

Summary
We analyzed molecular data on 2,579 TCGA tumors of four gynecological types plus breast. Our aims were to identify shared and unique molecular features, clinically significant subtypes, and potential therapeutic targets. We found 61 somatic copy number alterations (SCNAs) and 46 significantly mutated genes (SMGs). Eleven SCNAs and eleven SMGs had not been identified in previous TCGA studies of the individual tumor types. We found functionally significant estrogen receptor-regulated lncRNAs and gene/lncRNA interaction networks. Pathway analysis identified subtypes with high leukocyte infiltration, raising potential implications for immunotherapy. Using 16 key molecular features, we identified five prognostic subtypes and developed a decision tree that classified patients into the subtypes based on just six features that are assessable in clinical laboratories.
Keywords: Gynecologic cancer, breast cancer, ovarian cancer, uterine cancer, cervical cancer, uterine carcinosarcoma, TCGA, The Cancer Genome Atlas, molecular features, Pan-Gynecologic, omics
Introduction
Gynecologic cancers share a variety of characteristics: they arise from similar embryonic origins in the Müllerian ducts, their development is influenced by female hormones, and they are managed by a particular medical specialty, gynecologic oncology, as reflected in the departmental organizations of academic medical centers (Mullen & Behringer, 2014). Recently, similarities at the molecular level have been identified across gynecologic and breast cancers in a comprehensive analysis of all 33 TCGA tumor types (Hoadley et al., 2018). Despite the commonalities, however, the various gynecologic cancer types do differ from each other in a variety of intriguing and important ways. The principal aims of the present study are to highlight both similarities and differences among types and subtypes of gynecologic cancers, in addition to the ways in which they differ from non-gynecologic cancers. Because breast tumors share most of the generic characteristics listed above, we have chosen to include them in the analysis.
The study focuses on the following five TCGA tumor types: high-grade serous ovarian cystadenocarcinoma (OV), uterine corpus endometrial carcinoma (UCEC), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), uterine carcinosarcoma (UCS), and invasive breast carcinoma (BRCA). Although each Pan-Gyn organ site is subject to a variety of uncommon histologic cancer subtypes not studied by TCGA, the most frequent and/or aggressive tumors are represented. Despite impressive recent advances in diagnosis and management, these tumors share unmet needs for effective treatment. The analyses here can provide background biological information and prompt hypotheses about therapeutic choices or provide evidence for pre-existing hypotheses.
Taken together, the Pan-Gyn cohort reflects a projected incidence of more than 350,000 cases in the United States in 2017 (Siegel et al., 2017), with many more worldwide. Many of the commonalities and differences among cancer types and subtypes presented here were not identified in the individual TCGA disease-type projects (Cancer Genome Atlas Research Network, 2011; Cancer Genome Atlas Research Network, 2012; Cancer Genome Atlas Research Network et al., 2013; Cherniack et al., 2017; Cancer Genome Atlas Research Network, 2017).
Results
We used data generated from 2,579 TCGA patient samples (the “Pan-Gyn” cohort; n=1,087 BRCA, 308 CESC, 579 OV, 548 UCEC, and 57 UCS) using fresh-frozen primary samples prior to any chemo- or radiation therapy. All sample collections were approved by local Institutional Review Boards. We analyzed data of multiple types, including clinical, somatic copy number alterations (SCNAs), mutations, DNA methylation, and expression of mRNA, microRNA (miRNA), long non-coding RNA (lncRNA), and proteins. The data were adjusted for batch effects before further analysis (see STAR Methods). Here, we 1) present results that distinguish Pan-Gyn from the rest of the TCGA tumor types, 2) summarize platform-specific analysis results, and 3) propose cross-tumor type subtypes with potential prognostic and therapeutic value.
Molecular features that distinguish Pan-Gyn from other tumor types
We identified molecular features that differed in frequency among the five Pan-Gyn tumor types and the remaining 28 TCGA non-gynecologic (non-Gyn) tumor types (https://tcga-data.nci.nih.gov/docs/publications/tcga/). After adjusting for sample size per tumor type, we found 23 genes (including ARID1A, ERBB3, BRCA1, FBXW7, KMT2C, PIK3CA, PIK3R1, PPP2R1A, PTEN, and TP53) that were mutated at higher frequencies across the Pan-Gyn tumor types than across the non-Gyn types (FDR < 0.01, Fisher’s Exact test) (Figure 1A). Eighteen of those genes were found to be significantly mutated genes (SMGs) in the Pan-Gyn cohort (as described later).
Figure 1. Genomic features that distinguish Pan-Gyn from other tumor types.
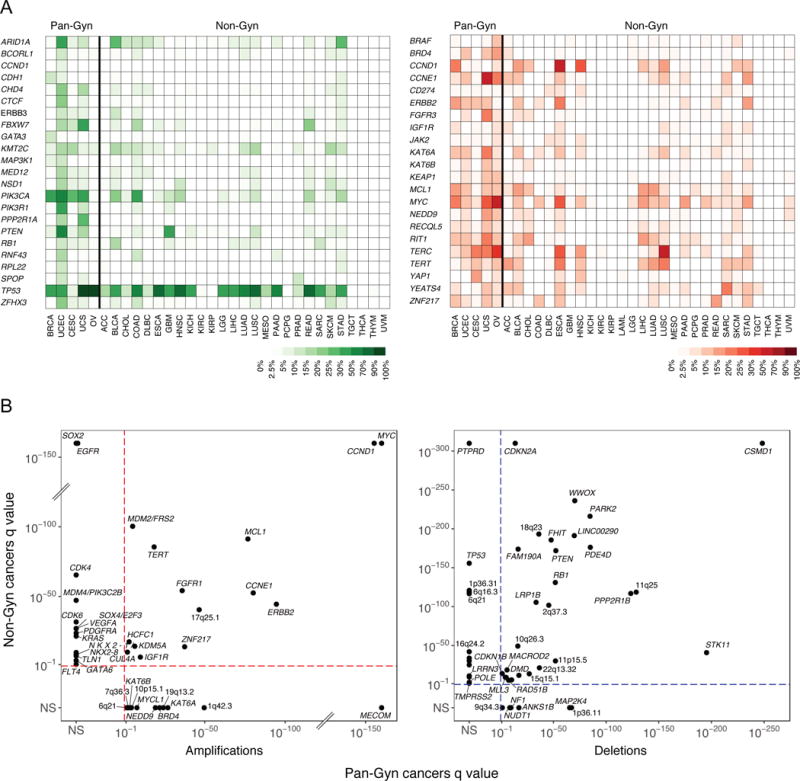
(A) Heat map showing the frequencies of mutations (green) in 23 genes across all 33 TCGA tumor types and frequencies of amplifications (red) in 23 genes across all 33 TCGA tumor types. (B) Amplification (red) and deletion (blue) q values from GISTIC2.0 for SCNA peaks of significant copy number gain and loss plotted for Pan-Gyn vs. non-Gyn cohorts. Genes named are the suspected targets of amplification or deletion, if identifiable. Otherwise, peaks are labeled with the nearest cytoband’s designation. Peaks found in only one cohort were assigned values of NS (not significant) in the other cohort. See also Figure S1 and Table S1.
Next, we used GISTIC2.0 (Mermel et al., 2011) to identify statistically significant recurring SCNAs in the Pan-Gyn cohort and, separately, in the non-Gyn cohort. We identified 61 significant regions in the Pan-Gyn tumors, 27 amplifications and 34 deletions, of which 12 amplifications and 6 deletions were not found in the non-Gyn cohort, suggesting relative specificity for Pan-Gyn tumors (Figure 1B, Figure S1A & Table S1). Two of the 12 uniquely Pan-Gyn amplifications and one of the six deletions had not previously been reported in single-disease TCGA studies of the same tumor types (Cancer Genome Atlas Research Network, 2011; Cancer Genome Atlas Research Network, 2012; Cancer Genome Atlas Research Network et al., 2013; Cherniack et al., 2017; Cancer Genome Atlas Research Network, 2017). One of the previously unreported amplifications was a focal region in 1q42.3 covering IRF2BP2, which encodes an interferon regulatory factor binding protein that is implicated in cellular differentiation, proliferation, and survival processes (Stadhouders et al., 2015). The other unreported amplification, located in 10p15.1, included an intergenic noncoding region downstream of KLF6 that bears striking resemblance to known oncogenic super-enhancer regions (Zhang et al., 2016) and PFKFB3, a gene which is being investigated as a therapeutic target in various cancers (Cantelmo et al., 2016; Li et al., 2017; Peng et al., 2018). The deletion consisted of a ~7 MB region in 9q34.3 that contains the tumor suppressor genes TSC1 and NOTCH1.
Figure 1B and Figure S1A depict suspected targets of the significant SCNAs in the Pan-Gyn and non-Gyn cohorts without adjusting for sample size per tumor type. MECOM, KAT6A, BRD4, NEDD9, MYCL1, and KAT6B were selectively amplified in the Pan-Gyn cohort, whereas SOX2, EGFR, CDK4, MDM4, and CDK6 were selectively amplified in the non-Gyn cohort. MAP2K4 and NF1 were notable tumor suppressor genes with recurring copy number losses specific to Pan-Gyn tumors, whereas PTPRD, RBFOX1, and TP53 were among the tumor suppressors more commonly deleted in non-Gyn samples. Significantly recurring deletions were found in known or putative fragile site genes, including LRRN3 (7q31.1) in non-Gyn, ANKS1B (12q23.1) in Pan-Gyn, and RAD51B (14q24) in both cohorts (McAvoy et al., 2007; Miron et al., 2015). Adjusting for sample size per tumor type, we identified 23 oncogenes among the genes in the 27 Pan-Gyn amplification regions that were consistently more frequently amplified across the five Pan-Gyn tumor types than across the non-Gyn types (FDR < 0.05, Fisher’s Exact test) (Figure 1A). We found no known tumor suppressors within the 34 somatic deletion regions that were more frequently deleted across the Pan-Gyn tumor types than across the non-Gyn types. In addition, we identified 197 genes that were statistically significantly hyper- or hypomethylated at different frequencies in the two cohorts (Figure S1B).
We performed bootstrapping-based analyses to investigate whether there were greater numbers of shared mutated or copy-number altered genes among the five Pan-Gyn tumor types vs. random sets of five tumor types. The results showed that 23 mutated genes were enriched in the Pan-Gyn tumor types vs. only 6 mutated genes expected by random chance (p = 0.10) (Figure S1C), whereas 122 SCNA genes were enriched in Pan-Gyn vs. 2 by random chance (p < 0.0001) (Figure S1D).
Individual data platform analyses
Mutation analysis
We analyzed 2,258 patient samples with mutation data from TCGA for SMGs and operative mutational processes across the Pan-Gyn tumor types. The types of mutations in the Pan-Gyn cohort are summarized in Table S2 The average mutation load varied widely by tumor type, with CESC samples having the highest median frequency (5.3 mutations/mbp). UCEC samples showed a bimodal distribution due to a subset of hyper-mutators described previously (Cancer Genome Atlas Research Network et al., 2013).
There were 46 SMGs based on the intersection of those genes identified by MutSigCV v1.4 (Lawrence et al., 2013) and those identified by previous methods (Vogelstein et al., 2013) (Figure 2A). The top five most frequently mutated genes were TP53 (44% of samples mutated), PIK3CA (32%), PTEN (20%), ARID1A (14%), and PIK3R1 (11%). Eleven of the 46 SMGs had not been previously reported in any of the TCGA gynecologic or breast marker papers (Cancer Genome Atlas Research Network, 2011; Cancer Genome Atlas Research Network, 2012; Cancer Genome Atlas Research Network et al., 2013; Cherniack et al., 2017; Cancer Genome Atlas Research Network, 2017) (Table S3). Among them, ACVR2A, a member of the TGF-β superfamily that functions in pathways implicated in both tumor progression and suppression (Ikushima & Miyazono, 2010), was the most frequently mutated (in 4.8% of the cohort). LATS1 was the next most frequently mutated (3.8%) and functions in the Hippo signaling pathway, which controls organ size, restricts proliferation, promotes apoptosis, and has been implicated in multiple cancer types (Yu et al. 2015; Deng et al. 2017). CCAR1 was mutated at 3.6%; its protein product functions as a p53 coactivator and plays roles in cell proliferation, apoptosis, and, in breast cancer, estrogen-dependent growth (Kim et al. 2008; Muthu et al. 2015). We found 220 patients (10%) that had no detectable SMGs.
Figure 2. Landscape of mutations in Pan-Gyn tumor types.

(A) Mutation profiles of 2,029 Pan-Gyn samples (columns) in which at least one somatic mutation occurred in at least one of the 46 significantly mutated genes (SMGs). (Top) Mutation burdens per sample, divided into synonymous and non-synonymous mutation types. (Middle) Types of mutations in each of the 46 SMGs per sample. (Bottom) Covariate bars showing the mutation cluster, genomic alterations in six genes from the DNA damage response pathway, and tumor type for each sample. (B) Clustered heat map showing correlations between 10 of our mutation signatures (rows labeled S1 to S10) and 30 COSMIC signatures (columns). (C) Clustered heat map of the mutation signatures (rows) present in each sample (columns) showing ten clusters. The dendrogram is color-coded by predominant COSMIC signature. See also Figure S2, and Tables S2-5.
