

Supplemental Digital Content is available in the text.
Keywords: acute kidney injury; sepsis; vasodilatory shock, angiotensin II, renal replacement therapy, acute renal disease
Abstract
Objective:
Acute kidney injury requiring renal replacement therapy in severe vasodilatory shock is associated with an unfavorable prognosis. Angiotensin II treatment may help these patients by potentially restoring renal function without decreasing intrarenal oxygenation. We analyzed the impact of angiotensin II on the outcomes of acute kidney injury requiring renal replacement therapy.
Design:
Post hoc analysis of the Angiotensin II for the Treatment of High-Output Shock 3 trial.
Setting:
ICUs.
Patients:
Patients with acute kidney injury treated with renal replacement therapy at initiation of angiotensin II or placebo (n = 45 and n = 60, respectively).
Interventions:
IV angiotensin II or placebo.
Measurements and Main Results:
Primary end point: survival through day 28; secondary outcomes included renal recovery through day 7 and increase in mean arterial pressure from baseline of ≥ 10 mm Hg or increase to ≥ 75 mm Hg at hour 3. Survival rates through day 28 were 53% (95% CI, 38%–67%) and 30% (95% CI, 19%–41%) in patients treated with angiotensin II and placebo (p = 0.012), respectively. By day 7, 38% (95% CI, 25%–54%) of angiotensin II patients discontinued RRT versus 15% (95% CI, 8%–27%) placebo (p = 0.007). Mean arterial pressure response was achieved in 53% (95% CI, 38%–68%) and 22% (95% CI, 12%–34%) of patients treated with angiotensin II and placebo (p = 0.001), respectively.
Conclusions:
In patients with acute kidney injury requiring renal replacement therapy at study drug initiation, 28-day survival and mean arterial pressure response were higher, and rate of renal replacement therapy liberation was greater in the angiotensin II group versus the placebo group. These findings suggest that patients with vasodilatory shock and acute kidney injury requiring renal replacement therapy may preferentially benefit from angiotensin II.
Vasodilatory shock is the most common form of shock, characterized by peripheral vasodilation and preserved or increased cardiac output (1, 2). Conditions that are associated with inflammation such as sepsis, pancreatitis, and major surgery are common causes of vasodilatory shock, with sepsis being the most frequent pathology (3). Acute kidney injury (AKI), defined as an increase in serum creatinine or a decrease in urine output due to a decline in glomerular filtration rate (GFR) (4), often complicates the course of vasodilatory shock and is associated with unfavorable outcomes (5). Sepsis also accounts for approximately 50% of AKI cases in critically ill patients (6, 7). The transition from sepsis to vasodilatory shock and AKI in an acute setting can progress quickly; however, the mechanisms that mediate the pathogenesis of these interconnected organ dysfunctions are not fully understood. In addition, patients who survive AKI are at risk for long-term morbidity that appears to be related to the severity of AKI (8).
In patients with both vasodilatory shock and AKI treated with renal replacement therapy (RRT; together referred to as AKI-RRT), prognosis tends to be worse (5). Such patients have a mortality rate between approximately 40% and 55% (9, 10). During vasodilatory shock, the classic paradigm is that AKI is caused by decreased renal perfusion due to hypotension associated with vasodilation as well as decreased blood flow potentiated by vasoconstrictive drugs (vasopressors) (11–13). However, the physiology of renal circulation in sepsis is likely complex (14), and studies utilizing vasodilators, inotropes, and natriuretic peptides have failed to demonstrate improved outcomes (5, 13, 15). Furthermore, recent evidence suggests that intrarenal vasodilatation and shunting may be important to the pathogenesis of AKI (16). Angiotensin II is known to have direct effects on intrarenal circulation, including vasoconstriction of efferent arterioles of the nephron, thereby increasing glomerular perfusion pressure (17). Such effect may help preserve or restore GFR in sepsis (18).
To assess angiotensin II treatment effect on survival and renal recovery, we conducted a post hoc analysis of data from the Angiotensin II for the Treatment of High-Output Shock 3 (ATHOS-3) trial (19, 20).
MATERIAL AND METHODS
Trial Design
The protocol for the international, randomized, double-blind, placebo-controlled ATHOS-3 trial has been previously described (19, 20) (Supplemental Digital Content 1, http://links.lww.com/CCM/D422). The writing committee for the present post hoc analysis included several ATHOS-3 investigators and the sponsor, each of whom vouch for the accuracy and completeness of the data and analysis. The ATHOS-3 trial was approved by a research ethics board at each participating institution. Written informed consent was obtained from all patients or their legal surrogates.
