.Galeano Nino JL, Kwan RY, Weninger W, et al. Antigen-specific T cells fully conserve antitumour function following cryopreservation. Immunol Cell Biol. 2016;94:411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zammit DJ, Cauley LS, Pham QM, et al. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity. 2005;22:561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hermans IF, Ritchie DS, Yang J, et al. CD8+ T cell-dependent elimination of dendritic cells in vivo limits the induction of antitumor immunity. J Immunol. 2000;164:3095–3101. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Huck SP, McHugh RS, et al. Perforin-dependent elimination of dendritic cells regulates the expansion of antigen-specific CD8+ T cells in vivo. Proc Natl Acad Sci U S A. 2006;103:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watchmaker PB, Urban JA, Berk E, et al. Memory CD8+ T cells protect dendritic cells from CTL killing. J Immunol. 2008;180:3857–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenblatt J, Glotzbecker B, Mills H, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother. 2011;34:409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stecher C, Battin C, Leitner J, et al. PD-1 blockade promotes emerging checkpoint inhibitors in enhancing T cell responses to allogeneic dendritic cells. Front Immunol. 2017;8:572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roeven MW, Hobo W, van der Voort R, et al. Efficient nontoxic delivery of PD-L1 and PD-L2 siRNA into dendritic cell vaccines using the cationic lipid SAINT-18. J Immunother. 2015;38:145–154. [DOI] [PubMed] [Google Scholar]
- 38.Nowicki TS, Anderson JL, Federman N. Prospective immunotherapies in childhood sarcomas: PD1/PDL1 blockade in combination with tumor vaccines. Pediatr Res. 2016;79:371–377. [DOI] [PubMed] [Google Scholar]
- 39.Bergh J, Smits E, Berneman ZN, et al. Monocyte-derived dendritic cells with silenced PD-1 ligands and transpresenting interleukin-15 stimulate strong tumor-reactive T-cell expansion. Cancer Immunol Res. 2017;5:710–715. [DOI] [PubMed] [Google Scholar]
- 40.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366:2517–2519. [DOI] [PubMed] [Google Scholar]
- 42.John LB, Devaud C, Duong CP, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636–5646. [DOI] [PubMed] [Google Scholar]
- 43.Yang L, Qin XF, Baltimore D, et al. Generation of functional antigen-specific T cells in defined genetic backgrounds by retrovirus-mediated expression of TCR cDNAs in hematopoietic precursor cells. Proc Natl Acad Sci U S A. 2002;99:6204–6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L, Baltimore D. Long-term in vivo provision of antigen-specific T cell immunity by programming hematopoietic stem cells. Proc Natl Acad Sci U S A. 2005;102:4518–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vatakis DN, Koya RC, Nixon CC, et al. Antitumor activity from antigen-specific CD8 T cells generated in vivo from genetically engineered human hematopoietic stem cells. Proc Natl Acad Sci U S A. 2011;108:E1408–E1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bendall SC, Simonds EF, Qiu P, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spitzer MH, Nolan GP. Mass cytometry: single cells, many features. Cell. 2016;165:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]