

Abstract
Rationale:
Protein S (PS) deficiency that can be inherited or acquired is an independent risk factor for venous thromboembolism (VTE).
Patient concerns:
In this report, we present a case of recurrent pulmonary embolism (PE) and deep venous thrombosis (DVT) due to PS deficiency.
Diagnoses:
A 32-year-old male patient with significant decrease in PS activity was detected by laboratory tests. Genetic examination of the PROS1 gene showed a transition of G to T in exon 14 (c.1792 G>T, p.E598X), which was a paternal inherited heterozygous G1792T substitution in the laminin G-type repeat domain, generating a premature stop codon at Glu598.
Interventions:
We considered that the inherited PS deficiency due to a PROS1 gene mutation may associate with recurrent VTE. The patient was suggested to have an extended anticoagulant therapy to avoid a severe VTE event.
Outcomes:
The patient was discharged home with continued oral anticoagulants and was still seen in clinic for follow-up.
Lessons:
It is necessary for the young patient with recurrent idiopathic thrombosis to perform an inherited PS deficiency test and receive anticoagulant therapy for an extended period.
Keywords: deep venous thrombosis, deficiency, protein S, pulmonary embolism
1. Introduction
Protein S (PS; gene symbol, PROS1; Gene ID, 5627; NM_000313.3) is a vitamin K-dependent plasma glycoprotein that functions as a non-enzymatic cofactor for activated protein C (APC) in the degradation of coagulation factors Va and VIIIa, thereby eliminating the prothrombinase and tenase complexes.[1–3] Independent of its role as a cofactor for APC, PS further acts as an anticoagulant by directly binding to factors Xa and Va and inhibiting factor Xa.[4] In addition, PS has prothrombinase inhibitory properties because of its high affinity for negatively charged phospholipid membranes.[5] PS deficiency is an inherited or acquired disorder associated with an increased risk of thrombosis.[6] The prevalence of PS deficiency ranges from 0.03% to 0.13% of the general population,[7,8] whereas it is approximately 2% of unselected patients and 1% to 13% of patients with venous thromboembolism.[9,10]
In human plasma, PS forms an equimolar complex with the complement regulatory protein C4b-binding protein (C4BP). The formation of this complex affects PS function, as only free PS is active as an APC cofactor.[11] PS bind C4BP with very high affinity, especially at physiological calcium concentrations. In plasma, free PS represents the molar excess of PS relative to C4BP binding sites, which are found in the beta chain of C4BP.[12–14] The constant concentration of free protein S is maintained and regulated by the molar balance between C4BP and total protein S.[15,16]
PS antigen and activity were detected using laboratory tests and specific tests for the free-form of PS have been developed.[17] According to plasma levels of total PS, free PS antigen and PS activity, PS deficiency is classified into 3 subtypes. Type I is a quantitative deficiency in the total PS and free PS. Type II, or qualitative PS deficiency, is characterized by normal PS levels but reduced PS activity due to a dysfunctional PS variant in plasma. Type III PS deficiency is characterized by low levels of free PS, though the total plasma concentration of PS is normal. Here we present a young patient with recurrent pulmonary embolism (PE) and deep vein thrombosis (DVT) and who was finally diagnosed as PS deficiency due to gene PROS1 mutation.
2. Case presentation
A 32-year-old male patient presented to the emergency room with sudden chest pain and dyspnea. One month ago he discontinued 1 year of anticoagulant therapy for the previous diagnosis of PE and DVT. His parents had no history of thromboembolic disease. Blood routine showed that the patient's hemoglobin was 15.9 g/dL, platelet count was 280 × 109/L, and the hematology-coagulation test showed that his prothrombin time (PT) was 12.1 s, activated partial thromboplastin time (APTT) was 37.9 s, fibrinogen was 5.25 g/L, factor Xa activity was 115%, and antithrombin III (AT III) activity was 92%. Owing to the symptoms and history, emergent CT angiography was requested and revealed recurrent massive PE (Fig. 1). The color Doppler ultrasound demonstrated evidence of DVT in the right lower leg.
Figure 1.

Computed tomography (CT) images showed bilaterally diffuse massive thrombus-filled pulmonary arteries (Arrows in A and B). The pulmonary thromboembolus is occluding the left pulmonary arterial trunk in the magnetic resonance pulmonary angiography (Arrows in C and D).
