

Abstract
HIF-1α is a transcriptional regulator that functions in the adaptation of cells to hypoxic conditions; it strongly impacts the prognosis of patients with cancer. High-dose, intravenous, pharmacological ascorbate (P-AscH−), induces cytotoxicity and oxidative stress selectively in cancer cells by acting as a pro-drug for the delivery of hydrogen peroxide (H2O2); early clinical data suggest improved survival and inhibition of metastasis in patients being actively treated with P-AscH−. Previous studies have demonstrated that activation of HIF-1α is necessary for P-AscH− sensitivity. We hypothesized that pancreatic cancer (PDAC) progression and metastasis could be be targeted by P-AscH− via H2O2-mediated inhibition of HIF-1α stabilization. Our study demonstrates an oxygen- and prolyl hydroxylase-independent regulation of HIF-1α by P-AscH−. Additionally, P-AscH− decreased VEGF secretion in a dose-dependent manner that was reversible with catalase, consistent with an H2O2-mediated mechanism. Pharmacological and genetic manipulations of HIF-1α did not alter P-AscH−-induced cytotoxicity. In vivo, P-AscH− inhibited tumor growth and VEGF expression. We conclude that P-AscH− suppresses the levels of HIF-1α protein in hypoxic conditions through a post-translational mechanism. These findings suggest potential new therapies specifically designed to inhibit the mechanisms that drive metastases as a part of PDAC treatment.
Keywords: Vitamin C, Hypoxia inducible factor, Metastasis, Ascorbate, Pancreatic adenocarcinoma
Introduction
The cellular response to hypoxia is primarily mediated by hypoxia-inducible transcription factors (HIF-1, 2, and 3), of which the HIF-1 isoform is well established to correlate with pancreatic cancer (PDAC) metastasis and prognosis [1–6] while the relationship with the other isoforms is inconclusive [7–9]. HIF-1 is active in its heterodimeric form, made up of HIF-1α and HIF-1β subunits. HIF-1β is a ubiquitously expressed nuclear protein, which binds to the HIF-1α subunit. The cytoplasmic HIF-1α subunit accumulates in hypoxia, translocating to the nucleus to dimerize with the HIF-1β subunit. The dimer then subsequently binds to Hypoxia Response Elements (HREs) that are responsible for the transcription of hundreds of genes involved in metabolism, angiogenic signaling, vasomotor regulation, matrix and barrier function, growth, apoptosis, and many other functions [10]. The angiogenic signaling pathways, including vascular endothelial growth factor (VEGF), have been the focus of various targeted therapies [11]. HIF-1α has two specific oxygen-dependent degradation domains that are targeted for hydroxylation by the 2-oxoglutarate-dependent prolyl hydroxylase (primarily PHD-2) under normoxic conditions, which is the primary mode of regulation of HIF-1α. Hydroxylated proline residues on HIF-1α have increased affinity for the von Hippel-Lindau-ubiquitin ligase complex, leading to the proteolysis of HIF-1α (3). Low levels of hydrogen peroxide (H2O2) stimulate HIF-1α protein accumulation while high levels inhibit HIF-1α accumulation [12].
While non-cancerous cells adapt to prolonged hypoxia via this signaling cascade, tumors create an aberrant, locally hypoxic environment due to loss of contact inhibition, outgrowth of vasculature, and increased oxidative phosphorylation. Pancreatic adenocarcinoma (PDAC) tumors expressing high levels of HIF-1α demonstrate poor overall survival, advanced tumor stage at diagnosis, increased micro-vessel density, and increased lymph node metastasis [13]. Recently, it has also been shown that the metabolic re-programming of HIF-1α upregulation makes metastatic cancer cells suited to particular metastatic sites [14].
Clinical data show that when ascorbate (vitamin C, ascorbic acid) is given orally, fasting plasma concentrations are tightly controlled at < 100 μM [15, 16]. In contrast, when ascorbate is administered intravenously, plasma concentrations as high as 1–30 mM are safely achieved with few side effects [17]. Thus, it is clear that intravenous administration of ascorbate can yield very high plasma levels, while oral treatment does not. When ascorbate is infused intravenously the resulting pharmacologic concentration distributes rapidly into the extracellular water space, generating ascorbate radical and H2O2 [18]. P-AscH− administered intravenously is proposed to serve as a pro-drug for delivery of H2O2 preferentially to tumor cells [19]. Recently, Tian and colleagues demonstrated that activation of HIF triggers a Warburg effect that renders cancer cells more sensitive to P-AscH−. In their study, down regulation of HIF-1α induced resistance to P-AscH−, while overexpression of constitutively active HIF subunits enhanced P-AscH− toxicity [20].
In our current study, we demonstrate that P-AscH− is cytotoxic to cancer cells in hypoxia, and that this cytotoxicity correlates with increased degradation of HIF-1α, which results in decreased expression of VEGF. These processes occur in a H2O2-dependent manner. We also show that, with manipulation of HIF-1α, both pharmacologically and through genome editing, these cancer cells remain sensitive to P-AscH−. In vivo, P-AscH− induced a decrease in VEGF expression that was accompanied by a reduction in the rate of tumor growth.
Materials and methods
Cell culture
Human PDAC cell lines MIA PaCa-2 and PANC-1 were purchased from the American Type Culture Collection (Manassas, VA, USA) and passaged for fewer than 6 months after receipt. MIA PaCa-2 and PANC-1 cells were maintained in Dulbecco’s modified Eagle media (DMEM) supplemented with 10% fetal bovine serum (FBS). All cells were maintained in a humidified atmosphere of 95% air/5% CO2 at 37 °C. Also, patient-derived PDAC cell line, 339, from the Medical College of Wisconsin surgical oncology tissue bank [21, 22] were cultured in Dulbecco’s Modified Eagle’s Media Nutrient Mixture F-12 supplemented with 5% FBS, penicillin/streptomycin, human recombinant EGF, bovine pituitary extract, hydrocortisone, and human recombinant insulin.
Reagents
Hypoxia inducible factor-1α inhibitor (CAS 934593-90-5) was purchased from Santa Cruz Biotechnology. MG-132 (CAS 133407-82-6) and cycloheximide (CAS 66-81-9) were purchased from EMD Millipore. L-Ascorbic acid was purchased from Macron Chemicals (Center Valley, PA). A stock solution of ascorbate (pH 7.0) was made under argon and stored in screw-top sealed test tubes at 4 °C. Ascorbate concentration was verified using, ε265 = 14,500 M−1 cm−123. The solution can be kept for several weeks without significant oxidation due to the lack of oxygen [23]. Intracellular ATP was measured using ENLITEN ATP assay system bio-luminescence detection kit from Promega (Madison, WI).
Catalase treatment and adenovirus transfection
To determine whether H2O2 was responsible for the cytotoxic effects of ascorbate and radiation, cells were treated with various forms of catalase including adenovirus catalase (AdCat) or bovine catalase (100 U/mL). Catalase was purchased from Sigma-Aldrich (St. Louis, MO). The AdCat construct used was a replication-defective, E1- and partial E3 deleted recombinant adenovirus [24]. Inserted into the E1 region of the adenovirus genome is the human catalase gene, which is driven by a cytomegalovirus promoter. For the adenovirus experiments approximately 106 cells were plated in 10 mL of complete media in a 100 mm2 tissue culture dish and allowed to attach for 24 h. Cells were then washed 3 times in serum- and antibiotic-free media. The adenovirus constructs were applied to cells in 4 mL of serum-and antibiotic-free media. Control cells were treated with the GFP adenovirus (AdGFP) construct. Cells were incubated with the adenovirus constructs for 12 h. Media was replaced with 4 mL of complete media for an additional 24 h before cells were harvested for Western blot or treated for clonogenic assay. In order to demonstrate sensitivity to P-AscH− in cells over-expressing HIF-1a, cells were transfected in the same manner to those described above with HIF-1α adenovirus construct.
Clonogenic survival assays
Clonogenic survival assays were performed as previously described [18]. Briefly, P-AscH− treatments were performed for 1 h in DMEM and 10% fetal bovine serum (FBS) at 37 °C and 4% O2, 24 h after initial seeding in 60 mm dishes. Cells were then trypsinized, counted, and seeded into 6-well plates at 300 cell well−1 with 4.0 mL growth media. The dishes were maintained in a 37 °C, 4% O2, for 10–14 days to allow colony formation. The colonies were then fixed with 70% ethanol, stained with Coomassie™ Blue (10% acetic acid, 50% methanol and 0.1% Coomassie Blue G-250) and counted (colonies containing > 50 cells were scored). Plating efficiency was determined by the formula: (number of colonies formed/number of cells inoculated) × 100.