Mutation signatures
Mutation signatures have provided insight into mechanisms underlying tumor development and have informed patient therapy (Helleday et al., 2014). Analysis by non-negative matrix factorization (NMF) on the Pan-Gyn data set suggested that 10 mutation signatures could explain nearly 90% of the variability observed in the original mutation/sample matrix (Figure S2A-B). The 10 Pan-Gyn signatures (S1 to S10) variably correlated with the 30 COSMIC signatures (http://cancer.sanger.ac.uk/cosmic/signatures) (Forbes et al., 2011) (Figure 2B). S1 correlated strongly with COSMIC signature 13 (r = 0.99) and S2 correlated with COSMIC signature 2 (r = 0.95); both signatures suggest activity of the AID/APOBEC family of cytidine deaminases. S3 correlated with COSMIC signature 1 (r = 0.94), indicating an endogenous process initiated by spontaneous deamination of 5-methylcytosine. S4 and the ultramutator COSMIC signature 10 were highly correlated (r = 0.97), presumably reflecting altered activity of POLE. A smaller correlation was found between S10 and COSMIC signature 3 (r = 0.58), associated with germline and somatic BRCA1 and BRCA2 mutations. All of the correlations were statistically significant (FDR < 0.05).
Unsupervised hierarchical clustering based on the contribution of each signature divided the Pan-Gyn samples into 10 clusters that showed associations with various molecular/clinical features (Figure 2C, Figure S2C & Table S4-5). Cluster C1 was highly enriched with OV samples (and basal BRCA and UCEC to a lesser extent) and contributed strongly to S10, a signature associated with germline and somatic BRCA1 and BRCA2 mutations that correlate with responsiveness to PARP inhibitors and platinum-based therapy (Konecny et al., 2016). C1 also had samples with frequent TP53 mutations and homozygous deletions, supporting the association with an ineffective DNA double-strand break repair COSMIC signature. C2, which contained BRCA, OV, and UCEC samples, was associated with transcriptional strand bias for T>C substitutions, whereas C3, which contained BRCA and OV samples, was associated with transcriptional strand bias for T>A mutations. C4 consisted principally of breast samples and contributed to S8, the signature most associated with COSMIC 5 (etiology unknown). C5, principally composed of UCEC tumors with high microsatellite instability and mutations in MLH1, MSH2, MSH3, or MSH6, contributed most strongly to signature S6. S6 is correlated with COSMIC signatures 6, 15, and 20, which are associated with defective DNA mismatch repair (suggesting possible sensitivity to immune checkpoint inhibitors). C9 comprised CESC and BRCA samples and represented the AID/APOBEC signatures S1 and S2, providing further evidence for enrichment of APOBEC mutagenesis in these cancers (Roberts et al., 2013). C10 was associated with POLE-mutant UCEC samples.
Somatic copy number alterations
Unsupervised hierarchical clustering of the Pan-Gyn cohort using ISAR-corrected (Zack et al., 2013) segmentation data revealed six clusters with distinct copy number profiles (Figure 3 & Table S4-5). Prominent features that distinguished the clusters included SCNAs in chr 8, 16q, and 1q, among others. OV, serous UCEC, UCS and basal-like, HER2+, and luminal B BRCA tumors clustered almost exclusively into C4 and C6. Conversely, luminal A BRCA and endometrioid UCEC samples were divided amongst all clusters, providing evidence for additional tumor subtypes beyond the traditional clinical classifications (Cancer Genome Atlas Research Network et al., 2013). C4 and C6 showed a high degree of genomic copy number instability, consistent with their prevailing TP53 mutation signatures (Ciriello et al., 2013), and contained the highest numbers of advanced stage cancers (Figure S3A). Unlike other clusters, more than 50% of the samples in C4 and C6 had undergone at least one whole genome-doubling event. C3 accounted for the largest proportion of CESC samples and uniquely exhibited a focal 11q22 amplification containing the oncogene YAP1. C2, with 74% endometrioid UCEC, contained a majority of the POLE-mutant cases and exhibited a quiet SCNA landscape with few broad-level gains or losses. C1 and C5 consisted primarily of endometrioid UCEC and luminal A BRCA tumors, accounting for 85% and 72% of the samples in the two clusters, respectively. Both clusters had similar alteration profiles genome-wide, except in the frequencies of 1q and chr 8 gains (p < 2.2e-16, Fisher’s Exact test); the former occurred twice as frequently in C1 and the latter seven times as frequently in C5. Overall, gain of 1q was the most frequent chromosomal arm-level event, occurring in 49.5% of samples across all five Pan-Gyn cancer types. Other frequently recurring arm-level events included gain of 3q, 8q, and chr 20, and loss of 4p, 13q, 16q, 17p, and 22q.
Figure 3. Clustered heat map of significantly recurring SCNAs as determined by GISTIC2.0 analysis across Pan-Gyn cancers.

The heat map shows SCNAs in tumor samples (columns) plotted by chromosomal location (rows). Red and blue indicate amplifications and deletions, respectively. See also Figure S3 and Tables S4-5.
DNA methylation
Unsupervised clustering of 2,586 cancer-specific, hyper-methylated loci across all Pan-Gyn tumors revealed heterogeneity of DNA methylation patterns (Figure S3B & Table S4-5). Unsurprisingly, tumor samples from the same tissue of origin (e.g., OV, UCS, or CESC) clustered together with the exception of two major groups, which were found to be highly robust via cluster stability analysis (83% and 90% for left and right branches respectively) (Figure S3C-D). The left branch with lower degrees of hypermethylation consisted of the majority of OV and UCS, normal and basal-like BRCA, and microsatellite stable UCECs (both endometrioid and serous subtype). The hypermethylator (right) cluster included most CESC tumors, the majority of BRCA, and microsatellite unstable UCEC. The 7 cluster resolution was retained when perturbing samples across all of the TCGA Pan-Can cohort (Figure S3E), with a small subset of UCEC samples reassigned. C7 (mostly CESC) had the highest degree of hypermethylation across all tumor types in the study, followed by a luminal B BRCA-rich C4, which also consisted of HER2+ and a small fraction of basal-like BRCA. Within tumor subtype (e.g., endometrioid UCEC), the heterogeneity of DNA methylation patterns identified samples that showed greater deficiency in DNA mismatch repair pathways (via MLH1 silencing). Hypermethylation and concomitant down-regulation of two genes in the Homologous Repair (HR) pathway, BRCA1 and RAD51C, were observed almost exclusively in OV (12.7% and 3.0%, respectively) and basal-like BRCA cancers (2.8% and 2.6%, respectively).
mRNA analysis
Unsupervised hierarchical clustering of 1,860 previously defined cancer genes (Sadelain et al., 2012) in 2,296 Pan-Gyn samples resulted in the identification of nine mRNA clusters with distinct clinicopathologic characteristics (Figure 4A & Table S4-5). Both C1 and C2 were BRCA-enriched, and C2 consisted of the majority of HER2+ and normal-like tumors. C2 was also significantly enriched with infiltrating lobular carcinomas, whereas over 95% of cases in C4 were basal-like ductal BRCA. C5 consisted mainly of OV and serous-like UCEC, a similarity previously noted (Cancer Genome Atlas Research Network et al., 2013). Over 50% of cases in C7 were UCS and, given its high EMT signature (Cherniack et al., 2017), C7 therefore likely exhibits EMT characteristics. Overall, the Pan-Gyn mRNA subtypes showed prognostic value, even after adjusting for lineage (p < 0.0001, chi-squared test) (Figure 4B). UCEC, in particular, appeared in five of the nine clusters and exhibited significant differences in overall survival, depending on cluster membership (Figure 4C).
Figure 4. mRNA expression clusters and their association with overall survival.
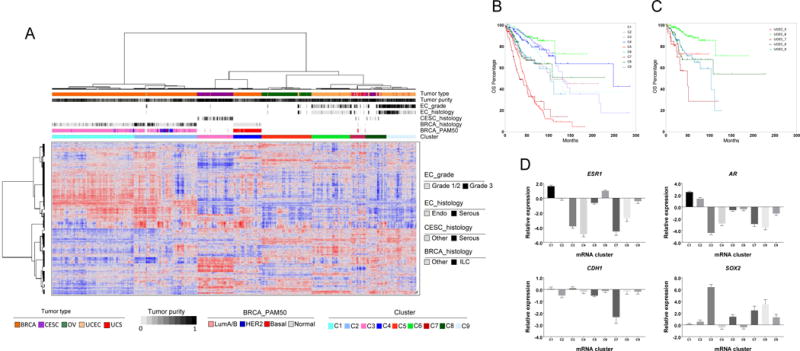
(A) Unsupervised hierarchical clustering of previously reported cancer genes identifies nine mRNA-based subtypes/clusters. Clinical and molecular features are indicated by the annotation bars above the heat map. (B) Overall survival for each of the gene expression clusters (chi-squared test p value < 0.0001, adjusted for differences in tumor type survival rates). (C) Overall survival for endometrial cancer (UCEC) patients in the gene expression clusters (log rank test p value < 0.0001). (D) Differential expression of ESR1, AR, SOX2, and CDH1 in different clusters (Kruskal-Wallis test p values < 0.0001 for all 4 genes). The bars represent mean expression of the gene (log2 scale) in each cluster, together with the upper or lower 95% confidence interval (whiskers above or below the bars, respectively). See also Tables S4-5.
We investigated which genes were differentially expressed among the clusters (Figure 4D). ESR1 and AR were significantly higher in C1 and C2 than in others, whereas C3 had high expression of SOX2. C3 consisted of cervical cancer samples with squamous histology, characterized by 3q26 amplification (the SOX2 gene loci). C7 had significantly lower expression of the classical epithelial marker CDH1, which is consistent with an EMT signature.
Proteomic analysis
Unsupervised hierarchical clustering of protein expression data for 1,967 samples across 216 proteins identified 5 clusters (Figure S4A & Table S4-5). C1 principally consisted of non-basal BRCA, C3 was enriched with endometrioid UCEC, and C4 was enriched with OV. Interestingly, C2 and C5 contained a mixture of samples across multiple disease types. C2 had high levels of caveolin1, MYH11, and HSP70 proteins, which have previously been identified as biomarkers for the reactive subtype found in luminal BRCA (Cancer Genome Atlas Research Network et al., 2012). In addition to luminal BRCA samples, C2 included some basal-like BRCA, CESC, OV, and UCEC samples (but not UCS). Cluster C5 contained most of the basal-like BRCA, squamous CESC, serous UCEC, UCS, and 10% of the serous OV samples. It had a low hormone receptor pathway score (Akbani et al., 2014) and high levels of cell cycle and DNA damage response activity, features that could indicate sensitivity to drugs that target DNA damage repair.
miRNA analysis
Unsupervised hierarchical clustering of the 293 most variable miRNAs in 2,417 samples grouped samples largely by disease type (Figure S4B-D & Table S4-5). The miRNA profile for OV, however, was especially distinct from other Pan-Gyn tumor types. Basal-like BRCA samples were more similar to CESC (C6), UCEC and UCS samples (C4 and C5) than they were to the non-basal BRCA subtypes in C2 and C3.
lncRNAs
We processed raw RNA-seq data to extract 1,986 lncRNAs that were predicted to regulate the 216 cancer-related proteins profiled by TCGA across four of the five tumor types (UCS did not have sufficient samples for the lncRNA extraction). An unsupervised consensus clustering of the data revealed six clusters (L1 to L6) that coincided significantly with protein-based clusters (C1 to C5) (p value < 0.05, Fisher’s Exact test) (Figures 5A, S4A and Table S4-5). BRCA and CESC had very similar lncRNA profiles and grouped together in clusters L2 and L3. UCEC (in L5) and OV (in L6) each had very distinct lncRNA profiles from those of BRCA and CESC. Portions of the OV (31%) and UCEC (11%) samples were both present in cluster L4.
Figure 5. lncRNA clusters and gene/lncRNA interaction networks.
