

High–energy density cathode materials for Li-ion batteries leverage oxygen and transition metal redox activity with reduced cost.
Abstract
Significant research effort has focused on improving the specific energy of lithium-ion batteries for emerging applications, such as electric vehicles. Recently, a rock salt–type Li4Mn2O5 cathode material with a large discharge capacity (~350 mA·hour g−1) was discovered. However, a full structural model of Li4Mn2O5 and its corresponding phase transformations, as well as the atomistic origins of the high capacity, warrants further investigation. We use first-principles density functional theory (DFT) calculations to investigate both the disordered rock salt–type Li4Mn2O5 structure and the ordered ground-state structure. The ionic ordering in the ground-state structure is determined via a DFT-based enumeration method. We use both the ordered and disordered structures to interrogate the delithiation process and find that it occurs via a three-step reaction pathway involving the complex interplay of cation and anion redox reactions: (i) an initial metal oxidation, Mn3+→Mn4+ (LixMn2O5, 4 > x > 2); (ii) followed by anion oxidation, O2−→O1− (2 > x > 1); and (iii) finally, further metal oxidation, Mn4+→Mn5+ (1 > x > 0). This final step is concomitant with the Mn migration from the original octahedral site to the adjacent tetrahedral site, introducing a kinetic barrier to reversible charge/discharge cycles. Armed with this knowledge of the charging process, we use high-throughput DFT calculations to study metal mixing in this compound, screening potential new materials for stability and kinetic reversibility. We predict that mixing with M = V and Cr in Li4(Mn,M)2O5 will produce new stable compounds with substantially improved electrochemical properties.
INTRODUCTION
Lithium-ion batteries (LIBs) have become one of the most widely used electrical energy storage technologies and have enabled the wireless revolution of consumer electronics (1). There has been a significant research effort to improve the specific energy of LIBs for emerging applications such as electric vehicles (2). Conventional cathode materials used in LIBs are typically Li-containing transition metal (TM) oxides or phosphates (for example, LiCoO2, LiFePO4, and LiMn2O4) (3–5) that can store (release) electrical energy via (de-)insertion of Li+ ions, accompanied by redox reactions of the TM cation. The specific capacity of the cathode is limited by the number of electrons per TM cation that can participate in the redox reaction. This exclusive dependence on the TM cations as the redox center in cathode materials typically used in LIBs has been challenged by the recent discovery of oxygen redox reactivity in Li-excess cathode materials. Koga et al. (6) and Sathiya et al. (7, 8) first reported that the enhanced capacities in Li1.20Mn0.54Co0.13Ni0.13O2 and Li2Ru1−ySnyO3 can be realized via reversible anionic redox: O2−→(O2)n−. In a number of further studies, the redox activity of the anionic species, oxygen, has been confirmed, for example, during the electrochemical cycling of the Li4FeSbO6 (9), Li3NbO4 (10), and β-Li2IrO3 (11) compounds. Understanding the origin of the oxygen redox, therefore, has become essential. McCalla et al. (12) and Grimaud et al. (13) proposed that the Li-driven reversible formation of peroxo-like (O2)n− species boosts the energy storage capacity of these Li-excess materials. Different explanations have been given by Luo et al. (14), Seo et al. (15), and Zhan et al. (16) who suggest that the formation of localized electron holes on O atoms with local Li-excess environments [contrary to (O2)n− dimers] is responsible for the excess capacity during Li removal. Exploration of these Li-rich compounds with combined cationic and anionic redox chemistry, high energy density, no O2 loss, and low cost is still ongoing and has drawn significant attentions from the electrochemical energy storage field.
Recently, Freire et al. (17) reported a new disordered rock salt–type Li-excess Li4Mn2O5 cathode material with partially occupied cation and anion sites that exhibits a discharge capacity of 355 mA·hour g−1 in the first cycle within an operating voltage window of 1.2 to 4.2 V versus Li/Li+. On subsequent cycling, the material is reported to preserve its rock salt structure with a discharge capacity of ~250 mA·hour g−1. A complex delithiation mechanism was proposed, consisting of three possible redox couples, Mn3+/Mn4+, O2−/O1−, and Mn4+/Mn5+, based on magnetic susceptibility measurements (17), as shown in the Eq. 1
| (1) |
However, the formation of an octahedrally coordinated Mn5+ (as in the rock salt structure) during electrochemical delithiation is rather rare. Mn5+ ions are usually tetrahedrally coordinated by oxygen atoms, such as in Li3MnO4 (18). Many factors can contribute to the difficulty in oxidizing Mn4+ to Mn5+, and in this study, we use the crystal field theory, which has been widely applied (19, 20), to analyze the preference of Mn5+ toward tetrahedral coordination. For a given metal/ligand, the energy level splitting (Δoct) of a metal d orbital by an octahedral field of ligands is larger than that caused by a tetrahedral field (Δtet), as schematically shown in Fig. 1 (19, 20). In addition, the ordering of the split energy levels, t2g and eg, in an octahedral field is opposite to that in a tetrahedral field. Consequently, the energy required to remove an electron from the low-lying t2g levels of an octahedrally coordinated metal ion (ΔEoct; see Fig. 1) is significantly larger than that needed to remove an electron from the high-lying t2g levels of a tetrahedrally coordinated metal ion (ΔEtet). Furthermore, the energy needed to remove an electron from the high-lying eg orbital (ΔEoct′) is much smaller compared to that needed to remove an electron from the t2g orbital (ΔEoct) in an octahedral field. As a result, the oxidation of octahedrally coordinated Mn4+ to Mn5+ would require an impractically high voltage compared to the oxidation of octahedrally coordinated Mn3+ to Mn4+. Hence, it is therefore worth examining the TM/O redox mechanism in Li4Mn2O5. Furthermore, the structural evolution and ionic coordination of redox active species in Li4Mn2O5 during the (de-)lithiation are not completely understood and also warrant further investigation. In addition, because of the difficulty of oxidizing Mn4+ to Mn5+, improving the structural stability and electrochemical properties of Li4Mn2O5 by doping or substitution of Mn with a different TM that can easily access an oxidation state greater than 4+ is an appealing prospect.
Fig. 1. Energy levels of the Mn d orbitals in octahedral and tetrahedral coordinations.
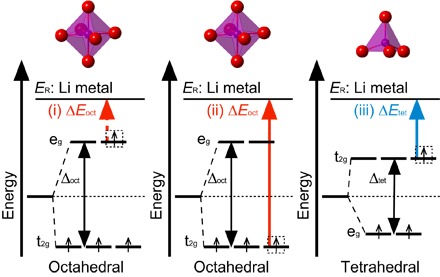
The energies needed to oxidize octahedrally coordinated (i) Mn3+(occupation, d4) to Mn4+(d3) and (ii) Mn4+(d3) to Mn5+(d2) to the reference state ER (Li metal) are indicated by the dashed and solid red arrows, respectively. The energy needed to oxidize tetrahedrally coordinated (iii) Mn4+(d3) to Mn5+(d2) is indicated by the solid blue arrow. The energy ΔEoct required to remove an electron from the t2g levels of octahedrally coordinated Mn4+ is significantly larger than that (ΔEtet) needed to remove an electron from t2g levels of tetrahedrally coordinated Mn4+. The oxidation of octahedrally coordinated Mn4+ to Mn5+ is rarely observed.