Immunoblot analysis
Protein (15–100 μg) was electrophoresed in a 4–20% Bio-Rad ready gel then electrotransferred to an Immobilon PVDF membrane (EMD Millipore, Billerica, MA). Membranes were blocked in 5% nonfat milk for 1 h, then treated with anti-HIF-1α antibody (1:1000; Abcam, Cambridge, MA) or with anti-catalase antibody (1:5000; Cell Signaling Technology, Danvers, MA). Horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse (1:50,000; Chemicon International, Temecula, CA) was used as a secondary antibody. Anti-GAPDH (1:10,000; EMD Millipore, Billerica, MA) was used as a loading control, followed by secondary antibodies conjugated to horseradish peroxidase (1:25,000, Millipore). Blots were treated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Rockford, IL) and exposed to autoradiography film. CoCl2, which blocks the active site of PHD-2, [25, 26] was used as a positive control in all western blots for HIF-1α. All Western blots were performed in triplicate.
CRISPR/Cas9 system
The CRISPR/Cas9 genome engineering system was utilized to create targeted disruption of human HIF-1a in MIA PaCa-2 cells. A pD1431-APuro Nickase expression vector containing Cas9 endonuclease, EF1 alpha promoter, and tandem chimeric gRNAs designed to target HIF-1a [GAAACCACCTATGACCTGCT and TGTCATTGGTTGGAGTCACA] was custom synthesized by DNA2.0 (Menlo Park, CA). Cells were transiently transfected in the absence of antibiotics or serum with Lipofectamine 3000 (Invitrogen, Carlsbad, CA) and 5 μg plasmid DNA for 48 h. Single cell clones were harvested after selection with 1.5 μg mL−1 puromycin for 3 days. Genomic DNA was isolated from clones displaying HIF1a disruption (based on decreased or absent protein expression on western blot analysis) utilizing Quick-Extract™ DNA extraction solution 1.0 (Epicentre, Madison, WI) as per manufacturer instructions. A 700 bp area encompassing the gRNAs targeting sites was PCR-amplified; PCR fragments of the appropriate size were excised and purified with the PureLink Quick Gel Extraction kit (Invitrogen, Carlsbad, CA). DNA sequencing analysis of PCR fragments was performed by the Genomics Division at The University of Iowa.
In vivo studies
All protocols were reviewed and approved by the Animal Care and Use Committee of The University of Iowa (Iowa City, IA). MIA PaCa-2 or PANC-1 human PDAC cells (2 × 106) were delivered subcutaneously into the flank region of nude mice with a 1 mL tuberculin syringe equipped with a 25-gauge needle. The tumors were allowed to grow until they reached between 3 and 4 mm in greatest dimension (2 weeks), at which time the mice were randomized and treatment was initiated. Mice were then treated with saline [1 M NaCl intraperitoneal (i.p.) daily] or P-AscH− (4 g kg−1 day−1 i.p.). Tumor size was measured twice a week using a digital caliper, and tumor volume was estimated according to the following formula: tumor volume = π/6 × L × W2, where L is the greatest dimension of the tumor, and W is the dimension of the tumor in the perpendicular direction [27]. Animals were euthanized when the tumors reached a predetermined size of 1000 mm3.
VEGFmeasurements
Relative amounts of secreted VEGF expressed were determined using a commercially available ELISA assay (R & D Systems, Minneapolis, MN). The VEGF165 isoform levels were quantified in the conditioned medium in accordance with the manufacturer’s protocol and measured spectrophotometrically at 450 nm with wavelength correction measured at 540 nm. Formalin fixed paraffin embedded tissues were routinely stained with HE and immunohistochemistry was also performed for VEGF. Antigen unmasking of all paraffin sections was performed (citrate buffer, pH 6) in a decloaker. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide and 1.5% horse serum was used to block non-specific staining. Sections were incubated with polyclonal rabbit anti-VEGF (Abcam ab46154) at 1:500 for 1 h. Slides were then incubated with the appropriate secondary antibody and detection reagent (DAKO Rabbit Envision HRP System reagent for 30 min). Slides were then developed with DAKO DAB plus for 5 min followed by DAB Enhancer for 3 min before being counterstained with hematoxylin. VEGF stained slides were evaluated in a blinded manner by a board-certified veterinary pathologist and graded using a semi-quantitative scoring system on a 0–3 scale. No immunostaining is given a 0; 1 = light, multifocal immunostaining; 2 = moderate, multifocal to coalescing immunostaining; 3 = dark, abundant coalescing to diffuse immunostaining. Twenty separate fields of view per slide were evaluated using the scale above and analyzed using an unpaired T test.
Statistical analysis
A one-way ANOVA followed by Tukey’s post-hoc test was used to determine statistical differences between means for multiple comparisons, or a student’s t-test was used when only two comparisons. All means were calculated from three or more experiments, with error bars representing the standard error of the mean (SEM). All Western blots were repeated at least twice. For the in vivo studies, the statistical analyses focused on the effects of different treatments on tumor progression. The primary outcome of interest was tumor growth over time. Linear mixed effects regression models were used to estimate and compare group-specific tumor growth curves. Tests of statistical significance were two-sided and performed using the GraphPad Prism (La Jolla, CA) software.
Results
P-AscH− inhibits pancreatic cancer cell growth in hypoxia
To determine the effect of P-AscH− on PDAC cell growth in hypoxia in vitro, MIA PaCa-2 and PANC-1 cells were treated with P-AscH− (2–10 mM) for 1 h in 4% O2 and subsequently grown in 4% O2. Increasing concentrations of P-AscH− led to significant inhibition of cell growth. Note that figures display P-AscH− dose in both molarity and on a mole cell− 1 basis. Recent studies have demonstrated that specifying the dose of xenobiotics, including P-AscH−, on a “mole per cell” basis results in improved correlations with toxicity markers as well as greater information content in experimental data [28, 29]. The doubling time of MIA PaCa-2 cells treated with P-AscH− was 59 ± 5 h versus 43 ± 0.2 h (means ± SEM, n = 3, p < 0.05) for controls when treated with the ED50 dose, 7 mM (7 pmol cell− 1) (Fig. 1a). A similar pattern was seen in PANC-1 cells where doubling time increased to 77 ± 8 h from 37 ± 2 h (means ± SEM, n = 3, p < 0.01, ED50 dose 20 mM, 20 pmol cell−1) (Fig. 1b). Likewise, clonogenic survival decreased in a dose-dependent manner in MIA PaCa-2 (Fig. 1c) PANC-1 (Fig. 1d), and 339 cells (Fig. 1e) treated with P-AscH− (2–10 mM). The presence of normoxia (i.e. 20% oxygen in cell culture) or varying degrees of hypoxia did not alter P-AscH−-induced cytotoxicity. As seen in Fig. 1f, there were no differences in the toxicity to cells when treated P-AscH− (7 pmol/cell, 7 mM) at 20, 4, or 1%, O2. As seen in normoxia [18], the decrease in clonogenic survival from P-AscH− in 1% O2 was reversed with addition of catalase (100 U/mL) to the media suggesting that H2O2 mediates ascorbate-induced cytotoxicity in hypoxia (Fig. 1g). Importantly, pre-incubation (conditioning) in hypoxia to overexpress HIF-1α conferred no increased resistance to P-AscH− relative to unconditioned controls (Fig. 1h).
Fig. 1.
