

Abstract
The recent years have seen significant progress in the development of systemic therapies to treat patients with advanced melanoma. Use of these new treatment modalities, which include immune checkpoint inhibitors and small molecule BRAF inhibitors, lead to increased overall survival and better outcomes. Although revolutionary, these therapies are often less effective against melanoma brain metastases, and frequently the CNS is the major site of treatment failure. The development of brain metastases remains a serious complication of advanced melanoma that is associated with significant morbidity and mortality. New approaches to both prevent the development of brain metastases and treat established disease are urgently needed. In this review we will outline the mechanisms underlying the development of melanoma brain metastases and will discuss how new insights into metastasis biology are driving the development of new therapeutic strategies. Finally, we will describe the latest data from the ongoing clinical trials for melanoma brain metastases patients.
Keywords: melanoma, metastasis, brain, BRAF, immunotherapy
Graphical abstract
1. Introduction
Among all tumor types, non-small cell lung cancer (NSCLC), melanoma and breast cancers have the highest propensity to metastasize to the brain1, 2. For melanoma, CNS involvement is clinically evident in over 40-60% of patients, with levels being as high as 75% at autopsy3. Without treatment, melanoma brain metastases (MBM) progress rapidly, with an average survival of approximately 3 months; approximately 50% of all melanoma deaths result from brain metastases 4. Brain metastatic melanoma cells disseminate through hematogenous spread, with the location of brain metastases being well correlated with the areas of the brain that receive the highest blood flow; 80% of brain metastases are located in the cerebral hemispheres, with much lower percentages being found in the cerebellum and brain stem (15% and 5%, respectively) 5. Risk factors for MBM development include male gender, head or neck primary disease site and the presences of visceral or nodal metastases 6. Other reported risk factors for the development of MBM include increased serum lactate dehydrogenase (LDH) levels, 3 or more visceral metastases and a high Clark's level/Breslow thickness of the primary disease 7, 8. The initial clinical presentation of melanoma brain metastases typically comprises of headache, seizures, and neurological impairment.
Over the recent years there has been speculation that the incidence of brain metastases is increasing; most likely a result of improved systemic therapies and better detection of brain-resident lesions through new imaging modalities. Until recently, no systemic therapies existed for MBM, with treatment largely consisting of surgery and radiation therapy (to manage symptoms and for palliation). The development of targeted therapies and immunotherapies for melanoma has improved overall survival (OS) for patients with stage IV disease9, 10. Despite these gains, treatment failure is common, and the brain is often the major site of disease progression; even when extracranial disease is well controlled 11. Indeed, in patients who achieve a complete response to the combination of dabrafenib and trametinib, 54% of eventual treatment failures are in the CNS. There is emerging evidence that patients with MBM respond to both immunotherapies and to targeted therapies, albeit with a reduced progression-free survival (PFS) compared to those with disease at extracranial sites 12. The reasons underlying these reduced durations of response remain to be determined, and may be a consequence of the unique microenvironment of the brain shaping the transcriptional and phenotypic behavior of brain-resident melanoma cells. In this review we will outline the latest findings on the biology that underlies MBM development. We will then discuss how this knowledge can help to define improved therapies for MBM.
2. The biology of melanoma brain metastases
2.1 Dissemination of melanoma cells to the brain: the blood brain barrier
Metastatic dissemination to the brain is a complex process that involves the escape of malignant cells from the primary tumor, their migration through the surrounding tissue into the vasculature, survival in the circulation, exit from the circulation and passage through the blood brain barrier (BBB). Of these steps, the movement of cancer cells across the blood brain barrier is particularly difficult. The BBB consists of a physical cellular barrier, extracellular matrix (ECM) and drug/solute transporters (Figure 1). The physical barrier of the BBB consists of contiguous endothelial cells linked by multiple tight junctions (modulated by components such as ZO-1, claudins and occludins) that securely restrict the passage of macromolecules, drugs, toxins and pathogens into the brain 13-16. Brain vascular endothelial cells are distinct from those that constitute the vasculature of other organs in expressing high levels of drug pumps and low expression of leukocyte adhesion molecules. They also show low rates of transcytosis. Together these serve to restrict the passage of macromolecules as well as preventing the adhesion (and therefore transmigration) of immune cells onto the surface of the brain. The behavior of brain-resident endothelial cells is tightly regulated by neighboring astrocytes that send out multiple protrusions (called astrocyte foot processes) which tightly wrap the endothelial cells (Figure 1). Signals from the astrocytes induce BBB-like properties in the brain endothelial cells with a role for development signaling pathways, such as Wnt, being suggested 16-18. Recent work has also shown that fibroblast-like pericytes can contribute to BBB integrity, with loss of pericytes in transgenic mouse models increasing the permeability of the BBB to water and high molecular weight tracers 19. Pericytes contribute to BBB function in other ways such as regulating adherens and tight junctions in brain endothelial cells and helping to polarize the astrocyte end feet that interact with the cerebral microvasculature 19. Other proposed functions of the pericytes in the brain vasculature include the regulation of transcytosis and control of extracellular matrix deposition 20. Multiple pericyte populations are known to exist within the brain microvasculature, and it has been suggested that distinct subsets of these may help facilitate metastasis development. In experimental breast cancer models, the development of brain metastases is associated with increased BBB permeability that is secondary to the selection of pericytes with high desmin expression and decreased expression of laminin α2 in the percytic basement membrane13. The development of brain metastases typically involves the breakdown of BBB integrity; a multi-step process that involves dilation of the cerebral blood vessel, increased VEGF release, decreased ZO-1 expression at the tight junctions and the initiation of an inflammatory response 21. There is evidence from preclinical models that the integrity of the blood brain barrier is an important regulator of the pattern of MBM growth. In cases where MBM growth was associated with breakdown of the BBB, the tumors grew as “ball shaped” masses that were demarcated from the brain parenchyma 14. In models where the BBB integrity was maintained (as demonstrated by lack of sodium fluorescein accumulation in the CNS space), the melanoma cells grew by co-opting existing blood vessels of the brain 14.