First-principles density functional theory (DFT) calculations have been widely used as effective tools to study battery materials to explore new, high-performance electrode materials and understand in detail the underlying mechanisms during electrochemical reactions (21–24). Freire et al. (17) reported that the charge and discharge processes of Li4Mn2O5 involve a series of intermediate phases and redox activities for both Mn and O. Here, we first give a detailed atomistic-level picture for the origin of the observed simultaneous anionic and cationic redox activity in this promising new high-capacity material. Moreover, we provide a mechanistic explanation behind the performance degradation/large polarization during the electrochemical cycling of Li4Mn2O5, address the thermodynamic phase stability of this compound, and give guidance to future experimental efforts in the synthesis of this Li4Mn2O5 electrode. Furthermore, we offer predictions for doping/alloying of this compound for improved electrochemical properties and cyclability by performing a comprehensive high-throughput DFT screening for Li4Mn2O5-derived cathode materials. Our predictions/discoveries shed light on simultaneous cation/anion redox in this high–energy density electrode and provide predictions of new compounds awaiting experimental validation.
RESULTS AND DISCUSSION
Determining the rock salt–type structure of Li4Mn2O5
Mechanical milling, as applied in the Li4Mn2O5 experimental study (17), has been proven to be a viable solid-state processing route for the synthesis of various equilibrium and nonequilibrium phases (25). Structural disordering and thermodynamic metastability can be introduced to the synthesized compounds through ball milling, such as in the intermetallic GdAl2 compound (26) or in cation-disordered oxides Li3NbO4 (10) and Li1.25Nb0.25Mn0.5O2 (27). According to the X-ray diffraction analysis of Freire et al. (17), Li4Mn2O5 forms adopt a disordered rock salt–type structure with Li/Mn mixed on the cation sites and O/vacancies (Vacs) mixed on the anion sites (see Fig. 2A). Here, we use the special quasi-random structure (SQS) method (28–30) using a rock salt–based 108-site supercell with Li/Mn occupying the 54 cation sites in a 2:1 ratio and O/Vac occupying the 54 anion sites in a 5:1 ratio. The SQS was generated (see Fig. 2B) using a Monte Carlo algorithm as implemented in the Alloy Theoretic Automated Toolkit (ATAT) package (28–30) with the pair and triplet correlation functions of the SQS constrained to be identical to those of the statistically random compound (Li/Mn occupying the cation sites and O/Vac occupying the anion sites) at least up to the third nearest neighbor.
Fig. 2. Determining the rock salt–type structure of Li4Mn2O5.
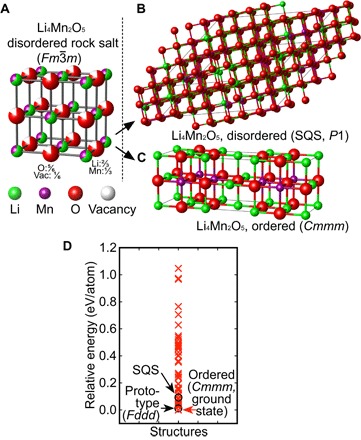
(A) A schematic illustration of the disordered rock salt Li4Mn2O5 structure with Li/Mn randomly mixed on the cationic sites and O/Vac randomly mixed on the anionic sites. (B) Simulated disordered structure using the SQS method. (C) Predicted ground-state structure of Li4Mn2O5, with all Mn octahedrally coordinated by O atoms and Li ions square-planarly or square-pyramidally coordinated because of O/Vac neighboring (space group Cmmm). (D) Total energy distribution of the 100 structures selected, the SQS structure, and the Na4Mn2O5 prototype structure from DFT calculations. The Cmmm structure as predicted in this study exhibits the lowest total energy.
In addition, we also study ionic ordering in the Li4Mn2O5 compound. We determine the lowest-energy, ground-state structure of Li4Mn2O5 by exploring a vast number of geometrically distinct Li/Mn/O ordered configurations using DFT calculations. Starting from the cubic rock salt primitive cell, we generate two sets of supercells: (i) containing 6 cations and 6 anions with all symmetrically distinct supercell shapes; and (ii) containing 12 cations and 12 anions with two specific shapes, given by 3 × 2 × 2 and 2 × 3 × 2 multiples of the primitive rock salt unit cell. We then populate the cation sites with Li and Mn atoms in the ratio of 2:1 and introduce Vacs on the anion sites with a O/Vac ratio of 1:5. A total of 616 geometrically different configurations were generated using the Enum code (31–33). We calculated the electrostatic total energy for all configurations using nominal charge states for the ions in the system as a quick energy sampling step (34). All structures were ranked by their normalized electrostatic energies, 100 Li4Mn2O5 structures with the lowest electrostatic energies were fully relaxed, and their energies were calculated using DFT. We find that the structure with the lowest DFT total energy, that is, the ground-state structure of Li4Mn2O5, has a space group Cmmm, with all Mn3+ ions octahedrally coordinated by six oxygen atoms (see Fig. 2C). Meanwhile, Li ions are square-planarly or square-pyramidally coordinated by four or five oxygen atoms as a result of O/Vac neighboring. The cation ordering between Li and Mn in our proposed Li4Mn2O5 ground-state structure is the ordering of the Ga2Zr compound (see fig. S1) (35) with the crystallographic information given in table S1. The fully relaxed DFT energy of the SQS is found to be higher than the ordered ground state by 119 meV per atom (34 ordered structures having lower total energies; see Fig. 2D). We note that the isoelectronic compound Na4Mn2O5 is known and adopts an ordered rock salt–type structure (Fddd), as shown in fig. S2 (36). Using this structure type for the Li compound, we find that Li4Mn2O5 in the Fddd structure type exhibits a total energy 4 meV per atom higher than the Cmmm structure we found here. The thermodynamic and dynamical stability of the ordered Cmmm Li4Mn2O5 structure is discussed in detail in the following section.