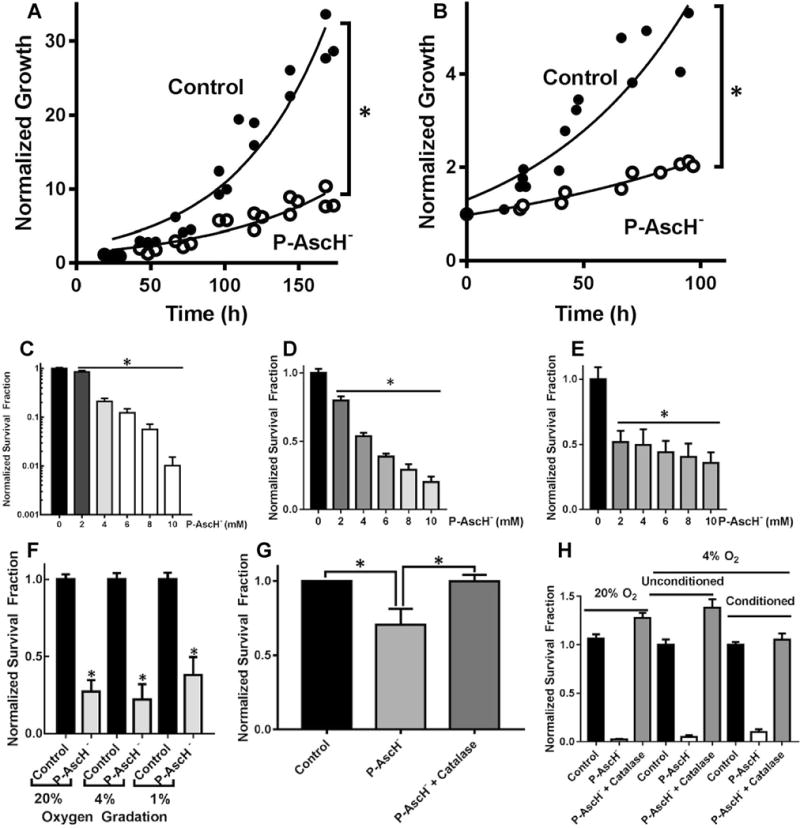
P-AscH−-induced cytotoxicity in hypoxia. a Treatment of MIA PaCa-2 cells with P-AscH− (7 mM; 7 pmol cell−1) significantly increased doubling time (n = 6, *p < 0.05 vs. control). b P-AscH− (10 mM; 10 pmol cell−1) significantly increased doubling time in PANC-1 cancer cell lines (n = 6, p < 0.05). c Clonogenic survival was decreased in a dose-dependent fashion in MIA-PaCa-2 cells treated and subsequently grown in 4% O2 (n = 3, *p < 0.01 vs. control). d Clonogenic survival was decreased in a dose-dependent fashion in PANC-1 cells treated and subsequently grown in 4% O2 (n = 3, *p < 0.01 vs. control). e Clonogenic survival was decreased in a dose-dependent fashion in 339 cells treated and subsequently grown in 4% O2 (n = 4, *p < 0.01 vs. control). f P-AscH−-induced decreases in clonogenic survival were similar in hypoxic environments (7 pmol/cell, 7 mM P-AscH−, means ± SEM, n = 3, *p < 0.05 vs. control). g Catalase reverses P-AscH−-induced cytotoxicity in 1% O2 (7 pmol/cell, 7 mM P-AscH−, means ± SEM, n = 3, *p < 0.05 vs. control). h Clonogenic survival of P-AscH− treated cells pre-conditioned to hypoxia by 16-h incubation in 4% O2 compared to unconditioned cells and cells treated in 20% O2. Cells conditioned to hypoxia were significantly sensitive to P-AscH− (7 pmol/cell, 7 mM P-AscH−, n = 3, p < 0.01), however, pre-conditioning to hypoxia yielded no significant resistance to P-AscH− relative to unconditioned controls, or cells treated in 20% O2
P-AscH−-induced suppression of HIF-1α protein and VEGF secretion correlate with decreased clonogenic survival
We hypothesized that alteration of the cellular redox environment associated with P-AscH− may lead to inhibition of HIF-1α signaling. As seen in Fig. 2a, Western blot analyses in MIA PaCa-2, PANC-1, and the patient-derived cell line, 339, demonstrated that 4% O2 induced HIF-1α immunoreactive protein when compared to 20% O2. Most importantly, there was a dose-dependent decrease in HIF-1α immunoreactive protein in MIA PaCa-2, PANC-1, and 339, human PDAC cells treated with P-AscH− (2–10 mM) (Fig. 2a), which was quantified by densitometric analysis in Fig. 2b.
Fig. 2.
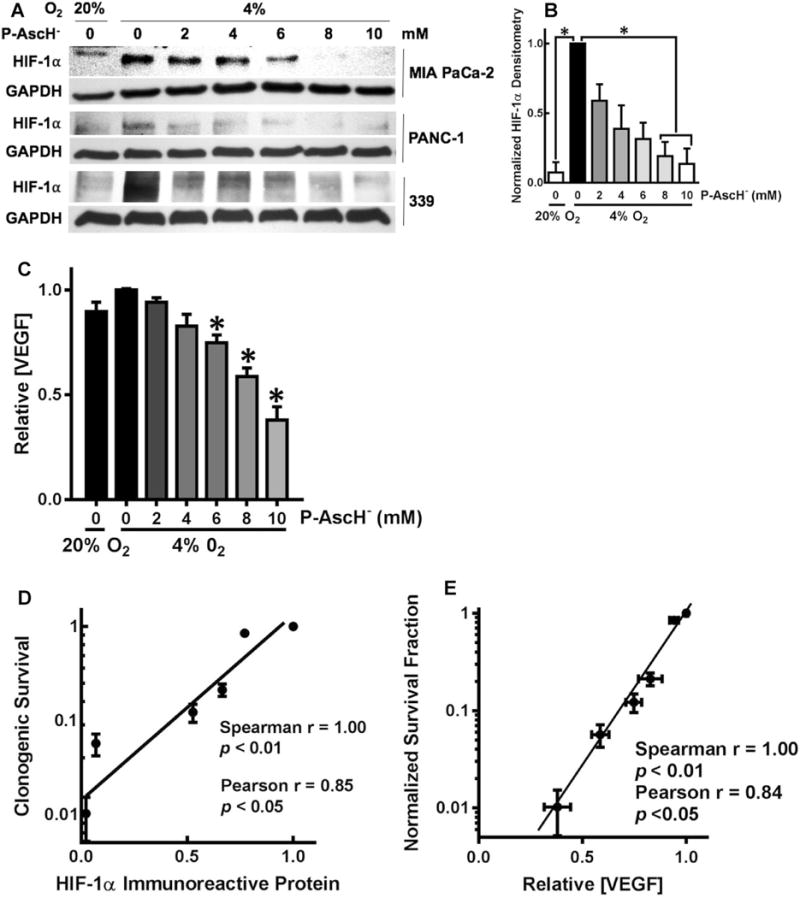
P-AscH− suppresses HIF-1α immunoreactive protein and VEGF secretion. a MIA PaCa-2, PANC-1, and 339 cell lines showed a dose-dependent decrease in HIF-1α expression after 1-h exposure to P-AscH−. Both cell lines had induction of HIF-1α immunoreactive protein in hypoxia. b In the MIA PaCa-2 cell line, densitometric analysis of HIF-1α demonstrates a decrease in HIF-1α expression with increasing doses of P-AscH− (n = 3, p < 0.05). c Secreted VEGF in media after 1-h P-AscH− and incubation in 4% O2 also demonstrated a dose-dependent decrease (means ± SEM, n = 6, *p < 0.05). d Clonogenic survival correlated with HIF-1α immunoreactive protein in MIA PaCa-2 cancer cells treated with P-AscH− (2–10 mM). e Clonogenic survival correlated with VEGF secretion in MIA PaCa-2 cancer cells with P-AscH− (2–10 mM)
Angiogenesis is a key step that supports tumor growth and may also provide a route for cells to metastasize [30]. VEGF is a potent angiogenic stimulant [31], and a mitogenic cytokine, that is secreted outside the cells to stimulate the proliferation of endothelial cells [32]. Exposure to hypoxia stimulates the production of VEGF mainly through the induction of the transcription factor HIF-1α [33]. Because P-AscH− suppressed hypoxic accumulation of HIF-1α protein, we hypothesized that P-AscH− would also modulate VEGF. VEGF secreted into the media was measured 24 h after treatment with P-AscH− (2–10 mM) in hypoxia. As seen with P-AscH−-induced decreases in HIF-1α, VEGF secretion decreased in a dose-dependent manner (Fig. 2c), which correlated with decreasing HIF-1α expression. Taken together, these results suggest a strong correlation between HIF-1α expression, VEGF secretion, and clonogenic survival, in cells treated with P-AscH− as demonstrated in Fig. 2d, e.
Our data demonstrate that P-AscH− suppresses HIF-1α stabilization within hours of treatment. The modulation of the physiological steady-state level or flux of reactive oxygen species can change the fundamental biology of cells. To determine if the results in Fig. 2 are simply reflecting the acute toxic effects of P-AscH−, we measured intracellular ATP as an indication of metabolic activity after ascorbate treatment (1.8 mM, 5.6 pmol cell− 1). Intracellular ATP levels remain stable 24 h after P-AscH− treatment in PDAC cells. Thus, P-AscH−-induced inhibition of HIF-1α is not due to overwhelming cell death (Supplementary Fig. 1).
