

Abstract
Rationale:
Dyskeratosis congenita (DC) is a rare inherited disease characterized by the classical mucocutaneous triad. Pulmonary fibrosis, bone marrow failure, and solid tumors are the main causes of mortality in DC. Pathogenic variants in TERT, TERC, and DKC1 have been identified in individuals with familial pulmonary fibrosis. Mutations in TINF2 gene have been reported to be associated with bone marrow failure in most cases. However, the relationship between TINF2 mutation and pulmonary fibrosis is not yet clear.
Patient concerns:
Here, we report the case of a 32-year-old woman presented with irritating cough for 2 years and progressive breathlessness for 6 months.
Diagnoses:
The patient was diagnosed with DC based on the following clinical evidences. Along with some family members, she had the typical mucocutaneous triad and pulmonary fibrosis. A heterozygous mutation (c.844C>T), located in exon 6 of TINF2 gene, that changed arginine to cysteine (Arg282Cys) was identified in this proband by whole exome sequencing.
Interventions:
The patient received corticosteroid therapy but refused to receive lung transplantation.
Outcomes:
The proband died of respiratory failure 4 months after the diagnosis. The missense mutation was located in the conserved region of TINF2 gene and predicted to be deleterious by altering the protein structure.
Lessons:
Lung transplantation should be considered for improved survival of patients with DC, and pulmonary fibrosis. Whole exome and whole genome sequencing should be widely used in the identification of such rare genetic variants for clinical diagnosis. The study of DC with pulmonary fibrosis can provide a more appropriate means of clinical research and therapy to the unfortunate patients who suffer from this rare disorder.
Keywords: dyskeratosis congenita, mutation, pulmonary fibrosis, TINF2 gene, whole exome sequencing
1. Introduction
Classic dyskeratosis congenita (DC) is a rare genetic disease, characterized by typical clinical manifestations of abnormal skin pigmentation, mucosal leukoplakia, and nail dystrophy. Bone marrow failure is the main cause of early death, and there are also malignant and fatal pulmonary complications.[1] Pulmonary fibrosis, which is a known complication of DC or any telomere biology disease, occurs in about 20% of patients.[2] The different kinds of molecular abnormalities are associated with the poor prognosis of the disease and the increased incidence of malignant tumor, resulting in the emerging concept of idiopathic pulmonary fibrosis (IPF) being a “clinically malignant pulmonary disorder.”[3] The unifying characteristics of all DC patients are abnormal short telomere lengths and telomere biological defects.[4,5] Up to now, CTC1, ACD, NHP2, DC1, PARN, NOP10, TERC, RTEL1, TINF2, TERT, and WRAP53 are the genes, pathogenic variants of which are known to cause DC and very short telomeres. Among these genes, a TINF2 gene mutation was first identified in DC in 2008. In most of the previously reported cases, mutations in the TINF2 gene have been reported to be associated with bone marrow failure. The functional consequences of TINF2 pathogenic variants are not yet known. Most patients with DC, having TINF2 gene mutations, have a high morbidity of aplastic anemia. This study analyzed the clinical phenotype of a pedigree with DC and detected pathogenic mutations by whole exome sequencing (WES) of a proband, affected by pulmonary fibrosis. We investigated whether TINF2 could be a disease-causing gene for pulmonary fibrosis, in order to increase our knowledge about the pathogenesis of pulmonary fibrosis.
2. Case presentation
Informed written consent was obtained from the patient for participation in the study. The institutional review board approved this study (conducted at the First Affiliated Hospital of Sun Yat-sen University). Her extended family had undergone genetic counseling, but declined genetic testing. This family was nonconsanguineous and of Chinese ancestry. At the time of admission, the proband was a 32-year-old woman. She had been complaining of a dry irritating cough for 2 years and progressive breathlessness on exertion for 6 months.