Li-Mn-O phase diagram and thermodynamic stability of ordered (Cmmm) Li4Mn2O5
Phase diagrams represent the thermodynamic phase equilibria of multicomponent systems and provide useful information on reactions of phases. Although the experimental determination of a phase diagram for specific system is significantly time- and labor-consuming, we can accelerate the phase diagram constructions by calculating energies of all known compounds in a specific chemical system using DFT and using them to construct a T = 0 K convex hull (37, 38). Here, we constructed ternary Li-M-O ground-state convex hulls using the structures with the lowest energy for each composition for M = Mn and all metal elements with possible oxidation states of 5+ or above: that is, M = Bi, Cr, Ir, Mo, Nb, Os, Pd, Pr, Pt, Re, Rh, Ru, Sb, Ta, V, and W (39). All compounds within each Li-M-O ternary system were adopted from the Inorganic Crystal Structure Database (ICSD) (40). The elemental reference states (Li, M, and nonsolid O2) were obtained by fitting (41) to experimental formation energies, mainly from two major databases, the SGTE (Scientific Group Thermodata Europe) substance database (SSUB) and a database constructed by Nash et al. (41–46). Calculations to construct equilibrium Li-M-O phase diagrams were carried out within the Open Quantum Materials Database (OQMD) framework (43, 47). We construct the convex hull of stable phases, that is, the set of compounds that have an energy lower than that of any other compound or linear combination of compounds at that composition, for each ternary Li-M-O system. Using these convex hulls or T = 0 K, phase diagrams, we can then evaluate the ground-state stability of TM oxides, for example, Li4M2O5 and Li4(Mn,M)2O5, by using the Grand Canonical Linear Programming (GCLP) technique (43, 48).
The Li-Mn-O phase diagram (T = 0 K) is shown in Fig. 3A with the ground-state, stable compounds marked by filled circles (that is, having lower energy than the linear combination of other structures). Li4Mn2O5 is shown as an empty circle, which is compositionally located on the tie line between Li2O and LiMnO2. Li4Mn2O5 at T = 0 K is predicted to have an energy only slightly higher (+13.6 meV per atom) than a two-phase mixture of Li2O and LiMnO2. The Li4Mn2O5 compound is therefore not a ground-state structure but rather is unstable at T = 0 K, albeit by a very small energy. The compound is very close to the convex hull and hence may be stabilized at elevated temperatures by entropic contributions, for example, vibrational entropy. Our DFT-calculated phonon dispersion of the Cmmm Li4Mn2O5 is provided in Fig. 3B. No imaginary phonons are shown in Fig. 3B, which indicates that our predicted Li4Mn2O5 compound is dynamically stable.
Fig. 3. Thermodynamic and dynamic stabilities of Li4Mn2O5.
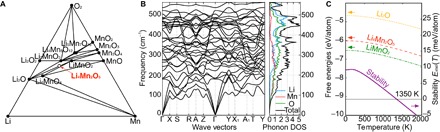
(A) Calculated Li-Mn-O (T = 0 K) phase diagram. The Li4Mn2O5 phase is slightly higher in energy (13.6 meV per atom) relative to that of the ground-state phases—a mixture of Li2O and LiMnO2. (B) Phonon dispersion of the ground-state Li4Mn2O5 and (C) calculated temperature-dependent free energy of Li4Mn2O5 (red dashed line), Li2O (orange dotted line), and LiMnO2 (green dotted line), as well as the stability (purple solid line) of Li4Mn2O5 versus temperature relative to Li2O and LiMnO2 phase mixtures. We find that Li4Mn2O5 is dynamically stable and can be entropically stabilized at ~1350 K.
By computing the harmonic phonons and vibrational entropies of Li4Mn2O5 (Fig. 3B), LiMnO2 (fig. S3), and Li2O (fig. S4), we can calculate the temperature dependence of the free energies between these three competing phases. Using the free energies for each of the three compounds, we can calculate the stability (formation free energy) for Li4Mn2O5 as Stability(Li4Mn2O5) = F (Li4Mn2O5) − F (Li2O) − 2F (LiMnO2). The stability of Li4Mn2O5 as the function of T is shown in Fig. 3C, with a temperature at which Li4Mn2O5 is stabilized of approximately 1350 K. The positive formation entropy is mainly due to the small entropy of Li2O, stemming from the light Li atom and strong bonding interaction between Li and O, as shown in fig. S4. Due to the relatively small entropy differences between phases, the uncertainty of this temperature (for example, due to an uncertainty of ±1 meV per atom in free energy) results in a range of transition temperatures of 1240 to 1450 K. Our calculations then suggest that the Li4Mn2O5 compound is stable at high temperatures, implying the favored formation of this compound above the transition temperature. However, the elevated temperature would also favor ionic disorder because of the greater configurational entropy contribution. Therefore, a competition occurs between (i) the stability of Li4Mn2O5 (either ordered or disordered) with respect to decomposition and (ii) the order-disorder transformation of cations in Li4Mn2O5. As a result, the synthesizabilities of the ordered and disordered phases depend on the thermodynamics associated with this competition. Depending on these competing free energies, a temperature window can exist where the ordered phase is stable with respect to both decomposition and the disordered phase and hence should be synthesizable (fig. S5A). If the entropy of the disordered phase is significantly larger than the ordered phase, the order-disorder transformation would occur at a lower temperature than the decomposition reaction; hence, only the disordered phase would be thermodynamically stable, and it might be possible to only synthesize the disordered phase (fig. S5B). As a result of these thermodynamic arguments, we suggest that the synthesis of the ordered Li4Mn2O5 compound might be difficult (although it has been accomplished for a disordered phase of Li4Mn2O5 and an ordered compound of Na4Mn2O5). The possible stable decomposition phases are LiMnO2, Li2O, Li2MnO3, and Li6MnO4, as predicted by the Li-Mn-O phase diagram, which might be observed as “impurity” phases during synthesis.
Electrochemical delithiation process of Li4Mn2O5 and TM/O redox competition
Having explored the structural ordering and thermodynamic stability of the Li4Mn2O5 phase, we next explore the electrochemical delithiation process of this compound. To examine delithiation, we calculated the energies of disordered SQS-Li4Mn2O5 and the fully delithiated SQS-Li0Mn2O5. Meanwhile, we consider compositions of LixMn2O5 (where x = 4, 3, 2, 1, and 0) in which (4 − x) Li+ ion(s) are removed from the original ordered Cmmm Li4Mn2O5 structure using many geometrically distinct configurations, and they are further relaxed using DFT. We evaluate the energies for these structures according to the following reaction: LixMn2O5→Mn2O5 + xLi+. The energies of these ordered/disordered delithiation products are plotted, and the delithiation convex hull of Li4Mn2O5-Mn2O5 is then constructed, as shown in Fig. 4A. In Fig. 4A, the delithiation convex hull of Li4Mn2O5-Mn2O5 is shown, where the ordered LixMn2O5 (where x = 3, 2, and 1) and disordered SQS-LixMn2O5 (where x = 0) structures are found to be on the hull. By converting the energies along this delithiation pathway into voltages, we find that voltage range obtained from our calculation (2.6 to 4.0 V) is in reasonable agreement with the voltage range measured in the first experimental delithiation (2.5 to 4.3 V) in Fig. 4B.
Fig. 4. Electrochemical delithiation process of Li4Mn2O5.
