

ABSTRACT
Lipid and cholesterol metabolism might play a role in the pathogenesis of Parkinson disease (PD). However, the association between cholesterol and PD is not clearly established. Cholesterol accumulation is closely related to the expression of multilamellar bodies (MLBs). Also, cholesterol controls autophagosome transport. Thus, impaired cholesterol and autophagosome trafficking might lead to robust autophagic vacuole accumulation. Our recent work provides the first evidence that the presence of the N370S GBA mutation produces an accumulation of cholesterol, which alters autophagy-lysosome function with the appearance of MLBs, rendering the cell more vulnerable and sensitive to apoptosis.
KEYWORDS: autophagosomes, cholesterol, GBA1, lysosomes, multilamellar bodies, Gaucher disease, Parkinson disease, lipid storage diseases
Parkinson disease is the most common multifactorial neurodegenerative movement disorder caused by the loss of the substantia nigra pars compacta dopaminergic neurons, which project to the striatum through the medial forebrain bundle. The majority of PD cases are idiopathic, although some are associated with gene mutations. In this sense, there is a clear relationship between PD and mutations in the GBA gene. Heterozygous GBA mutations have been estimated to increase the risk of PD 20–30-fold, while 7–10% of PD cases contain a GBA mutation. GBA-associated PD (GBA-PD) is practically identical to idiopathic PD, with slightly earlier onset and a tendency for cognitive impairment and dementia. Although heterozygous mutations are the most common genetic risk factor for PD, the homozygous mutations cause Gaucher disease (GD), the most prevalent lysosomal storage disorder. Although the precise molecular basis of GBA-mediated PD and the impact on cellular homeostasis remains unknown, clinical and experimental evidence indicates that GBA haploinsufficiency can contribute to pathological aggregation of SNCA/α-synuclein. Indeed, SNCA is a major component of Lewy bodies, which represent the histopathological hallmark of PD.
The GBA gene encodes the lysosomal enzyme glucosylceramidase beta/β-glucocerebrosidase, which hydrolyses the glycolipid glucocerebroside to ceramide and glucose (Figure 1A). Approximately 300 mutations have been detected in the GBA gene sequence. Our study focuses on the N370S mutation that is the second most frequent in the population. This point mutation replaces the amino acid asparagine, localized at protein position 370, with the amino acid serine, affecting its conformation and stability without altering the catalytic site.
Figure 1.
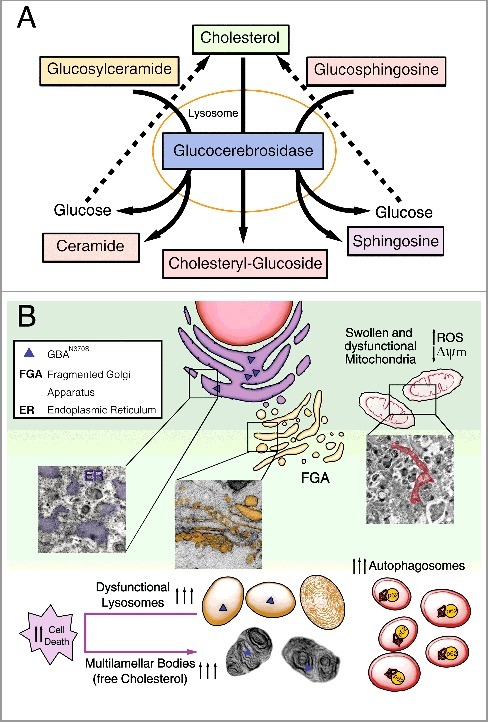
Diagram summarizing the primary altered features caused by GBAN370S. (A) In physiological conditions GBA hydrolyzes its primary substrate glucocerebroside into glucose and ceramide. An alternate substrate, glucosylsphingosine, is also degraded into glucose and sphingosine. GBA also acts as a glycosyltransferase, catalyzing the transfer of glucose from glucocerebroside to cholesterol and leading to the formation of cholesteryl-glucoside. (B) Normal lysosomal function is required for the autophagic clearance of defective cellular organelles and misfolded proteins. The N370S mutation results in GBA loss of function in the lysosomes (caused by its retention in the ER) and accumulation of its substrates. This defect leads to stress, enlargement and disorganization of the ER, and Golgi apparatus fragmentation (FGA), along with subsequent defects in autophagy shown as an autophagosome accumulation. Accordingly, SQSTM1 accumulates due to dysfunctional lysosomes likely caused by cholesterol accumulation that promotes MLB formation. These alterations hamper the removal of damaged mitochondria, inducing ROS production. All this together renders these cells vulnerable to stress-induced apoptosis.