Suppression of HIF-1α,VEGF, and clonogenic survival is mediated by P-AscH−-induced H2O2 generation
P-AscH− will increase extracellular H2O2, which is transported across the plasma membrane into cancer cells [18, 29, 34]. To determine whether the P-AscH−-induced HIF-1α suppression, VEGF suppression, and decreased clonogenic survival in hypoxia were due to H2O2, cells were co-treated with catalase (100 U/mL in the culture medium) or transfected with either a control adenoviral vector, AdGFP (50 MOI), or the adenoviral vector containing the catalase gene, AdCat (50 MOI), to increase intracellular catalase activity. Catalase supplemented in the media (Fig. 3a) reversed the P-AscH−-induced (2 mM, 5.4 pmol cell−1) inhibition of HIF-1α. Intracellular catalase overexpression with the AdCat vector alone decreased HIF-1α protein (Fig. 3b). However, the AdCat vector also reversed the P-AscH−-induced (2 mM, 5.4 pmole cell−1) inhibition of HIF-1α. To further substantiate the importance of the reduced form of vitamin C (ascorbate) in establishing the need for a flux of H2O2 to suppress HIF-1α, cells were treated with the two-electron oxidized form of vitamin C (dehydroascorbic acid, DHA). DHA did not suppress HIF-1α in hypoxia (Supplemental Fig. 2). DHA can be taken up by cells, reduced to ascorbate and then participate in the cellular activities as vitamin C. However, it will not be reduced in the extracellular space of the cell culture medium and thus will not generate a flux of H2O2. Inclusion of catalase in the media also reversed the P-AscH−-induced suppression of VEGF secretion (Fig. 3c). Lastly, the P-AscH−-induced decrease in clonogenic survival was reversed with transfection with the AdCat vector in MIA PaCa-2 (Fig. 3d) and PANC-1 cells (Fig. 3e). These results suggest that HIF-1α and VEGF suppression, along with decreased clonogenic survival by P-AscH−-treatment, are due to a mechanism involving increased levels of H2O2 and not the chemistry or biochemistry of DHA.
Fig. 3.
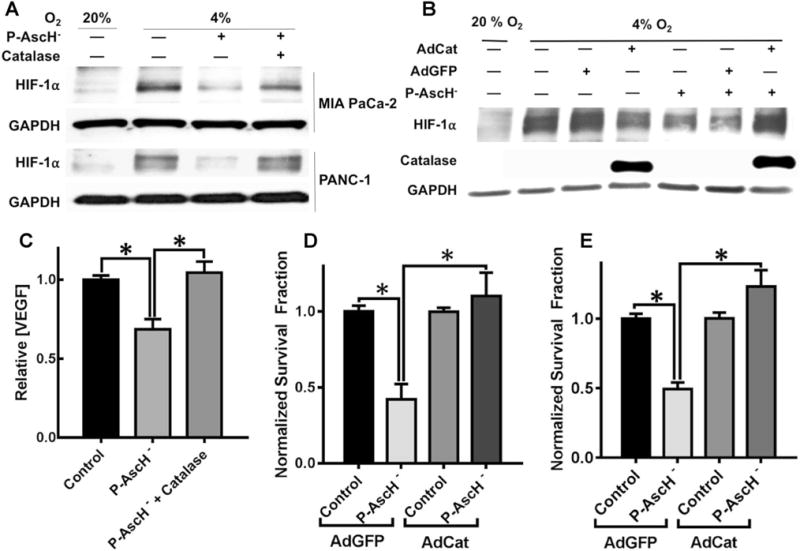
Catalase reverses the hypoxic P-AscH−-induced suppression of clonogenic survival, HIF-1α and VEGF. a Western Blot demonstrates the reversal of P-AscH−-induced (2 mM, 5.4 pmol cell−1) decrease in HIF-1α in 4% O2 with extracellular catalase in both MIA PaCa-2 and PANC-1 cell lines. Again, hypoxia (4% O2) induced HIF-1α in both cell lines. P-AscH− decreased HIF-1α immunoreactive protein which was then reversed with addition of exogenous catalase. b Over-expression of intracellular catalase reverses the P-AscH−-induced (2 mM, 5.4 pmol cell−1) HIF-1α suppression. HIF-1α was induced with hypoxia while cells were infected with an adenovirus containing the cDNA for catalase or containing GFP (control vector). Catalase immunoreactivity demonstrated robust increase in catalase protein with the AdCat vector. Catalase overexpression had no effect of HIF-1α in hypoxia; however, catalase overexpression reversed the P-AscH−-induced HIF-1α suppression. c P-AscH−-induced decreases in VEGF secretion were reversed with extracellular scavenging of H2O2 by catalase (MIA PaCa-2, n = 6, *p < 0.05, Mean ± SEM). d P-AscH−-induced (5 mM, 7 pmol cell−1) clonogenic cell death which was reversed with overexpression of intracellular catalase in MIA PaCa-2 cells (means ± SEM, n = 3, *p < 0.05). e P-AscH−-induced (5 mM, 7 pmol cell−1) clonogenic cell death was reversed with over-expression of intracellular catalase in PANC-1cells (means ± SEM, n = 3, *p < 0.05)
P-AscH− increases the rate of HIF-1α protein degradation
To further investigate the mechanism of P-AscH− inhibition of HIF-1α expression, we tested whether the degradation of HIF-1α protein was affected by P-AscH− in the presence of cycloheximide, an inhibitor of protein translation. MIA PaCa-2 cells were pre-incubated in hypoxia for 6 h to increase HIF-1α levels and then treated with cycloheximide with and without P-AscH−. In hypoxia, HIF-1α immunore-active protein degraded more rapidly with P-AscH− treatment, as seen in the Western blot in Fig. 4a and demonstrated by densitometry in Fig. 4b. These results suggest that P-AscH− suppresses the levels of HIF-1α protein under hypoxic conditions through a posttranslational mechanism [35]. Next we examined whether P-AscH− could regulate HIF-1α protein synthesis. MIA PaCa-2 cells were treated with MG-132, a proteasome inhibitor, to interrupt HIF-1α proteasomal degradation. Cells were pre-incubated in hypoxia for 4 h to induce HIF-1α and MG-132 was added to cell culture medium. Protein was collected over a period of 3 h. In hypoxia, P-AscH− in the presence of MG-132, resulted in no changes in HIF-1α protein expression over 3 h, as seen in the Western blot in Fig. 4c and demonstrated by densitometric analysis in Fig. 4d, suggesting that inhibition of the proteasome prevents P-AscH−-mediated HIF-1α inhibition. Overall, these data suggest that P-AscH− decreases HIF-1α levels via degradation as opposed to decreased translation. Prolyl hydroxylase (PHD-2) regulates HIF-1α expression. PHD-2 is an iron-containing enzyme that hydroxylates HIF-1α at specific proline sites, ultimately marking it for degradation [12, 36]. To have activity, the iron moiety must be in its reduced form, and theoretically P-AscH− could recycle the iron, increasing PHD-2 activity. However, as shown in Fig. 4e, when cells were treated with P-AscH− in combination with cobalt chloride (CoCl2, an inhibitor of PHD-2), HIF-1α levels were still diminished, suggesting a mechanism independent of the role PHD-2 has in the degradation of HIF-1α.
Fig. 4.