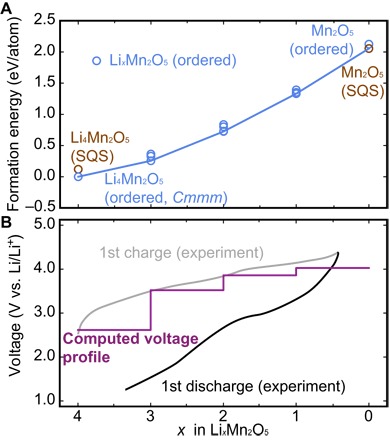
(A) Li4Mn2O5-Mn2O5 convex hull with calculated delithiated structures generated from ordered and disordered (SQS) Li4Mn2O5 phase. (B) Corresponding voltage profile during the delithiation process in Li4Mn2O5, the voltage range obtained from our calculation reasonably matches the interval as measured in the first experimental delithiation (17).
We next use our calculations of the Li4Mn2O5 phase and its delithiation products to interrogate in detail the TM/O redox sequence. We examine the oxidation states of Mn and O ions during the delithiation process and investigate the local atomistic environments for cations and anions. The oxidation states can be determined by comparing calculated magnetizations of Mn and O ions with the number of unpaired electrons of the corresponding ions with known oxidation states. The numbers of unpaired electrons for Mn3+ (octahedrally coordinated), Mn4+ (octahedrally coordinated), and Mn5+ (tetrahedrally coordinated) are 4, 3, and 2, respectively, as shown in fig. S6A; and the numbers of unpaired electrons for O2− and O1− (octahedrally coordinated) are 0 and 1, respectively (fig. S6B). We find that the electrochemical delithiation of Li4Mn2O5 can be categorized in three different reaction steps, where each step contains a dominant redox of either TM or O ions:
Cationic redox Mn3+/Mn4+ delithiation (LixMn2O5, 4 > x > 2)
During the delithiation process of Li4Mn2O5→Li3Mn2O5→Li2Mn2O5, we find that the Mn magnetizations decrease from 3.94 bohr magneton (μB)→3.56 μB→3.14 μB (see fig. S6A), indicating an overall oxidation of Mn3+ to Mn4+. Meanwhile, the O magnetizations retain a value between 0.01 and 0.14 μB (see fig. S6B), implying a constant anion oxidation state of O2−. The initial energetic preference of TM redox over O redox is confirmed by examining the projected density of states (p-DOS) of O 2p and Mn 3d orbitals (eg and t2g) of O2− and Mn3+ ions in both ordered and disordered Li4Mn2O5. As shown in Fig. 5C, the contribution from Mn eg to the valence band immediately below the Fermi level (EF) is larger than that from O, which shows a preference for electron extraction from Mn (Fig. 5B) during the initial stages of the charging process, as discussed in the Introduction. As a result, the first delithiation step is dominated by the cationic redox of Mn3+/Mn4+. It is interesting to connect the competition between cation and anion redox to the local ionic environments in the LixMn2O5 structures. Recently, Seo et al. (15) proposed that a specific local Li-excess environment around the oxygen atoms (that is, a Li-O-Li linear configuration) is a key structural signature indicating the feasibility of both cationic (TM) and anodic (oxygen) redox process in Li-rich cathode materials. In other words, the electrons from the oxygen atom in this local Li-O-Li configuration can more easily contribute to the redox process because of the overlapping TM states and O 2p states. Our examination of the local environments of oxygen shows that many O ions are in this Li-O-Li configuration in the ordered (Cmmm) and disordered structures, as shown in Fig. 5A. However, we find that Mn3+-to-Mn4+ cation oxidation is still the main redox contribution during the initial charging process. After two Li+ ions are removed (that is, Li2Mn2O5), a large fraction of O ions (four-fifth) still remain in these linear Li-excess environments (see Fig. 5A). The p-DOS of O 2p and Mn 3d orbitals (eg and t2g) for O2− ions in the Li-excess environment and nearest neighbor Mn4+ ions is shown in Fig. 5D. The contribution from oxygen in the valence band immediately below EF is significantly larger than that coming from Mn t2g, implying the possibility of oxygen redox participation in the second delithiation step, as described below. Taking extra electrons out from the t2g orbital of Mn is significantly more difficult compared with the eg orbital (Fig. 5B), as discussed in Introduction.
Fig. 5. Cationic and anionic redox sequence during the delithiation of Li4Mn2O5.
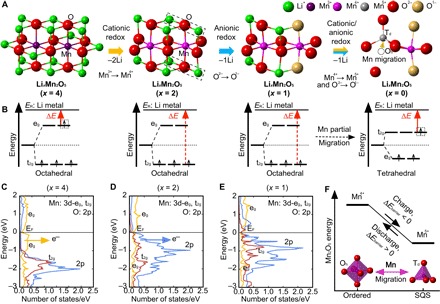
(A) Local atomistic environments for Mn and O ions in LixMn2O5 (where x = 4, 3, 2, 1, and 0). (B) Energies needed to oxidize octahedrally coordinated Mn3+(d4) and Mn4+(d3) and tetrahedrally coordinated Mn4+(d3) to the reference state ER (Li metal) (indicated by the red arrows). p-DOS of the O 2p and Mn 3d orbitals (eg and t2g) of O2− ions in the “Li-O-Li” configurations and the nearest Mn ions in (C) Li4Mn2O5, (D) Li2Mn2O5, and (E) LiMn2O5. (F) Energy difference between ordered and disordered Mn2O5 with the partial Mn migration from octahedral to tetrahedral sites. The redox reaction along with the Li4Mn2O5 delithiation proceeds in three steps: (i) cationic Mn3+/Mn4+ (4 > x > 2), (ii) anionic O2−/O1− (2 > x > 1), and (iii) mixed cationic Mn4+/Mn5+ and anionic O2−/O1− (1 > x > 0). The oxidation of Mn4+ to Mn5+ necessitates the migration of the Mn ion from its octahedral site to a nearby unoccupied tetrahedral site and impairs the reaction reversibility.
Anionic redox O2−/O1− dominant delithiation (LixMn2O5, 2 > x > 1)
Upon further delithiation of Li2Mn2O5 into LiMn2O5, we find that the observed Mn magnetizations are largely constant in the range of 3.14 to 3.30 μB, indicative of Mn4+, consistent with the experimental findings (17). Here, the Mn ions are still octahedrally coordinated. We find that one-fifth of the O ions exhibit magnetic moments around 0.69 μB, implying the partial oxidation of O2− toward O1−. By examining the local atomistic environments of all O1− ions in Li1Mn2O5 and by comparing to their previous local environments in Li2Mn2O5, we notice that all O1− ions participating in redox during this step are located in the Li-O-Li Li-excess environments (Fig. 5A), validating Seo et al.’s hypothesis (15). Our calculations thus indicate that the delithiation step from Li2Mn2O5 to LiMn2O5 is dominated by anionic redox processes (that is, with O2− being partially oxidized to O1−). For the LiMn2O5 phase, as shown in Fig. 5E, the contribution from Mn orbitals (mainly t2g) to the valence band immediately below the EF is still slightly lower than that from O, implying a preference for electron extraction from O (Fig. 5E) during the final stages of the charging process (LixMn2O5, 1 > x > 0). However, in the experimental studies, further oxidation of Mn4+ to Mn5+ was observed during this stage (17). Therefore, we suggest that some additional reaction mechanisms must account for the Mn oxidation during the final stage.