Patients
A detailed description of the study eligibility in the parent trial is reported in Supplemental Digital Content 1 (http://links.lww.com/CCM/D422). In brief, ATHOS-3 patients had vasodilatory shock (cardiac index > 2.3 L/min/m2, or central venous oxygen saturation > 70% coupled with central venous pressure > 8 mm Hg) and were on high doses of vasopressors (as > 0.2 µg/kg/min of norepinephrine-equivalent dose [Supplemental Table 1, Supplemental Digital Content 1. http://links.lww.com/CCM/D422]). Exclusion criteria included, among others, 20% body surface area burns, acute occlusive coronary syndrome, and active bleeding. No exclusion criteria in the parent study directly addressed renal function. We enrolled patients from sites in North America, Australasia, and Europe (Supplemental Table 2, Supplemental Digital Content 1, http://links.lww.com/CCM/D422).
In the present post hoc analysis, we identified patients who were treated with RRT at the time of study drug initiation, and excluded those with a history of end-stage renal disease (ESRD).
Treatment Assignment
In ATHOS-3, we randomized patients (1:1) to receive treatment with either La Jolla Pharmaceutical Company’s formulation of IV synthetic human angiotensin II (LJPC-501) or saline placebo administered by blinded study personnel at participating institutions. Randomization was stratified by mean arterial pressure (MAP) at screening (< 65 mm Hg or ≥ 65 mm Hg) and Acute Physiology and Chronic Health Evaluation II (APACHE II) score (≤ 30, 31 to 40, or ≥ 41). The form of human angiotensin II used in ATHOS-3 (LJPC-501) has an identical amino acid sequence to naturally occurring human angiotensin II.
Clinical Regimen
In the ATHOS-3 trial, infusions were initiated at a rate equivalent to 20 ng/kg/min of angiotensin II and titrated to increase MAP to ≥ 75 mm Hg during the first 3 hours (Supplemental Table 3, Supplemental Digital Content 1, http://links.lww.com/CCM/D422). During this titration, doses of standard-of-care vasopressors were held constant and could not be increased except for safety reasons. If vasopressor doses were increased, the patient was designated a nonresponder. The maximum rate of administration of angiotensin II or placebo allowed during the first 3 hours was equivalent to a dose of 200 ng/kg/min.
Between hours 3:15 and 48, investigators could titrate study infusions to a rate (mL/hour) equivalent to a dose of 1.25 to 40 ng/kg/min in patients assigned to angiotensin II. During this period, investigators titrated the combination of angiotensin II or placebo and other vasopressors to maintain MAP between 65 and 75 mm Hg; the protocol included a nonbinding titration scheme (Supplemental Table 4, Supplemental Digital Content 1, http://links.lww.com/CCM/D422).
At hour 48, investigators discontinued the study infusion per a protocol-defined tapering process. If the standard-of-care vasopressor dose subsequently required a norepinephrine-equivalent dose increase to > 0.1 µg/kg/min, or if the patient became unstable, study infusion could be resumed for up to 7 days. However, once stopped for > 3 hours, study infusion could not be restarted. RRT discontinuation was at the discretion of the investigator.
Assessments and End Points
In the present post hoc analysis, patients were assessed for the primary, patient-centered effectiveness outcome of survival through day 28; secondary, patient-centered effectiveness outcomes of time to discontinuation of RRT, vasopressor(s), and ventilator through day 7; and secondary, physiologic efficacy end point of MAP response (defined as an increase from baseline of ≥ 10 mm Hg or an increase to ≥ 75 mm Hg) at hour 3.
Statistical Analyses
The present post hoc analysis was not a priori although all analysis end points and covariates were prespecified for the ATHOS-3 trial. The present post hoc analysis was conducted to explore the potential treatment effects of angiotensin II that may otherwise go unnoticed in a heterogeneous population, in the subgroup of AKI-RRT patients receiving RRT at study drug initiation. We based the post hoc analyses on the modified intention-to-treat population that included all randomized patients who initiated treatment. We analyzed data using SAS software (version 9.4). We used descriptive statistics with 95% confidence intervals to summarize data by treatment group; differences between treatment groups were analyzed by the Wilcoxon rank-sum test or analysis of variance for continuous or ordinal variables, and the χ2 or Fisher exact test for discrete variables. A 2-tailed alpha of 0.05 was used to test the hypothesis of treatment difference. No adjustments for multiple comparisons were included in the results. An independent statistician verified the appropriateness of the analyses and inferences made from the data.
We compared time-to-event data by log-rank test and characterized the data by Kaplan-Meier estimates. We estimated hazard rates from the proportional hazards model. For RRT, vasopressor, and ventilator, the analyses were based on the cause-specific model censoring data at time of death. In addition, cumulative incidence estimates were performed accounting for death as a competing risk. For analyses of death, dates and times were included. For analyses of RRT, vasopressor, and ventilator, dates were used because exact times were not reported; censored deaths occurring on the same day as an event were assumed to have occurred after an event. We included baseline factors that may have influenced end points in 3 multivariate logistic regression and proportional hazards models as sensitivity analyses. Covariates used for such models included dichotomized baseline: age ≥ 65 years, MAP < 65 mm Hg, APACHE II score > 30, albumin < 2.5 g/dL, angiotensin I/II ratio < 1.63 (the median angiotensin I/II ratio in the parent trial), Model for End-stage Liver Disease (MELD) score ≥ 30, chest radiograph finding of acute respiratory distress syndrome, and norepinephrine-equivalent dose ≥ 0.5μg/kg/min. Model A was based on known factors that influence outcomes in patients with vasodilatory shock. Model B was based on covariates that were not well balanced between patients treated with angiotensin II and placebo. Model C was a combination of the variables from Model A or B that predicted outcomes placed together.