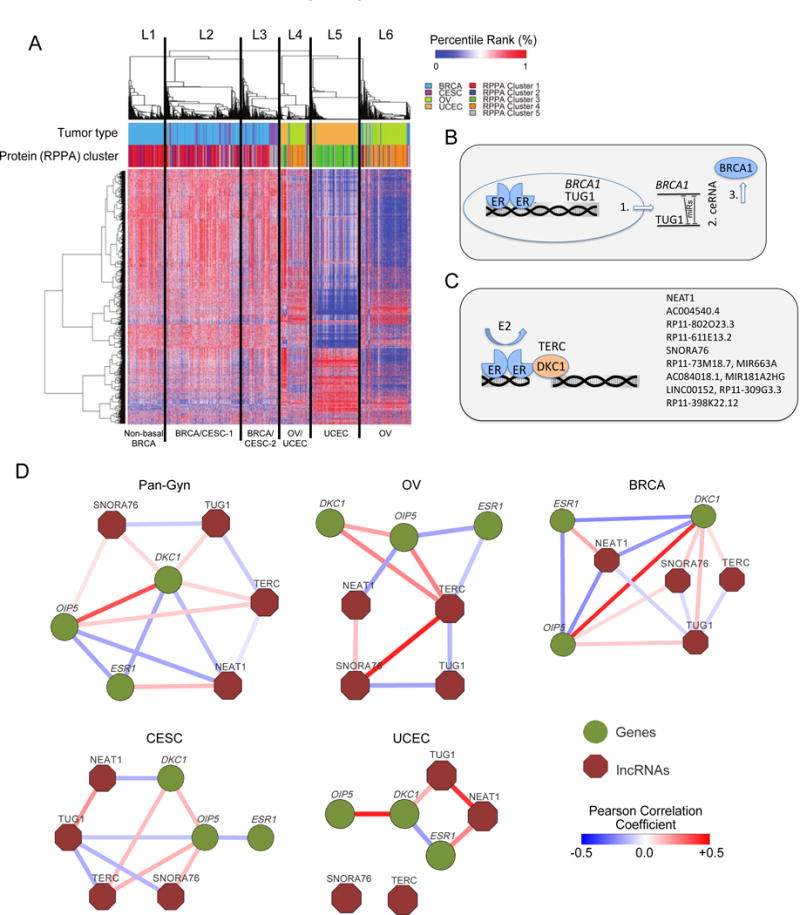
(A) Clustered heat map based on expression of cancer lncRNA regulators. The rows have 1,986 lncRNAs, whereas the columns have 1,597 samples. L1-L6 indicate the six clusters and their association with protein clusters is shown (p value < 0.05, Fisher’s Exact test). (B) Schematic illustration of dual-layer ER-ceRNA regulation of BRCA1. ER transcriptionally regulates both BRCA1 and non-coding TUG1 in ER-positive breast cancer. Those RNAs subsequently compete for miRNA binding. (C) ER modulates the TERC-DKC1 complex and its transcriptional activity. Estradiol (E2)-activated ER binds to cis-regulatory DNA regions of both DKC1 and TERC and regulates their activity. Further, ER binds to regulatory regions of DKC1-regulated lncRNAs (listed on the right) and modulates their expression. (D) Gene/lncRNA interaction networks in the overall Pan-Gyn lncRNA cohort and each of the four individual disease types. The nodes represent genes (green) or lncRNAs (burgundy), whereas each edge represents statistically significant Pearson’s correlation coefficient between the connected nodes. See also Figure S4-5, and Tables S4-5.
Previous studies have suggested that estrogen receptor (ER) regulates BRCA1 expression, Dyskerin (DKC1) expression (a binding partner of the lncRNA TERC), and the lncRNA TUG1 (Figure 5B) (Jonsson et al., 2015; Hurtado et al., 2011). ER binds to regulatory regions of DKC1, either to induce or to repress multiple lncRNAs (Figure 5C). In the present study, our analysis has revealed significant Pearson’s correlation (t-test p value < 0.05) between key lncRNAs and their regulator genes’ transcripts, ESR1, OIP5, and DKC1, in a context-specific manner (Figure 5D). Using Gene Set Enrichment Analysis (GSEA), we found 12.04% of the 1,537 Gene Ontology gene sets to be significantly enriched (FDR < 0.05) with TERC-correlated genes across all four cancer types (Figure S5). Included were gene sets associated with TERT and telomere maintenance and packaging as well as gene sets linked to MYC. The latter result supports earlier findings of TERC binding peaks in the MYC promoter region (Chu et al., 2011).
Pathway analysis
We performed PARADIGM pathway analysis (Vaske et al., 2010) followed by unsupervised consensus clustering of pathway scores that clustered samples primarily by tissue type, with a few notable exceptions (Figure 6A-B, Table S4-5). A subset of basal-like BRCA cancers co-clustered with a subset of UCEC and UCS in C2, whereas the remaining basal-like BRCA samples clustered with non-basal BRCA in C4. Contrary to transcriptomic analysis, pathway analysis clustered approximately half of the basal-like BRCA cancer samples together with the HER2+ and luminal B samples.
Figure 6. Pathways-based clusters.
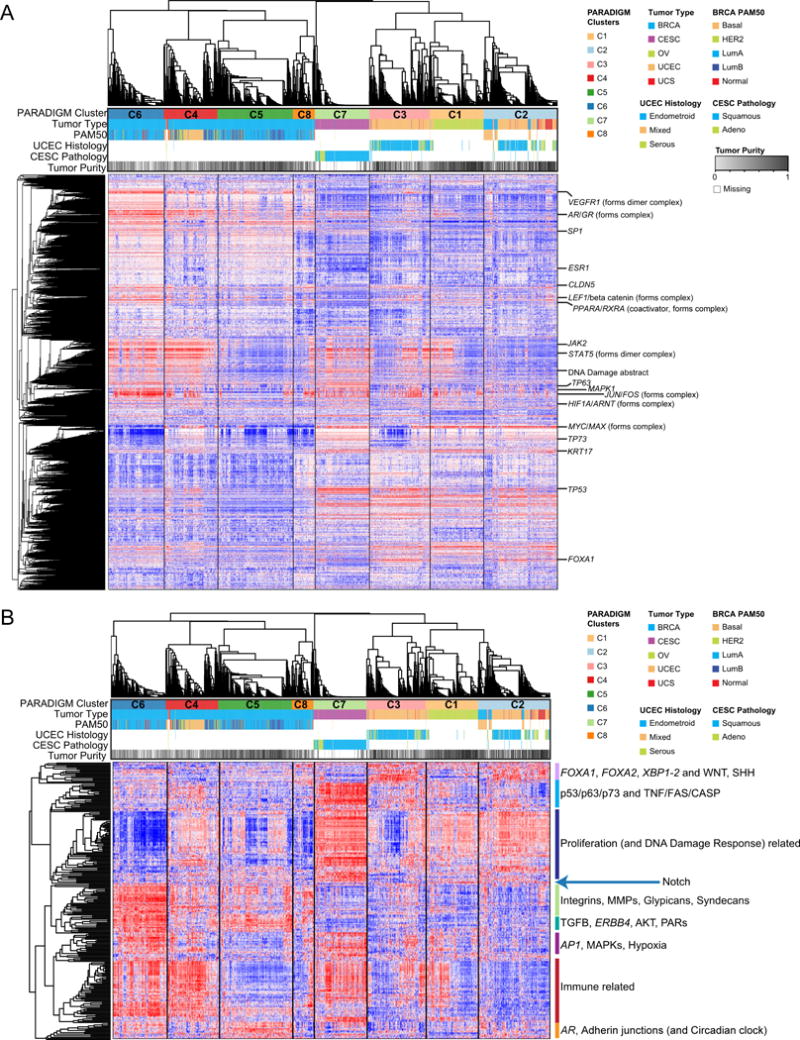
(A) Consensus Clustered heat map based on PARADIGM integrated pathway levels (IPLs). Selected pathway features with characteristic patterns of inferred activation across clusters are labeled on the rows. Samples are in columns. (B) Constituent pathways with differential single-sample gene set enrichment analysis (ssGSEA) scores across PARADIGM clusters. A comparison of ssGSEA scores of constituent pathways integrated by the PARADIGM algorithm identified 263 differentially enriched pathways across clusters. Samples are arranged in the same order as (A) and differentially expressed pathways are arranged based on unsupervised clustering of their ssGSEA scores. Dominant themes within sub-groupings of differential pathways across PARADIGM clusters are labeled. Examples of immune-related pathways include IL12, IL23, IL27, IFNG, STAT and T-cell receptor signaling pathways. Proliferation and DNA damage repair related pathways include FOXM1, PLK2, Cyclins, MYC, E2F, ATM, ATR, BARD1, and Fanconi anemia pathways. See also Tables S4-5.
All PARADIGM clusters had distinct patterns of high or low immune-related signaling, assessed by inferred activation (Figure 6A) and pathway enrichment (Figure 6B), suggesting an important role for immune response in subsets of Pan-Gyn cancers. Interestingly, the two basal-like BRCA subtypes differed between inferred activation of immune-related signaling pathways. Enrichment with adhesion-related proteins, such as the integrins, matrix metalloproteinases, and syndecans, were also distinguished between the two basal-like subtypes, suggesting distinctive tumor microenvironments. As with basal-like BRCA, UCEC split into two clusters (C2 and C3) that did not correspond to obvious variations in UCEC histology. These clusters were mainly differentiated by proliferation, Notch signaling, and immune activity levels.
Integrated analysis across Pan-Gyn tumor types
We used cluster assignments from the six major TCGA platforms (mutations, SCNA, DNA methylation, mRNA, miRNA, and protein) to perform integrated clustering across the Pan-Gyn cohort using the CoCA algorithm (Figure S6A). The resulting CoCA clusters were heavily dominated by tumor type because the intrinsic gene expression patterns were lineage dependent. The association with tumor type was especially prominent in the DNA methylation, mRNA, miRNA, and protein clusters. Therefore, we turned to an alternative method (described next) to define subtypes that would span the Pan-Gyn tumor types and emphasize high-level similarities among them.
Subtypes across the Pan-Gyn tumors
We present molecular subtypes that illuminate commonalities and distinguishing features across the Pan-Gyn tumor types, with the potential to inform future cross-tumor type therapies. We first identified 16 features (listed in STAR Methods) across 1,956 samples that were either (i) currently used in the clinic for at least one of the five tumor types, or (ii) identified as informative in previous TCGA gynecologic and breast cancer studies. Next, we clustered the feature matrix and obtained five clusters (Figure 7A, Table S4-5). SCNA load was the predominant feature and produced the first division. In the low SCNA-load-group, we found two clusters, non-hyper-mutator (C1) and hyper-mutator (C2). The non-hyper-mutator cluster had virtually no hyper-mutators but had high levels of ER+, PR+, and/or AR+ samples, indicating potential susceptibility to hormone therapies. C2, the hyper-mutator cluster, could be further subdivided into four sub-clusters (clusters C2A-C2D). C2A was enriched with POLE mutations, which have previously been associated with “ultramutators” and their extremely high mutation rates (>100 mutations/mbp) (Cancer Genome Atlas Research Network, 2013). C2B showed enrichment with MSI-high samples and C2C showed high immune-infiltration levels. C2D was depleted of hyper-mutators and showed enrichment with high immune-infiltration and HPV-positive samples. The high SCNA-load group consisted of three clusters: immune high (C3), AR- or PR-low (C4), and AR or PR high (C5). The immune high cluster showed low levels of hormone receptors and enrichment with HPV-positive samples. Interestingly, samples with ERBB2 amplification fell into two main clusters; those in clusters C3 (n = 39) and C4 (n = 30) showed high and low immune infiltration levels, respectively (purple and black rectangles in Figure 7A). C3 displayed a tendency towards better survival than C4 (hazard ratio = 2.8), with a p value that trended towards significance (p = 0.087) (Figure S6B). C4 showed low levels of AR and PR and had a sub-cluster with BRCA1 or BRCA2 somatic mutations. C5 had high levels of at least one of the three hormone receptors, again suggesting sensitivity to hormone therapies. Each cluster had varying levels of representation of samples from each disease, mitigating tissue-specificity (Figure 7B).
Figure 7. Cross-tumor type Pan-Gyn subtypes with prognostic significance.
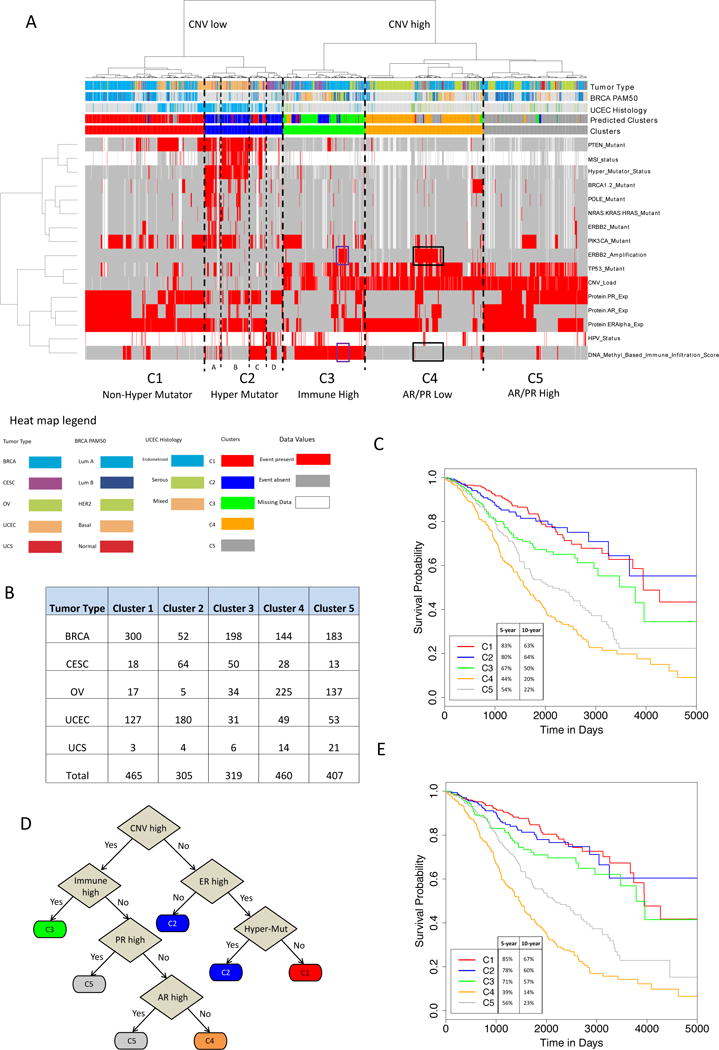
(A) Clustered heat map of 16 features across 1,956 Pan-Gyn samples. Cluster 2 is split further into four subclusters, 2A-2D. Purple rectangles highlight HER2+ samples that have high immune infiltration scores; black rectangles highlight HER2+ samples with low immune infiltration scores. (B) Cross-tabulation showing the distribution of Pan-Gyn tumor types across the five clusters. (C) Kaplan-Meier curves showing differences in overall survival among the five clusters (with 5-and 10-year survival rates shown). Before adjusting for tumor type differences in overall survival rates, the log rank test p value < 0.0001, and after adjusting for tumor type differences, p value = 0.0006 (chi-squared test). (D) Decision tree that predicts clusters using just six of the 16 features. The predicted clusters are shown in a covariate bar in the heat map in A. (E) Kaplan-Meier curves showing differences in overall survival among the five decision tree-based predicted clusters (with 5- and 10-year survival rates shown). Log rank test p values are less than 0.0001, before (log rank test) and after (chi-squared test) adjusting for tumor type differences in overall survival rates. See also Figure S6 and Tables S4-5.