She had no exudative retinopathy, growth retardation, immune deficiency, or cancer. Physical examination revealed reticulated hypopigmentation of the body, nail dystrophy, and tongue leukoplakia (Fig. 1A–C). Computed tomography of chest showed subpleural, basal predominance of reticular abnormalities and honeycombing with traction bronchiectasis (Fig. 1D). Her pulmonary function test showed severe restrictive ventilator dysfunction. Forced vital capacity was 32.46% of the predicted value, and the forced expiratory volume in first second was 36.81% of the predicted value. A right heel rash biopsy showed obvious pathological hyperkeratosis, focal acantholysis, squamous epithelial hyperplasia, and epithelial foot extension. Some members of her family were also found to have all 3 features of the mucocutaneous triad.
Figure 1.
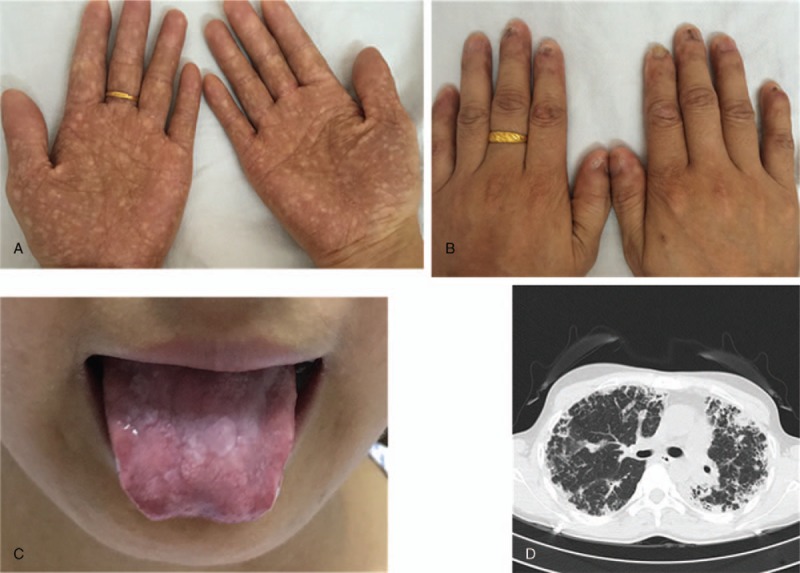
Clinical features of the proband with dyskeratosis congenita. (A) Abnormal skin pigmentation, (B) nail dystrophy, (C) oral leukoplakia, (D) chest computed tomography showing subpleural, basal predominance of reticular abnormalities and honeycombing with traction bronchiectasis.
We conducted genetic counseling for the kindred with DC. The DC disorder clearly followed a pattern of autosomal dominant inheritance in this family (Fig. 2). She recalled her father, I-1, having reticulated hypopigmentation, nail dystrophy, and eventually dying of respiratory failure at the age of 41. Medical files of I-1 were not available for review. Her brother and sister were diagnosed with DC and pulmonary fibrosis. The son of her brother (III-1) and the son of her sister (III-2) were diagnosed with DC without pulmonary fibrosis. Subject III-2 was diagnosed with aplastic anemia at the age of 5. Normal blood counts were found in other family members.
Figure 2.