RESULTS
Patients
In the ATHOS-3 trial, we enrolled 321 patients to receive either angiotensin II or placebo; of these, 167 patients received RRT. In the present post hoc analysis, 105 patients with AKI (n = 45 angiotensin II, n = 60 placebo) treated with RRT at study drug initiation were included. We excluded 12 patients who were previously diagnosed with ESRD. Patients with AKI who initiated RRT after study drug initiation (n = 50) were excluded to ensure that the variable that defines the subgroup is not affected by the treatment response of RRT. Sensitivity analyses were performed on these 50 patients to evaluate outcomes on survival through day 28, RRT discontinuation, and MAP response. Outcomes are reported in Supplemental Figures 1, 2, and 3 (Supplemental Digital Content 1, http://links.lww.com/CCM/D422), respectively. Overall, there were no safety signals observed in either treatment group.
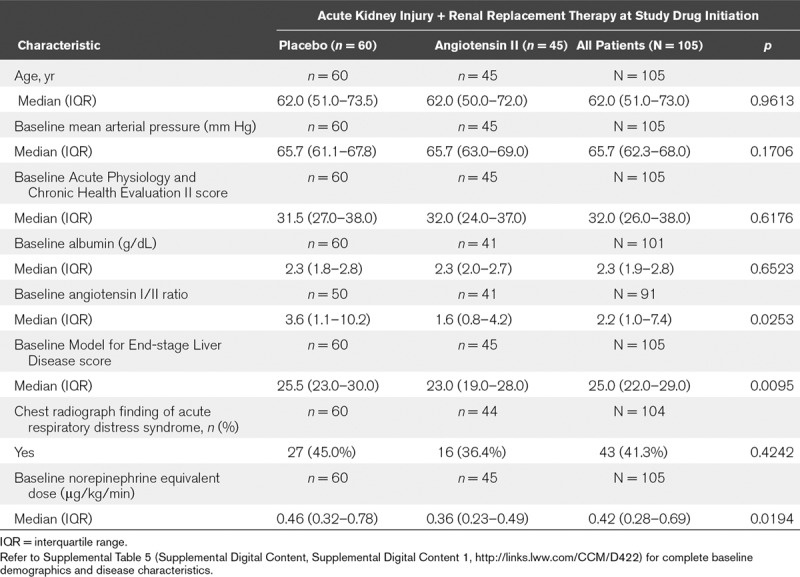
Table 1 summarizes the baseline characteristics (for the full results, refer to Supplemental Table 5, Supplemental Digital Content 1, http://links.lww.com/CCM/D422). Overall, patients in this analysis were severely ill as indicated by elevated baseline norepinephrine-equivalent dose and high MELD score, which were higher in the placebo group. The angiotensin I/II ratio was also higher in the placebo group (Table 1).
TABLE 1.
Summary of Baseline Demographic and Disease Characteristics
Outcomes
At day 28, 24 (53%) and 18 (30%) AKI-RRT patients were alive in the angiotensin II and placebo groups, respectively (Table 2); survival through day 28 was significantly longer in the angiotensin II group than in the placebo group (unadjusted hazard ratio [HR], 0.52; 95% CI, 0.30–0.87; p = 0.012 [Fig. 1]). A multivariate analysis was performed to account for clinically relevant covariates of interest at baseline and confirmed the findings of the unadjusted comparison (adjusted HR, 0.44; 95% CI, 0.24–0.80; p = 0.007) (Table 2).
TABLE 2.
Outcomes – Patients With Severe Acute Kidney Injury and Renal Replacement Therapy at Study Drug Initiation
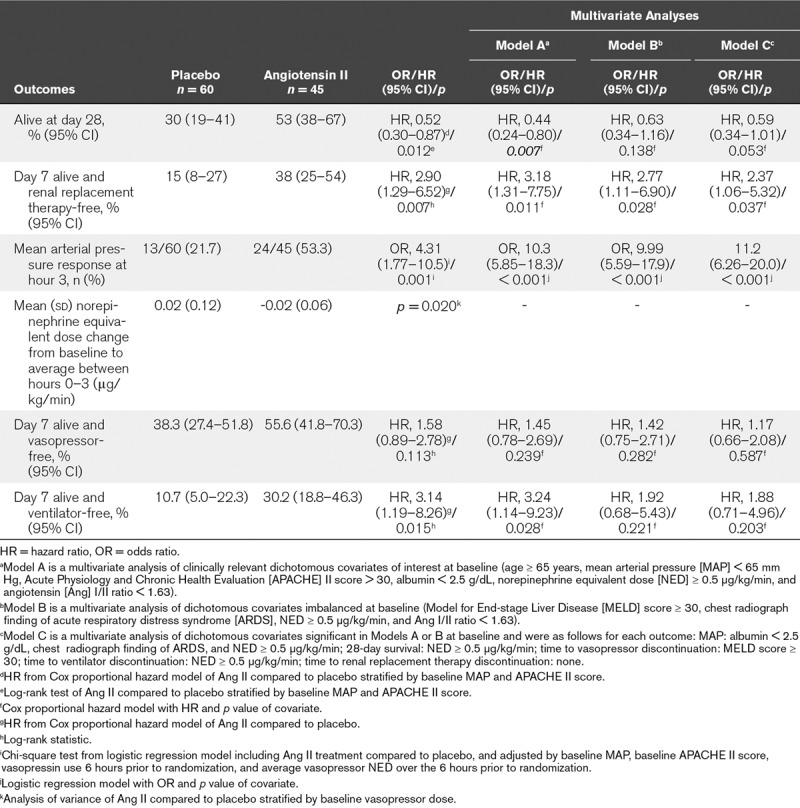
Figure 1.