We then performed overall survival analysis on the five clusters and obtained very significant survival differences among them (p < 0.0001, log-rank test) (Figure 7C). The 5-year survival rate ranged from 83% (C1) to 44% (C4), and the 10-year survival rate ranged from 64% (C2) to 20% (C4). We assessed the statistical significance of the added prognostic value of the 16-feature clusters after accounting for tumor type differences to control for effects that may be due to individual tumor type contributions; the resulting p value was still significant (p = 0.0006, log-rank test).
Finally, we used dichotomous decision tree methodology (Quinlan, 1983) to reduce the number of assessed molecular variables needed to classify patients into one of the five subtypes. The resulting tree required specification of only six of the original 16 features (Figure 7D). The tree had an accuracy of 82% predicting the original 16-feature based clusters, with a receiver-operator characteristic (ROC) area under the curve (AUC) of 0.94.
We repeated the same type of survival analysis for the clusters predicted by the decision tree as we did for the original clusters (Figure 7E). Log-rank test p values for the tumor type-unadjusted and adjusted methods were both highly significant (p < 0.0001), showing that the decision tree-based clusters retained prognostic value despite not having 100% accuracy. These survival rates were comparable to the original clusters, with a 5-year survival rate ranging from 85% (C1) to 39% (C4), and a 10-year survival rate ranging from 67% (C1) to 14% (C4).
Discussion
We performed an integrative, multi-platform analysis of the TCGA Pan-Gyn tumors based on 2,579 clinical cases. In addition to confirming the robustness of many observations cited in previous TCGA publications on the individual tumor types, our approaches also provided a considerable number of additional findings: (i) Multiple genomic and epigenomic features that help to distinguish gynecologic and breast tumors from the other 28 TCGA tumor types; (ii) 61 somatic copy number peaks in the Pan-Gyn cohort, 11 not previously reported by TCGA; (iii) three somatic copy number alterations (containing genes of potential therapeutic relevance) unique to gynecologic cancers among the 33 TCGA tumor types; (iv) 46 SMGs in the Pan-Gyn cohort, 11 not previously reported by TCGA; (v) 10 predominant mutation signatures, with 10% of the samples lacking identified SMGs; (vi) analyses of the 10 mutation signatures in relation to the 30 COSMIC signatures, demonstrating relationships between the two sets of signatures; (vii) shared similar miRNA profiles between most of the Pan-Gyn tumor types; the exception, OV, was extremely different from the rest, and, unexpectedly, the miRNA profiles of basal-like BRCA cancers closely resembled those of CESC cancers; (viii) some OV and UCEC samples exhibited the “reactive” proteomic signature previously identified and shown to be prognostically relevant in BRCA; (ix) identification of a subtype with low protein expression of ER and AR (important markers for hormone therapy) that spanned all five tumor types; (x) large-scale lncRNA analysis not performed previously for any of the TCGA gynecologic or breast marker papers; our findings included several ER-regulated lncRNAs and an ER-TERC/DKC1-NEAT1/OIP5-AS1-TUG1 gene/lncRNA network; (xi) similar lncRNA profiles in BRCA and CESC, in contrast to the very distinct profiles in UCEC and OV; (xii) lineage-specific gene expression patterns and lineage-related (but not always cancer type-specific) features revealed by multi-platform clustering of tumor samples; (xiii) pathway analyses that revealed subsets of BRCA, OV, and UCEC samples with high levels of leukocyte infiltration, a primary marker of immune response and possible susceptibility to immunotherapy; most of the CESC samples, but virtually none of the UCS samples, showed high leukocyte infiltration; (xiv) roughly half of the basal-like BRCA samples resembled luminal/HER2+ BRCA samples at the pathway level (but not the gene expression level); this pattern suggests convergence of independent gene expression changes to drive a limited number of pathway outputs and could prove useful with respect to development and selection of therapies across BRCA subtypes; (xv) five cross-Pan-Gyn subtypes defined by multi-platform clustering of 16 molecular features; these five clusters have possible clinical implications and predictive value for survival beyond that of tumor type alone; (xvi) reduction of the 16 molecular features to six in the form of a binary decision tree that retained prognostic value.
From a potential therapeutic perspective, two of the Pan-Gyn clusters (C1 and C5) in (xv) showed high levels of hormone receptors (ER, PR, and/or AR), suggesting possible responsiveness to hormone therapy. C3 showed high levels of immune markers, warranting further exploration for possible value in selecting patients for immunotherapy. C2 included hyper-mutators and ultramutators, which have been associated with relatively good survival on conventional therapy. A subset of C4 showed ERBB2 amplification, suggesting possible responsiveness to HER2-targeted therapy. ERBB2 mutation and amplification were mutually exclusive, but both sets of tumors might benefit from HER2-targeted therapy.
The decision tree we propose could potentially enable clinicians to classify patients more easily into one of the five Pan-Gyn subtypes. The tree is based on six features, three of which (ER, PR, and AR status) are already routinely used in the clinic. Widely available CLIA-certified gene-panel assays can estimate SCNA and mutation loads, and immune infiltration can be assessed by standard immunohistochemistry or new imaging technologies. Therefore, after further study and validation, our decision tree might be able to aid in assignment of patients to treatment groups. It should be understood, however, that all of the clinically interesting possibilities illuminated by a project like Pan-Gyn should be considered as hypothesis-generators, yielding clues to be tested and, if possible, validated in follow-up studies.
DNA methylation data revealed large high- and low-methylation clusters. CESC, as well as luminal B and HER2+ BRCA tumors, showed high levels of DNA methylation, suggesting epigenetics as a driving force in those tumor types. Clustering based on DNA methylation separated MLH1-silenced (i.e., hypermutator) endometrioid UCEC samples from the non-MLH1-silenced ones, suggesting that MLH1 may not be specifically targeted for epigenetic silencing but, instead, may be silenced by a more generic mechanism that silences multiple genes.
Gene sets associated with myeloid and stem cell development suggest that TERC activity, initially identified in zebrafish, might play a role in human development as well (Chiu et al., 2017). In the present study, CESC and OV showed positive correlation of TERC with MYC, TERT, telomere maintenance targets, miR-21, and CTNNB1 gene targets. However, serous UCEC showed a unique pattern of negative correlation with TERT targets, positive correlation with miR-21 targets, and no correlation with MYC, CTNNB1, or telomere maintenance targets. In luminal A BRCA, miR-21 targets were positively correlated with TERC.
Pathway and subtype analyses revealed an important role for immune markers. OV, basal-like BRCA, luminal BRCA, and HER2+ BRCA cancer samples split into immune-high and immune-low subtypes. Immune-high HER2+ tumors showed a trend toward longer survival than their immune-low counterpart, but the difference was not quite statistically significant for the sample size available. Most of the CESC samples showed high immune marker signatures, likely due to their almost 100% prevalence of HPV. In contrast, most of the UCEC and UCS samples showed little immune infiltration. The high-immune subsets might potentially benefit from immunotherapy.
Pathway analysis unexpectedly showed that approximately half of the basal-like BRCA cancers clustered together with the HER2 and luminal B samples, whereas the other half did not, suggesting pathway-level similarities not detected at the level of single RNAs. The similarities included higher inferred activation of AR signaling and lower enrichment of FOXA1, FOXA2, and XBP1/2, as well as the WNT and SHH pathways. Those observations are consistent with convergence of diverse transcriptional events on a limited number of functional pathways. Additional study will be required to test the robustness of those observations.
In summary, this integrative, multiplatform Pan-Gyn analysis has confirmed similarities previously identified across the five tumor types and identified relationships not observed in previous studies of the individual diseases. A number of the observations have possible prognostic and/or therapeutic relevance. Our capture of major molecular information content using a simple six-parameter binary decision tree could facilitate the clinical use of Pan-Gyn molecular subtypes and may help in selection for and administration of therapeutic trials across the Pan-Gyn spectrum. However, all of the clinical possibilities illuminated by this study will require extensive additional research, particularly functional validation (which is beyond the intended scope of TCGA studies), before they would be ready for practical application. In addition to its particular observations, this study presents a broad-based, curated atlas of Pan-Gyn molecular features that we believe will be useful as a starting point for many researchers in the field.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| RPPA antibodies | RPPA Core Facility, MD Anderson Cancer Center; Tibes et al., 2006; Gonzalez-Angulo et al., 2011 | https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core.html |
| Bacterial and Virus Strains | ||
| N/A | N/A | N/A |
| Biological Samples | ||
| Primary tumor samples | Multiple tissue source sites, processed through the Biospecimen Core Resource | See Methods: SUBJECT DETAILS, METHOD DETAILS |
| Chemicals, Peptides, and Recombinant Proteins | ||
| N/A | N/A | N/A |
| Critical Commercial Assays | ||
| AmpFISTR Identifier kit | Applied Biosystems | Cat: A30737 |
| DNA/RNA AllPrep kit | Qiagen | Cat: 80204 |
| Genome-Wide Human SNP Array 6.0 | ThermoFisher Scientific | Cat: 901153 |
| HumanMethylation450 | Illumina | Cat: HM450 |
| Illumina Barcoded Paired-End Library Preparation Kit | Illumina | https://www.illumina.com/techniques/sequencing/ngs-library-prep.html |
| Infinium HumanMethylation450 BeadChip Kit | Illumina | Cat: WG-314-1002 |
| mirVana miRNA Isolation kit | Ambion | |
| Phusion High-Fidelity PCR Master Mix with HF Buffer | New England Biolabs | Cat: M0531L |
| QiaAmp blood midi kit | Qiagen | Cat: 51185 |
| RNA6000 Nano Assay | Agilent | Cat: 5067-1511 |
| SureSelect Human All Exon 50 Mb | Agilent | Cat: G3370J |
| TruSeq PE Cluster Generation Kit | Illumina | Cat: PE-401-3001 |
| TruSeq RNA Library Prep Kit | Illumina | Cat: RS-122-2001 |
| VECTASTAIN Elite ABC HRP Kit (Peroxidase, Standard) | Vector Lab | Cat: PK-6100 |
| Deposited Data | ||
| Digital pathology images | Genomic Data Commons; Cancer Digital Slide Archive | https://gdc-portal.nci.nih.gov/legacy-archive/; http://cancer.digitalslidearchive.net/ |
| Raw and processed clinical, array, and sequencing data | Genomic Data Commons | https://portal.gdc.cancer.gov/legacy-archive/ |
| Experimental Models: Cell Lines | ||
| N/A | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| N/A | N/A | N/A |
| Oligonucleotides | ||
| N/A | N/A | N/A |
| Recombinant DNA | ||
| N/A | N/A | N/A |
| Software and Algorithms | ||
| ABSOLUTE | Carter et al., 2012 | PMID: 22544022 |
| ABySS v1.3.4 | Simpson et al., 2009 | PMID: 19251739 |
| ABySS v1.4.8 | Robertson et al., 2010 | PMID: 20935650 |
| BioBloomTools(v1.2.4.b) | Chu et al., 2014 | PMID: 25143290 |
| Birdseed | Korn et al., 2008 | PMID: 18776909 |
| Blastn (v2.2.23) | Altshul et al., 1997 | PMID: 9254694 |
| CARNAC | Totoki et al., 2014 | PMID: 25362482 |
| Circular Binary Segmentation | Olshen et al., 2004 | PMID: 15475419 |
| ConsensusClusterPlus | Wilkerson and Hayes, 2010 | PMID: 20427518 |
| ContEst | Cibulskis et al., 2011 | PMID: 21803805 |
| deconstructSigs | Rosenthal et al., 2016 | PMID: 26899170 |
| deFuse | McPherson et al., 2011 | PMID: 21625565 |
| FireHose | The Broad Institute of MIT & Harvard | https://www.broadinstitute.org/cancer/cga/Firehose |
| GenePattern | Reich et al., 2006 | PMID: 16642009 |
| GISTIC2.0 | Mermel et al., 2011 | PMID: 21527027 |
| Indelocator | Ratan et al., 2015 | PMID: 25879703 |
| MAP-RSeq | https://bioinformaticstools.mayo.edu/research/maprseq | |
| MapSplice 0.7.4 | Wang et al., 2010 | PMID: 20802226 |
| MuTect | Cibulskis et al., 2013 | PMID: 23396013 |
| MutSigCV v1.4 | Lawrence et al., 2013 | PMID: 23770567 |
| Next-Generation Clustered Heatmap | MD Anderson Cancer Center | https://bioinformatics.mdanderson.org/TCGA/NGCHMPortal/ |
| Oncotator | Ramos et al., 2015 | PMID: 25703262 |
| PARADIGM | Vaske et al., 2010 | PMID: 20529912 |
| PathSeq | Kostic et al., 2011 | PMID: 21552235 |
| Picard | The Broad Institute of MIT & Harvard | https://picard.sourceforge.net/ |
| PRADA | Torres-Garcia et al., 2014 | PMID: 24695405 |
| RADIA | Radenbaugh et al., 2014 | PMID: 25405470 |
| RSEM | Li and Dewey, 2011 | PMID: 21816040 |
| SNPFileCreator | Li and Hung Wong, 2001 | PMID: 11532216 |
| SomaticSignatures | Gehring et al., 2015 | PMID: 26163694 |
| STAR | Dobin et al., 2013 | PMID: 23104886 |
| Strelka | Saunders et al., 2012 | PMID: 22581179 |
| Tophat v2.0.8 | Trapnell et al., 2009 | PMID: 19289445 |
| WEKA | Smith and Frank, 2016 | PMID: 27008023 |
| Other | ||
| N/A | N/A | N/A |
CONTACT FOR RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Rehan Akbani (rakbani@mdanderson.org).