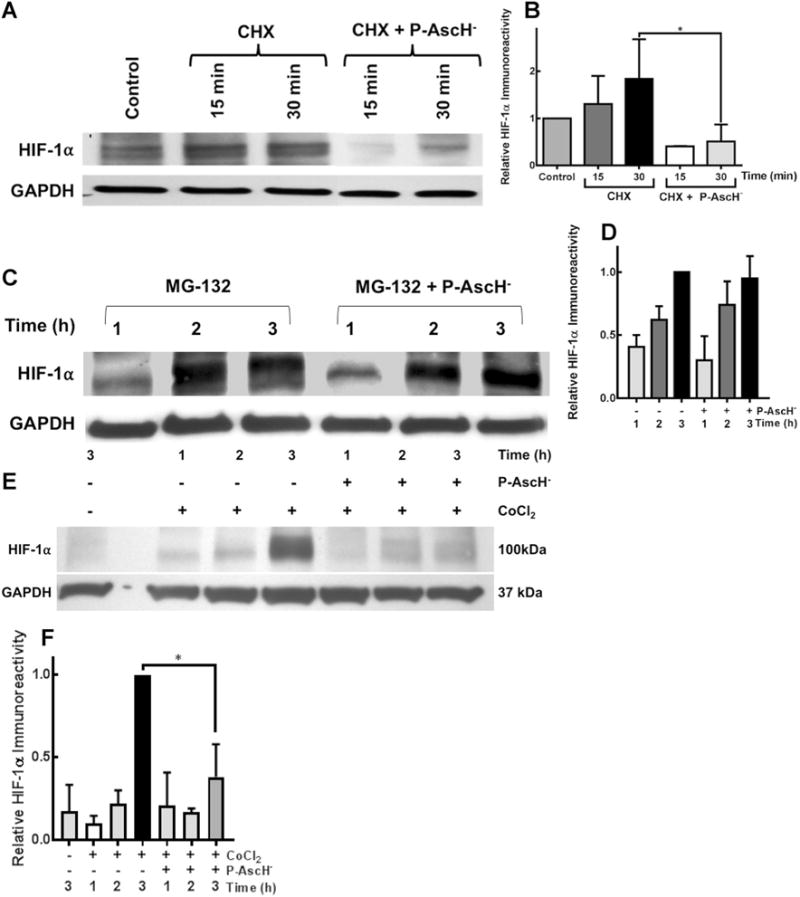
P-AscH−-induced HIF-1α suppression is due to a prolyl hydroxylase-independent increase in degradation. a Ribosomal inhibition in hypoxia with cycloheximide (CHX) and subsequent treatment with P-AscH− (5 mM, 7 pmol cell− 1) results in decreased HIF-1α as demonstrated by Western blot. b Densitometric analysis demonstrates decreased HIF-1α with CHX and P-AscH− (*p < 0.1, n = 3, One-Way ANOVA, Dunn’s multiple comparisons test). c Proteasomal inhibition utilizing MG-132 reverses P-AscH− induced (5 mM, 7 pmol cell−1) suppression of HIF-1α protein as demonstrated by Western blot. d Densitometric analysis, suggesting P-AscH− increases degradation of HIF-1α as opposed to inhibiting synthesis. e Representative Western blot demonstrating inhibition of prolyl hydroxylase utilizing cobalt chloride (CoCl2) fails to reverse P-AscH−-induced (7 mM, 7 pmol cell−1) HIF-1α protein suppression. f Densitometric analysis demonstrated that CoCl2 does not reverse P-AscH− HIF-1α suppression (*p < 0.05, n = 3)
Inhibition of HIF-1α has no effect on P-AscH−-induced cytotoxicity
Previous studies have demonstrated that cells absent HIF-1α are resistant to P-AscH−-induced toxicity [20]. To determine the role of HIF-1α in P-AscH−-toxicity, we used both pharmacological and genetic inhibition of HIF-1α. First, HIF-1α was inhibited in MIA PaCa-2 cells using the small molecule inhibitor sc-205346. As seen in Fig. 5a, Western blotting demonstrated a dose-dependent inhibition of HIF-1α immunoreactive protein with sc-205346, which was quantified using densitometry as demonstrated in Fig. 5b. However, chemical inhibition of HIF-1α did not reverse P-AscH−-induced decreases in clonogenic survival, Fig. 5c.
Fig. 5.
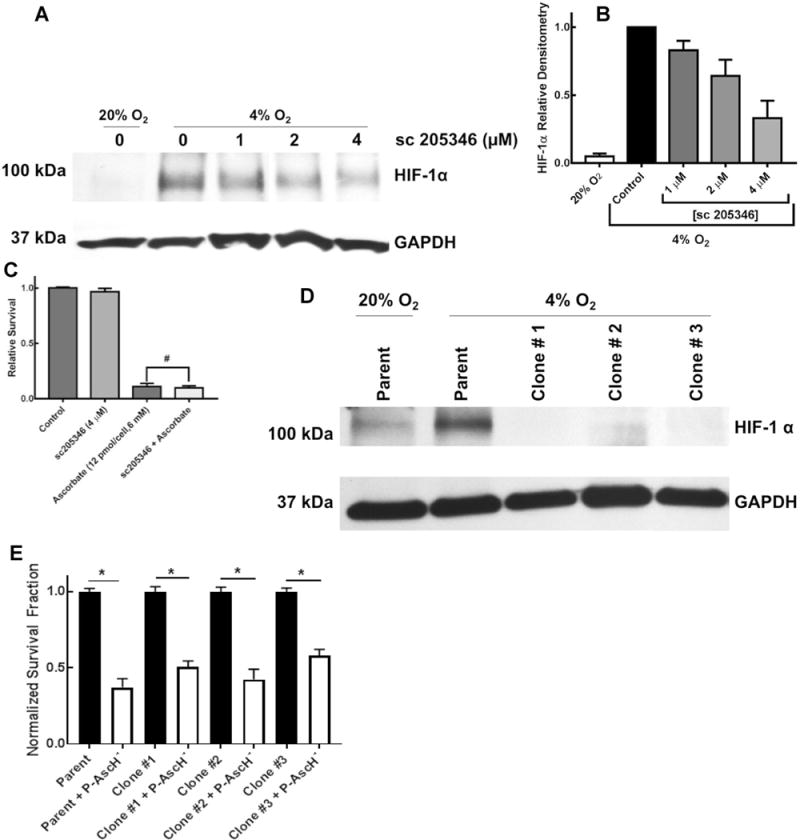
Selective HIF-1α suppression does not reverse P-AscH−-induced cytotoxicity. a Western blotting demonstrates induction of HIF-1α protein in hypoxia. Pharmacologic inhibition with sc-205346 suppressed HIF-1α as demonstrated by Western blotting. b Densitometry from Western blots demonstrating decreasing HIF-1α with pharmacologic inhibition using the compound sc-205356. c Pharmacological inhibition of HIF-1α does not alter P-AscH− (6 mM, 12 pmol cell−1) decreases in clonogenic survival (means ± SEM, n = 3 * p < 0.05 vs. control). d Three clones of MIA PaCa-2 cancer cells subjected to CRISPR/Cas9 HIF-1α genome editing demonstrated decreased expression of HIF-1α in hypoxia (4% O2, 8 h). e Decreased HIF-1α expression did not alter P-AscH− (10 mM) induced decreases in clonogenic survival (means ± SEM, *p < 0.0001 comparing each respective untreated cell line vs. P-AscH− treated cell lines. The untreated cell lines were normalized to 1.0, n = 3)
To genetically disrupt HIF-1α expression in pancreatic cancer cells, we employed the CRISPR/Cas9 genome editing system with dual gRNAs in a single vector. The CRISPR/Cas9 system produces a chromosomal break in DNA resulting in insertions and/or deletions when the genomic site is repaired through non-homologous end joining. Utilizing tandem chimeric gRNAs in this process allows for increased specificity and minimal off-target effects. The CRISPR/Cas9 system utilized caused sufficient genetic disruption of HIF1α in MIA PaCa-2 cells resulting in the generation of three distinct clones (clone 1, 2, and 3).
Each clone displayed significant decreases in detectable immunoreactive HIF-1α protein, Fig. 5d. The clonogenic survival assay was performed utilizing the CRISPR/Cas9 HIF-1α knocked down clones. Consistent with the previous studies using the chemical inhibitor of HIF-1α, the data presented in Fig. 5e demonstrate that there are no significant differences in clonogenic survival when comparing the effect of P-AscH− (10 mM) on the parent cell line and the three HIF-1α knocked down clones. Thus, inhibition of or absence of HIF-1α does not concur resistance to P-AscH−-induced toxicity.
Overexpression of HIF-1α does not change P-AscH−-induced cytotoxicity
HIF-1α expression in PDAC is associated with increased micro-vessel density, advanced tumor stage, lymph node metastasis, and poor overall survival [37]. It is unclear whether HIF-1α overexpression confers resistance to P-AscH−. In order to investigate this, HIF-1α was overexpressed in MIA-PaCa-2 cells via chemical inhibitors and genetic manipulation. As seen in Fig. 6a, CoCl2, which inhibits prolyl hydroxylase-2, resulted in increases in HIF-1α protein. In addition, P-AscH− treatment resulted in a significant decrease in clonogenic survival that was not significantly different from non-CoCl2 treated controls, Fig. 6b. Transfection with the AdHIF vector also increased HIF-1α protein at 100 MOI, Fig. 6c. Likewise, after transfection with 100 MOI AdHIF, P-AscH− treatment resulted in a significant decrease in clonogenic survival which was not significantly different from control vector (AdGFP), Fig. 6d. Thus, overexpression of HIF-1α by either chemical inhibition of prolyl hydroxylase-2 or genetic manipulations, does not confer resistance to P-AscH−-induced cytotoxicity.
Fig. 6.