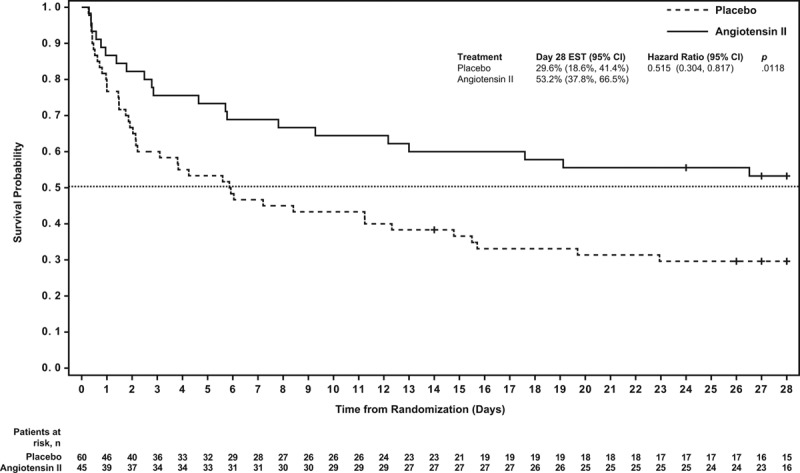
Survival probability through day 28. Patients in the angiotensin II group were more likely to survive to day 28 than those in the placebo group (p = 0.012).
Using cumulative incidence estimates to adjust for death as a competing risk, patients in the angiotensin II group were more likely to discontinue RRT within 7 days (adjusted HR, 2.90; 95% CI, 1.29–6.52; p = 0.007) (Table 2; and Fig. 2). By day 7, 38% (95% CI, 25%–54%) of patients who received angiotensin II discontinued RRT versus 15% (95% CI, 8%–27%) who received placebo. Multivariate analyses accounting for baseline covariates confirmed this difference (adjusted HR, 3.18; 95% CI, 1.31–7.75; p = 0.011) (Table 2).
Figure 2.
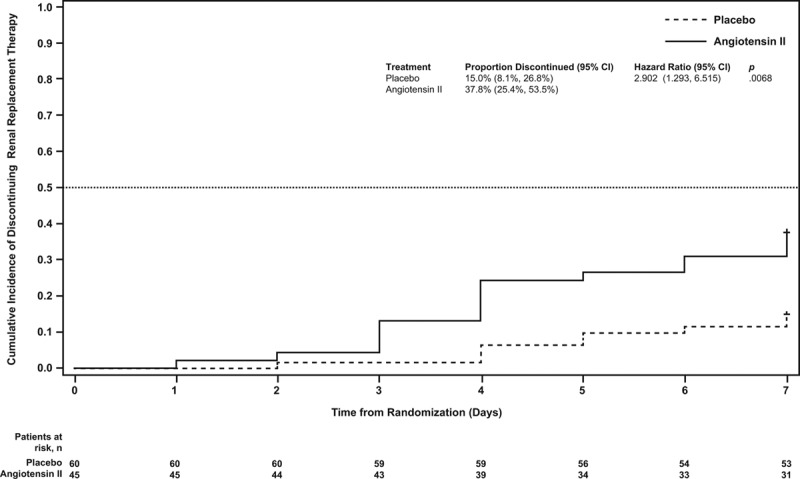
Cumulative incidence of time to discontinuation of renal replacement therapy through day 7. Subjects with death prior to day 7 are censored at day 7. Patients in the angiotensin II group were more likely to discontinue renal replacement therapy within 7 days than those in the placebo group (p = 0.0068).
At hour 3, patients in the angiotensin II group treated with RRT at study drug initiation achieved a significantly (p = 0.001) greater MAP response than the placebo group (Table 2; and Fig. 3). Several multivariate analyses were performed to account for potential baseline imbalances and confirmed these findings (p < 0.001) (Table 2). MAP increased rapidly in the angiotensin II group (Supplemental Table 6, Supplemental Digital Content 1, http://links.lww.com/CCM/D422), allowing the angiotensin II dose to be down-titrated from the original 20 ng/kg/min in 53% of patients at 30 minutes, as well as permitting decreased doses of concomitant vasopressors.