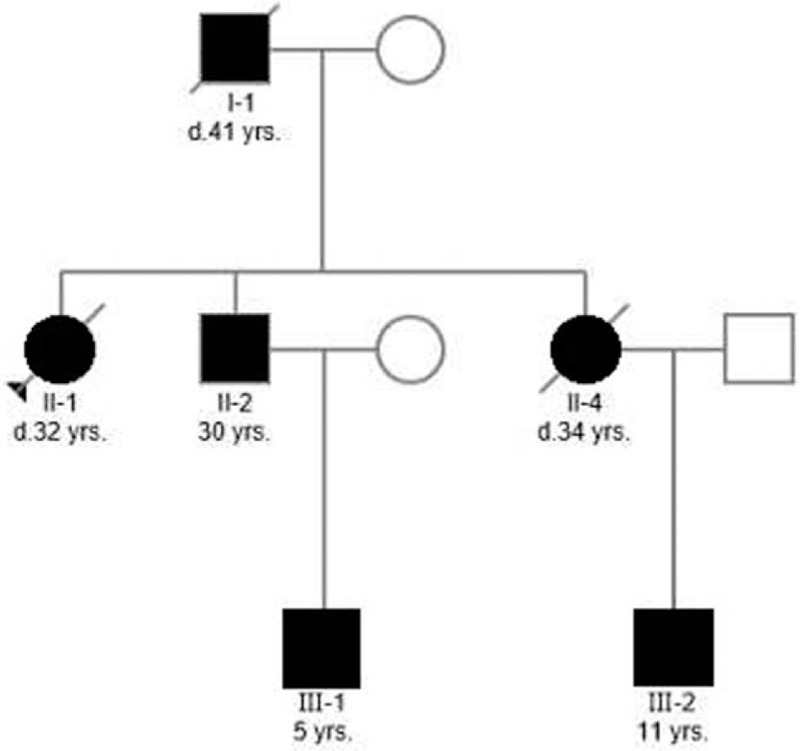
Pedigree of a Chinese family with dyskeratosis congenita. This study represents a 3-generation pedigree in which individuals from 2 generations (I-1, II-1, II-2, and II-4) had pulmonary fibrosis. Individuals III-1 and III-2 had abnormal skin pigmentation, nail dystrophy, and oral leukoplakia, but no pulmonary fibrosis; III-2 was diagnosed with aplastic anemia. Affected subjects are denoted in black. The proband is indicated by an arrow. Strikethrough symbol (/) denotes individuals who are no longer living.
A peripheral blood sample was collected from the proband (II-1), and genomic DNA was extracted with phenol–chloroform method. A total of 20 unrelated Chinese employees were included as normal controls, who neither had clinical symptoms nor family history of DC. Next, WES was performed with an Illumina HiSeq 2500 instrument by Macrogen Inc (Seoul, Korea). The preparation of samples was according to the guide of Agilent SureSelect Target Enrichment Kit (Agilent Technologies, California). Sequence reads obtained were mapped to the human genome GRCh37/hg19 assembly using the BWA software and analyzed by the Picard-tools-1.118, GATK.v4, and SnpEff_v4.1.software.[6–8] The mean depth of target regions was 141X. Variants were annotated using dbSNP142, 1000 Genome, ESP, ClinVar, and our in-house database. Finally, candidate pathogenic variants identified by WES were verified with Sanger sequencing.
Three-dimensional structures’ templates of wild type (WT) and mutant TINF2 were searched, and models were constructed with SWISS-MODEL. Three-dimensional structure estimation indicated a decrease in the area of β-strand in the TINF2 mutant protein. Furthermore, its α-helical content increased by 1% compared with the WT TINF2 (Fig. 3). This structural analysis further supported the fact that the TINF2 gene mutation, identified in the proband, strongly affected the function and/or stability of the protein.
Figure 3.
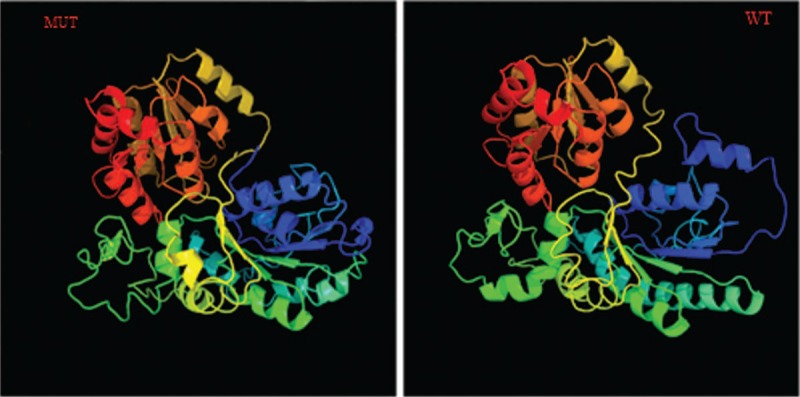
Three-dimensional structure prediction of the wild type and mutant TINF2.
The proband (II-1) was heterozygous for a C-to-T mutation at exon 6 in TINF2 gene (Fig. 4), resulting in an amino acid change from arginine to cysteine at amino acid 282. DKC1, TERC, and TERT mutations were not identified in the proband. This identified mutation (c.844C>T) was not found in the controls. The proband received corticosteroid therapy but refused to receive lung transplantation. Other family members did not receive corticosteroid therapy. The proband died of respiratory failure 4 months after the diagnosis. Patient II-4 died of respiratory failure half a month earlier than the proband.