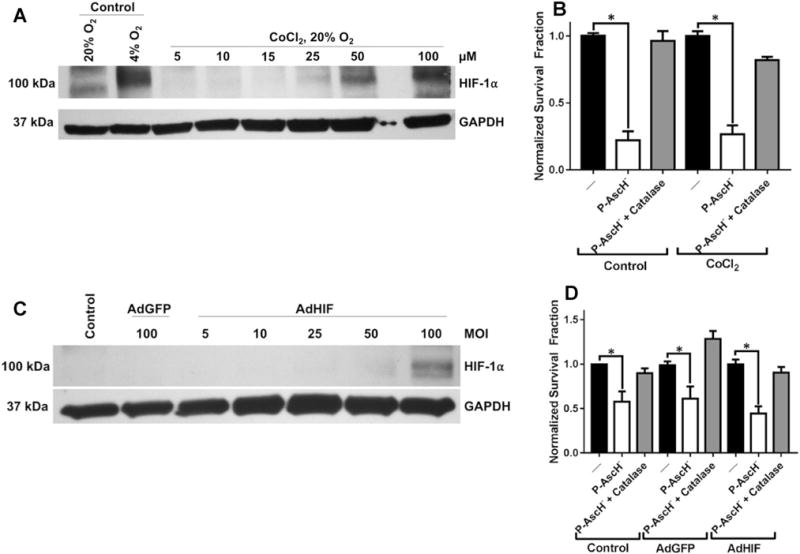
HIF-1α overexpression confers no resistance to P-AscH−. a Western blot of protein collected from MIA PaCa-2 cells incubated in normoxia with increasing concentration of CoCl2 (5–100 μM). Significant increases in HIF-1α protein are demonstrated at 50–100 μM CoCl2. b Clonogenic survival of MIA PaCa-2 cells pre-incubated for 8 h with CoCl2 (50 μM) prior to treatment with P-AscH− for 1 h demonstrates significant cytotoxicity (n = 3, p < 0.01) relative to non-P-AscH− controls. There were no significant differences in survival between P-AscH−-treated cells incubated with CoCl2 compared to untreated cells. c Western blot of protein collected from MIA PaCa-2 cells transfected with increasing MOI of AdHIF adenovirus. Significant overexpression of HIF-1α was demonstrated at 100 MOI. d Clonogenic survival of MIA PaCa-2 cells transfected with AdHIF adenovirus (100 MOI) treated with P-AscH− for 1 h in hypoxia. P-AscH− induced a significant cytotoxicity to AdHIF transfected cells (n = 3, p < 0.01) that was not significantly different from AdGFP and non-transfected controls
P-AscH− inhibits tumor growth and VEGF expression in vivo
P-AscH− inhibits hypoxic growth of PDAC cells, which is accompanied by suppression of HIF-1α protein accumulation and a decrease in VEGF. To determine if P-AscH− could also modulate tumor growth and affect VEGF expression in vivo, mice with pre-established xenografts of pancreatic tumors were treated with saline (1 M, i.p., b.i.d) or P-AscH− (4 g kg−1 i.p. b.i.d.). As demonstrated in Fig. 7, P-AscH− inhibited tumor growth in both MIA PaCa-2 and PANC-1 human PDAC xenografts. Comparison of normalized mean tumor size at 14 days demonstrated significantly decreased growth in P-AscH− treated mice compared to control mice. In MIA PaCa-2 tumor xenografts, there was a greater than fourfold decrease in growth when compared to controls at two weeks of treatment (Fig. 7a, n = 8, p < 0.05). In PANC-1 xenografts, there was a greater than twofold decrease in tumor growth compared to saline treated animals (Fig. 7b, n = 8, p < 0.05). Our in vitro data demonstrate that P-AscH− decreases HIF-1α expression and VEGF secretion. To determine if P-AscH− would alter VEGF in vivo, in separate groups of animals, tumors were excised to determine VEGF immunostaining after treatment with saline or P-AscH−. Immunohistochemistry analysis was determined using semi-quantitation by a comparative pathologist blinded to the treatment groups (Supplementary Fig. 3). Semi-quantitation of immunohistochemistry demonstrated decreases in VEGF staining in mice treated with P-AscH− in both MIA PaCa-2 (Fig. 7c) and in PANC-1 (Fig. 7d) tumor xenografts.
Fig. 7.
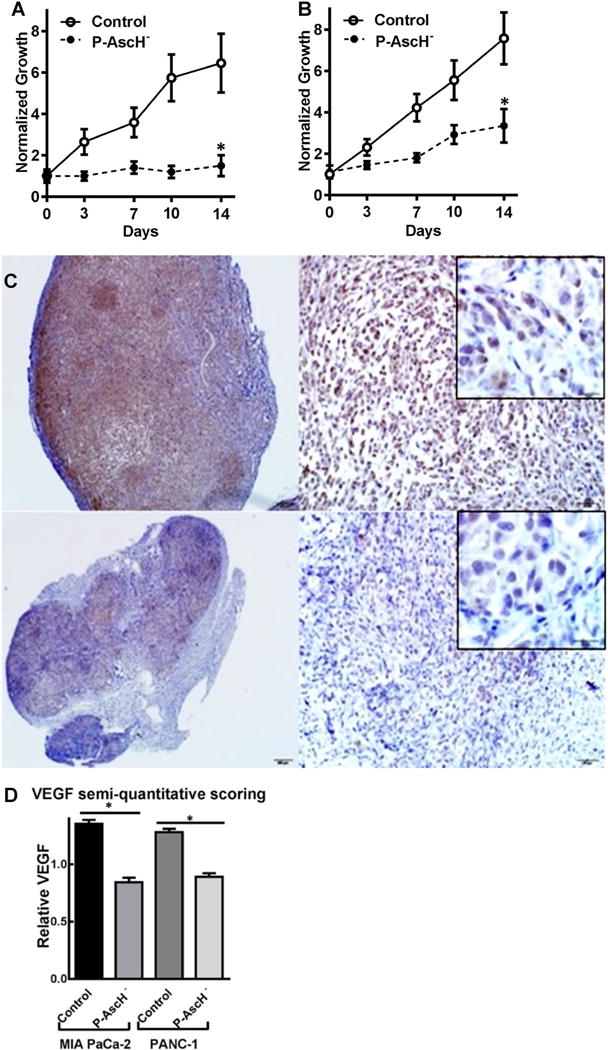
P-AscH− inhibits in vivo tumor growth and VEGF expression. A MIA PaCa-2 human tumor xenografts in athymic nude mice have significantly decreased tumor growth in mice treated with daily I.P. P-AscH− (4 g kg−1, b.i.d.) compared to mice receiving saline (control). Comparison of normalized mean tumor size at 14 days demonstrates significantly decreased growth of control mice and P-AscH−-treated mice (1.5-fold relative growth vs. 6.5-fold relative growth at 2 weeks, means ± SEM, n = 8, *p < 0.05). b Comparison of normalized mean tumor growth at 14 days demonstrated significantly decreased growth in P-AscH− treated mice compared to control mice in PANC-1 xenografts (3.7-fold relative growth vs. 9.1-fold relative growth at 2 weeks, means ± SEM, n = 8, *p < 0.05.). C. Representative VEGF immunohistochemistry (brown cytoplasmic staining) of MIA PaCa-2 tumor xenograft treated with saline (top panels) and P-AscH− (bottom panels). d Immunohistochemistry for VEGF. Sections were incubated with polyclonal rabbit anti-VEGF. VEGF stained slides were blindly evaluated by a board-certified veterinary pathologist (KGC) and graded using a semi-quantitative scoring system on a 0–3 scale. Twenty separate fields of view per slide were evaluated and analyzed. VEGF Semi-quantitative immunohistochemistry of all xenografts demonstrate significant decreased VEGF expression (n = 8, means ± SEM, *p < 0.01) in tumors of mice treated with P-AscH−
Discussion
Multiple phase I trials have shown that P-AscH− is well tolerated with few adverse events and some suggestion of efficacy. Previous studies have demonstrated that P-AscH− has synergistic or additive effects with many radio-chemotherapy regimens in cancer cells, and radio-protective effects in normal cells [17, 38–42]. Our current study extends these observations to clearly demonstrate the efficacy of P-AscH− in the hypoxic environment of PDAC. Metastases are a major cause of cancer mortality; over 80% of PDACs are diagnosed after metastasis or vascular invasion have occurred [43]. Recent studies suggest that metastasis occurs very early in the development of PDAC [44]. Most importantly, we have demonstrated that P-AscH−-induced cytotoxicity correlates with increased degradation of HIF-1α and decreased expression of VEGF, via a peroxide-dependent manner. Since HIF-1α is a major transcription factor in the development of metastatic disease, therapies specifically designed to inhibit the mechanisms that drive metastases would be another benefit of P-AscH− in treatment of malignancies.