Figure 3.
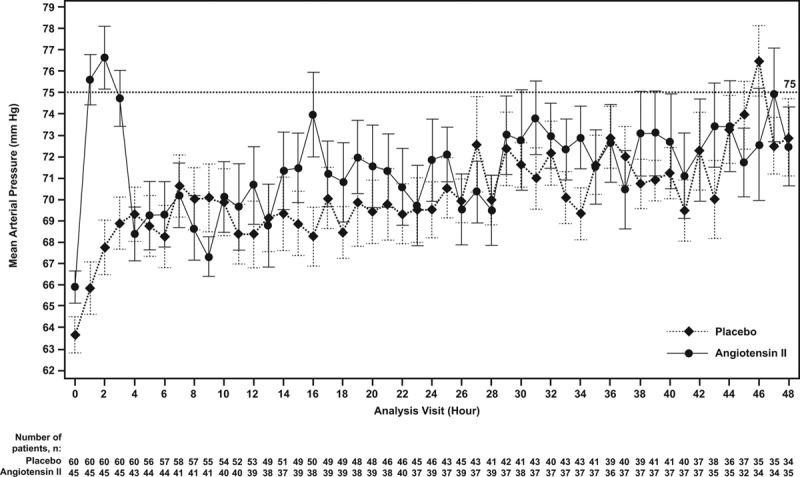
Mean (se) of mean arterial pressure by hour. Mean arterial pressure (mm Hg) is shown from initiation of angiotensin II or placebo through hour 48 by study group. By hour 3, the angiotensin II group achieved a significantly (p = 0.001) greater mean arterial pressure response than the placebo group.
Compared to baseline, the average norepinephrine-equivalent dose received between hours 0 and 3 was significantly (p = 0.020) lower in AKI-RRT patients in the angiotensin II group (–0.02 [0.06] μg/kg/min) than in the placebo group (0.02 [0.12] μg/kg/min) (Table 2; and Supplemental Table 7, Supplemental Digital Content 1, http://links.lww.com/CCM/D422).
Using cumulative incidence estimates to adjust for death as a competing risk, the proportion of patients who discontinued vasopressor(s) within 7 days did not significantly differ between groups (p = 0.113) (Table 2; and Supplemental Fig. 4, Supplemental Digital Content 1, http://links.lww.com/CCM/D422); however, there was a significant difference in the proportion of patients who discontinued ventilation within 7 days (p = 0.015) (Table 2; and Supplemental Fig. 5, Supplemental Digital Content 1, http://links.lww.com/CCM/D422). These findings were dependent on the covariates included in the model (Table 2).
DISCUSSION
In this post hoc analysis of patients with vasodilatory shock and AKI-RRT at study drug initiation, patients treated with angiotensin II were more likely to survive to 28 days, and they recovered renal function more rapidly as assessed by being alive and off RRT at day 7. Moreover, correction of hypotension was achieved in significantly more patients treated with angiotensin II.
This post hoc analysis illustrated a potential beneficial effect of angiotensin II in patients with AKI-RRT at study drug initiation. This could be due in part to the fact that the kidneys may be particularly susceptible to the effects of perfusion pressure to maintain blood flow (21). In this regard, ATHOS-3 (19), the parent trial of this analysis, and previous clinical and case studies (8, 22–25) consistently demonstrated that synthetic angiotensin II can increase blood pressure in patients with shock. However, the salutary effect of angiotensin II may also be due to a mechanistic link beyond perfusion alone. One of the most damaging impacts of vasodilatory shock due to inflammation is endothelial cell injury, which leads to capillary leak, coagulation defects, and reduced organ perfusion. The endothelial cell injury may also cause loss of an important membrane-bound endothelial enzyme, angiotensin-converting enzyme (ACE) (26). ACE capacity may be estimated by measuring the ratio of the precursor and product of ACE, angiotensin I, and angiotensin II, respectively. Healthy individuals have an angiotensin I/II ratio of approximately 0.5, whereas patients in the ATHOS-3 trial had a median angiotensin I/II ratio of 1.63 (19). These data suggest that ACE may be highly dysregulated in vasodilatory shock. Thus, angiotensin II infusion may compensate for such dysregulation.