Figure 1. The migration of melanoma cells through the blood brain barrier.
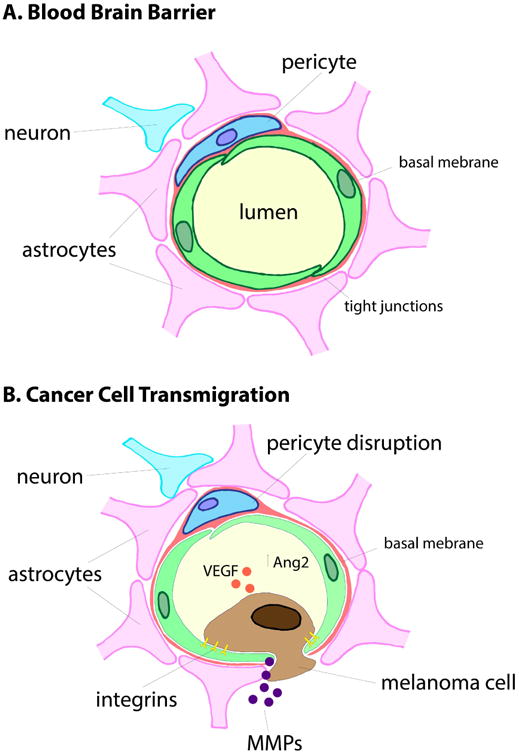
To invade into the brain, the melanoma cells must migrate through the blood brain barrier. Under normal physiology the BBB is formed by tightly interlocking endothelial cells that are surrounded by a basement membrane and astrocyte foot processes. Pericytes also contribute to BBB integrity by regulating brain endothelial cell behavior. Movement of melanoma cells through the BBB is a multi-step process that involves pro-invasive integrins, the loosening of the endothelial cell tight junctions, degradation of the basement membrane (through the release of MMPs and other proteases) and mechanical forces that push the endothelial cells apart.
2.2 Migration into the brain
For many years the mechanisms that underlie the migration of melanoma cells into the brain have remained obscure. Dramatic new insights into this process have been made through the development of in vivo, live-cell imaging techniques that allow the fates of individual cancer cells to be followed through cranial window imaging and 2-photon microscopy22. These studies have demonstrated the migration of melanoma cells into the brain to be a multi-step process. In the initial phase, melanoma cells in the circulation move passively through the brain vasculature and are carried along by the blood flow (Figure 2). Upon reaching the narrow capillaries of the microvasculature, the melanoma cells arrested and remained in a quiescent state for 1-9 days, before beginning to extravasate 22. The migration of cancer cells into the brain was significantly slower than into any other organ (∼6 hrs into the liver), perhaps explaining the long latency of MBM development compared to that of other organs 23. The process of extravasation was dependent, in part, upon mechanical forces, with the cancer cells becoming rounded and developing cytoplasmic protrusions that served to push the endothelial cells apart (Figure 1). The loosening of the tight junctions between endothelial cells preceded the migration of the cancer cells through the BBB and was dependent upon multiple mechanisms including release of angiopoetin-2 and the expression of multiple pro-invasive integrins on the cancer cells (including integrin α3β1, αvβ3 and α4β1) 24-26. At the same time, the invading melanoma cells also expressed proteases (such as MMP-9 and heparanase) that degraded the basement membrane of the BBB 15, 27 (Figure 1). Other proteases, such as cathepsin-S, were also critical for BBB transmigration with studies showing it to be secreted by multiple cell types including both macrophages and tumor cells. Mechanistically, cathepsin-S promoted brain metastasis development through its proteolytic activity, that cleaved the BBB tight junction protein JAM-B 25. Strategies to maintain BBB integrity, such as inhibition of cathepsin-S, using the small molecule inhibitor (VBY-999) reduced experimental brain metastasis formation 25. Other proteolytic enzymes, such as heparanase, also contribute to loss of BBB integrity, with studies showing heparanase expression to be increased in melanoma cells following exposure to neurotrophins 28.
Figure 2. The process of MBM development.
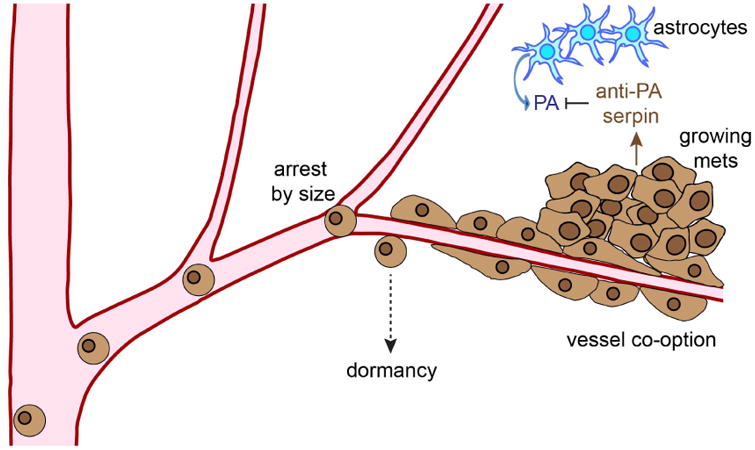
Melanoma cells move passively through the vasculature until they reach the smaller vessels of the brain. Here they arrest due to size restriction until moving through the blood vessel wall and then remaining in contact with the abluminal surface. Successful brain metastases often secrete protease inhibitors that abrogate the anti-tumor effects of serpins secreted by the host astrocytes.
Once melanoma cells have migrated through the BBB, they stay associated with the endothelial cells at the abluminal surface, in a manner analogous to pericytes (Figure 2). The melanoma cells that did not maintain contact with the blood vessels typically died 22. Initial growth of the melanoma micrometastases typically occurred along side the vasculature, with the vessels being frequently co-opted 22. Macrometastases eventually grew from the smaller micrometastases and remained associated with the co-opted blood vessels. Interestingly, some individual melanoma cells stayed dormant while associated with the brain microvasculature but maintained the ability to migrate along the blood vessels 22 (Figure 2). Whether these represent a potential reservoir of dormant melanoma cells remains to be determined.