Many tumors rely on HIF-1α to mediate adaptation to a relatively hypoxic environment, and tumors that can produce HIF-1α in abundance are particularly malignant. Our current study demonstrates that in a hypoxic environment, P-AscH− acts as a pro-drug for the production of H2O2 to cytotoxic levels, and that this correlates with increases in the degradation of HIF-1α in a dose-dependent manner. When the H2O2, produced by the two-electron reduction of O2, mediated by P-AscH−, is degraded by supplementation with intracellular or extracellular catalase, the effects on HIF-1α and cell cytotoxicity are reversed. Downstream from HIF-1α, VEGF is inhibited in a similar dose-dependent manner. The slowed tumor growth and inhibition of VEGF production translates to our in vivo xenograft mouse model as well. These results are consistent with the studies of Campbell et al. who demonstrated that expression of HIF-1α and its target proteins were down-regulated with P-AscH−. In addition, elevated tumor ascorbate levels could be maintained with daily administration of P-AscH− resulting in reduction of both HIF-1α and VEGF protein levels. P-AscH− slowed tumor growth and also reduced tumor microvessel density [45].
Lastly, while HIF-1α degradation correlates strongly with clonogenic cell death, our present study demonstrates that both pharmacologic and genetic inhibition of HIF-1α is not essential for sensitivity to P-AscH−. Previous studies have demonstrated increased malignant features of tumors expressing high levels of HIF-1α, and the suppression of these features with selective inhibition of HIF-1α with natural degradation pathways that bypass the more common PHD-2/VHL pathways [46]. In agreement with these studies, here we show the decreased clonogenic survival in vitro and tumor growth in vivo correlate with HIF-1α suppression. Also, in tandem with these studies, we show a pathway of increased degradation that appears to be independent of the PHD-2 pathway of proline hydroxylation marking HIF-1α for degradation.
The increased oxidative stress placed on cells by the production of abundant H2O2 and its byproducts, including hydroxyl radical, could potentially inhibit PHD-2 by oxidation of the iron moiety or essential thiol groups and subsequent increase in HIF-1α [47, 48]. Indeed, past studies have shown induction of HIF-1α by reactive oxygen species can be reversed by N-acetylcysteine [49]. In contrast, we’ve shown here that the oxidative stress induced by the production of H2O2, mediated by P-AscH−, results in increased degradation of HIF-1α independent of the function of prolyl hydroxylase. The disparity between this study, and those that precede it, are likely explained by the relatively large fluxes of H2O2 produced by P-AscH− compared to the low levels of oxidant stress produced in prior studies [50]. While HIF-1α is a major response factor to hypoxia for cancer cells, the overlap in response by other transcription factors likely explains the ability of these cells to continue to survive in hypoxia [51]. Previously published reports have shown decreased sensitivity to P-AscH− when HIF-1α is inhibited, suggesting that HIF-1α is necessary for P-AscH−-induced decreases in cell survival [20]. These studies modulated HIF-1α pharmacologically, instead of treating the cells in a hypoxic environment, as they were performed to study pVHL-deficient cancers [20]. Our current study differs from Tian and colleagues as we demonstrated that treating cancer cells in hypoxia did not reverse P-AscH−-induced decreases in clonogenic survival when HIF-1α was inhibited pharmacologically or genetically.
Unlike the other isoforms of HIF [7–9], clinically HIF-1α appears to be important in PDAC survival. Using immunohistochemical methods, Sun et al. demonstrated that HIF-1α in PDAC specimens has a strong association with prognosis and correlates with expression of vascular endothelial growth factor (VEGF) [2]. Hoffman et al. demonstrated that PDAC specimens with high HIF-1α expression were associated with decreased patient survival when compared to those with low HIF-1α expression [3]. They also revealed a positive correlation of HIF-1α with VEGF levels, lymph node metastasis, and tumor size. Similarly, Shibaji et al. demonstrated that in HIF-1α correlated positivity with metastatic status [4]. Buchler and colleagues demonstrated that in hypoxia, HIF-1α binds to the VEGF promoter with high specificity; this binding correlated with levels of VEGF [5]. Under hypoxic conditions cancer cells undergo the process of epithelial to mesenchymal transition (EMT) and acquire invasive and drug-resistant phenotypes [6]. Hypoxia or over-expression of HIF-1α promotes EMT in cancer. Molecular or pharmacologic inhibition of HIF-1α resulted in hypoxic cells regaining expression of E-cadherin, lost expression of N-cadherin, and attenuation of the invasive and drug-resistant phenotype [4]. These results suggest that HIF-1α activation may be a specific target to reverse EMT in pancreatic cancer.
In conclusion, P-AscH− decreased hypoxic clonogenic survival, suppressed HIF-1α expression via increased degradation, and decreased VEGF expression transcription and secretion in a dose-dependent manner via a peroxide-mediated mechanism. P-AscH−-induced cytotoxicity was independent of HIF-1α status, while in vivo, P-AscH− inhibited tumor growth and VEGF expression. Thus, P-AscH− suppresses the levels of HIF-1α protein in hypoxic conditions through a post-translational mechanism providing a potential therapy specifically designed to inhibit the mechanisms that drive metastases as a part of PDAC treatment.
Supplementary Material
Acknowledgments
HIF-1α Adenovirus construct obtained from Patrick H Maxwell, FMedSciRegius Professor of Physic & Head of the School of Clinical Medicine, University of Cambridge. Supported by NIH Grants CA184051, CA148062, CA169046, CA086862, and a Merit Review grant from the Medical Research Service, Department of Veterans Affairs 1I01BX001318-01A2.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s10585-018-9876-z) contains supplementary material, which is available to authorized users.
Author contributions
JC and JW conceived the hypothesis, designed the study, and performed the majority of the experiments. JW, BO, JD and KGC, performed experiments. JW, JC, and BO wrote the manuscript. JD, ZS, AK, KGC, and ST contributed reagents and tools for the study. JW, BO, GB, and JC contributed to discussion and edited the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Compliance with ethical standards
Conflict of interest The Authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Matsuo Y, Ding Q, Desaki R, et al. Hypoxia inducible factor-1 alpha plays a pivotal role in hepatic metastasis of pancreatic cancer: an immunohistochemical study. J Hepatobiliary Pancreat Sci. 2014;21(2):105–112. doi: 10.1002/jhbp.6. [DOI] [PubMed] [Google Scholar]
- 2.Sun HC, Qiu ZJ, Liu J, et al. Expression of hypoxia-inducible factor-1 alpha and associated proteins in pancreatic ductal adenocarcinoma and their impact on prognosis. Int J Oncol. 2007;30(6):1359–1367. [PubMed] [Google Scholar]
- 3.Hoffmann AC, Mori R, Vallbohmer D, et al. High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia. 2008;10(7):674–679. doi: 10.1593/neo.08292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shibaji T, Nagao M, Ikeda N, et al. Prognostic significance of HIF-1 alpha overexpression in human pancreatic cancer. Anticancer Res. 2003;23(6C):4721–4727. [PubMed] [Google Scholar]
- 5.Buchler P, Reber HA, Buchler M, et al. Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer. Pancreas. 2003;26(1):56–64. doi: 10.1097/00006676-200301000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Cheng ZX, Sun B, Wang SJ, et al. Nuclear factor-kappaB-dependent epithelial to mesenchymal transition induced by HIF-1alpha activation in pancreatic cancer cells under hypoxic conditions. PloS ONE. 2011;6(8):e23752. doi: 10.1371/journal.pone.0023752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12(1):9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang M, Chen MY, Guo XJ, Jiang JX. Expression and significance of HIF-1alpha and HIF-2alpha in pancreatic cancer. J Huazhong Univ Sci Technol Med Sci. 2015;35(6):874–879. doi: 10.1007/s11596-015-1521-3. [DOI] [PubMed] [Google Scholar]
- 9.Yang J, Zhang X, Zhang Y, et al. HIF-2alpha promotes epithelial-mesenchymal transition through regulating Twist2 binding to the promoter of E-cadherin in pancreatic cancer. J Exp Clin Cancer Res. 2016;26:35. doi: 10.1186/s13046-016-0298-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sendoel A, Kohler I, Fellmann C, Lowe SW, Hengartner MO. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature. 2010;465(7298):577–583. doi: 10.1038/nature09141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma WW, Adjei AA. Novel agents on the horizon for cancer therapy. CA Cancer J Clin. 2009;59(2):111–137. doi: 10.3322/caac.20003. [DOI] [PubMed] [Google Scholar]