SUBJECT DETAILS
Human data and tumor data selection
Molecular data were obtained from patients that had not received prior treatment for their disease (ablation, chemotherapy, or radiation therapy) and had provided informed consent as part of The Cancer Genome Atlas Project (TCGA). Local Institutional Review Boards (IRBs) at the tissue source sites reviewed protocols to approve submission of cases.
We selected samples from five TCGA projects to represent the gynecologic cancers: breast invasive carcinoma (BRCA), endocervical adenocarcinoma (CESC), high-grade serous ovarian cystadenocarcinoma (OV), uterine corpus endometrial carcinoma (UCEC), and uterine carcinosarcoma (UCS). Sample selection was based on availability of data and propriety of genomic features. Eight CESC samples designated as UCEC-like using mRNA data and 14 OV cases lacking TP53 mutations were excluded. The Pan-Gyn cohort was eventually comprised of 2579 cases, consisting of 1087 BRCA cases, 579 OV cases, 548 UCEC cases, 308 CESC cases, and 57 UCS cases.
TCGA Project Management collected necessary human subjects documentation to ensure the project complies with 45-CFR-46 (the “Common Rule”). The program has obtained documentation from every contributing clinical site to verify that IRB approval has been obtained to participate in TCGA. Such documented approval may include one or more of the following:
An IRB-approved protocol with Informed Consent specific to TCGA or a substantially similar program. In the latter case, if the protocol was not TCGA-specific, the clinical site PI provided a further finding from the IRB that the already-approved protocol is sufficient to participate in TCGA.
A TCGA-specific IRB waiver has been granted.
A TCGA-specific letter that the IRB considers one of the exemptions in 45-CFR-46 applicable. The two most common exemptions cited were that the research falls under 46.102(f)(2) or 46.101(b)(4). Both exempt requirements for informed consent, because the received data and material do not contain directly identifiable private information.
A TCGA-specific letter that the IRB does not consider the use of these data and materials to be human subjects research. This was most common for collections in which the donors were deceased.
METHOD DETAILS
Sample processing
Cases were staged according to the American Joint Committee on Cancer (AJCC). Each frozen primary tumor specimen had a companion normal tissue specimen (blood or blood components, including DNA extracted at the tissue source site). Adjacent tissue was submitted for some cases. Specimens were shipped overnight using a cryoport that maintained an average temperature of less than −180°C.
RNA and DNA were extracted from tumor and adjacent normal tissue specimens using a modification of the DNA/RNA AllPrep kit (Qiagen). The flow-through from the Qiagen DNA column was processed using a mirVana miRNA Isolation Kit (Ambion). This latter step generated RNA preparations that included RNA <200 nt suitable for miRNA analysis. DNA was extracted from blood using the QiaAmp blood midi kit (Qiagen). Each specimen was quantified by measuring Abs260 with a UV spectrophotometer or by PicoGreen assay. DNA specimens were resolved by 1% agarose gel electrophoresis to confirm high molecular weight fragments. A custom Sequenom SNP panel or the AmpFISTR Identifiler (Applied Biosystems) was utilized to verify tumor DNA and germline DNA were derived from the same patient. Five hundred nanograms of each tumor and normal DNA were sent to Qiagen for REPLI-g whole genome amplification using a 100 g reaction scale. Only specimens yielding a minimum of 6.9 g of tumor DNA, 5.15 g RNA, and 4.9 g of germline DNA were included in this study. RNA was analyzed via the RNA6000 nano assay (Agilent) for determination of an RNA Integrity Number (RIN), and only the cases with RIN >7.0 were included in this study. Reasons for rejection are described at https://tcga-data.nci.nih.gov/datareports.
DNA sequencing data
Exome capture was performed using Agilent SureSelect Human All Exon 50 Mb according to the manufacturers’ instructions. Briefly, 0.5–3 micrograms of DNA from each sample were used to prepare the sequencing library through shearing of the DNA followed by ligation of sequencing adaptors. All whole exome (WES) and whole genome (WGS) sequencing was performed on the Illumina HiSeq platform. Paired-end sequencing (2 × 101 bp for WGS and 2 × 76 bp for WE) was carried out using HiSeq sequencing instruments; the resulting data was analyzed with the current Illumina pipeline. Basic alignment and sequence QC was done on the Picard and Firehose pipelines at the Broad Institute. Sequencing data were processed using two consecutive pipelines:
Sequencing data processing pipeline (“Picard pipeline”). Picard (http://picard.sourceforge.net/) uses the reads and qualities produced by the Illumina software for all lanes and libraries generated for a single sample (either tumor or normal) and produces a single BAM file (http://samtools.sourceforge.net/SAM1.pdf) representing the sample. The final BAM file stores all reads and calibrated qualities along with their alignments to the genome.
- Cancer genome analysis pipeline (“Firehose pipeline”). Firehose (http://www.broadinstitute.org/cancer/cga/Firehose) takes the BAM files for the tumor and patient-matched normal samples and performs analyses including quality control, local realignment, mutation calling, small insertion and deletion identification, rearrangement detection, coverage calculations and others as described briefly below. The pipeline represents a set of tools for analyzing massively parallel sequencing data for both tumor DNA samples and their patient-matched normal DNA samples. Firehose uses GenePattern (Reich et al., 2006) as its execution engine for pipelines and modules based on input files specified by Firehose. The pipeline contains the following steps:
- Quality control. This step confirms identity of individual tumor and normal to avoid mix-ups between tumor and normal data for the same individual.
- Local realignment of reads. This step realigns reads at sites that potentially harbor small insertions or deletions in either the tumor or the matched normal, to decrease the number of false positive single nucleotide variations caused by misaligned reads.
- Identification of somatic single nucleotide variations (SSNVs). This step detects candidate SSNVs using a statistical analysis of the bases and qualities in the tumor and normal BAMs, using Mutect (Cibulskis et al., 2013).
- Identification of somatic small insertions and deletions. In this step, putative somatic events were first identified within the tumor BAM file and then filtered out using the corresponding normal data, using Indellocator (Ratan et al., 2015).
Molecular features that distinguished Pan-Gyn from other tumor types
Mutations and CNVs
We identified the mutation and CNV events, which are enriched in gynecologic cancers (BRCA, UCEC, CESC, UCS, and OV) compared to all other cancers. 617 oncogenes listed in the COSMIC census list are included in the analysis. A multi-step statistical enrichment analysis method is devised for this purpose and applied to mutation and CNV data separately (see methods subsection dedicated to CNV data below for reference). The analysis involves creating a contingency table for altered vs. unaltered cases in Pan-Gyn vs. other cancers. First, the bias from sample sizes in different cancer types are removed by normalizing the alteration counts in each cancer type with sample sizes. For this purpose, an expected gynecological alteration/gene is calculated. For each gene, the mutation or a CNV high-level amplification (i.e. GISTIC thresholded CN value of +2) count in each gynecological cancer type is divided by the number of samples in the associated disease type and multiplied by the total number of samples (after normalization to hundred samples/disease for mutation counts) in the gynecological cancers. The same normalization is performed for non-gynecological cancers. This is critical to avoid cancers with large sample size (e.g. BRCA, N = 982 vs. UCS, N = 57) dominate the whole analysis. The genes with p value adjusted < 0.01 for mutation and p value adjusted < 0.05 for CNV are visualized in Figure 1A-B (see Statistical Analysis section for details on calculation of p values).
We addressed the question of whether the Pan-Gyn tumor types (BRCA, OV, USC, UCEC, CESC) share a significantly larger number of enriched mutated genes compared to a null distribution of enriched mutated genes in randomly selected 5 disease types. The bootstrapping analysis in Figure S1C-D involves an iterative process of randomly selecting 5 cancer types out of non-gyn cancers (N=28), calculating number of enriched genes in the randomly selected group using the same criteria (Fisher exact test with FDR-adjusted p value < 0.01) we used for generating Figure 1A. The iterations were performed 10,000 times to generate the null distribution. Following the same strategy, we performed CNA analysis using gene level CNA results from GISTIC2.0 (Mermel et al., 2011).
DNA methylation
Aim of this section was to identify genes that are differentially methylated in gynecological tumors (BRCA, UCEC, CESC, UCS, OV) versus the other tumor types. For this purpose we used two different approaches. We first mapped all the probes from the sequencing platforms to unique genes. For genes having more than one probe mapping to its promoter, median beta value was considered. For the first analysis, a threshold beta value of 0.3 was used to call methylation status of genes. Having converted our data to binary form, we then counted the percentage of samples of each tumor type in whom the gene was in methylated state. By taking percentages instead of just the number of cases for each tumor, we could correct for variation in number of samples that were available for each type. For example, whereas 966 cases of breast cancer (BRCA) was available, only 36 cases were available for cholangiocarcinoma (CHOL). To make sure that our analysis does not get skewed by this variation in sample sizes, we normalized number of samples for each tumor type to 100. We then grouped samples into Gyn vs non-Gyn cancers, and again adjusted size of each population to 100. Refer to Statistical Analysis section for details on the identification of significant genes.
In order to get more robust results, we performed a second kind of analysis to identify significant differentially methylated genes. We logit transformed the beta values into M-values, z-normalized the scores across all samples for a given gene, and took median across all member samples as the methylation score for each tumor type. We then dichotomized the dataset into gyn and non-gyn populations and identified the statistically significant genes between the two populations (see Statistical Analysis section for details).
We then compared the lists of statistically significant genes from the two analyses. A total of 197 genes were called significantly differentially methylated between the two populations by both our analyses. The median beta values of these genes across member samples of each tumor type were then plotted into a heatmap, with the Z-normalized M values being used for hierarchical clustering of genes using Euclidean distances and Ward’s linkage.
Mutation analysis
We used clinical information from 2579 women with gynecological (Pan-Gyn) cancer in TCGA database (1097 breast carcinomas (BRCA), 579 ovary carcinomas (OV), 308 uterine cervical carcinomas (CESC), 548 endometrial carcinomas (UCEC) and 57 uterine carcinosarcomas (UCS). The mutation data include 2,271 gynecologic tumor samples. We used the pancan.merged.V0.2.4.filtered.maf and applied two different approaches to identify the most significantly mutated genes across all Pan-Gyn samples (see Statistical Analysis section for details). The mutation calls used in all of our analyses were somatic mutations only, not germline, so tumor purity differences had minimal impact. We considered as driver mutation the intersection between the two methods and the mutation classification as a potential oncogene or a tumor suppressor gene was based on the inferred scores.
Generation of mutational signatures
We used the pancanmerged.v0.2.4.sorted.maf file to analyze the operative mutational processes in PanGyn samples. We selected all SNVs and created a Grange object in R for every substitution and converted all mutations into a matrix made up of all substitution contexts. For every pyrimidine substitution (C>A, C>G, C>T, T>A, T>C and T>G) we used the 5′ and 3′ base according to the hg19 human reference genome (http://hgdownload.cse.ucsc.edu/) creating 96 possible mutation contexts as described by Alexandrov et al (Alexandrov et al., 2013). We used the SomaticSignatures package for R to implemented an algorithm that uses the non-negative matrix factorization (NMF) to decompose the original matrix to the minimal set of mutation signatures. This algorithm estimates the contribution of each signature across the samples. This last information was used to perform an unsupervised hierarchical clustering to identify samples that share similar mutational spectra (Gehring et al., 2015).