2.3 The role of the brain microenvironment in the development of brain metastases
Once in the brain, tumor cells interact with multiple cell types; primarily the glia, which includes microglia, oligodendrocytes and astrocytes. The interaction of cancer cells with normal brain cells is complex with both tumor suppressive and tumor supportive effects being reported. For the most part, the brain is a hostile environment for cancer cells, with imaging studies demonstrating that the majority of cancer cells reaching the brain rapidly die 22, 29. This protection is mediated, in part, through the release of proteases such as plasmin within the brain, that activate the death-inducing FAS ligand on astrocytes that trigger apoptosis in the cancer cells and inactivate L1-CAM 30. Successful brain metastatic cancer cells counteract these measures through the expression of protease inhibitors (such as the Serpins) and by increasing their activity in cell survival pathways 30, 31. Under physiological conditions, healthy brain cell are protected from this proteolytic activity through expression of brain-specific protease inhibitors, such as the neuroserpins 30. Conversely, the host brain cells can also contribute to tumor progression, and it is known that both microglia and astrocytes become activated upon contact with cancer cells and often infiltrate the tumor mass 32-34. Co-culture of cancer cells with glia in vitro leads to increased tumor proliferation, suggesting that the glia are permissive for brain metastasis growth 32, 34-36. Microglia are the brain-resident macrophages and frequently become activated following interaction with tumor cells 37. There is evidence that the microglia, like tissue-resident macrophages, are involved in metastasis, with studies implicating them in the increased invasion and colonization of breast cancer cells into the brain through a mechanism involving Wnt signaling 32, 37, 38. Intriguingly, the activation of the microglia occurred following the arrest of the cancer cells in the cerebral blood vessels, long before they actually extravasate into the brain parenchyma 39.
The major non-neuronal cell type in the brain are the astrocytes, which comprise ∼50% of the total cells in the brain. Their primary function is to maintain homeostasis and they play roles in ionic balance, pH, metabolism, and the integrity of the BBB 40. Astrocytes are also the stromal cell most frequently implicated in brain metastasis development and become activated upon interaction with cancer cells (so-called reactive astrocytes), secreting many growth factors, chemokines and cytokines (factors including IL-6, TNF-α and IL-1β) that contribute to tumor cell survival 32,31, 35, 36 (Figure 3). Reactive astrocytes can induce the expression of multiple pro-survival genes in brain-resident cancer cells including BCL2L1, TWIST 35, as well as pro-invasive matrix metalloproteinases such as MMP-2 and MMP-9 38. Recent studies have suggested that brain metastatic cancer cells may also communicate directly with astrocytes through protocadherins and gap junctions, such as Connexin-43, through which the second messenger 2′3′-cyclic GAMP-AMP (cGAMP) is transferred 41 (Figure 3). This, in turn, activates the STING pathway in the astrocytes, leading to the release of interferon (IFN)-α and tumor necrosis factor (TNF)-α, which acts in a paracrine fashion to increase STAT1 and NFκB signaling in the cancer cells, increasing their survival and drug resistance 41.
Figure 3. Cross-talk between cancer cells and astrocytes contributes to the progression of brain metastases.
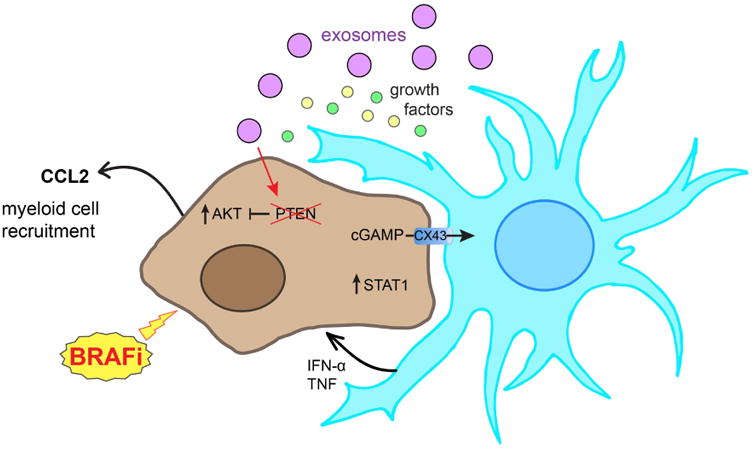
The interaction of cancer cells and astrocytes frequently causes astrocyte activation and the release of multiple growth factors that contribute to tumor progression. Astrocytes can also secrete exosomes containing microRNAs that silence key tumor suppressors such as PTEN. Co-culture of astrocytes with melanoma cells also limits responses to BRAF inhibition through increased PI3K/AKT activity in the melanoma cells. At the same time astrocytes connect directly to cancer cells through Connexin-43 (Cx43) mediated gap junctions, where the transfer of the second messenger cGAMP activates the STING pathway in astrocytes leading to the release of IFNα and TNFα to stimulate STAT1 signaling in the cancer cells, leading to enhanced survival and chemoresistance.
Crosstalk between brain metastatic cancer cells and astrocytes also seems critical for the maintenance of stemness and self-renewal in breast cancer brain metastases 33. Recent work has shown that the release of IL-1β from breast cancer brain metastases stimulates the expression of the Notch ligand JAG1 in nearby astrocytes, which increases self-renewal in the cancer stem cell compartment 33. Further evidence for a critical role of reactive astrocytes comes from direct targeting studies in which inhibition of PDGF receptor-β (a key growth factor receptor for astrocytes) prevents the growth of breast cancer brain metastases 42. As well as directly influencing tumor growth, astrocytes can also modulate drug sensitivity, with studies suggesting that astrocytes protect multiple cancer types including melanoma, breast cancer and lung cancer from chemotherapy, through direct cell-cell contact 36.