Figure 4.
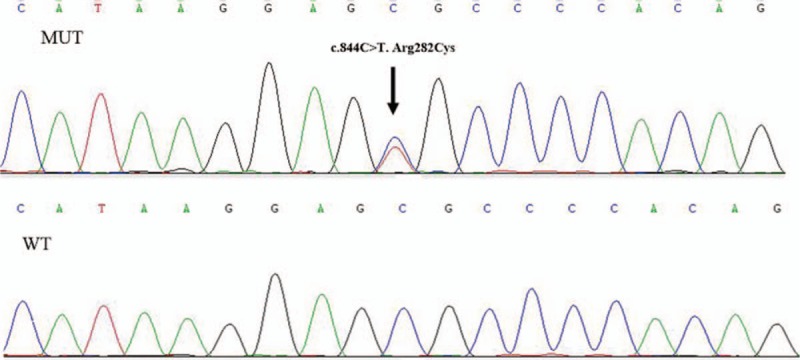
DNA sequences of wild type (left) and mutant (right) were validated by Sanger sequencing. A heterozygous missense mutation (c.844C>T), located in exon 6 of TINF2 gene, was identified in the proband.
3. Discussion
We report the clinical features of a Chinese pedigree affected by DC, with pulmonary fibrosis, followed by WES analysis of the proband. A heterozygous mutation (c.844C>T), located in exon 6 of the TINF2 gene which changes arginine to cysteine (Arg282Cys), was identified. The diagnosis of DC was established in the proband with identification of a TINF2 gene mutation by WES and the presence of typical mucocutaneous triad. In the proband, the TINF2 gene mutation was inherited from her father, because the latter had abnormal skin pigmentation and lung involvement. In this pedigree, DC caused by mutation of TINF2 gene was inherited in an autosomal dominant manner. The risk to the siblings of the proband was 50%, although, all the siblings of the proband were clinically affected. Their offsprings also had a 50% chance of inheriting the DC-related pathogenic variant. Due to the lack of understanding of this disease and the absence of prenatal diagnosis, all the offsprings of the affected father (I-1) along with some of his grandchildren are suffering from DC.
More than 50% of the patients with DC have mutations in genes identified for telomere maintenance (TERT, MIM 187270; DC1, MIM 30126; TERC, MIM602322; TINF2, MIM 604319; NOP10, MIM 606471; NHP2, MIM 606470; or WRAP53, MIM 612661).[1]TINF2 gene encodes TINF2, which is a part of the shelterin complex. Without the protection of shelterin, telomeres are no longer hidden by DNA repair mechanism, and the ends of chromosome are mishandled by the DNA repair pathways. About 11% of all cases with DC have been reported to be related to TINF2 gene.[9] TINF2, like other important telomere genes, is limited in nucleotide diversity and is conserved in evolution.[10] Mutations located in highly conserved genomic regions are believed to be pathogenic.[11] The functional consequences of pathogenic TINF2 variants are yet to be revealed. Mutations in TINF2 gene, in most of the previously reported cases, have been found associated with bone marrow failure. Only Fukuhara et al had reported a patient with DC along with pulmonary fibrosis, who was identified with a different TINF2 gene mutation (n871–874 tetranucleotide AGGA deletion).[12] Most patients with DC with TINF2 gene mutations are reported to have severe consequences, and compared with other DC genes, patients with TINF2 gene mutations have a higher morbidity of aplastic anemia before the age of 10.[9] However, in this Chinese pedigree with DC, adult patients had pulmonary fibrosis, rather than aplastic anemia; only 1 boy in the next generation was diagnosed with aplastic anemia. This report proves that the heterozygous mutation (c.844C>T) in TINF2 cannot only cause aplastic anemia but also IPF in DC.