- 12.Wang M, Kirk JS, Venkataraman S, et al. Manganese superoxide dismutase suppresses hypoxic induction of hypoxia-inducible factor-1alpha and vascular endothelial growth factor. Oncogene. 2005;24(55):8154–8166. doi: 10.1038/sj.onc.1208986. [DOI] [PubMed] [Google Scholar]
- 13.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5(5):343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 14.Dupuy F, Tabaries S, Andrzejewski S, et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015;22(4):577–589. doi: 10.1016/j.cmet.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Graumlich JF, Ludden TM, Conry-Cantilena C, Cantilena LR, Jr, Wang Y, Levine M. Pharmacokinetic model of ascorbic acid in healthy male volunteers during depletion and repletion. Pharm Res. 1997;14(9):1133–1139. doi: 10.1023/a:1012186203165. [DOI] [PubMed] [Google Scholar]
- 16.Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93(8):3704–3709. doi: 10.1073/pnas.93.8.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welsh JL, Wagner BA, van’t Erve TJ, et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother Pharmacol. 2013;71(3):765–775. doi: 10.1007/s00280-013-2070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du J, Martin SM, Levine M, et al. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res. 2010;16(2):509–520. doi: 10.1158/1078-0432.CCR-09-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Q, Espey MG, Sun AY, et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci USA. 2007;104(21):8749–8754. doi: 10.1073/pnas.0702854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian W, Wang Y, Xu Y, et al. The hypoxia-inducible factor renders cancer cells more sensitive to vitamin C-induced toxicity. J Biol Chem. 2014;289(6):3339–3351. doi: 10.1074/jbc.M113.538157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy I, Zimmerman NP, Mackinnon AC, Tsai S, Evans DB, Dwinell MB. CXCL12 chemokine expression suppresses human pancreatic cancer growth and metastasis. PloS ONE. 2014;9(3):e90400. doi: 10.1371/journal.pone.0090400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc. 2009;4(11):1670–1680. doi: 10.1038/nprot.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buettner GR. In the absence of catalytic metals ascorbate does not autoxidize at pH 7: ascorbate as a test for catalytic metals. J Biochem Biophys Methods. 1988;16(1):27–40. doi: 10.1016/0165-022x(88)90100-5. [DOI] [PubMed] [Google Scholar]
- 24.Du J, Daniels DH, Asbury C, et al. Mitochondrial production of reactive oxygen species mediate dicumarol-induced cytotoxicity in cancer cells. J Biol Chem. 2006;281(49):37416–37426. doi: 10.1074/jbc.M605063200. [DOI] [PubMed] [Google Scholar]
- 25.Matsuura H, Ichiki T, Ikeda J, et al. Inhibition of prolyl hydroxylase domain-containing protein downregulates vascular angiotensin II type 1 receptor. Hypertension (Dallas Tex: 1979) 2011 Sep;58(3):386–393. doi: 10.1161/HYPERTENSIONAHA.110.167106. [DOI] [PubMed] [Google Scholar]
- 26.Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;05(1):43–54 107. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 27.Euhus DM, Hudd C, LaRegina MC, Johnson FE. Tumor measurement in the nude mouse. J Surg Oncol. 1986;31(4):229–234. doi: 10.1002/jso.2930310402. [DOI] [PubMed] [Google Scholar]
- 28.Doskey CM, van ‘t Erve TJ, Wagner BA, Buettner GR. Moles of a substance per cell is a highly informative dosing metric in cell culture. PloS ONE. 2015;10(7):e0132572. doi: 10.1371/journal.pone.0132572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doskey CM, Buranasudja V, Wagner BA, et al. Tumor cells have decreased ability to metabolize H2O2: implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016;10:274–284. doi: 10.1016/j.redox.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown LF, Detmar M, Claffey K, et al. Vascular permeability factor/vascular endothelial growth factor: a multifunctional angiogenic cytokine. EXS. 1997;79:233–269. doi: 10.1007/978-3-0348-9006-9_10. [DOI] [PubMed] [Google Scholar]
- 31.Nagy JA, Vasile E, Feng D, et al. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J Exp Med. 2002;196(11):1497–1506. doi: 10.1084/jem.20021244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dvorak HF, Detmar M, Claffey KP, Nagy JA, van de Water L, Senger DR. Vascular permeability factor/vascular endothelial growth factor: an important mediator of angiogenesis in malignancy and inflammation. Int Arch Allergy Immunol. 1995;107(1–3):233–235. doi: 10.1159/000236988. [DOI] [PubMed] [Google Scholar]
- 33.Choi KS, Bae MK, Jeong JW, Moon HE, Kim KW. Hypoxia-induced angiogenesis during carcinogenesis. J Biochem Mol Biol. 2003;36(1):120–127. doi: 10.5483/bmbrep.2003.36.1.120. [DOI] [PubMed] [Google Scholar]
- 34.Erudaitius D, Huang A, Kazmi S, Buettner GR, Rodgers VG. Peroxiporin expression is an important factor for cancer cell susceptibility to therapeutic H2O2: implications for pharmacological ascorbate therapy. PloS ONE. 2017;12(1):e0170442. doi: 10.1371/journal.pone.0170442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol Cell. 2007;25(2):207–217. doi: 10.1016/j.molcel.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaewpila S, Venkataraman S, Buettner GR, Oberley LW. Manganese superoxide dismutase modulates hypoxia-inducible factor-1 alpha induction via superoxide. Cancer Res. 2008;68(8):2781–2788. doi: 10.1158/0008-5472.CAN-07-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maes C, Carmeliet G, Schipani E. Hypoxia-driven pathways in bone development, regeneration and disease. Nat Rev Rheumatol. 2012;8(6):358–366. doi: 10.1038/nrrheum.2012.36. [DOI] [PubMed] [Google Scholar]
- 38.Du J, Cieslak JA, 3rd, Welsh JL, et al. Pharmacological ascorbate radiosensitizes pancreatic cancer. Cancer Res. 2015;75(16):3314–3326. doi: 10.1158/0008-5472.CAN-14-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serrano OK, Parrow NL, Violet PC, et al. Antitumor effect of pharmacologic ascorbate in the B16 murine melanoma model. Free Radic Biol Med. 2015;87:193–203. doi: 10.1016/j.freeradbiomed.2015.06.032. [DOI] [PubMed] [Google Scholar]
- 40.Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med. 2014;6(222):222ra218. doi: 10.1126/scitranslmed.3007154. [DOI] [PubMed] [Google Scholar]
- 41.Hoffer LJ, Robitaille L, Zakarian R, et al. High-dose intravenous vitamin C combined with cytotoxic chemotherapy in patients with advanced cancer: a phase I-II clinical trial. PloS ONE. 2015;10(4):e0120228. doi: 10.1371/journal.pone.0120228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rouleau L, Antony AN, Bisetto S, et al. Synergistic effects of ascorbate and sorafenib in hepatocellular carcinoma: New insights into ascorbate cytotoxicity. Free Radic Biol Med. 2016;95:308–322. doi: 10.1016/j.freeradbiomed.2016.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malik NK, May KS, Chandrasekhar R, et al. Treatment of locally advanced unresectable pancreatic cancer: a 10-year experience. J Gastrointest Oncol. 2012;3(4):326–334. doi: 10.3978/j.issn.2078-6891.2012.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campbell EJ, Vissers MC, Wohlrab C, et al. Pharmacokinetic and anti-cancer properties of high dose ascorbate in solid tumours of ascorbate-dependent mice. Free Radic Biol Med. 2016;99:451–462. doi: 10.1016/j.freeradbiomed.2016.08.027. [DOI] [PubMed] [Google Scholar]
- 46.Montagner M, Enzo E, Forcato M, et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature. 2012;487(7407):380–384. doi: 10.1038/nature11207. [DOI] [PubMed] [Google Scholar]
- 47.Briggs KJ, Koivunen P, Cao S, et al. Paracrine induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell. 2016;166(1):126–139. doi: 10.1016/j.cell.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerald D, Berra E, Frapart YM, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118(6):781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 49.Daijo H, Hoshino Y, Kai S, et al. Cigarette smoke reversibly activates hypoxia-inducible factor 1 in a reactive oxygen species-dependent manner. Sci Rep. 2016;6:34424. doi: 10.1038/srep34424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rawal M, Schroeder SR, Wagner BA, et al. Manganoporphyrins increase ascorbate-induced cytotoxicity by enhancing H2O2 generation. Cancer Res. 2013;73(16):5232–5241. doi: 10.1158/0008-5472.CAN-13-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee DC, Sohn HA, Park ZY, et al. A lactate-induced response to hypoxia. Cell. 2015;161(3):595–609. doi: 10.1016/j.cell.2015.03.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.