In addition to influencing the behavior of brain metastatic cells through the release of soluble factors and through cell-cell adhesion, astrocytes also regulate cancer cells through epigenetic means. One major target of astrocyte mediated epigenetic regulation is expression of the tumor suppressor (and PI3K/AKT pathway regulator) PTEN in brain metastatic cancer cells. Studies on breast cancer cells xenografted into mice have demonstrated that PTEN expression is typically lost only when the cells metastasize to the brain, but not to other organs 43. The loss of PTEN expression was reversible, being restored when the cancer cells were removed from the brain microenvironment 43. Mechanistic analyses showed this effect to be mediated by neighboring astrocytes that secreted microRNAs in exosomes that were then taken up by the cancer cells, leading to the epigenetic silencing of both PTEN RNA and protein (Figure 3). Depletion of the PTEN-targeting microRNA from the astrocytes, reversed the loss of PTEN in the neighboring tumor cells 43. Further experiments showed that PTEN loss was critical for the progression of brain metastases through a mechanism involving increased CCL2 release from the tumor cells that led to the recruitment of myeloid cells, resulting in increased growth and reduced the apoptotic levels in the brain metastases 43.
Other studies in melanoma have supported the idea that the CNS microenvironment shapes the transcriptional profile of brain metastases, with studies showing that melanoma cells in the brain adopt a more neuronal-like transcriptional state44,45. Genes showing altered expression in melanoma cells in the brain microenvironment included those involved in neuropathic pain signaling, synaptic long-term potentiation, glutamate signaling and axonal guidance 45. The most significantly down-regulated genes were those involved in metabolism such as oxidative phosphorylation and mitochondrial function 45. Again, the effects of the brain environment upon the transcriptional profiles of MBM cells appeared to be epigenetic with methylation profiling showing a tight clustering between all of the brain metastases samples 43. These effects upon methylation could be partly recapitulated when the cancer cell lines were co-cultured with astrocytes.
The role of oligodendrocytes, whose primary function is to produce myelin that wraps axons and ensures efficient neurotransmission, in melanoma brain metastases development and progression has been little studied 46. It is however known that oligodendrocytes are highly sensitive to many of the chemotherapeutics commonly used to treat brain metastases and that the loss of oligodendrocytes following drug treatment is thought to underlie some of the neurotoxicities associated with chemotherapy use 47.
2.4 Signaling in melanoma brain metastases
The brain microenvironment is quite distinct from that of other organs raising the possibility that brain metastatic cancer cells may be dependent upon a unique series of signaling pathways. Early work, in which a series of non-matched cranial and extracranial melanoma metastases were compared by reverse phase protein array (RPPA) and immunohistochemistry, revealed a preferential role for PI3K/AKT signaling in the brain metastases samples 48. Specifically, it was shown that 60% of the brain metastasis samples had reduced PTEN expression and elevated PI3K/AKT pathway signaling; including S473-AKT, T308-AKT and phospho-GSKα/β 48. These initial findings were later confirmed in a small immunohistochemistry (IHC) study of nine matched sets of cranial and extracranial metastases 49, as well as a further study of matched samples in which AKT pathway activity was measured using RPPA 50. In the latter study, loss of PTEN expression was only observed infrequently, even in specimens with high levels of AKT signaling 50.
Recent preclinical animal modeling studies have demonstrated that AKT may also be required for the initiation of melanoma brain metastases. It was found that introduction of myristolated AKT1 into mouse melanocytes that were BRAF V600E mutant/Cdkn2a-null led to the development of melanomas that metastasized to the lung and brain in 67% and 17% of cases, respectively 51. In this instance, the effects of AKT1 activation were not recapitulated by the silencing of PTEN, but PTEN loss did co-operate with myristolated AKT1 to reduce the latency of tumor development and the appearance of distant metastases 51. Significant differences in PI3K/AKT/mTOR pathway utilization was observed between the PTEN-null and PTEN/myr-AKT groups detected by RPPA, with the myr-AKT1 group showing greater mTOR signaling activity 51. Although the role of PTEN loss in brain metastasis development from animal models has been somewhat conflicting, and only a subset of established brain metastases seemed to lack PTEN protein expression 50, there is evidence that its complete loss can be prognostic for MBM development at earlier disease stages 52. A retrospective analysis of 136 patients with stage IIIB/C melanoma showed that loss of PTEN expression (as determined by IHC staining) in conjunction with a BRAF V600E mutation predicted for a reduced time to brain metastasis development and a shorter overall survival 52.
In other recent work, gene expression profiling approaches have been utilized to identify putative drivers of MBM development. One such study identified the phospho-inositide binding protein PLEKHA5 as being associated with increased risk of MBM development 53. It was found that silencing of PLEKHA5 decreased the viability of the A375BR cerebrotropic melanoma cells and prevented their migration through BBB-like structures in vitro. Similar results were also observed in a cell line derived from a melanoma patient with extensive brain metastases 53. A major question facing the field is whether the melanoma cells that metastasize to the brain are genetically or epigenetically distinct from those that metastasize to other organs. There is already evidence from other cancers (including breast, lung and kidney cancers) that up to 53% of brain metastases have distinct, and possibly therapeutically tractable, mutations in the PI3K/AKT and EGFR/HER2 signaling cascades that are not present in matched metastases from extra-cranial sites 54. Other studies, characterizing circulating tumor cells (CTCs) from patients with and without breast cancer brain metastases, identified a unique gene expression signature in CTCs competent for brain metastases characterized by increased Notch, innate/adaptive immunity, pluripotent stem cell behavior, and immunosuppression 55.
