

ABSTRACT
In the atherosclerotic plaque, macrophages are the key catabolic workhorse responsible for clearing lipid and dead cell debris. To survive the highly proinflammatory and lipotoxic plaque environment, macrophages must adopt strategies for maintaining tight homeostasis and self-renewal. Macroautophagy/autophagy is a pro-survival cellular pathway wherein damaged or excess cellular cargoes are encapsulated by a double-membrane compartment and delivered to the lysosome for hydrolysis. Previously, macrophage-specific autophagy deficiency has been shown to be atherogenic through several complementary mechanisms including hyperactivation of the inflammasome, defective efferocytosis, accumulation of cytotoxic protein aggregates, and impaired lipid degradation. Conversely, in a recent study we hypothesized that enhancing the macrophage autophagy-lysosomal system through genetic or pharmacological means could protect against atherosclerosis. We demonstrated that TFEB, a transcription factor master regulator of autophagy and lysosome biogenesis, coordinately enhances the function of this system to reduce atherosclerotic plaque burden. Further, we characterized the disaccharide trehalose as a novel inducer of TFEB with similar atheroprotective effects. Overall, these findings mechanistically interrogate the importance and therapeutic promise of a functional autophagy-lysosome degradation system in plaque macrophage biology.
KEYWORDS: Apoptosis, atherosclerosis, autophagy, inflammation, lysosome, macrophage, p62, protein aggregation, TFEB, trehalose
Atherosclerosis is the progressive buildup of plaque in the arterial wall ultimately resulting in rupture and thrombosis manifesting as potentially lethal myocardial infarction or stroke. In response to inflammatory cues, macrophages are recruited to the growing plaque and attempt to clear deposited lipids and apoptotic cells. This highly pro-inflammatory and toxic environment renders the macrophage itself susceptible to undergoing apoptotic or necrotic cell death and tragically contributing to, rather than ameliorating, plaque instability. One promising pathway for maintaining plaque macrophage function and survival is autophagy—a cellular catabolic process wherein toxic or damaged intracellular cargo is engulfed and delivered to the lysosome for hydrolysis.
In our recent study, we first characterized the functional status of the autophagy-lysosome system in both murine and human atherosclerotic plaques. We focused on 2 key autophagy markers (i.e., LC3, a protein coating autophagosomes and involved in maturation, and SQSTM1/p62, a receptor involved in protein aggregation and linking of polyubiquitinated targets to LC3 for degradation). Early lesions feature high levels of LC3 colocalized with SQSTM1, suggesting a robust recruitment of autophagy as a stress response to the burgeoning plaque. Advanced lesions, however, feature reductions in LC3 and dissociation from SQSTM1, indicative of a broad dysfunction in the autophagic process. Future studies will be required to investigate this apparently dynamic and heterogeneous nature of autophagy dysfunction in the plaque. For example, mouse models are available to track specific macrophage populations, and autophagic flux can be more precisely assessed using pH-sensitive fluorescent tags in vivo.
Given these observations, we assessed whether enhancing the autophagy-lysosome system could be atheroprotective. To accomplish this, we generated a murine model with macrophage-specific overexpression of TFEB, a transcription factor master regulator of autophagy and lysosome genes. TFEB successfully induces this system in vitro and in vivo, resulting in restored autophagic function (Figure 1). This increase in autophagy activity translates into broad atheroprotection including reductions in overall plaque burden as well as features of plaque complexity such as apoptotic and necrotic areas. Mechanistically, TFEB reduces accumulation of ubiquitinated and SQSTM1-enriched protein aggregates, IL1B/IL-1β levels, and macrophage apoptosis (Figure 1). Notably, these beneficial phenotypes are dependent on autophagy: Macrophage TFEB overexpression in the absence of ATG5 or SQSTM1 is no longer able to ameliorate atherosclerosis.
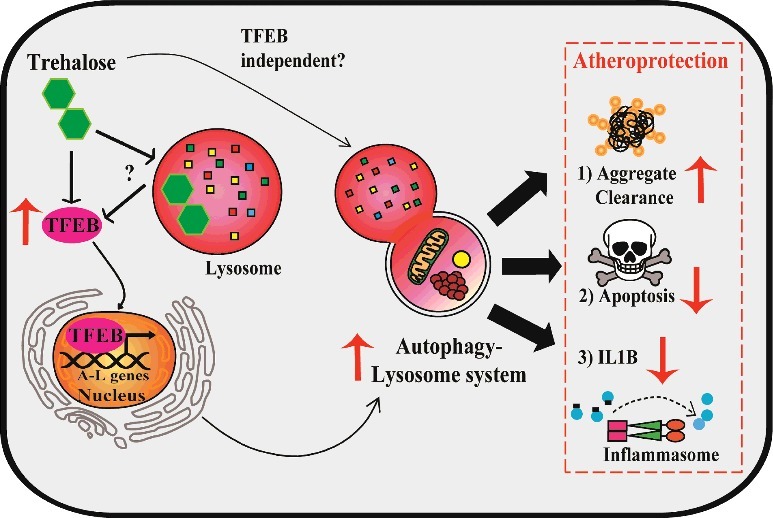
Model of TFEB- and trehalose-induced atheroprotection in macrophages. Left: Overexpression of TFEB, a transcription factor master regulator of autophagy-lysosome biogenesis, functionally drives this system in plaque macrophages. Trehalose is an autophagy-inducing disaccharide capable of inducing TFEB through mechanisms potentially involving acute perturbation of lysosomal function. Right: Enhanced function of the autophagy-lysosome system in macrophages attained with TFEB or trehalose results in atheroprotection associated with increased protein aggregate clearance, reduced apoptosis, and reduced IL1B production.