From the basic scientific standpoint, DC may be considered a direct example of the impact of dysfunctional telomere biology on humans. It is difficult to replicate even in ordinary animal models such as mice.[13] The prognosis of patients with IPF is similar to that of stages 1 and 2 nonsmall cell lung cancer. The median survival time was <3 years after diagnosis.[14,15] In that context, the pedigree with DC and pulmonary fibrosis provides a good system for studying the intricacies and functional roles of telomere biology.
Herein, we described a unique pedigree with DC, whose main clinical presentations were pulmonary fibrosis and mucosal changes. We identified a missense mutation (c.844C>T), located in exon 6 of TINF2 gene, that changes an arginine to cysteine (Arg282Cys) in the proband. This identified heterozygous mutation was responsible for the pathogenesis of DC with pulmonary fibrosis. WES and whole genome sequencing should be widely used in the identification of such rare genetic variants for clinical diagnosis. The study of DC with pulmonary fibrosis can provide a more appropriate means of clinical research and therapy to those patients who suffer from this rare disorder.
Furthermore, both clinicians and patients should realize the importance of genetic counseling. Individuals with DC or with a family history of DC should be followed closely by a perinatologist during pregnancy. Genetic counseling and testing should be provided to help couples make informed medical and personal decisions. Lung transplantation should be considered for improved survival in patients with DC and IPF. We believe that more such pedigrees should be explored and studied to shed light on this devastating disease.
Author contributions
Conceptualization: Canmao Xie, Hongchun Du, Yubiao Guo.
Data curation: Canmao Xie, Hongchun Du, Yubiao Guo, Kejing Tang.
Investigation: Canmao Xie, Hongchun Du, Yifeng Luo.
Methodology: Hongchun Du, Yifeng Luo.
Project administration: Hongchun Du, Di Ma.
Software: Decheng Cai.
Writing – original draft: Hongchun Du, Canmao Xie.
Writing – review and editing: Hongchun Du, Canmao Xie, Di Ma, Yubiao Guo, Kejing Tang.
Footnotes
Abbreviations: DC = dyskeratosis congenita, IPF = idiopathic pulmonary fibrosis, WES = whole exome sequencing, WT = wild type.
The authors have no funding and conflicts of interest to disclose.
References
- [1].Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol 2000;110:768–79. [DOI] [PubMed] [Google Scholar]
- [2].Ballew BJ, Savage SA. Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol 2013;6:327–37. [DOI] [PubMed] [Google Scholar]
- [3].Vancheri C, Failla M, Crimi N, et al. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J 2010;35:496–504. [DOI] [PubMed] [Google Scholar]
- [4].Walne AJ, Marrone A, Dokal I. Dyskeratosis congenita: a disorder of defective telomere maintenance? Int J Hematol 2005;82:184–9. [DOI] [PubMed] [Google Scholar]
- [5].Alter BP, Baerlocher GM, Savage SA, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood 2007;110:1439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAM tools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Walne AJ, Vulliamy T, Beswick R, et al. TINF2 gene mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood 2008;112:3594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Savage SA, Stewart BJ, Eckert A, et al. Genetic variation, nucleotide diversity, and linkage disequilibrium in seven telomere stability genes suggest that these genes may be under constraint. Hum Mutat 2005;26:343–50. [DOI] [PubMed] [Google Scholar]
- [11].Zhu Y, Spitz MR, Amos CI, et al. An evolutionary perspective on single-nucleotide polymorphism screening in molecular cancer epidemiology. Cancer Res 2004;64:2251–7. [DOI] [PubMed] [Google Scholar]
- [12].Fukuhara A, Tanino Y, Ishii T, et al. Pulmonary fibrosis in dyskeratosis congenita with TINF2 gene mutation. Eur Respir J 2013;42:1757–9. [DOI] [PubMed] [Google Scholar]
- [13].Kirwan M, Dokal I. Dyskeratosis congenita, stem cells and telomeres. Biochim Biophys Acta 2009;1792:371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Raghu G, Weycker D, Edelsberg J, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006;174:810–6. [DOI] [PubMed] [Google Scholar]
- [15].Hubbard R, Johnston I, Britton J. Survival in patients with cryptogenic fibrosing alveolitis: a population-based cohort study. Chest 1998;113:396–400. [DOI] [PubMed] [Google Scholar]