Although much of the MBM research focus to date has been on the role of the PI3K/AKT/mTOR pathway, other signal transduction cascades have also been implicated. One such signaling pathway is JAK/STAT3; a key driver of both cell growth and angiogenesis in multiple melanoma models. STAT3 functions as a transcription factor with direct effects upon the expression of cyclin D1, MMP-2, c-Myc and VEGF 56. Expression of STAT3 in melanoma cells increased their potential to develop brain metastases in nude mice, whereas expression of a dominant negative STAT3 reduced MBM formation 56. Further studies showed that increased STAT3 expression led to increased angiogenesis in vivo and increased melanoma cell invasion in vitro, associated with increased expression of VEGR, bFGF and MMP-2 56. Attempts have also been made to determine whether growth factors can increase the homing of melanoma cells to the brain. A number of potential factors have been identified, including expression of endothelin receptor B (EDRB) on brain-metastatic melanoma cells 57. The brain is known to express high levels of the ligand ET-3, suggesting a possible brain homing mechanism 57. Melanomas also express high levels of receptors for neurotrophins, with levels of p75NTR being associated with increased risk of MBM development 28. Multiple ligands for the neurotrophin receptors, such as neurotrophin-3 (NT3) are expressed in astrocytes, and their expression is typically increased upon astrocyte activation; again suggesting a possible homing signal for melanoma cells to the brain 28, 58.
Another unique feature of the brain is its metabolic environment, which is characterized by high levels of glucose oxidation that are required to meet the energy demands of neuronal function 59, 60. There has been some suggestion that the metabolic microenvironment of the brain may influence both tumor progression and drug responses in MBM. Preclinical studies in which CTCs from stage IV breast cancer patients were seeded into either cranial or extra cranial sites of immunodeficient mice showed that the cancer cells that adapted to grow in the brain underwent metabolic rearrangement characterized by increased dependency upon glycolysis, the TCA cycle and oxidative phosphorylation 61. These brain-adapted cancer cells also showed increased activity in the pentose phosphate pathway and the glutathione system which provide protection under conditions of high-level ROS generation 61. Other work showed that breast cancer cells growing in the brain had higher expression of glycolytic enzymes but did not show signs of increased glucose uptake. Instead it was found that these metastatic breast cancer cells proliferated independently of glucose, and showed enhanced gluconeogenesis as well as metabolism of glutamine and branched chain amino acids. Silencing of key metabolic enzymes such as fructose 1,6-bisphosphatase reduced the growth of brain metastatic cells, leading to increased overall survival of the tumor bearing animals 62.
3. Therapeutic strategies for Melanoma Brain Metastases
3.1 Radiation therapy
Whole brain radiation therapy (WBRT) has been a mainstay of treatment for brain metastases since the 1950s, when it was first shown to improve neurological function of MBM patients. Despite its widespread use, WBRT has not been shown to improve long-term survival and most patients develop recurrences. An analysis of patients treated with WBRT during the early 2000s demonstrated the median OS to be 3.4 months, compared to 2.1 months in patients receiving best supportive care. Melanomas are known to be radiation resistant - perhaps explaining the lack of long term clinical benefit following WBRT. At this time use of WBRT is restricted to patients who are inoperable, or have large volume tumors or diffuse, symptomatic disease. A more effective approach, particularly in the treatment of smaller lesions, has been stereotactic radiosurgery (SRS). This approach utilizes a Gamma knife or a linear accelerator and involves use of a small, focused beam of high-energy radiation to directly ablate the lesion while leaving the surrounding brain tissue intact. In patients with low numbers (less than 5-7) of small MBM, SRS can be associated with a high rate of local control that has been associated with a median OS of 5-11 months in single-institution studies 8. Although SRS is now the standard of care for patients with limited numbers of MBM, its impact on PFS and OS has never been formally evaluated in randomized clinical trials.
3.2 Targeted therapy approaches to brain metastasis treatment
The discovery of activating mutations in the serine/threonine kinase BRAF as a driver oncogene in ∼50% of all cutaneous melanomas has revolutionized the treatment of melanoma 63. At the molecular level, mutations in BRAF contribute to the oncogenic behavior of melanoma cells through activation of the mitogen activated protein kinase (MAPK) pathway leading to uncontrolled cell growth, increased cell survival and invasion 64-66. To date, two BRAF inhibitors (vemurafenib, dabrafenib) have been FDA-approved for the treatment of stage IV melanoma with a third (encorafenib) being submitted to the FDA for review (in combination with the MEK inhibitor binimetinib). All three of these drugs show good single-agent activity and rapidly shrink established melanomas at multiple extracranial sites. Although initial responses to BRAF inhibitors are highly impressive, resistance is commonplace, with reactivation of the MAPK pathway through multiple mechanisms (NRAS-mutations, BRAF-splice mutants, MEK mutations, adaptive RTK signaling) being implicated 67-70.
BRAF-mutant melanomas frequently metastasize to the brain, with 58% of MBM patients being BRAF-mutant 71. Patients on BRAF inhibitor therapy show an increased risk of progression in the brain, even when the extracranial disease is well-controlled 72-75. Even among patients who respond well to BRAF inhibitor therapy and have complete responses, the CNS is the most frequent site of treatment failure. It has long been believed that the brain is a sanctuary site for drug resistant disease because of poor CNS drug penetration. It is known that BRAF inhibitor levels are lower in the CSF of mice compared to plasma, with both dabrafenib and vemurafenib being substrates for the drug efflux pumps ABCB1 and ABCG2 76, 77. Of the two FDA-approved BRAF inhibitors, vemurafenib has a much lower brain penetration (typically a 3-log lower concentration in mouse CSF than plasma) than dabrafenib 76, 77. Despite this, the BBB is frequently compromised in MBM patients and therapeutics with extracranial activity frequently show intracranial activity75, 78-80. Analysis of MBM patients on vemurafenib therapy showed drug levels in the CSF to be highly variable (a likely reflection of differences in BBB integrity), but consistently lower than that of the plasma 81. Routine MBM treatments such as SRS and surgery frequently compromise the BBB and likely increase the concentrations of drug reaching the brain. Of note, the MBM of two patients who showed the highest reported vemurafenib levels in their CSF had prior SRS 81.