Many open questions remain regarding the complex set of genes TFEB targets and their impact on the macrophage phenotype. First, TFEB also stimulates genes involved in endocytosis, phagocytosis, and immune responses, with established functional relevance to bacterial phagocytosis. Enhancing these processes may also help macrophages find, dock on, and engulf apoptotic cells, a process known as efferocytosis. Second, TFEB drives expression of mitochondrial and lipid metabolic genes, a process that in part depends on the transcriptional coactivator PPARGC1A/PGC-1α. While unexamined in macrophages, this arm of TFEB function may assist in metabolizing the massive lipid loads present in the plaque. Though these possibilities add complexity to how TFEB regulates the overall macrophage phenotype, the clear theme is coordinated induction of catabolic pathways that in concert with autophagy are beneficial in the context of atherosclerosis.
We next sought to therapeutically harness the autophagy-lysosome system in macrophages using trehalose, a naturally occurring disaccharide with reported autophagy-inducing properties. We discovered a previously uncharacterized transcriptional component of trehalose-dependent autophagy-lysosome induction in macrophages that is associated with increased levels and nuclear translocation of TFEB (Figure 1). Similar to TFEB overexpression, trehalose is able to reduce polyubiquitinated protein aggregates, IL1B levels, and apoptosis in cultured macrophages. In vivo, trehalose also reduces atherosclerosis and plaque complexity in a macrophage ATG5- and SQSTM1-dependent manner.
Basic aspects of trehalose pharmacokinetics warrant consideration for therapeutic use. First, many previous studies have administered oral trehalose assuming adequate tissue distribution for therapeutic effect. However, we show oral trehalose does not appreciably enter the blood stream and is largely ineffective in our atherosclerosis studies compared to parenteral (intraperitoneal) administration. Several factors largely prohibit substantial oral trehalose bioavailability: It must first escape intestinal hydrolysis by the trehalose-specific enzyme called trehalase, then transit both the intestinal barrier and portal circulation. In fact, direct action on the gut with accompanying endocrine/enterokine effects may be a more plausible mechanism of action in settings where oral trehalose is effective. Future therapeutic strategies could employ degradation-resistant trehalose analogs, concurrent administration of trehalase inhibitors, and/or intravenous administration.
Layered with these pharmacokinetic challenges is the ongoing mystery of how trehalose actually induces TFEB and autophagy in cells, which is likely to be a complex integration of several intra- and extracellular actions. First, most previous studies have used 100 mM trehalose to study its mechanism of action in vitro. We showed that such doses diverge significantly from what is attainable in vivo and may poorly represent the physiologically relevant mechanisms contributing to autophagy induction. For example, intraperitoneal injections of trehalose at a high dose of 2 g/kg only translate to maximal transient circulating levels of 10 mM with an average of ∼1 mM. In this regard, we have shown that more therapeutically realistic doses (e.g., 1 mM and in some cases even 100 μM trehalose) are a sufficient stimulus for TFEB and the related transcription factor TFE3 to activate a transcriptional program of autophagy-lysosome biogenesis. However, precisely how trehalose activates TFEB or TFE3 and whether its autophagy-inducing actions are purely TFEB dependent is unknown (Figure 1). Due to the relative resistance of trehalose to acid hydrolysis, it is possible trehalose accumulates in the low pH environment of lysosomes and perturbs their function to activate pathways such as PRKC/PKC, MTOR, and PPP3/Calcineurin, all regulators of TFEB. Trehalose could also affect the autophagy-lysosome system independent of cell entry through binding surface receptors or modulating cellular glucose transport. Overall, continued study of the mechanistic and therapeutic aspects of autophagy-lysosome induction holds great promise in treating cardiovascular disease.
Funding Statement
American Diabetes Association [grant number 1-18-IBS-029]; National Heart, Lung, and Blood Institute [grant number F31 HL132434]; National Heart, Lung, and Blood Institute [grant number R01 HL125838]; National Institute of Diabetes and Digestive and Kidney Diseases [grant number P30 DK020579]; U.S. Department of Veterans Affairs [grant number I01 BX003415].
Disclosure of potential conflict of interest
The authors have nothing relevant to disclose.