The finding that exogenous angiotensin II was associated with improved outcomes, particularly renal outcomes, is plausible. Preclinical data in vasodilatory shock models endorse the notion that an IV infusion of angiotensin II can restore GFR and increase urine output without decreasing intrarenal oxygenation (16, 18, 27, 28). In contrast, the use of catecholamines is associated with an unfavorable microcirculatory oxygenation profile (29). Unlike catecholamines, angiotensin II preferentially vasoconstricts efferent arterioles (16, 18, 27), which increases glomerular capillary pressure and augments GFR (14). The effects from angiotensin II have the potential to mitigate the microcirculatory defects from intrarenal factors such as endothelial injury, microvascular thrombosis, and inflammation that can cause AKI (30). Preclinical studies have demonstrated that septic shock creates a form of AKI that is responsive to angiotensin II infusion (16, 18, 27, 31). This phenotype of sepsis-associated AKI appears to create efferent vasodilation at the level of the glomerulus that results in decreased GFR, reduced creatinine clearance, and increased sodium resorption despite an increase in renal blood flow. The administration of exogenous angiotensin II restored GFR and increased urine output.
In addition, multiple studies of sepsis have implicated both ACE polymorphisms and other genetic variations related to the renin-angiotensin-aldosterone system (RAAS) that appear to impact both survival and AKI (32–34). In a prospective 6-month observational study in critically ill patients (n = 180) (35), patients with AKI had significantly higher mortality rates in the ICU and hospital overall compared with those without AKI. Moreover, a specific ACE II genotype frequency was significantly higher in patients with AKI than in those without AKI. Furthermore, septic patients with less effective ACE activity, as measured by elevated angiotensin I/II ratios, were more likely to die than those with more intact ACE activity (36). Drug-induced alterations in the renin-angiotensin system from ACE inhibitors and angiotensin receptor blockers (which, contrary to angiotensin II, causes efferent arteriolar dilatation) that are used to treat chronic kidney disease have been shown to increase the risk of AKI in patients with septic shock (37). This further substantiates the role angiotensin II may have in treating patients at risk for AKI due to sepsis. Additional evidence suggests that modulation of vascular tone by the renin-angiotensin-aldosterone axis may be important to optimizing renal vascular function in this setting (38).
Importantly, there are no proven therapies available for the treatment of severe AKI. The potential capacity for angiotensin II to increase survival and improve time to RRT liberation are novel (39).
This study had multiple strengths. The parent ATHOS-3 trial, on which the data for this current post hoc analysis rely, was a placebo-controlled, double-blind randomized study with patient-centered, clinically relevant outcomes. In addition, there is a mechanistic rationale for the observed effect, supported by preclinical and clinical studies that have previously investigated the role of RAAS in AKI. In addition, the hypothesis that angiotensin I/II ratio is a key risk measure of AKI that could be ameliorated with exogenous angiotensin II was published prior to the completion of the ATHOS-3 clinical trial (40). Lastly, the results of our analysis were consistent when adjusted for clinically relevant covariates, adding credence to our conclusions.
This study had several limitations, including all the inherent limitations of a post hoc analysis. Follow-up was limited to 7 days for most outcomes as opposed to the full 30 days, and survival was limited to 28 days. We could not test whether angiotensin II increased survival at the expense of creating more patients with ESRD. Thus, any findings need to be considered only as hypothesis generating. Moreover, volume status was not analyzed in this post hoc analysis, and the absence of a premorbid serum creatinine precludes an AKI staging assessment and precludes any analysis of the impact of preexisting chronic kidney disease; however, all patients in the analysis were similar in that their renal function was sufficiently reduced to require RRT. Finally, we excluded from analysis any patients with ESRD as well as any patients who were treated with RRT after the initiation of the study drug, both of which are common scenarios. As such, this may limit the generalizability of our results.
CONCLUSIONS
In this post hoc analysis, restricted to patients with AKI-RRT at study drug initiation, unadjusted survival was improved with angiotensin II compared with placebo. Covariate-adjusted survival was also improved in some models but not others. In addition, patients were more likely to be alive and liberated from RRT by day 7 with angiotensin II in both unadjusted and adjusted analyses.
ACKNOWLEDGMENTS
We thank the patients and their families for entrusting us to conduct the ATHOS-3 trial, and the study investigators, study coordinators, and support staff for their compassion, dedication, and diligence in the conduct of the ATHOS-3 trial.
Supplementary Material
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website (http://journals.lww.com/ccmjournal).
A full list of the Angiotensin II for the Treatment of High-Output Shock 3 (ATHOS-3) Investigators and study sites are listed in Supplemental Digital Content 1 (http://links.lww.com/CCM/D422).