In the largest brain-specific single agent BRAF inhibitor trial to date, dabrafenib had an intracranial response rate of 39% 82. Despite these promising results, PFS and OS levels remain lower for brain metastasis patients than for patients without CNS involvement, and intracranial progression is frequently the major factor that limits the overall systemic responses. More recent studies have focused on the BRAF-MEK inhibitor combination, with the COMBI-MB trial of dabrafenib-trametinib reporting response rates of up to 58% in patients with asymptomatic MBM who had not received prior therapy for brain metastases 12. Encouragingly, the rate of response to the BRAF-MEK inhibitor combination in the brain was similar to that observed at extracranial sites and no unique brain-specific toxicities were observed 12. Despite these promising results, responses in the brain were more short-lived than those at non-CNS sites (6.5 months vs 12.9 months), and the brain was the major site of treatment failure 12. The finding that responses were lower in the brain, suggested a role for the brain microenvironment in therapeutic escape, and there is growing evidence that the brain microenvironment plays an active role in BRAF-MEK inhibitor resistance 83. This phenomenon has been most extensively explored in vitro with studies demonstrating that secreted factors in the CSF limit vemurafenib-induced apoptosis in melanoma cells through increased PI3K/AKT signaling49, 73, 84. Other work showed that growth of melanoma cells in astrocyte conditioned media also reduced sensitivity to vemurafenib 49 (Figure 3). The protective effects of both CSF and astrocyte-conditioned media were reversed through PI3K inhibition 49. Other evidence of brain microenvironment-driven resistance comes from studies in breast cancer in which CNS metastases exhibited HER3-mediated resistance to brain-penetrant PI3K inhibitors that was lacking at extracranial metastatic sites 85. More recent in vivo studies demonstrated that the PI3K inhibitor buparlisib was effective at treating human melanoma cells xenografted into the brains of mice 84. In this instance, PI3K inhibition was more effective against NRAS-mutant rather than BRAF-mutant melanoma brain xenografts 84. Other studies showed that the combination of buparlisib with the BRAF inhibitor encorafenib improved survival of mice with intracranial melanomas, compared to encorafenib alone 50. There may be circumstances in which brain-resident melanoma cells show resistance to PI3K/mTOR inhibition. Studies using cranial window chamber assays showed melanoma cells located within the perivascular niche of the cerebral vasculature to undergo less apoptosis following PI3K inhibitor treatment than melanoma cells further away from blood vessels 14. These data suggest that brain endothelial cells could regulate melanoma cell survival following drug treatment. Whether or not the vascular niche also conveys “stemness” to brain resident melanoma cells – as has been reported for glioblastoma – remains to be determined 86. Stemness in melanoma remains a controversial issue. Multiple studies have convincingly shown that melanomas lack a classical “cancer stem cell” population and instead harbor a high percentage of cells (∼25%) that possess tumor-initiating qualities 87. Another potential therapeutic target in brain metastases and brain tumors is the autophagy pathway. Autophagy is a process in which cellular material is transported to lysosomes for degradation, recycling and the maintenance of cell metabolism. There is evidence that astrocytes upregulate autophagy in breast cancer cells that are metastatic to the brain, and that autophagy inhibition, using chloroquine can resensitize BRAF mutant brain tumor cells to BRAF inhibitors 88, 89.
3.3 Immunological approaches to brain metastasis treatment
Although the brain is generally considered to be an immunologically privileged site, the BBB is frequently impaired in patients with MBM, making immune infiltration likely. An analysis of multiple brain specimens using immunohistochemical staining revealed that lymphocyte infiltration was correlated positively with overall survival. Among the various T-cell subsets, increased levels of CD8+ effector T-cells was predictive of a better prognosis. The inhibitory ligand programmed death ligand-1 (PD-L1) was expressed in the tumor, infiltrating lymphocytes and the reactive glia of the brain 90-92.
PD-L1 is the ligand of an immune inhibitory receptor called programmed death (PD)-1, which is expressed on the surface of activated (and exhausted) T-cells 93. This axis constitutes the most pertinent example of an immune checkpoint; a complex array of ligands and receptors that serve to tone down immune responses that would otherwise damage healthy tissues. It is now well established that tumors have the ability to hijack this mechanism of immune control, in order to prevent tumor immune destruction. Therefore, they constitute an attractive target for the design of cancer therapies aimed at stimulating an anti-tumor immune response94. The first therapy targeting immune checkpoint molecules was an antibody that blocked the interaction of cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) with its ligands 95. Several reports, including anecdotal cases96 and retrospective analyses of clinical trial data 97-100, have documented that patients benefited from anti-CTLA-4 therapies based upon the systemic administration of ipilimumab. The first prospective phase II clinical trial of this immune checkpoint inhibitor in patients with MBM was published in 2012. The authors reported that, within a cohort of neurologically asymptomatic patients, intravenous anti-CTLA-4 treatment resulted in disease control in 18% of the patients. It is important to note that they include complete responders, partial responders, and patients with stable disease in the category “disease control”. Within a parallel cohort that included patients displaying neurological symptoms and receiving corticosteroid therapy, the rate of disease control dropped to 5% 101. It was concluded that systemic anti-CTLA-4 treatment provided a therapeutic benefit to MBM patients, in particular to those with a small disease burden.
In a recent publication, Kluger et al. showed that high PD-L1 expression correlated with greater CD8+ T-cell infiltration which, in turn, correlated with a delayed onset of melanoma brain metastases and improved survival 102. These findings showed that cytotoxic T-cells have the ability to accumulate, and presumably recognize, melanoma lesions located in the brain. Moreover, they suggested that patients with spontaneous CD8+ T-cell infiltrates could benefit from anti-PD-L1 or anti-PD-1 therapies. Consistently, Anderson and collaborators described, in a small retrospective study, that patients receiving radiotherapy in combination with anti-PD-1 therapy (pembrolizumab) experienced a higher rate of objective responses than those receiving radiotherapy alone. Importantly, this effect was not associated with severe adverse events, suggesting that immune checkpoint inhibition may represent an effective and safe option for patients receiving radiotherapy103.