Copy Number Alteration (CNA) analysis
Data generation and processing
Tumor sample DNA was hybridized to Affymetrix SNP6.0 arrays by the Genome Analysis Platform at the Broad Institute as previously described (McCarroll et al., 2008). The resulting probe intensities were normalized and combined using SNPFileCreator (Li and Hung Wong, 2001) and then processed with Birdseed (Korn et al., 2008) to yield preliminary copy-number estimates. The preliminary copy-number estimates were refined using tangent normalization (Tabak et al., unpublished) and then underwent Circular Binary Segmentation (Olshen et al., 2004) to yield segmented relative copy-number profiles. The processed SNP intensities, Birdseed clusters files, and segmented copy-number profiles were input to HAPSEG to create haplotyped copy-number data, which was then utilized with MC3 mutation calls (https://www.synapse.org/MC3) to obtain tumor heterogeneity and ploidy estimates from ABSOLUTE (Carter et al., 2012). CNAs were assessed as deviations in the tumor sample from the paired normal tissue sample, so they only reflected somatic changes. However, the amplitude of CNA signals can be suppressed in tumor samples with normal cell contamination. We thus utilized ABSOLUTE-derived tumor purity and ploidy estimates for In Silico Admixture Removal (ISAR) of the segmentation data (Zack et al., 2013) in order to correct for any signal dampening that may have occurred before proceeding to analyze somatic copy number alterations.
Identification and analysis of significant somatic copy number alterations
There were 2,246 gynecologic samples and 7,707 non-gynecologic samples used for downstream copy-number analyses. To adjust for tumor heterogeneity and ploidy in both the gynecologic and non-gynecologic cohorts, the segmented relative copy-number data was ISAR-corrected (Zack et al., 2013). GISTIC2.0 (Mermel et al., 2011) was ran on the resulting purity and ploidy-adjusted data for both cohorts to obtain genome-wide estimates for significant broad and focal somatic copy number alterations. The frequency of high-level copy-number amplifications in the amplification lesion gene targets (i.e. gene targets with thresholded values of +2 produced by GISTIC) were calculated for each tumor type and plotted (Figure 1A) to visualize the differences between the gynecologic and non-gynecologic cancers. The q-values of all of the significant GISTIC amplification and deletion alterations in the gynecologic and non-gynecologic cohorts were plotted against each other (Figure 1B), and the alterations that were exclusive to each cohort were also visualized by plotting the amplification and deletion lesion region boundaries in genomic coordinates and using the lesion q-values as lesion amplitudes (Figure S1A).
The unsupervised hierarchical clustering, utilizing Ward’s objective function and a Euclidean distance metric, was performed on the amplification and deletion lesions predicted by GISTIC2.0 across the gynecologic cancers. The six resulting cluster groups were visualized with copy number data (Figure 3) and with various other metrics such as gene-level mutations (Figure S3A). P values were calculated to determine significant differences across the various metrics between the cluster groups (see Statistical Analysis section for details). GISTIC2.0 was also performed on the ISAR-corrected copy data within each cluster group in order to compare amplification and deletion lesions between groups.
DNA methylation
Data preprocessing
Illumina infinium DNA methylation arrays (including both HumanMethylation27 (HM27) and HumanMethylation450 (HM450)) were used to assay 2,566 pan-gynecological tumor and 167 normal samples in total, which includes 1,074 BRCA, 573 OV, 555 UCEC, 307 CESC and 57 UCS primary tumor samples. Level 3 data from two generations of Illumina infinium DNA methylation arrays were combined and further normalized between platforms using a probe-by-probe proportional rescaling method as outlined below to yield a final common set of 22,601 probes with comparative methylation levels between platforms. During data generation a single technical replicate of the same cell line control sample from either of two different DNA extractions (TCGA-07-0227/TCGA-AV-A03D) was included on each plate as a control, and measured 44/198 times and 12/169 times on HM27 and HM450 respectively. These repeated measurements were therefore used for rescaling of the HM27 data to be comparable to HM450. For each probe within each platform, we computed the median beta value across all technical replicates of each of the two TCGA IDs. We then combined the two extractions by taking the mean of the two medians obtained for each of the two replicate TCGA IDs, and obtained a single summarized DNA methylation read out (beta value) for the corresponding probe i for each platform, noted as hm27,i, and hm450,i, respectively. We then applied a constrained (within the rage of 0 to 1 for beta values) linear rescaling of the HM27 data for each probe and for each patient sample using hm27,i and hm450,i. When the HM27 beta value of a patient sample j for probe i was smaller than the mean of median replicate samples on the HM27 for that probe, we linearly rescaled the HM27 beta value Betahm27,i,j in the (0, Betahm27,i,j) space; and when Betahm27,i,j is greater, we linearly rescaled the HM27 beta value Betahm27,i,j in the (Betahm27,i,j,1) space; This translates into the following mathematical computation: Betahm450,i,j = Betahm27,i,j*(hm450,i/hm27,i), if Betahm27,i,j < hm27,i; and Betahm450,i,j = 1-(1-Betahm27,i,j)*((1-hm450,i)/(1-hm27,i)), if Betahm27,i,j > hm27,i.
After the between-platform normalization, we further excluded 779 probes that still showed a consistent platform difference (mean beta value difference greater than or equal to 0.1) in six or more tumor types. To minimize the influence of normal tissue contamination and leukocytes infiltration in DNA methylation data, we chose probes not methylated in all relevant normal tissues and blood cells, to get rid of methylation signals from possible confounding factors. In order to deal with tumor samples with low tumor purity, we further chose cancer-specific probes by requiring those unmethylated probes to be methylated (defined as beta value > 0.3) in more than 5% samples per tumor type, and then applied dichotomized clustering methodology to run cluster analysis.
DNA methylation analysis
Unsupervised and dichotomized clustering was performed based on a set of cancer-specific autosomal loci, which were defined as unmethylated in normal tissues and blood cells (mean beta value < 0.2 for each tissue types), but methylated in more than 5% samples of each tumor type included in this analysis (beta value > 0.3). For tumor type with less than 100 samples, we require the portion of methylated samples to be greater than 10% instead. In order to generate a set of high-confident probes, we further removed 3373 probes showing standard deviations bigger than 0.05 using 97 technical replicates run along with the breast and gynecological samples. To minimize the influence of tumor purity, we dichotomize the data into 0’s and 1’s with a beta value cut off of 0.3, and used Ward’s method to cluster the distance matrix computed with the Jaccard Index. Heatmaps are colored using methylation beta values but ordered according to the above clustering procedures. Pre-defined clusters (k=7) were generated based on cutree function using R program.
Epigenetic silencing status for gene BRCA1 (measured by probe cg04658354 for both platforms), MLH1 (measured by probe cg00893636 for both platforms) and RAD51C (measured by probes cg14837411 and cg27221688 for platform HM27 and HM450 separately) was computed based on an experiential beta value cutoff of 0.3, 0.1 and 0.15, with beta values higher than 0.3, 0.1 and 0.15 considered as silenced, separately.
mRNA analysis
Identification of mRNA gene expression-based subtypes and analysis
The combination of available functionally defined cancer genes was first obtained from the literature (Sadelain et al., 2012). The previously-reported cancer gene expression profiling of total 2296 breast and gynecological tumors (1097 BRCA, 305 CESC, 305 OV, 532 UCEC and 57 UCS) was further filtered to eliminate unreliably measured genes and to limit the clustering to relevant genes (Cancer Genome Atlas Research Network et al., 2013). Genes that are not present in the TCGA data set were first removed. We then filtered out genes having missing values in any of the samples. Next we filtered out genes that have small expression values in at least one-third of the samples. Implementation of these filters resulted in 1,860 unique genes with reliably measured expression and with cancer characteristics. The gene expression data were then median centered and log transformed. Next we applied the hierarchical unsupervised clustering analysis with the preprocessed gene expression data. The distance metric was one minus the Pearson’s correlation coefficient and Ward was used as a linkage algorithm. This unsupervised approach clustered samples and identified nine robust gene expression-based subtypes. The nine subtypes and their gene expression patterns were viewed by using the next-generation clustered heat map (NG-CHM), a tool that was developed at the University of Texas MD Anderson Cancer Center. See Statistical Analysis section for details on calculation of statistically significant correlations and differences between the subtypes.
miRNA analysis
To identify samples from different tissues that had similar miRNA expression profiles, we used hierarchical clustering with pheatmap v1.0.2 in R. The input was a batch-corrected, miRNA-Seq mature strand data matrix that contained normalized (RPM) abundance for the 293 mature strands that were the union of the most-variant 200 mature strands for each cancer, in 2417 tumor samples from UCEC (n = 524), UCS (56), CESC (306), BRCA (1057), and OV (474). We transformed each row of the matrix by log10(RPM + 1), then used pheatmap to scale the rows. We used Ward.D2 for the clustering method, and correlation and Euclidean as the distance measures for clustering the columns and rows, respectively.
Proteomic analysis
Batch effects corrected protein expression data (generated using the RPPA platform) were clustered using the hierarchical clustering function hclust() in the R language. We used 1-Pearson’s correlation coefficient as our distance metric with Ward linkage to cluster both the rows and the columns. The data matrix consisted of a total of 1967 samples across 217 antibodies. The matrix was median-centered in both directions prior to clustering. Clusters were separated by using the cutree() function with k=5 clusters.
lncRNA analysis
We used ConsensusClusterPlus (Wilkerson and Hayes, 2010) package in R to perform consensus clustering (Monti et al., 2003) and discover the best partition of samples. The K-medoids method, a modification of the K-means algorithm, first randomly selects k data points (or medoids) that are used to form k clusters, where k is a user supplied variable. Then, all remaining data points are iteratively partitions to minimize the distance between the medoids and all other data points in the same cluster. Once all data points are assigned, a medioid is selected for each cluster and the process is repeated until it converges or until a maximum number of iterations is reached. We used Partitioning Around Medoids (PAM) algorithm to implement the K-medoids method, with the Pearson’s correlation coefficient as a measure similarity between data points. We used bootstrapping to select k. For each of 1,000 bootstraps, we selected 80% of the samples and 80% of lncRNA genes to investigate how frequent they are grouped in the same cluster for each k. The best k value between 2 and 15 was selected by the Silhouette index, a clustering validation measure used to evaluate the level of similarity within a cluster and dissimilarity between the clusters. Standard deviation produced from this bootstrapping computation was used to compare the Silhouette index across choices of k.
Batch effects analysis
We investigated batch effects first within individual disease types, and then across tumor types. Specifically, we investigated the effects of multiple confounding factors, including differences in: (i) batches in which the samples were processed, (ii) tissue source sites from where the samples were obtained, (iii) the date on which the samples were shipped to the data generation centers, (iv) the instrument on which the samples were processed, (v) the centers that generated the data. The results from individual tumor type analyses can be found online at: (http://bioinformatics.mdanderson.org/tcgambatch/). We assessed the magnitude of batch effects using the following algorithms, (i) clustered heat maps, (ii) enhanced PCA plots, and (iii) box plots. Whenever batch effects were observed, we corrected them using (i) ComBat (Johnson et al., 2007), or an enhanced version of it, (ii) Replicates Based Normalization (RBN) (Akbani et al., 2014), or (iii) removal of bad gene/probe data. Using those methods, we corrected the mRNA, miRNA, DNA methylation and protein expression data. The mutations and copy number data were already discretized and corrected for background loads.
Pathways analysis
PARADIGM integrated pathway analysis from copy number and expression data
We used the PARADIGM algorithm (Vaske et al., 2010; Sedgewick et al., 2013) to infer the activities of ~19K pathway features based on expression, copy number and pathway interaction data for 9829 tumor samples, including 2173 Pan-Gynecological cancers.
Platform corrected expression data and gene-level copy number data were obtained from Synapse (syn4976369 and syn5049520). Whitelisted samples assayed on both platforms were identified. One was added to all expression values, which were then log2 transformed and median-centered across samples for each gene. The log2 transformed, median-centered mRNA data were rank transformed based on the global ranking across all samples and all genes and discretized (+1 for values with ranks in the highest tertile, −1 for values with ranks in the lowest tertile, and 0 otherwise) prior to PARADIGM analysis.
Pathways were obtained in BioPax Level 3 format, and included the NCIPID and BioCarta databases from http://pid.nci.nih.gov and the Reactome database from http://reactome.org. Gene identifiers were unified by UniProt ID then converted to Human Genome Nomenclature Committee’s HUGO symbols using mappings provided by HGNC (http://www.genenames.org/). Altogether, 1524 pathways were obtained. Interactions from all of these sources were then combined into a merged Superimposed Pathway (SuperPathway). Genes, complexes, and abstract processes (e.g. “cell cycle” and “apoptosis”) were retained and henceforth referred to collectively as pathway features. The resulting pathway structure contained a total of 19504 features, representing 7369 proteins, 9354 complexes, 2092 families, 82 RNAs, 15 miRNAs and 592 abstract processes.