Shannan Lynch, Jeff Jensen, Stew Kroll, Lakhmir Chawla, and George Tidmarsh are employees of La Jolla Pharmaceutical Company. The Angiotensin II for the Treatment of High-Output Shock 3 (ATHOS-3) trial was funded and supported by La Jolla Pharmaceutical Company. All other authors participated in the ATHOS-3 trial as investigators and work(ed) at institutions that were funded by La Jolla Pharmaceutical Company in support of the ATHOS-3 trial. Additionally, John R. Prowle has received consultancy fees and speaker honoraria from Nikkiso Europe GmbH and speaker honoraria from Baxter, Inc. Raghavan Murugan was awarded a research grant from La Jolla Pharmaceutical Company, and has received consulting fees from Beckman and Coulter, Inc and Bioporto, Inc. Kianoush Kashani has received a travel grant from La Jolla Pharmaceutical Company for the ATHOS-3 investigator meeting. Marlies Ostermann has received research funding and speaker honoraria from Fresenius Medical Care. Alexander Zarbock has received consulting fees from Astellas and Quark Pharmaceutical; speaker honoraria from Astute Medical, Baxter, Frensenius, and Braun; and grant support from Astute Medical and Quark Pharmaceutical. James Tumlin has received research grant support from La Jolla Pharmaceutical Company. Kevin Finkel will be a member of the La Jolla Pharmaceutical Company Speakers Bureau in 2018. Lawrence Busse has received consulting fees from La Jolla Pharmaceutical Company. Lui Forni has received research funding and honoraria from Fresenius, Baxter Gambro Renal, OrthoClinical Diagnostics, and La Jolla Pharmaceutical Company. Drs. Tumlin, Murugan, Deane, Ham, Szerlip, Prowle, Bihorac, Zarbock, and Bellomo’s institutions received funding from La Jolla Pharmaceuticals. Drs. Tumlin and Kroll disclosed off-label product use of angiotensin II for circulatory shock. Drs. Busse, Forni, and Kroll received funding from La Jolla Pharmaceutical Company. Dr. Szerlip’s institution also received funding from Baylor Research Institute. Dr. Finkel received funding from Alexion Pharmaceuticals (speakers bureau). Dr. Zarbock received funding from Baxter, Astute Medical, Fresenius, Braun, Astellas, and DFG. Dr. Forni also received funding from Ortho Clinical Diagnostics (honoraria). Drs. Lynch, Jensen, Kroll, Chawla, and Tidmarsh disclosed that they are employees of the trial sponsor (La Jolla Pharmaceutical Company). Dr. Tidmarsh disclosed work for hire. The remaining authors have disclosed that they do not have any potential conflicts of interest.
REFERENCES
- 1.Vincent JL, De Backer D. Circulatory shock. N Engl J Med 2013; 369:1726–1734. [DOI] [PubMed] [Google Scholar]
- 2.De Backer D, Biston P, Devriendt J, et al. SOAP II Investigators: Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789. [DOI] [PubMed] [Google Scholar]
- 3.Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med 2001; 345:588–595. [DOI] [PubMed] [Google Scholar]
- 4.Chawla LS, Bellomo R, Bihorac A, et al. Acute Disease Quality Initiative Workgroup 16: Acute kidney disease and renal recovery: consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat Rev Nephrol 2017; 13:241–257. [DOI] [PubMed] [Google Scholar]
- 5.Joannidis M, Druml W, Forni LG, et al. Prevention of acute kidney injury and protection of renal function in the intensive care unit: update 2017: Expert opinion of the Working Group on Prevention, AKI section, European Society of Intensive Care Medicine. Intensive Care Med 2017; 43:730–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol 2011; 22:999–1006. [DOI] [PubMed] [Google Scholar]
- 7.Uchino S, Kellum JA, Bellomo R, et al. Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators: Acute renal failure in critically ill patients a multinational, multicenter study. JAMA 2005; 294:813–818. [DOI] [PubMed] [Google Scholar]
- 8.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 2014; 371:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaudry S, Hajage D, Schortgen F, et al. AKIKI Study Group: Initiation strategies for renal-replacement therapy in the intensive care unit. N Engl J Med 2016; 375:122–133. [DOI] [PubMed] [Google Scholar]
- 10.Zarbock A, Kellum JA, Schmidt C, et al. Effect of early vs delayed initiation of renal replacement therapy on mortality in critically ill patients with acute kidney injury: the ELAIN randomized clinical trial. JAMA 2016; 315:2190–2199. [DOI] [PubMed] [Google Scholar]
- 11.Lim HA, Lee HH, Kim AJ, et al. Renin-angiotensin-aldosterone system blockade in critically ill patients is associated with increased risk for acute kidney injury. Tohoku J Exp Med 2016; 238:17–23. [DOI] [PubMed] [Google Scholar]
- 12.Langenberg C, Bagshaw SM, May CN, Bellomo R. The histopathology of septic acute kidney injury: a systematic review. Crit Care 2008; 12:R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest 2004; 114:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prowle JR, Bellomo R. Sepsis-associated acute kidney injury: macrohemodynamic and microhemodynamic alterations in the renal circulation. Semin Nephrol 2015; 35:64–74. [DOI] [PubMed] [Google Scholar]