As no other etiologies were found, the patient's plasma taken before heparin treatment was analyzed for PS activity by a clotting assay (Staclot Protein S; Diagnostica Stago, Asnieres, France). The results revealed a significant decrease in PS activity at 22% (male reference range, 77%–143%). The activity of protein C and antithrombin was within the reference ranges (108% and 99%, respectively). Therefore, a diagnosis of protein S deficiency was established. The patient's father had been thrombosis-free, but he was also found to have PS deficiency with low PS activity (23%) (Fig. 2A). Sequence analysis of the PROS1 gene from this patient showed a heterozygous transition of G to T at nucleotide 1792 in exon 14 (NM_000313.3 c.1792G>T) in the laminin G-type repeat domain, generating a premature stop codon at Glu598 in exon 14 (Fig. 2B and C). No additional abnormalities were found in any of the subjects after testing for anticardiolipin antibodies and DNA testing for protein C (PROC), protein C receptor (PROCR), factor V Leiden, and prothrombin G20210A.
Figure 2.
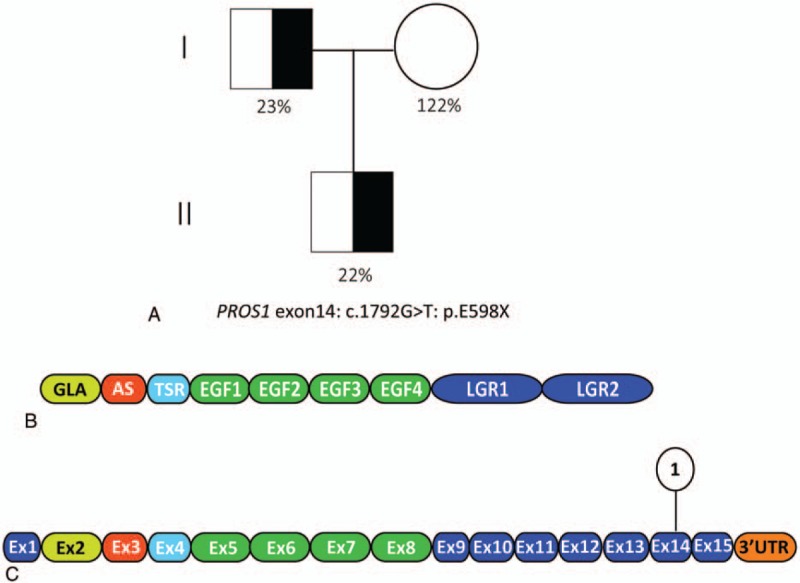
A, Molecular analysis of the protein S deficient family. Pedigree is shown with the PS functional activities and genetic determination where know DNA sequence analysis of PROS1 exon 14 results in a premature stop at Glu598. Paternal PS activity 23%, maternal PS activity 122%, the patient PS activity 22%; (B) domain structure of mature PS. Color-coding corresponds to mRNA exons encoding each PS domain. C, mRNA encoding PS. Upper symbol represent PROS1 mutation causing PS deficiency in this case. AS = aromatic stack domain, EGF = epidermal growth factor-like domain, GLA = γ-carboxyglutamic acid domain, LGR= laminin G-type repeat, PS = protein S, TSR = thrombin-sensitive region domian.
The analysis of possible functional effect of the sequence variation was performed using 2 prediction programs, Polymorphism Phenotyping (PolyPhen, http://genetics.bwh.harvard.edu/pph2/) and Sort Intolerant from Tolerant (SIFT, http://sift.jcvi.org/). This variant was predicted to be ‘probably non-detrimental’ by PolyPhen and ‘tolerated’ by SIFT program.
The patient signed informed consent and this study was approved by the Ethics Committee of the Second Affiliated Hospital of Zhejiang University.
3. Discussion
Hereditary PS deficiency is an autosomal dominant disease, which can increase the risk of thrombosis because of the failure to deactivate the coagulation.[8] Patients with PS deficiency usually present with unprovoked or recurrent venous thromboembolism (VTE).[18,19] Exposure to precipitating factors such as surgery and trauma may trigger thrombosis in these individuals.[20] Interestingly, in the case described above, the patient suffered from recurrent PE and DVT after ceasing anti-coagulation treatment without any other acquired risk factors, indicating a potential abnormality in anticoagulation mechanisms. In our opinion, young patients with recurrent venous thromboembolism in the absence of precipitating factors need to be tested to identify whether there is a deficiency of PC, PS, or antithrombin III (AT III). In this case, the patient was found to be PS deficient.
To our knowledge, quite a lot of genetic mutations in PROS1 have been reported in the Human Genome Mutation Database. But we are the first to identify a mutation in exon 14, which is predicted to result in a truncated form of PS, due to a premature stop codon at residue 598. As PS deficiency is inherited as an autosomal dominant disorder, the individual described here has severe PS deficiency due to haploinsufficiency. The truncated protein expressed in both propositi probably result in protein misfolding or nonsense-mediated RNA decay (NMD); both mechanisms would cause low PS activity.[21] This patient is the first reported case of heterozygous qualitative PS deficiency with the mutation site on exon 14 (NM_000313.3 c.1792G>T p.E598X) and recurrent venous thromboembolism. The mechanism underlying the association between low PS activity and clinical manifestations of VTE requires further investigation.