Currently, four clinical trials are listed in clinicaltrials.gov, that match the search for “immunotherapy AND melanoma brain metastasis”: NCT03340129, NCT02374242 (Australia), NCT02460068 (Italy), and NCT03297463 (USA). All of these involve multimodal treatments: combining two types of immunotherapies (NCT02374242, anti-PD-1 and anti-CTLA4; NCT03297463, IL-2 plus anti-CTLA-4) or checkpoint blockade with radiotherapy (NCT03340129, anti-PD-1 and anti-CTLA-4) or with chemotherapy (anti-PD-1 and anti-CTL4, plus Fotemustine). We anticipate that the number of immunotherapy clinical trials including patients with melanoma brain metastases will continue to increase, as the notion of the CNS being an immune sanctuary continues to be challenged. Moreover, the description of multiple immune checkpoint receptors beyond CTLA-4 and PD-1 will likely lead to the development of newer and hopefully more effective combinatorial approaches. A careful analysis of the specific features of intracranial melanoma lesions, and their immunological properties, will be instrumental for the design of the appropriate therapies.
Two Phase 2 studies that combined anti-CTLA treatment (Ipilimumab) with anti-PD1 inhibition (Nivolumab) were recently completed and presented in abstract form 104, 105. Both studies, which consisted of patients with mostly asymptomatic MBMs along with a small cohort of symptomatic or previously irradiated MBMs, showed very similar results. The Checkmate 204 Study (NCT02320058) comprised short-term follow up of 75 patients. In the asymptomatic cohort there was an overall intracranial response rate of 56% (95% CI=44-68%) with approximately 20% of the patients experiencing a CR. In general, the response rate of systemic (i.e. non-intracranial) disease was concordant with that of intracranial brain disease. The results were remarkable (Figure 4), but came at a cost. Approximately half of the patients had Grade 3-4 toxicities - which were manageable. Together these studies suggest that the ipilumumab-nivolumab combination has good activity against asymptomatic MBM along with a tolerable safety profile. Future adoption of this regimen may change clinical practice and could avoid or delay the use of WBRT or SRT.
Figure 4. Impressive responses to ipilimumab and nivolumab in the brain.
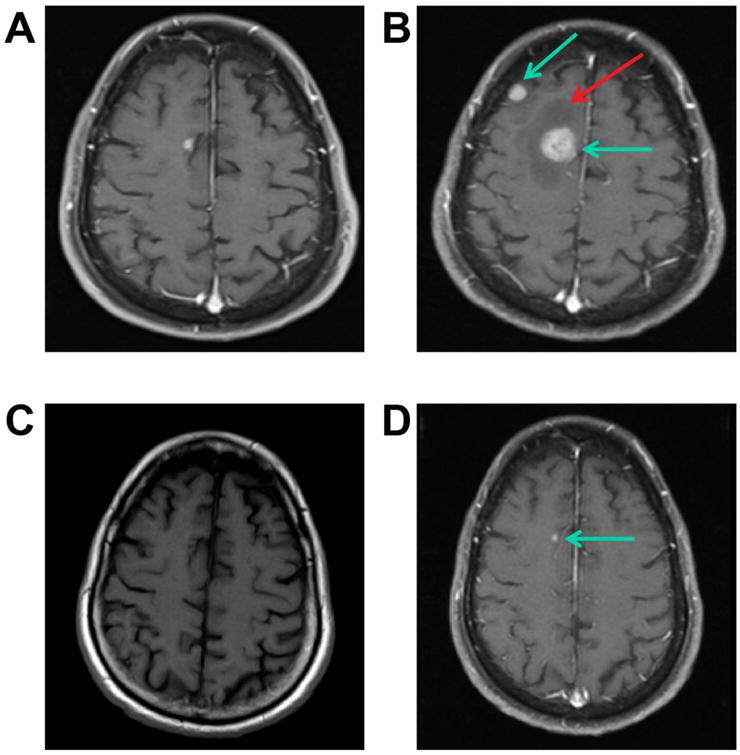
A CT scan of a 71 y.o. man with melanoma lung metastases and approximately 12 asymptomatic brain metastases (Panels A pre-gadolinium scan & B post-gadolinium, blue arrows). Panel B shows considerable surrounding edema (red arrow). After one cycle of Ipilimumab and Nivolumab he had a PR in his lungs and a major PR in his brain (not shown). Four months later, he had a near CR in his lungs and near CR in his brain (Panel C pre-gadolinium scan shows no abnormalities or hemorrhage) and a single tiny area of enhancement in his brain (Panel D, post-gadolinium, blue arrow). The other 11 brain lesions resolved completely. In spite of the edema on his baseline scan (Panel B solid arrow) this was never symptomatic nor had any “pseudoprogression”.
Both studies had small cohorts of poorer prognosis patients who were symptomatic, receiving corticosteroids, had leptomeningeal disease or had previously been irradiated 105. These patients had a poorer response rate and possibly a worse OS. The current challenge for the field is to understand the biology that distinguishes responders and non-responders allowing better predictions to be made as to who should receive conventional therapy for MBMs (such as SRT).
In addition to immune checkpoint blockade, adoptive immunotherapy has gained momentum over the past few years, leading to the recent FDA approval of two T-cell products for the treatment of B-cell malignancies106, 107. This group of therapies involves the isolation of immune cells from the patient, ex vivo expansion and manipulation of the T-cell compartment, and reinfusion of activated tumor-targeted T-cells. Open-repertoire T-cells isolated from the peripheral blood can be retargeted to tumor-associated antigens through genetic manipulation, by induced expression of a cloned T-cell receptor (TCR) or a synthetic immune receptor (CAR, zetakine, etc.)108. Alternatively, naturally occurring tumor-targeting T-cells can be isolated from the tumors109. Tumor-infiltrating lymphocytes (TIL) can be removed from the suppressive tumor microenvironment and stimulated to regain their effector function. Once administered systemically, TILs are expected to traffic back to the tumors and unleash their cytolytic potential. Although TILs have been successfully tested in clinical trials of advanced melanoma110, there is limited information on their efficacy in the control of intracranial lesions. Patients with MBM have been typically excluded from most clinical trials. However, anecdotal cases suggesting that TILs can induce objective responses in intracranial lesions have been reported. In a retrospective analysis, Hong and collaborators showed that 41% of patients treated systemically with TIL experienced complete regression of their melanoma brain metastases and an additional 35% experienced partial responses. In the same report, the authors described that 22% of patients treated with TCR-transgenic T-cells also underwent complete responses 111. In a separate retrospective study, including MBM patients treated with a wider range of immunotherapy regimens (high-dose IL-2, vaccines, adoptive immunotherapy, monoclonal antibodies, and combinations thereof), 9.8% of the patients experienced a complete response to systemic immunotherapy. In addition, 26.8% experienced partial responses and 4.9% had stable disease following treatment 112. While loco-regional cellular therapies have not been tested for MBM, a recent case report on the treatment of glioblastoma multiforme, with intraventricular administration of autologous T-cells targeting IL13RA2, resulted in a complete response with no evidence of toxicity113. These results suggest that intracranial immunotherapy may be an option for MBM as well, provided that the correct tumor-associated antigen is targeted.