The PARADIGM algorithm infers an integrated pathway level (IPL) for each feature that reflects the log likelihood of the probability that it is activated (vs. inactivated). PARADIGM IPLs of the 19504 features within the SuperPathway is available on Synapse (syn6171376).
We also computed the single sample gene set enrichment (ssGSEA) score, as described by Barbie et al (Barbie et al., 2009), of the constituent pathways forming the SuperPathway structure from the PARADIGM IPL data using the GSVA package in R (Hänzelmann et al., 2013). Of the 1524 pathways obtained, only 1387 have pathway members within the interconnected SuperPathway structure; and their ssGSEA scores are available on Synapse (syn10184122).
Consensus clustering based on PARADIGM inferred pathway activation
Consensus clustering based on the 4876 most varying features (i.e. IPLs with variance within the highest quartile) was used to identify Pan-Gynecological subtypes implicated from shared patterns of pathway inference. Consensus clustering was implemented with the ConsensusClusterPlus package in R (Wilkerson and Hayes, 2010). Specifically, median-centered IPLs were used to compute the squared Euclidean distance between samples; and this metric was used as the input to the ConsensusClusterPlus algorithm. Hierarchical clustering using the Ward’s minimum variance method (i.e. ward inner linkage option) with 80% subsampling was performed over 1000 iterations; and the final consensus matrix was clustered using average linkage. The number of clusters was selected by considering the relative change in the area under the empirical cumulative distribution function (CDF) curve. We selected k=8 as further separation provides minimal change. Heatmap display of the top varying IPLs was generated using the heatmap.plus package in R. See Statistical Analysis section for details on identification of significant pathway differences between the resulting clusters.
Integrated analysis across Pan-Gyn tumor types
Cluster of Cluster Assignments (CoCA) analysis was performed using the cluster assignments from each of the 6 major platforms (mutations, CNV, DNA methylation, mRNA, miRNA, and protein). Clusters assignments defined from each platform were coded into a series of indicator variables for each platform of the form <platform>-<cluster number>, with samples belonging to the particular platform/cluster having a value of 1, and other samples having a value of 0. The matrix of 1 and 0s was then clustered using hierarchical clustering from the hclust() function in R, with Euclidean distance and Ward linkage.
Subtypes across the Pan-Gyn tumors
For the subtype analysis, we identified features that were either (i) currently used in the clinic for at least one of the five tumor types or (ii) identified as informative in previous TCGA gynecologic and breast cancer studies (Cancer Genome Atlas Research Network, 2011; Cancer Genome Atlas Research Network, 2012; Cancer Genome Atlas Research Network et al., 2013; Cherniack et al., 2017; Cancer Genome Atlas Research Network, 2017; Hoadley et al., 2014; Akbani et al., 2014; Cherniack et al., 2017). Features belonging to the former group were (i) protein expression of ER and PR, BRCA1/BRCA2 mutation status, ERBB2 amplification status, and HPV status. Features in the latter group were (ii) MSI status, hyper-mutator status (>10 mutations/mbp), SCNA load, AR protein expression, leukocyte infiltration score based on DNA methylation, and mutation status of PTEN, TP53, H-RAS/K-RAS/N-RAS, ERBB2, PIK3CA, and POLE. We initially selected a total of 19 features for the analysis. We then combined N-RAS, H-RAS, and K-RAS mutations into a single feature (using OR logic), and BRCA1 and BRCA2 mutations into another single feature (again using OR logic), yielding a final tally of 16 features. MSI status was available only for UCEC and UCS, and HPV status was available only for CESC, so we treated the features as missing for the remaining tumor types.
We dichotomized those 16 features into present/absent (for discrete features like mutations) and high/low (for continuous features) in each sample. Eleven of those features were discrete (all the mutations, MSI status, hyper-mutator status, ERBB2 amplifications, and HPV status), whereas the remaining 5 features (CNV load, immune score, ER, PR, and AR protein expressions) were continuous. The CNV load and immune score thresholds were obtained by modeling the expression with a bimodal Gaussian distribution and using the value between the two modes as the threshold. For ER and PR, the thresholds were identified by maximizing the area under curve (using the Youden index), using the continuous-valued expression value to determine the binary valued-ER/PR status obtained from immunohistochemistry (IHC) in BRCA. AR cutoff was obtained similarly, using the continuous-valued AR protein expression level to model the binary valued AR status between TCGA prostate cancer (PRAD) data vs. UCEC, BRCA, OVA, CESC, and UCS. Samples without protein expression data were removed, leaving 1,956 samples out of an original 2,579. Once all the features were binarized, we constructed a matrix of samples × features where each cell had a 1 if the sample had that feature (or had high levels of that feature), and 0 otherwise. The resulting matrix was clustered using hierarchical clustering from the hclust() function in R, with 1 - Pearson’s correlation and Ward linkage. The clusters were separated using the cutree() function with k=5 clusters.
We used the J48 decision tree function in the Weka package (Frank et al., 2016) to construct a pruned decision tree using the feature matrix as input and cluster assignments as the class variable. The software output the decision tree shown in Figure 7D.
For the survival analysis shown in Figures 7C and 7E, we used the “survival” library in R and used the survfit() function followed by plot() to generate the figures. The survival data were fitted using the “cluster” variable. For p value computation see the “Quantification and Statistical Analysis” section.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests for distinguishing Pan-Gyn from other tumor types
For both mutations and high-level amplifications, the normalized mutated vs. non-mutated counts in gynecological vs. non-gynecological cancers formed the 2×2 contingency table for each gene. A Fisher’s exact test was applied for each gene to determine significant differences of enrichment between the two populations and the resulting p values were adjusted for false discovery rates with the Benjamini-Hochberg method. For DNA methylation, we used a combination of two approaches. For the first approach, we used the proportions test to see if any genes were significantly differentially methylated in one population versus the other. We performed FDR correction using BH method, and considered genes having adjusted one-sided p values of less than 0.05 to be significant. For the second approach, a Mann Whitney U test was utilized to identify genes with significant differences between the median methylation levels of genes in the gyn vs. non-gyn populations, with the resulting p values being adjusted for FDR using BH correction.
Mutation analysis
We applied two different approaches to identify the most significantly mutated genes across all PanGyn samples. First, we used the method described by Vogelstein et al to estimate the oncogene (ONG) and tumor suppressor gene (TSG) scores (Vogelstein et al., 2013). ONG score was estimated by the ratio of recurrent mutations (defined as missense and in-frame mutations that affected the same codon of the annotated transcript). The TSG score used the ratio of inactivating mutations (nonsense and frameshift mutations, and variants that affected splice sites) in a specific transcript. Genes with an ONC or TSG scores > 0.2 were classified as putative driver mutation (Table S3). For the second method, we used the MutSigCV v1.4 (www.broadinstitute.org) to infer significant cancer mutated genes across all PanGyn samples (Lawrence et al., 2013). We found 46 significantly mutated genes based on the intersection of those genes identified by MutSigCV v1.4 and those identified by the Vogelstein et al. method. For the ten mutation signatures identified by NMF, we calculated correlations between the ten mutation signatures and the 30 COSMIC gene signatures (http://cancer.sanger.ac.uk/cosmic/signatures). P values were calculated for these correlations and corrected to FDR values.
Determining significant patterns of somatic copy number aberration
Identification of genomic regions undergoing significant levels of copy number arrangements and identification of the significant targets of these somatic copy number alterations along with their q-values was accomplished using GISTIC2.0 (Mermel et al., 2011) for both gyn and non-gyn sample cohorts. High-level amplifications for a gene were defined as having a thresholded copy number value of +2 as estimated by GISTIC. Broad-level copy number contributions estimated by GISTIC of having a value of greater than +1 or less than −1 were classified as broad-level amplifications and broad-level deletions, respectively. Testing for significant differences between the six resulting Pan-Gyn SCNA cluster groups was done using binomial tests for broad-level chromosomal alterations (identified through GISTIC), Kruskal-Wallis tests for continuous variables such as number of segments and tumor purity (identified through ABSOLUTE), and Chi-squared tests for independence comparing discrete variables such as gene mutations and tumor pathologic stage.
mRNA analysis
This unsupervised approach clustered samples and identified nine robust gene expression-based subtypes. Chi-squared test were used to evaluate the correlation between mRNA clusters and tumor type, grade, histology, or molecular subtypes as determined by individual diseases. Log-rank test and Kaplan-Meier survival curves were used to compare overall survival (OS) between different clusters of patients (Cancer Genome Atlas Research Network et al., 2013). To adjust for lineage differences, the log-likelihood ratio (LR) statistic was calculated for a Cox proportional hazards model built using just tumor type information. We then added mRNA cluster information to the model and recomputed the LR statistic. We calculated the difference between the two LR statistics and computed its p value using the chi-squared test (Hoadley et al, Cell, 158:929-944, 2014). For the discriminatory genes analysis, we used the Kruskal-Wallis test to identify the top genes that discriminated between the mRNA clusters.
lncRNA statistical analysis
We used Pearson’s correlation as the metric when we performed unsupervised consensus clustering of the lncRNA data by K-medoid with bootstrapping. Silhouette analysis suggested 6 as the optimal number of clusters (L1 to L6). We compared cluster membership with membership of the five protein-based clusters by performing Fisher’s Exact tests and corrected the resulting p values to FDR values. We determined significance using a FDR-corrected p value of < 0.05. We also calculated p values for Pearson’s correlations between expression of key lncRNAs and their regulators and used a p value of 0.05 as the cutoff for significance. Gene Set Enrichment Analysis (GSEA) was utilized to determine significant enrichment (with a cutoff of FDR < 0.05) of gene sets containing TERC-correlated genes.
Pathway differences between Pan-Gynecological PARADIGM clusters
Pathway biomarkers of each PARADIGM clusters were identified by comparing one cluster vs. all others using the t-test and Wilcoxon Rank sum test with Benjamini-Hochberg (BH) false discovery rate (FDR) correction. An initial minimum variation filter (at least 1 sample with absolute activity > 0.05) was applied; and the 15502 features passing the minimum variation feature were considered in this analysis. Features deemed significant (FDR corrected p value <0.05) by both tests and showing an absolute difference in group means > 0.05 were selected. The selected pathway features were assessed for interconnectivity; and constituent pathways enriched among interconnected differential features were identified using a modified Fisher’s test with BH FDR correction. We also compare ssGSEA scores of the constituent pathways in one cluster vs. all other comparisons; and pathways with differential ssGSEA scores and are enriched among the interconnected differential features are selected for display in a heatmap.
Subtypes across the Pan-Gyn tumors survival analysis
Survival analysis of the subtype groups was done using the R package “survival.” Log-rank test was used to compute the p value (unadjusted for tumor type). The p value adjusted for tumor type was computed by first constructing a Cox proportional hazards model using both “cluster” and “tumor type” as the fitting variables. Then, a second Cox proportional hazards model was constructed using just the “tumor type” variable. The test statistic from the second model was subtracted from the test statistic from the first model. The resulting difference in the test statistics followed a X2 distribution with 4 degrees of freedom (because there were 5 clusters), and was a measure of the additional prognostic value provided by the clusters above and beyond the information provided by tumor type alone. The p value for the difference in the test statistics is shown in Figures 7C and 7E as the tumor type adjusted p value.
DATA AND SOFTWARE AVAILABILITY
The raw data, processed data and clinical data can be found at the legacy archive of the GDC (https://portal.gdc.cancer.gov/legacy-archive/search/f) and the PancanAtlas publication page (https://gdc.cancer.gov/about-data/publications/pancanatlas). The mutation data can be found here (https://gdc.cancer.gov/about-data/publications/mc3-2017). TCGA data can also be explored through the Broad Institute FireBrowse portal (http://gdac.broadinstitute.org) and the Memorial Sloan Kettering Cancer Center cBioPortal (http://www.cbioportal.org). Details for software availability are in the Key Resources Table.
Supplementary Material
Table S1. (Related to Figure 1) Amplification and deletion peaks identified by GISTIC in the Pan-Gyn cohort.
Table S3. (Related to Figure 2) Table showing 46 statistically significantly mutated genes across the Pan-Gyn cohort.
Table S4. (Related to Figures 2-7) Clinical data and cluster memberships of the samples in the study across all of the platforms and analysis types.
Table S5. (Related to Figures 2-7) Contingency tables showing numbers of samples from tumor types vs. clusters for various platforms.
Significance.
Gynecologic and breast (Pan-Gyn) cancers have a projected incidence of more than 350,000 cases in the United States in 2017, with much larger numbers worldwide. Despite recent clinical advances, more comprehensive information on molecular characteristics of the tumors is a priority. As part of The Cancer Genome Atlas (TCGA) Pan-Cancer Atlas project, we present here an integrated analysis of 2,579 patients’ Pan-Gyn cancers at the DNA, RNA, protein, histopathological, and clinical levels. We highlight shared characteristics and unique molecular features of the tumors, identifying clinically significant subtypes and suggesting potential therapeutic targets. Finally, we present a practical decision tree with only six laboratory-assessable molecular features that classifies patient samples into one of five prognostic molecular subtypes.