- 15.Herget-Rosenthal S, Saner F, Chawla LS. Approach to hemodynamic shock and vasopressors. Clin J Am Soc Nephrol 2008; 3:546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lankadeva YR, Kosaka J, Evans RG, Bailey SR, Bellomo R, May CN. Intrarenal and urinary oxygenation during norepinephrine resuscitation in ovine septic acute kidney injury. Kidney Int 2016; 90:100–108. [DOI] [PubMed] [Google Scholar]
- 17.Abuelo GJ. Normotensive ischemic acute renal failure. N Engl J Med 2007; 357:797–805. [DOI] [PubMed] [Google Scholar]
- 18.Wan L, Langenberg C, Bellomo R, May CN. Angiotensin II in experimental hyperdynamic sepsis. Crit Care 2009; 13:R190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khanna A, English SW, Wang XS, et al. ATHOS-3 Investigators: Angiotensin II for the treatment of vasodilatory shock. N Engl J Med 2017; 337:419–430. [DOI] [PubMed] [Google Scholar]
- 20.Chawla LS, Russell JA, Bagshaw SM, et al. Angiotensin II for the treatment of high-output shock 3 (ATHOS-3): protocol for a phase III, double-blind, randomised controlled trial. Crit Care Resusc 2017; 19:43–49. [PubMed] [Google Scholar]
- 21.Kato R, Pinsky MR. Personalizing blood pressure management in septic shock. Ann. Intensive Care 2015; 5:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Busse LW, McCurdy MT, Ali O, Hall A, Chen H, Ostermann M. The effect of angiotensin II on blood pressure in patients with circulatory shock: a structured review of the literature. Crit Care 2017; 21:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yunge M, Petros A. Angiotensin for septic shock unresponsive to noradrenaline. Arch Dis Child 2000; 82:388–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wray GM, Coakley JH. Severe septic shock unresponsive to noradrenaline. Lancet 1995; 346:1604. [DOI] [PubMed] [Google Scholar]
- 25.Thomas VL, Nielsen MS. Administration of angiotensin II in refractory septic shock. Crit Care Med 1991; 19:1084–1086. [DOI] [PubMed] [Google Scholar]
- 26.Orfanos SE, Armaganidis A, Glynos C, et al. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in acute lung injury. Circulation 2000; 102:2011–2018. [DOI] [PubMed] [Google Scholar]
- 27.Lankadeva YR, Kosaka J, Evans RG, Bellomo R, May CN. Urinary oxygenation as a surrogate measure of medullary oxygenation during angiotensin II therapy in septic acute kidney injury. Crit Care Med 2018; 46:e41–e48. [DOI] [PubMed] [Google Scholar]
- 28.Brenner M, Schaer GL, Mallory DL, Suffredini AF, Parrillo JE. Detection of renal blood flow abnormalities in septic and critically ill patients using a newly designed indwelling thermodilution renal vein catheter. Chest 1990; 98:170–179. [DOI] [PubMed] [Google Scholar]
- 29.Calzavacca P, Evans RG, Bailey M, Bellomo R, May CN. Variable responses of regional renal oxygenation and perfusion to vasoactive agents in awake sheep. Am J Physiol Regul Integr Comp Physiol 2015; 309:R1226–R1233. [DOI] [PubMed] [Google Scholar]
- 30.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 2011; 121:4210–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.May CN, Ishikawa K, Wan L, et al. Renal bioenergetics during early gram-negative mammalian sepsis and angiotensin II infusion. Intensive Care Med 2012; 38:886–893. [DOI] [PubMed] [Google Scholar]
- 32.Nakada T, Russell JA, Boyd JH, et al. Association of angiotensin II type 1 receptor-associated protein gene polymorphism with increased mortality in septic shock. Crit Care Med 2011; 39:1641–1648. [DOI] [PubMed] [Google Scholar]
- 33.Yang H, Wang Y, Liu L, Hu Q. Increased susceptibility of sepsis associated with CD143 deletion/insertion polymorphism in Caucasians: a meta-analysis. Int J Clin Exp Pathol 2014; 7:6551–6558. [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang W, Chen X, Huang L, et al. Severe sepsis: low expression of the renin-angiotensin system is associated with poor prognosis. Exp Ther Med 2014; 7:1342–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.du Cheyron D, Fradin S, Ramakers M, et al. Angiotensin converting enzyme insertion/deletion genetic polymorphism: its impact on renal function in critically ill patients. Crit Care Med 2008; 36:3178–3183. [DOI] [PubMed] [Google Scholar]
- 36.Wunderink RG, Alberton TE, Busse LW. Baseline angiotensin levels and ACE effects in patients with vasodilatory shock treated with angiotensin II. Intensive Care Med Exp 2017; 5:0703 [Google Scholar]
- 37.Suberviola B, Rodrigo E, González-Castro A, Serrano M, Heras M. Castellanos-Ortega A: Association between exposure to angiotensin-converting enzyme inhibitors and angiotensin receptor blockers prior to septic shock and acute kidney injury. Med Intensiva 2017; 41:21–27. [DOI] [PubMed] [Google Scholar]
- 38.Corrêa TD, Takala J, Jakob SM. Angiotensin II in septic shock. Crit Care 2015; 19:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen A, Busse LW. Novel therapies for acute kidney injury. Kidney Int Rep 2017; 2:785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chawla LS, Busse LW, Brasha-Mitchell E, Alotaibi Z. The use of angiotensin II in distributive shock. Crit Care 2016; 20:137. [DOI] [PMC free article] [PubMed] [Google Scholar]