Mixed cationic Mn4+/Mn5+ and anionic O2−/O1− redox delithiation (LixMn2O5, 1 > x > 0)
During the final delithiation step, that is, LiMn2O5 to Mn2O5 (here, we examined the disordered SQS-Mn2O5 with the lowest DFT energy), we find that the Mn magnetizations are distributed from 3.3 to 1.9 μB (see fig. S6A), indicating that Mn4+ ions have been partially (one-sixth) oxidized to Mn5+. At the same time, the magnetizations of one-third of the O ions are found to be 0.71 to 0.82 μB (fig. S6B), implying a further oxidation of O2− to O1− (remember that we find that one-fifth of anions are O1− in LiMn2O5). The coexistence of Mn5+ and O1− is consistent with the experimental observations in the study of Freire et al. (17). As depicted in Fig. 5A, we find that all the Mn5+ ions are tetrahedrally coordinated in this Mn/Li Vac disordered configuration. The observation of tetrahedrally coordinated Mn5+ confirms our discussion above that the oxidation of tetrahedrally coordinated Mn4+ to Mn5+ is energetically favored compared to the octahedrally coordinated Mn4+ because of the crystal field t2g/eg effects (Fig. 5B). Therefore, we suggest that the Mn ion migration to tetrahedral positions is necessary in the final delithiation process toward Mn2O5, which corresponds to the oxidation of Mn from 4+ to 5+. At the same time, all the Mn ions in our ordered Mn2O5 (blue circle in Fig. 4A; 0.85 eV per Mn higher in energy than the disordered configuration), however, are still located in the octahedral sites, where the oxidation states are preserved at 4+, as shown in fig. S6A. As a result, the Mn migration not only enables the Mn4+ oxidation to Mn5+ but also lowers the energy of the system at this stoichiometry by 0.85 eV per Mn (see Fig. 5F). To achieve a reversible redox reaction, these Mn5+ ions would need to migrate back to their original octahedral sites during the following lithiation (that is, the discharge process). The large reverse migration barrier (that is, at least 0.85 eV per Mn, the difference in DFT energetic stability between these two structures) will result in a significant kinetic barrier for the tetrahedral Mn5+ to migrate back to its original octahedral position; therefore, we suggest that this metal migration will impair the reversibility of the reaction. After the extended cycling of Li4Mn2O5 cathodes, it is likely that more Mn ions will migrate into the tetrahedral sites and get trapped. We suggest that the phase transformation caused by the Mn ion migrations could be one significant factor underlying the polarization between charge and discharge, as well as the capacity fade observed experimentally after the first cycle (17). We also suggest that improved performance and reversibility should be achieved by limiting charging to avoid the formation of Mn5+ and hence the migration of these metal cations.
The above results imply that a design strategy to improve the extended cyclability of the rock salt–type Li4Mn2O5 cathodes would be to avoid Mn migration to the tetrahedral sites during the Mn4+/Mn5+ redox process. The electrochemical cycling of Li4Mn2O5 can be confined to a smaller range: LixMn2O5, 4 > x > 1, without removing all Li from the system and oxidizing Mn to 5+. Thus, improved cyclability could be achieved by sacrificing a limited amount of capacity. An alternate strategy to achieve this goal of improved reversibility would be to partially substitute Mn in Li4Mn2O5 with other TM elements that can access the oxidation state of 5+ or above, thereby eliminating the need for oxidation of Mn to 5+. In the following section, we present a high-throughput DFT screening strategy to determine stable metal dopants (M) in Li4(Mn,M)2O5 compounds.
TM doping in Li4Mn2−xMxO5 with accessible 5+ oxidation state or above
We first start with all the metal elements (M) with possible oxidation states of 5+ or above: that is, M = Bi, Cr, Ir, Mo, Nb, Os, Pd, Pr, Pt, Re, Rh, Ru, Sb, Ta, V, and W (39). For each of these elements, we compute the properties of mixed-metal Li4(Mn,M)2O5 compounds, specifically focusing on stability and mixing energy. The mixing energies between Li4Mn2O5 and Li4M2O5 in Li4(Mn,M)2O5 help to determine the stability of metal mixing in this structure. When the mixing energy (Emix) is found to be slightly negative or positive (near-zero, that is, −30 to 30 meV per site), the mixing entropy at finite temperatures will overcome the mixing energy, and hence, there will be a tendency for metal mixing in a solid solution. A larger positive mixing energy (>30 meV per site) or a larger (in magnitude) negative mixing energy (<−30 meV per site) would lead to phase separation in the former case and a quaternary ordered compound in the latter. These cases may have undesired phase transformations or possible mass transport kinetic limitations. As a result, we narrow down the list of our candidates to those with near-zero mixing energies between −30 and 30 meV per site in our study (Fig. 6). Similar to Li4Mn2O5, all Li4M2O5 compounds are unstable at T = 0 K, with the potential to be entropically stabilized at finite temperatures. For cases where the Li4M2O5 convex hull distance (that is, stability) is significantly positive, it will lead to an instability of the corresponding Li4(Mn,M)2O5 compound. Here, we also exclude the Li4(Mn,M)2O5 compounds with related Li4M2O5 convex hull distance larger than 50 meV per atom. In Fig. 6, we suggest the top Li4(Mn,M)2O5 candidates with stability near the convex hull and a small mixing energy in the Mn sublattice (favoring solid solution formation). We predict that mixing with M = V and Cr as particularly promising additives. Predicted gravimetric capacities (theoretical) and average voltages of recommended candidates are listed in table S2. We expect that the doping/substitution of these elements into Li4(Mn,M)2O5 cathodes will lead to reduced phase transformation and controlled anionic oxygen chemistry for further improved electrochemical performance, therefore calling for imminent experimental validations.
Fig. 6. HT-DFT screening for doping into the Mn sublattice in the Li4(Mn,M)2O5 cathode system.
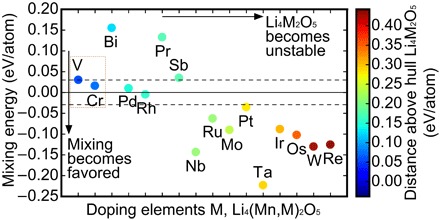
We performed computational screening of mixing on the Mn sites with metal cations (M) that produce energetically stable Li4(Mn,M)2O5 mixtures and have stable 5+ oxidation states by examining the mixing energy and Li4M2O5 stability. The top candidates with −30 < Emix < 30 meV per site and the lowest formation energies are shown. Our top two TM dopant candidates in the Li4(Mn,M)2O5 system are located in the left center of the plot: M = V and Cr.