Highlights.
Integrated analysis finds molecular features characteristic of gynecologic tumors
Subtypes with high leukocyte infiltration, a marker for immune response, identified
Gene-lncRNA interaction network of ESR1, DKC1 & lncRNAs TERC, NEAT1, TUG1 identified
Decision tree to group patients into clinically relevant prognostic subtypes proposed
Acknowledgments
This work was supported by the following NCI grants: U54 HG003273, U54 HG003067, U54 HG003079, U24 CA143799, U24 CA143835, U24 CA143840, U24 CA143843, U24 CA143845, U24 CA143848, U24 CA143858, U24 CA143866, U24 CA143867, U24 CA143882, U24 CA143883, U24 CA144025, U24 CA210949, U24 CA210950, P30 CA016672.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Please see the supplementary document “authors list” for complete listing of authors.
Sample and Data providers: AJL, AKG, CA, DAL, JR, NP, RRB, REZ, CSS, SOe, SOr, MLA, JMdA, HHAC, JB, KL, BW. Sample freeze list: RSK, WL, CPV, TML, DAL, RA. Pan-Gyn vs. non-Gyn analysis: AK, ACB, RA, VR, AR, GM. Mutations: DGT. SCNA: ACB, CW, ADC. DNA methylation: HF, HS, PWL. mRNA: YL, WL. miRNA: RB, GR. RPPA: AMH, RSK, CPV, GBM, RA. lncRNA: CC, DY, PS, CW, PM, LL, AKS, TWC, HSC, SL, PHG. PARADIGM: CY. Pan-Gyn Subtyping: WL, ADC, AKG, NMK, JSR, REZ, DAL, AR, RA. Clinical and Pathology: AKG, AIO, AJG, RDB, NMK, JSR, REZ, AKS, AJL, JNW, GBM, DAL. Editing team: ACB, AK, AKS, AJL, GBM, DAL, JNW, NMK, RA. Project leadership: GBM, DAL, RA.
TCGA PanCanAtlas Declaration of Interest:
Michael Seiler, Peter G. Smith, Ping Zhu, Silvia Buonamici, and Lihua Yu are employees of H3 Biomedicine, Inc. Parts of this work are the subject of a patent application: WO2017040526 titled “Splice variants associated with neomorphic sf3b1 mutants.” Shouyoung Peng, Anant A. Agrawal, James Palacino, and Teng Teng are employees of H3 Biomedicine, Inc. Andrew D. Cherniack, Ashton C. Berger, and Galen F. Gao receive research support from Bayer Pharmaceuticals. Gordon B. Mills serves on the External Scientific Review Board of Astrazeneca. Anil Sood is on the Scientific Advisory Board for Kiyatec and is a shareholder in BioPath. Jonathan S. Serody receives funding from Merck, Inc. Kyle R. Covington is an employee of Castle Biosciences, Inc. Preethi H. Gunaratne is founder, CSO, and shareholder of NextmiRNA Therapeutics. Christina Yau is a part-time employee/consultant at NantOmics. Franz X. Schaub is an employee and shareholder of SEngine Precision Medicine, Inc. Carla Grandori is an employee, founder, and shareholder of SEngine Precision Medicine, Inc. Robert N. Eisenman is a member of the Scientific Advisory Boards and shareholder of Shenogen Pharma and Kronos Bio. Daniel J. Weisenberger is a consultant for Zymo Research Corporation. Joshua M. Stuart is the founder of Five3 Genomics and shareholder of NantOmics. Marc T. Goodman receives research support from Merck, Inc. Andrew J. Gentles is a consultant for Cibermed. Charles M. Perou is an equity stock holder, consultant, and Board of Directors member of BioClassifier and GeneCentric Diagnostics and is also listed as an inventor on patent applications on the Breast PAM50 and Lung Cancer Subtyping assays. Matthew Meyerson receives research support from Bayer Pharmaceuticals; is an equity holder in, consultant for, and Scientific Advisory Board chair for OrigiMed; and is an inventor of a patent for EGFR mutation diagnosis in lung cancer, licensed to LabCorp. Eduard Porta-Pardo is an inventor of a patent for domainXplorer. Han Liang is a shareholder and scientific advisor of Precision Scientific and Eagle Nebula. Da Yang is an inventor on a pending patent application describing the use of antisense oligonucleotides against specific lncRNA sequence as diagnostic and therapeutic tools. Yonghong Xiao was an employee and shareholder of TESARO, Inc. Bin Feng is an employee and shareholder of TESARO, Inc. Carter Van Waes received research funding for the study of IAP inhibitor ASTX660 through a Cooperative Agreement between NIDCD, NIH, and Astex Pharmaceuticals. Raunaq Malhotra is an employee and shareholder of Seven Bridges, Inc. Peter W. Laird serves on the Scientific Advisory Board for AnchorDx. Joel Tepper is a consultant at EMD Serono. Kenneth Wang serves on the Advisory Board for Boston Scientific, Microtech, and Olympus. Andrea Califano is a founder, shareholder, and advisory board member of DarwinHealth, Inc. and a shareholder and advisory board member of Tempus, Inc. Toni K. Choueiri serves as needed on advisory boards for Bristol-Myers Squibb, Merck, and Roche. Lawrence Kwong receives research support from Array BioPharma. Sharon E. Plon is a member of the Scientific Advisory Board for Baylor Genetics Laboratory. Beth Y. Karlan serves on the Advisory Board of Invitae.
References
- Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J, Ling S, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun. 2014;5:3887. doi: 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378–384. doi: 10.1038/nature21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, Chaturvedi P, Hol J, Thienpont B, Teuwen LA, et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell. 2016;30:968–985. doi: 10.1016/j.ccell.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nature Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherniack AD, Shen H, Walter V, Stewart C, Murray BA, Bowlby R, Hu X, Ling S, Soslow RA, Broaddus RR, et al. Integrated Molecular Characterization of Uterine Carcinosarcoma. Cancer Cell. 2017;31:411–423. doi: 10.1016/j.ccell.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667–678. doi: 10.1016/j.molcel.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Sadeghi S, Raymond A, Jackman SD, Nip KM, Mar R, Mohamadi H, Butterfield YS, Robertson AG, Birol I. BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics. 2014;30:3402–3404. doi: 10.1093/bioinformatics/btu558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, McKenna A, Fennell T, Banks E, DePristo M, Getz G. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics. 2011;27:2601–2602. doi: 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Zhang W, Liu S, An H, Tan L, Ma L. LATS1 suppresses proliferation and invasion of cervical cancer. Mol Med Rep. 2017;15:1654–1660. doi: 10.3892/mmr.2017.6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E, Hall MA, Witten IH. The WEKA Workbench Online Appendix for “Data Mining: Practical Machine Learning Tools and Techniques”. Fourth Morgan Kaufmann; 2016. [Google Scholar]
- Gehring JS, Fischer B, Lawrence M, Huber W. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics. 2015;31:3673–3675. doi: 10.1093/bioinformatics/btv408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Angulo AM, Hennessy BT, Meric-Bernstam F, Sahin A, Liu W, Ju Z, Carey MS, Myhre S, Speers C, Deng L, et al. Functional proteomics can define prognosis and predict pathologic complete response in patients with breast cancer. Clin Proteomics. 2011;8:11. doi: 10.1186/1559-0275-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15:585–598. doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MD, Niu B, McLellan MD, Uzunangelov V, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, et al. Cell-of-origin patterns dominate molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018 doi: 10.1016/j.cell.2018.03.022. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Rabinovic A, Li C. Adjusting batch effects in microarray expression data using Empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Jonsson P, Coarfa C, Mesmar F, Raz T, Rajapakshe K, Thompson JF, Gunaratne PH, Williams C. Single-Molecule Sequencing Reveals Estrogen-Regulated Clinically Relevant lncRNAs in Breast Cancer. Mol Endocrinol. 2015;29:1634–1645. doi: 10.1210/me.2015-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Yang CK, Heo K, Roeder RG, An W, Stallcup MR. CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol Cell. 2008;22:510–9. doi: 10.1016/j.molcel.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konecny GE, Kristeleit RS. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: current practice and future directions. Br J Cancer. 2016;115:1157–1173. doi: 10.1038/bjc.2016.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nature Genetics. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RG, Getz G, Meyerson M. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat Biotechnol. 2011;29:393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Dai W, Mo W, Li J, Feng J, Wu L, Liu T, Yu Q, Xu S, Wang W, et al. By inhibiting PFKFB3, aspirin overcomes sorafenib resistance in hepatocellular carcinoma. Int J Cancer. 2017;141:2571–2584. doi: 10.1002/ijc.31022. [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biology. 2001;2:research0032.1–research0032.11. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAvoy S, Ganapathiraju SC, Ducharme-Smith AL, Pritchett JR, Kosari F, Perez DS, Zhu Y, James CD, Smith DI. Non-random inactivation of large common fragile site genes in different cancers. Cytogenet Genome Res. 2007;118:260–269. doi: 10.1159/000108309. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nature Genetics. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M, Heravi Moussavi A, Senz J, Melnyk N, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron K, Golan-Lev T, Dvir R, Ben-David E, Kerem B. Oncogenes create a unique landscape of fragile sites. Nat Commun. 2015;6:7094. doi: 10.1038/ncomms8094. [DOI] [PubMed] [Google Scholar]
- Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Machine Learning. 2003;52:91–118. [Google Scholar]
- Mullen RD, Behringer RR. Molecular genetics of Müllerian duct formation, regression, and differentiation. Sex Dev. 2014;8:281–296. doi: 10.1159/000364935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthu M, Cheriyan VT, Rishi AK. CARP-1/CCAR1: a biphasic regulator of cancer cell growth and apoptosis. Oncotarget. 2015;6:6499–6510. doi: 10.18632/oncotarget.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Peng F, Li Q, Sun JY, Luo Y, Chen M, Bao Y. PFKFB3 is involved in breast cancer proliferation, migration, invasion and angiogenesis. Int J Oncol. 2018;52:945–954. doi: 10.3892/ijo.2018.4257. [DOI] [PubMed] [Google Scholar]
- Quinlan R. Learning efficient classification procedures. In: Michalski, Carbonell, Mitchell, editors. Machine Learning: an artificial intelligence approach. Morgan Kaufmann Publishers Inc; 1983. pp. 463–482. [Google Scholar]
- Radenbaugh AJ, Ma S, Ewing A, Stuart JM, Collisson E, Zhu J, Haussler D. RADIA: RNA and DNA Integrated Analysis for Somatic Mutation Detection. PLoS One. 2014;9:e111516. doi: 10.1371/journal.pone.0111516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, Getz G. Oncotator: cancer variant annotation tool. Hum Mutat. 2015;36:E2423–2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratan A, Olson TL, Loughran TP, Jr, Miller W. Identification of indels in next-generation sequencing data. BMC Bioinformatics. 2015;16:42. doi: 10.1186/s12859-015-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, et al. De novo assembly and analysis of RNA-seq data. Nat Methods. 2010;7:909–912. doi: 10.1038/nmeth.1517. [DOI] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Papapetrou EP, Bushman FD. Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer. 2012;12:51–58. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Sedgewick AJ, Benz SC, Rabizadeh S, Soon-Shiong P, Vaske CJ. Learning subgroup-specific regulatory interactions and regulator independence with PARADIGM. Bioinformatics. 2013;29:i62–i70. doi: 10.1093/bioinformatics/btt229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TC, Frank E. Introducing Machine Learning Concepts with WEKA. In: Mathé, Ewy, Davis, Sean, editors. Statistical Genomics: Methods and Protocols. Humana Press Inc; 2016. pp. 353–378. [DOI] [PubMed] [Google Scholar]
- Stadhouders R, Cico A, Tharshana S, Thongjuea S, Kolovos P, Baymaz HI, Yu X, Demmers J, Bezstarosti K, Maas A, et al. Control of developmentally primed erythroid genes by combinatorial co-repressor actions. Nat Commun. 2015;6:8893. doi: 10.1038/ncomms9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, Kornblau SM. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- Torres-Garcia W, Zheng S, Sivachenko A, Vegesna R, Wang Q, Yao R, Berger MF, Weinstein JN, Getz G, Verhaak RG. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics. 2014;30:2224–2226. doi: 10.1093/bioinformatics/btu169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, Stuart JM. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26:i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Bachman J, Lai ZC. Mutation analysis of large tumor suppressor genes LATS1 and LATS2 supports a tumor suppressor role in human cancer. Protein Cell. 2015;6:6–11. doi: 10.1007/s13238-014-0122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, Meyerson M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet. 2016;48:176–182. doi: 10.1038/ng.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. (Related to Figure 1) Amplification and deletion peaks identified by GISTIC in the Pan-Gyn cohort.
Table S3. (Related to Figure 2) Table showing 46 statistically significantly mutated genes across the Pan-Gyn cohort.
Table S4. (Related to Figures 2-7) Clinical data and cluster memberships of the samples in the study across all of the platforms and analysis types.
Table S5. (Related to Figures 2-7) Contingency tables showing numbers of samples from tumor types vs. clusters for various platforms.