PS testing and PROS1 testing should not be considered in unselected patients with venous thromboembolism, because inherited PS deficiency is rare in the general population.[22] With regard to the many difficulties the diagnosis of PS deficiency implies, Marlar and Gausman[23] recently proposed a diagnostic algorithm for the assessment of PS abnormalities. Current evidence indicates that screening for inherited thrombophilia is appropriate in cases of VTE without obvious cause < 45 to 50 years; VTE in patients with a family history of thrombosis; recurrent VTE; thrombosis at an unusual location; and developing VTE during pregnancy, use of oral contraceptives, or hormone replacement therapy.[24]
Testing should be done at least in several weeks after an acute clotting event to allow acute-phase reactant proteins to return to baseline.[25] PS deficiency should not be diagnosed or excluded on the basis of assays performed when the patient is taking vitamin K antagonist. To avoid false-positive test results, plasma samples should be taken after temporary interruption of oral anticoagulant therapy for at least 10 days, and, in some cases, bridging anticoagulation with alternative agents such as LMWH positive test results should be confirmed by a second blood sample. In pregnant women, positive test results should be established after the postpartum period.[26] To prove inherited deficiency, testing of family members is recommended. Both patient and family members should receive genetic counseling prior to genetic testing, and such testing should only be performed after obtaining consent. Interestingly, the patient's father had also a severe PS deficiency, but was thrombosis-free, suggesting that a triggering event might have been involved in the patient's thrombus formation or the existence of a protective mechanism in the patient's father.
Patients with the known PROS1 mutation and low PS levels are at high risk of recurrent thrombosis and require lifelong oral anticoagulation even after the first episode. This patient underwent sudden chest pain and dyspnea just 1 month after discontinuing a year-long anticoagulant regimen, which means prolonged anticoagulant therapy should be suggested for this condition. There are some recommended indications for extended anticoagulation after 1 episode of VTE: anticoagulant therapy for up to 2 years without other thrombophilia; and life-long if other thrombophilia is present (coexistence of PC/PS heterozygous deficiency with FV Leiden or prothrombin 20210A).
4. Conclusion
In summary, our report supports the view that PS deficiency should be taken into consideration for young people with recurred PE and DVT without predisposing causes. Genetic studies could be valuable in the identification of PROS1 mutations for the PS deficient patients. As in this case, the described mutation in the PROS1 (c.1792 G>T, p.E598X) leads to a functional PS deficiency and appears to have clinical relevance with thrombosis. For this reason, the patient was suggested to have an extended anticoagulant therapy to avoid a severe VTE event.
Author contributions
Conceptualization: Laigen Shen, Bing Chen.
Data curation: Xiaojie Huang.
Investigation: Fangfang Xu.
Resources: Laigen Shen, Bing Chen.
Software: Carmel Rebecca Assa.
Supervision: Zhenjie Liu.
Footnotes
Abbreviations: APC = activated protein C, APTT = activated partial thromboplastin time, AT III = antithrombin III, DVT = deep venous thrombosis, NMD = nonsense-mediated RNA decay, PE = pulmonary embolism, PS = protein S, PT = prothrombin time, VTE = venous thromboembolism.
This study was supported by National Natural Scientific Foundation of China (No 81300236, 81670433).
The authors declare no conflict of interest.