CONCLUSION
Here, we exploit the structural and electrochemical properties of Li-rich rock salt Li4Mn2O5 cathode materials using the first-principles DFT calculations. We simulate the disordered rock salt–type Li4Mn2O5 structure using the SQS method. Then, we identify the ground-state structure of Li4Mn2O5 using an enumeration method by exploring geometrically distinct Li/Mn and O/Vac configurations on cation and anion sites of the rock salt structure, respectively. The ordered structure is predicted to have a much lower energy than the disordered structure. We reveal the underlying mechanism in the electrochemical delithiation process of Li4Mn2O5 that proceeds through a three-step reaction with a different dominant cationic/anionic redox process in each step: (i) LixMn2O5 (4 > x > 2; Mn3+/Mn4+), (ii) LixMn2O5 (2 > x > 1; O2−/O1−), and (iii) LixMn2O5 (1 > x > 0; Mn4+/Mn5+). We find that the oxidation of Mn4+ to Mn5+ can be achieved by the Mn ion migrations from the octahedral to the adjacent tetrahedral sites. A large barrier for Mn migration back to the octahedral site in the following lithiation process impairs the reversibility of the redox processes, which directly explains the capacity fades observed in Li4Mn2O5 after the first cycle in experiment. Last, to improve the extended cyclability of Li4Mn2O5 cathode system, we search for a suitable dopant that can access the oxidation state of 5+ or above by evaluating the mixing energies and stabilities. We recommend the dopant candidates, including M = V and Cr in the Li4(Mn,M)2O5 system, for further experimental investigations. Our theoretical findings provide valuable insights into the structural and electrochemical behavior of Li-rich Li4Mn2O5 cathode materials and could help in designing the next-generation high–energy density LIB cathode materials for future research and development.
MATERIALS AND METHODS
DFT calculations
All DFT calculations reported in this study were performed using the Vienna Ab initio Simulation Package (49–52) with the projector augmented-wave potentials (53) and the Perdew-Burke-Ernzerhof (54) exchange correlation. A plane-wave basis with a cutoff energy of 520 eV and Γ-centered k-meshes with a density of 8000 k-points per reciprocal atom were used for all calculations. All calculations were spin-polarized, with Mn atoms initialized in a high-spin configuration and relaxed to self-consistency. Both ferromagnetic (FM) and antiferromagnetic (AFM) configurations of the Mn ions were used to explore the ground state of Li4Mn2O5, with the FM arrangement exhibiting a slightly lower energy (0.2 meV per atom). Therefore, the FM configuration was applied in all subsequent calculations. The DFT + U method introduced by Dudarev et al. (55) was used to treat the localized 3d electrons of Mn with a U of 3.8, obtained by fitting it to experimental and calculated formation enthalpies in a previous study (42). Phonon calculations were carried out with the frozen phonon approach as implemented in the Phonopy package (56), and phonon density of states was computed using a dense 30 × 30 × 30 mesh in the irreducible Brillouin zone. Furthermore, Heyd-Scuseria-Ernzerhof screened hybrid functional (HSE06) (57) was used to accurately determine the energies and magnetic and electronic states of Mn and O in the delithiated phases with structures relaxed using DFT + U: Li4−xMn2O5 (where x = 0, 1, 2, 3, and 4).
Voltage profile calculations
The average lithiation/delithiation voltage (relative to Li/Li+) can be computed using the negative of the reaction free energy per Li added/removed, as shown in Eq. 2 (58)
| (2) |
where F is the Faraday constant, ΔNLi is the amount of Li added/removed, and ΔGf is the (molar) change in free energy of the reaction. Considering a two-phase reaction between LixMO and LiyMO, LixMO + (y − x)Li➝LiyMO, ΔGf can be approximated by the total internal energies from DFT calculations neglecting the entropic contributions (T = 0 K)
| (3) |
where E(LixMO) and E(LiyMO) are the DFT energies at the respective compositions. The neglect of entropic contributions means that the lithiation voltage profiles will follow the T = 0 K ground-state convex hull and consist of a series of constant voltage steps along the two-phase regions of the convex hull, separated by discontinuities that indicate the single-phase compounds on the hull. It is worth mentioning here that, in practice, lithiation/delithiation do not necessarily proceed through two-phase reactions. Thus, the calculated T = 0 K voltage profiles should be viewed as an approximation to the actual voltage profiles (59). At finite temperatures (for example, room temperature), the “steps” in the voltage profile became more rounded because of entropic effects (60).
Mixing energy
The tendency of two ordered rock salt Li4M2O5 and Li4M2′O5 (space group Cmmm) materials to mix and form a mixed-metal rock salt Li4MM′O5 structure can be evaluated by calculating the mixing energy as shown in Eq. 4
| (4) |
where E(Li4(M, M′)2O5), E(Li4M2O5), and E(Li4M′2O5) are the total energies of the Cmmm structure with two geometrically identical TM sites occupied by metal atoms M and M′, M alone, and M′ alone, respectively.
Supplementary Material
Acknowledgments
Funding: Z.Y. and C.W. were supported as part of the Center for Electrochemical Energy Science, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Science under award number DE-AC02-06CH11357. S.K. was supported by the financial assistance award 70NANB14H012 from the U.S. Department of Commerce, National Institute of Standards and Technology as part of the Center for Hierarchical Materials Design (CHiMaD). J.H. was supported by the Office of Naval Research STTR N00014-13-P-1056. V.I.H. was supported by the NSF through grant DMR-1309957. We acknowledge the computing resources from (i) the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the DOE under contract DE-AC02-05CH11231; and (ii) Blues, a high-performance computing cluster operated by the Laboratory Computing Resource Center at Argonne National Laboratory. Author contributions: Z.Y. conceived the overall project and DFT calculations of the ground-state structure prediction, structural pathway, voltage, and magnetizations. C.W. provided the overall direction and advice for project. S.K. performed the initial voltage calculations and helped conceive the overall project. J.H. performed the calculations of phonon dispersions. V.I.H. performed high-throughput calculation of doping options. Competing interests: Z.Y., S.K., J.H., V.I.H., and C.W. are inventors on a provisional patent held by Northwestern University related to this work (no. 62/609,620; 22 December 2017). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaao6754/DC1
fig. S1. Cationic ordering between Li and Mn in Li4Mn2O5 and between Ga and Zr in Ga2Zr.
fig. S2. Na4Mn2O5 prototype structure with a space group Fddd.
fig. S3. Phonon dispersion of the ground-state LiMnO2.
fig. S4. Phonon dispersion of the ground-state Li2O.
fig. S5. Schematic illustration of the relative stabilities between the disordered and ordered Li4Mn2O5, as well as the decomposed combination, 2LiMnO2 + Li2O.
fig. S6. The magnetization and oxidation state evolution of Mn and O ions during delithiation.
table S1. Structure information of the Li4Mn2O5 ground state.
table S2. Top Li4(Mn,M)2O5 cathode candidates from the HT-DFT screening with predicted gravimetric capacities (theoretical) and averaged voltages using Li4Mn2O5 as the benchmark.