Using fibroblasts obtained from PD patients with the N370S GBA mutation, we performed a complete characterization of the autophagy-lysosome pathway. We demonstrate that this mutation decreases GBA protein levels and activity due to the retention of GBAN370S within the ER, interrupting its cellular traffic without altering GBA mRNA. Moreover, the misfolded GBA trapped in the ER triggers ER stress and the unfolded protein response (UPR) along with Golgi apparatus fragmentation. The prolonged activation of the UPR and ER stress subsequently may lead to autophagy impairment and increased apoptosis (Figure 1B).
Our study shows the presence of a higher number of autophagic structures and accumulation of SQSTM1/p62 in cells with the N370S GBA mutation (Figure 1B). Our induction and blockade of autophagy experiments using various treatments indicate that, in the N370S fibroblasts, there is an increase of autophagosome synthesis, which appears to be a primary defect in GBA-PD fibroblasts. However, accumulation of glycosphingolipids, including glucocerebroside, could be another cause by which autophagy is altered. Particularly, our study reveals (for the first time) that cholesterol accumulates in lysosomes of GBA-PD patients. As reported, cholesterol can increase the accumulation of autophagosomes impairing their transport and position within the cell. Future research will be needed to resolve whether cholesterol is the primary storage product in the disease process, or if its accumulation occurs rather secondarily to that of other glycosphingolipids.
Another main highlight of our study is that the N370S mutation results in the accumulation of MLBs (Figure 1(B)). In pathological conditions, these concentric bodies are typical of lysosomal storage diseases, such as the Niemann-Pick disease type I, as well as drug-induced phospholipidosis, as we show in chloroquine-treated control fibroblasts. MLBs are generated as lysosomal structures, which frequently appear as multiple intracellular concentric membrane structures containing primarily undegraded phospholipids and cholesterol. MLBs are formed via cellular autophagy, and their lysosomal nature is suggestive of the involvement of various lysosomal enzymes. Initially, single or multiple foci of lamella appear within an autophagic vacuole, and then transform into multilamellar structures. We propose that cholesterol accumulation in N370S lysosomes traps lipid raft components, promoting the formation of MLBs, or that they are formed indirectly through autophagy impairment via the ER. We further propose that these MLBs may correspond to degenerating autophagosomes and/or autolysosomes supported by the lysosomal dysfunction we found as a consequence of decreased GBA and build-up of free cholesterol. This cholesterol accumulation could also reduce the ability of lysosomes to efficiently fuse with endocytic and autophagic vesicles by affecting SNARE function.
Cholesterol is an essential component of cellular membranes and is critical for many cellular functions, including maintenance of membrane integrity and fluidity and signal transduction. However, the mechanisms of cholesterol accumulation and altered lipid trafficking remain unknown. Lysosomal cholesterol can mediate changes in membrane lipid and protein trafficking either through a change in intralumenal membrane fluidity or the coalescence of cholesterol-sphingolipid microdomains. These changes in membrane physical properties could be modulated by cholesteryl-glucoside, which is the resulting product of the glucosyltransferase activity of GBA (Figure 1A). Future research should determine whether secondary abnormalities in cholesteryl-glucoside or other glucosylated metabolites could be due to abnormal glucocerebroside metabolism contributing to particular symptoms related to PD. In our study, the N370S mutation increases apoptosis associated with high reactive oxygen species (ROS) levels. Thus, it is possible that the cell vulnerability associated with N370S might be caused by lysosomal membrane permeability induced by ROS or cholesterol build-up. Accordingly, all this might contribute to triggering apoptotic cell death by the release of cathepsins into the cytosol.
We show that the presence of these MLBs is associated with GBA-PD, thus providing further evidence of lysosomal dysfunction in PD. We propose that misfolded GBA might contribute to Parkinsonism by leading to lysosomal insufficiency, thereby impairing the ability of autophagy to prevent synucleinopathies. In conclusion, our integrated molecular landscape yields detailed insights into the mechanisms underlying PD pathogenesis and highlights the involvement of cholesterol and lipid metabolism. Finally, our data suggest the use of cholesterol reduction as a therapeutic option for the treatment of GBA-PD.
Funding Statement
This work was supported by Grants from the Spanish Ministeries of Economía y Competitividad (Secretarıa de Estado de Investigación, Desarrollo e Innovación), Sanidad Política Social e Igualdad and ISCIII, Centro de Investigación Biomédica en Red Enfermedades Neurodegenerativas (CIBERNED): SAF2016-78207-R, PCIN2015-098, CB06/05/0055 and PI2015-2/02 to RM; SAF2013-4759R and CB06/05/0065 to CV; Ramón Areces Fundation (172275) to RM and CV; CB06/05/004 and PI15/00034 and Junta de Extremadura (Consejería de Educación y Empleo):GR15045 to JMF.
Acknowledgments
We thank Enrique Sahagun PhD (Scixel, Madrid, Spain) for his assistance with the Figure 1.
Disclosure of potential conflicts of interest
There were no potential conflicts of interest to be disclosed.