References
- [1].Walker FJ. Regulation of activated protein c by protein s. The role of phospholipid in factor Va inactivation. J Biol Chem 1981;256:11128–31. [PubMed] [Google Scholar]
- [2].Dahlback B. Protein S and C4b-binding protein: components involved in the regulation of the protein c anticoagulant system. Thromb Haemost 1991;66:49–61. [PubMed] [Google Scholar]
- [3].Ten Kate MK, Platteel M, Mulder R, et al. Pros1 analysis in 87 pedigrees with hereditary protein s deficiency demonstrates striking genotype-phenotype associations. Hum Mutat 2008;29:939–47. [DOI] [PubMed] [Google Scholar]
- [4].Heeb MJ, Rosing J, Bakker HM, et al. Protein S binds to and inhibits factor Xa. Proc Natl Acad Sci U S A 1994;91:2728–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].van Wijnen M, Stam JG, van’t Veer C, et al. The interaction of protein S with the phospholipid surface is essential for the activated protein C-independent activity of protein S. Thromb Haemost 1996;76:397–403. [PubMed] [Google Scholar]
- [6].ten Kate MK, Van d MJ. Protein S deficiency: a clinical perspective. Hemophilia 2008;14:1222–8. [DOI] [PubMed] [Google Scholar]
- [7].Dykes AC, Walker ID, McMahon AD, et al. A study of protein S antigen levels in 3788 healthy volunteers: Influence of age, sex and hormone use, and estimate for prevalence of deficiency state. Br J Haematol 2001;113:636–41. [DOI] [PubMed] [Google Scholar]
- [8].Engesser L, Broekmans AW, Briet E, et al. Hereditary protein S deficiency: clinical manifestations. Ann Intern Med 1987;106:677–82. [DOI] [PubMed] [Google Scholar]
- [9].Lane DA, Mannucci PM, Bauer KA, et al. Inherited thrombophilia: part 1. Thromb Haemost 1996;76:651–62. [PubMed] [Google Scholar]
- [10].Seligsohn U, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med 2001;344:1222–31. [DOI] [PubMed] [Google Scholar]
- [11].Dahlback B. Inhibition of protein Ca cofactor function of human and bovine protein S by C4b-binding protein. J Biol Chem 1986;261:12022–7. [PubMed] [Google Scholar]
- [12].Griffin JH, Gruber A, Fernandez JA. Reevaluation of total, free, and bound protein S and C4b-binding protein levels in plasma anticoagulated with citrate or hirudin. Blood 1992;79:3203–11. [PubMed] [Google Scholar]
- [13].Hillarp A, Dahlback B. Novel subunit in C4b-binding protein required for protein s binding. J Biol Chem 1988;263:12759–64. [PubMed] [Google Scholar]
- [14].Hardig Y, Garcia d FP, Dahlback B. Expression and characterization of a recombinant C4b-binding protein lacking the beta-chain. Biochem J 1995;308(Pt 3):795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Garcia d FP, Alim RI, Hardig Y, et al. Differential regulation of alpha and beta chains of C4b-binding protein during acute-phase response resulting in stable plasma levels of free anticoagulant protein S. Blood 1994;84:815–22. [PubMed] [Google Scholar]
- [16].Zoller B, Garcia d FP, Dahlback B. Evaluation of the relationship between protein S and C4b-binding protein isoforms in hereditary protein S deficiency demonstrating type I and type III deficiencies to be phenotypic variants of the same genetic disease. Blood 1995;85:3524–31. [PubMed] [Google Scholar]
- [17].Comp PC. Laboratory evaluation of protein s status. Semin Thromb Hemost 1990;16:177–81. [DOI] [PubMed] [Google Scholar]
- [18].Llovera I, Roit Z, Kiriaki S, et al. Mesenteric ischemia and protein s deficiency: a rare case report. J Emerg Med 2010;39:579–82. [DOI] [PubMed] [Google Scholar]
- [19].Porter J, Vesely M, Jane S, et al. Mesenteric venous thrombosis with protein S deficiency. Am J Gastroenterol 1993;88:2143. [PubMed] [Google Scholar]
- [20].Ji M, Yoon SN, Lee W, et al. Protein S deficiency with a PROS1 gene mutation in a patient presenting with mesenteric venous thrombosis following total colectomy. Blood Coagul Fibrinolysis 2011;22:619–21. [DOI] [PubMed] [Google Scholar]
- [21].Garcia d FP, Fuentes-Prior P, Hurtado B, et al. Molecular basis of protein S deficiency. Thromb Haemost 2007;98:543–56. [PubMed] [Google Scholar]
- [22].Pintao MC, Ribeiro DD, Bezemer ID, et al. Protein S levels and the risk of venous thrombosis: results from the mega case-control study. Blood 2013;122:3210–9. [DOI] [PubMed] [Google Scholar]
- [23].Marlar RA, Gausman JN. Protein s abnormalities: a diagnostic nightmare. Am J Hematol 2011;86:418–21. [DOI] [PubMed] [Google Scholar]
- [24].Wypasek E, Undas A. Protein C and protein S deficiency: practical diagnostic issues. Adv Clin Exp Med 2013;22:459–67. [PubMed] [Google Scholar]
- [25].Lipe B, Ornstein DL. Deficiencies of natural anticoagulants, protein C, protein S, and antithrombin. Circulation 2011;124:e365–8. [DOI] [PubMed] [Google Scholar]
- [26].Heit JA. Thrombophilia: common questions on laboratory assessment and management. Hematology Am Soc Hematol Educ Program 2007;127–35. [DOI] [PubMed] [Google Scholar]