REFERENCES AND NOTES
- 1.Goodenough J. B., Park K.-S., The Li-ion rechargeable battery: A perspective. J. Am. Chem. Soc. 135, 1167–1176 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Thackeray M. M., Wolverton C., Isaacs E. D., Electrical energy storage for transportation—Approaching the limits of, and going beyond, lithium-ion batteries. Energy Environ. Sci. 5, 7854–7863 (2012). [Google Scholar]
- 3.Gummow R. J., de Kock A., Thackeray M. M., Improved capacity retention in rechargeable 4 V lithium/lithium-manganese oxide (spinel) cells. Solid State Ion. 69, 59–67 (1994). [Google Scholar]
- 4.Padhi A. K., Nanjundaswamy K. S., Goodenough J. B., Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144, 1188–1194 (1997). [Google Scholar]
- 5.Mizushima K., Jones P. C., Wiseman P. J., Goodenough J. B., LixCoO2 (0<x<-1): A new cathode material for batteries of high energy density. Mater. Res. Bull. 15, 783–789 (1980). [Google Scholar]
- 6.Koga H., Croguennec L., Ménétrier M., Douhil K., Belin S., Bourgeois L., Suard E., Weill F., Delmas C., Reversible oxygen participation to the redox processes revealed for Li1.20Mn0.54Co0.13Ni0.13O2. J. Electrochem. Soc. 160, A786–A792 (2013). [Google Scholar]
- 7.Sathiya M., Rousse G., Ramesha K., Laisa C. P., Vezin H., Sougrati M. T., Doublet M.-L., Foix D., Gonbeau D., Walker W., Prakash A. S., Ben Hassine M., Dupont L., Tarascon J.-M., Reversible anionic redox chemistry in high-capacity layered-oxide electrodes. Nat. Mater. 12, 827–835 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Sathiya M., Leriche J.-B., Salager E., Gourier D., Tarascon J.-M., Vezin H., Electron paramagnetic resonance imaging for real-time monitoring of Li-ion batteries. Nat. Commun. 6, 6276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCalla E., Sougrati M. T., Rousse G., Berg E. J., Abakumov A., Recham N., Ramesha K., Sathiya M., Dominko R., Van Tendeloo G., Novák P., Tarascon J.-M., Understanding the roles of anionic redox and oxygen release during electrochemical cycling of lithium-rich layered Li4FeSbO6. J. Am. Chem. Soc. 137, 4804–4814 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Yabuuchi N., Takeuchi M., Nakayama M., Shiiba H., Ogawa M., Nakayama K., Ohta T., Endo D., Ozaki T., Inamasu T., Sato K., Komaba S., High-capacity electrode materials for rechargeable lithium batteries: Li3NbO4-based system with cation-disordered rocksalt structure. Proc. Natl. Acad. Sci. U.S.A. 112, 7650–7655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pearce P. E., Perez A. J., Rousse G., Saubanère M., Batuk D., Foix D., Abakumov E. A. M., Van Tendeloo G., Doublet M.-L., Tarascon J.-M., Evidence for anionic redox activity in a tridimensional-ordered Li-rich positive electrode β-Li2IrO3. Nat. Mater. 16, 580–586 (2017). [DOI] [PubMed] [Google Scholar]
- 12.McCalla E., Abakumov A. M., Saubanère M., Foix D., Berg E. J., Rousse G., Doublet M.-L., Gonbeau D., Novák P., Van Tendeloo G., Dominko R., Tarascon J.-M., Visualization of O-O peroxo-like dimers in high-capacity layered oxides for Li-ion batteries. Science 350, 1516–1521 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Grimaud A., Hong W. T., Shao-Horn Y., Tarascon J.-M., Anionic redox processes for electrochemical devices. Nat. Mater. 15, 121–126 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Luo K., Roberts M. R., Hao R., Guerrini N., Pickup D. M., Liu Y.-S., Edström K., Guo J., Chadwick A. V., Duda L. C., Bruce P. G., Charge-compensation in 3d-transition-metal-oxide intercalation cathodes through the generation of localized electron holes on oxygen. Nat. Chem. 8, 684–691 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Seo D.-H., Lee J., Urban A., Malik R., Kang S., Ceder G., The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials. Nat. Chem. 8, 692–697 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Zhan C., Yao Z., Lu J., Ma L., Maroni V. A., Li L., Lee E., Alp E. E., Wu T., Wen J., Ren Y., Johnson C., Thackeray M. M., Chan M. K. Y., Wolverton C., Amine K., Enabling the high capacity of lithium-rich anti-fluorite lithium iron oxide by simultaneous anionic and cationic redox. Nat. Energy. 2, 963–971 (2017). [Google Scholar]
- 17.Freire M., Kosova N. V., Jordy C., Chateigner D., Lebedev O. I., Maignan A., Pralong V., A new active Li–Mn–O compound for high energy density Li-ion batteries. Nat. Mater. 15, 173–177 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Meyer H., Hoppe R., Zum thermischen Verhalten von Li3MnO4 I. [1]. Über α- und β-Li3MnO4. Z. Anorg. Allg. Chem. 424, 249–256 (1976). [Google Scholar]
- 19.Saint J. A., Doeff M. M., Reed J., Synthesis and electrochemistry of Li3MnO4: Mn in the +5 oxidation state. J. Power Sources 172, 189–197 (2007). [Google Scholar]
- 20.R. G. Burns, Mineralogical Applications of Crystal Field Theory (Cambridge Univ. Press, 1993). [Google Scholar]
- 21.Morgan D., Van der Ven A., Ceder G., Li conductivity in LixMPO4 (M = Mn, Fe, Co, Ni) olivine materials. Electrochem. Solid State Lett. 7, A30–A32 (2004). [Google Scholar]
- 22.Kang K., Meng Y. S., Bréger J., Grey C. P., Ceder G., Electrodes with high power and high capacity for rechargeable lithium batteries. Science 311, 977–980 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Curtarolo S., Hart G. L. W., Nardelli M. B., Mingo N., Sanvito S., Levy O., The high-throughput highway to computational materials design. Nat. Mater. 12, 191–201 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Yao Z., Kim S., Aykol M., Li Q., Wu J., He J., Wolverton C., Revealing the conversion mechanism of transition metal oxide electrodes during lithiation from first-principles. Chem. Mater. 29, 9011–9022 (2017). [Google Scholar]
- 25.Murty B. S., Ranganathan S., Novel materials synthesis by mechanical alloying/milling. Int. Mater. Rev. 43, 101–141 (1998). [Google Scholar]
- 26.Zhou G. F., Bakker H., Mechanically induced structural and magnetic changes in the GdAl2 Laves phase. Phys. Rev. B Condens. Matter 52, 9437–9445 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Wang R., Li X., Liu L., Lee J., Seo D.-H., Bo S.-H., Urban A., Ceder G., A disordered rock-salt Li-excess cathode material with high capacity and substantial oxygen redox activity: Li1.25Nb0.25Mn0.5O2. Electrochem. Commun. 60, 70–73 (2015). [Google Scholar]
- 28.Cockayne E., van de Walle A., Building effective models from scarce but accurate data: Application to an alloy cluster expansion model. Phys. Rev. B Condens. Matter 81, 12104–12113 (2010). [Google Scholar]
- 29.van de Walle A., Multicomponent multisublattice alloys, nonconfigurational entropy and other additions to the alloy theoretic automated toolkit. Calphad 33, 266–278 (2009). [Google Scholar]
- 30.van de Walle A., Methods for first-principles alloy thermodynamics. JOM 65, 1523–1532 (2013). [Google Scholar]
- 31.Hart G. L. W., Forcade R. W., Algorithm for generating derivative structures. Phys. Rev. B Condens. Matter 77, 224115 (2008). [Google Scholar]
- 32.Hart G. L. W., Forcade R. W., Generating derivative structures from multilattices: Algorithm and application to hcp alloys. Phys. Rev. B Condens. Matter 80, 014120 (2009). [Google Scholar]
- 33.Hart G. L. W., Nelson L. J., Forcade R. W., Generating derivative structures at a fixed concentration. Comput. Mater. Sci. 59, 101–107 (2012). [Google Scholar]
- 34.Michel K. J., Zhang Y., Wolverton C., Fast mass transport kinetics in B20H16: A high-capacity hydrogen storage material. J. Phys. Chem. C 117, 19295–19301 (2013). [Google Scholar]
- 35.Schubert K., Poetzschke M., Zum aufbau einiger T(4)-B(3) homologer und quasihomologer systeme. I. Die systeme Ti-Ga, Zr-Ga und Hf-Ga. Naturwissenschaften 53, 474–488 (1962). [Google Scholar]
- 36.Brachtel G., Hoppe R., Die koordinationszahl 5 bei MnIII: Na4Mn2O5 [1,2]. Z. Anorg. Allg. Chem. 468, 130–136 (1980). [Google Scholar]
- 37.Akbarzadeh A. R., Ozoliņš V., Wolverton C., First-principles determination of multicomponent hydride phase diagrams: Application to the Li-Mg-N-H system. Adv. Mater. 19, 3233–3239 (2007). [Google Scholar]
- 38.Wolverton C., Yan X.-Y., Vijayaraghavan R., Ozoliņš V., Incorporating first-principles energetics in computational thermodynamics approaches. Acta Mater. 50, 2187–2197 (2002). [Google Scholar]
- 39.N. N. Greenwood, A. Earnshaw, Chemistry of the Elements (Butterworth-Heinemann, 1997). [Google Scholar]
- 40.Belsky A., Hellenbrandt M., Karen V. L., Luksch P., New developments in the inorganic crystal structure database (ICSD): Accessibility in support of materials research and design. Acta Crystallogr. B 58, 364–369 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Grindy S., Meredig B., Kirklin S., Saal J. E., Wolverton C., Approaching chemical accuracy with density functional calculations: Diatomic energy corrections. Phys. Rev. B Condens. Matter 87, 075150 (2013). [Google Scholar]
- 42.Wang L., Maxisch T., Ceder G., Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. B Condens. Matter 73, 195107 (2006). [Google Scholar]
- 43.Kirklin S., Saal J. E., Meredig B., Thompson A., Doak J. W., Aykol M., Rühl S., Wolverton C., The open quantum materials database (OQMD): Assessing the accuracy of DFT formation energies. npj Comput. Mater. 1, 15010 (2015). [Google Scholar]
- 44.Stevanović V., Lany S., Zhang X., Zunger A., Correcting density functional theory for accurate predictions of compound enthalpies of formation: Fitted elemental-phase reference energies. Phys. Rev. B Condens. Matter 85, 115104 (2012). [Google Scholar]
- 45.SGTE, Thermodynamic Properties of Inorganic Materials (Springer-Verlag Berlin Heidelberg, 1999). [Google Scholar]
- 46.P. Nash, Thermodynamic database (2013).
- 47.Saal J. E., Kirklin S., Aykol M., Meredig B., Wolverton C., Materials design and discovery with high-throughput density functional theory: The open quantum materials database (OQMD). JOM 65, 1501–1509 (2013). [Google Scholar]
- 48.Jain A., Ong S. P., Hautier G., Chen W., Richards W. D., Dacek S., Cholia S., Gunter D., Skinner D., Ceder G., Persson K. A., Commentary: The materials project: A materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013). [Google Scholar]
- 49.Kresse G., Hafner J., Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens. Matter 47, 558–561 (1993). [DOI] [PubMed] [Google Scholar]
- 50.Kresse G., Hafner J., Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B Condens. Matter 49, 14251–14269 (1994). [DOI] [PubMed] [Google Scholar]
- 51.Kresse G., Furthmüller J., Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996). [DOI] [PubMed] [Google Scholar]
- 52.Kresse G., Furthmüller J., Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Blöchl P. E., Projector augmented-wave method. Phys. Rev. B Condens. Matter 50, 17953–17979 (1994). [DOI] [PubMed] [Google Scholar]
- 54.Perdew J. P., Ernzerhof M., Burke K., Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 105, 9982–9985 (1996). [Google Scholar]
- 55.Dudarev S. L., Botton G. A., Savrasov S. Y., Humphreys C. J., Sutton A. P., Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B Condens. Matter 57, 1505–1509 (1998). [Google Scholar]
- 56.Togo A., Oba F., Tanaka I., First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B Condens. Matter 78, 134106 (2008). [Google Scholar]
- 57.Heyd J., Scuseria G. E., Ernzerhof M., Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003). [Google Scholar]
- 58.Aydinol M. K., Kohan A. F., Ceder G., Cho K., Joannopoulos J., Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides. Phys. Rev. B Condens. Matter 56, 1354–1365 (1997). [Google Scholar]
- 59.Chan M. K. Y., Wolverton C., Greeley J. P., First principles simulations of the electrochemical lithiation and delithiation of faceted crystalline silicon. J. Am. Chem. Soc. 134, 14362–14374 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Wolverton C., Zunger A., First-principles prediction of vacancy order-disorder and intercalation battery voltages in LixCoO2. Phys. Rev. Lett. 81, 606–609 (1998). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaao6754/DC1
fig. S1. Cationic ordering between Li and Mn in Li4Mn2O5 and between Ga and Zr in Ga2Zr.
fig. S2. Na4Mn2O5 prototype structure with a space group Fddd.
fig. S3. Phonon dispersion of the ground-state LiMnO2.
fig. S4. Phonon dispersion of the ground-state Li2O.
fig. S5. Schematic illustration of the relative stabilities between the disordered and ordered Li4Mn2O5, as well as the decomposed combination, 2LiMnO2 + Li2O.
fig. S6. The magnetization and oxidation state evolution of Mn and O ions during delithiation.
table S1. Structure information of the Li4Mn2O5 ground state.
table S2. Top Li4(Mn,M)2O5 cathode candidates from the HT-DFT screening with predicted gravimetric capacities (theoretical) and averaged voltages using Li4Mn2O5 as the benchmark.