4. Future perspectives: are we doing better and what next?
Targeted therapies and immune checkpoint inhibitors have proven transformative in the treatment of advanced melanoma. It is an open question to whether these have also impacted the survival of patients with MBM. Prior to the widespread use of these new therapeutic modalities (2011), 44% of melanoma patients with distant metastases had brain involvement during the course of their disease. The median OS for this patient cohort from time of first brain metastases diagnosis was 4.6 months 4. Other large studies, such as that from the Sydney Melanoma Unit reported similar levels of OS, at 4.1 months 6. In these analyses, the numbers of brain metastases were prognostic, with individuals having >3 lesions having a worse outcome than those with less than 3 lesions. The presence of disease in the leptomeninges was associated with significantly worse outcome and associated with a median OS of less than 2 months. A more recent analysis by our group on a cohort of 610 stage III/IV melanoma patients evaluated between 2000-2012 highlighted the impact of new targeted and immunotherapies upon MBM patient survival 114. It was found that although the incidence of MBM development has remained constant at 40% of patients with advanced melanoma, the median OS improved from 7.5 months (2000-2008) to 22.7 months (2011-last date of follow up) 114. No significant differences were noted with regards to BRAF mutational status, sex or type of therapy received.
Although there are clear improvements in outcomes for MBM patients, we do not yet have therapies that are equi-effective against extracranial and cranial disease. A number of clinical trials are currently underway, evaluating novel drugs and drug combinations such as anti-CTLA-4/anti-PD-1 (ipilimumab-nivolumab), anti-PD-1/VEGFR inhibitor (pembrolizumab-bevacizumab), anti-CTLA-4/SRS, single-agent PI3K inhibition (buparlisib) and CDK4/6 inhibition (abemaciclib). What is needed are new therapeutics, defined by both the biology of the brain microenvironment and the metastatic cascade. Routine tissue interrogation of the MBMs of patients treated with novel therapies would help uncover novel targets and mechanisms of resistance. Strategies that can predict which patients are risk for MBMs (e.g. serum CTCs that are neurotrophic), prevent and treat resistance mechanism in the brain microenvironment, and the abrogate the emergence of brain-specific MBM clones are expected to make major inroads towards the long term management of melanoma.
Acknowledgments
Grant support: Supported by R21 CA198550, R21 CA216756, P50 CA168536 from the National Institutes of Health and a Career Development Award from the Melanoma Research Foundation
Footnotes
Conflict of interest: The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fife KM, Colman MH, Stevens GN, Firth IC, Moon D, Shannon KF, Harman R, Petersen-Schaefer K, Zacest AC, Besser M, Milton GW, McCarthy WH, et al. Determinants of outcome in melanoma patients with cerebral metastases. Journal of Clinical Oncology. 2004;22:1293–300. doi: 10.1200/JCO.2004.08.140. [DOI] [PubMed] [Google Scholar]
- 2.Patchell RA. The management of brain metastases. Cancer Treat Rev. 2003;29:533–40. doi: 10.1016/s0305-7372(03)00105-1. [DOI] [PubMed] [Google Scholar]
- 3.Kenchappa RS, Tran N, Rao NG, Smalley KS, Gibney GT, Sondak VK, Forsyth PA. Novel Treatments for Melanoma Brain Metastases. Cancer Control. 2013;20:298–306. doi: 10.1177/107327481302000407. [DOI] [PubMed] [Google Scholar]
- 4.Davies MA, Liu P, McIntyre S, Kim KB, Papadopoulos N, Hwu WJ, Hwu P, Bedikian A. Prognostic Factors for Survival in Melanoma Patients With Brain Metastases. Cancer. 2011;117:1687–96. doi: 10.1002/cncr.25634. [DOI] [PubMed] [Google Scholar]
- 5.Eichler AF, Chung E, Kodack DP, Loeffler JS, Fukumura D, Jain RK. The biology of brain metastases-translation to new therapies. Nat Rev Clin Oncol. 2011;8:344–56. doi: 10.1038/nrclinonc.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fife KM, Colman MH, Stevens GN, Firth IC, Moon D, Shannon KF, Harman R, Petersen-Schaefer K, Zacest AC, Besser M, Milton GW, McCarthy WH, et al. Determinants of outcome in melanoma patients with cerebral metastases. J Clin Oncol. 2004;22:1293–300. doi: 10.1200/JCO.2004.08.140. [DOI] [PubMed] [Google Scholar]
- 7.Bedikian AY, Wei CM, Detry M, Kim KB, Papadopoulos NE, Hwu WJ, Homsi J, Davies M, McIntyre S, Hwu P. Predictive Factors for the Development of Brain Metastasis in Advanced Unresectable Metastatic Melanoma. Am J Clin Oncol-Canc. 2011;34:603–10. doi: 10.1097/COC.0b013e3181f9456a. [DOI] [PubMed] [Google Scholar]
- 8.Ajithkumar T, Parkinson C, Fife K, Corrie P, Jefferies S. Evolving treatment options for melanoma brain metastases. Lancet Oncol. 2015;16:e486–97. doi: 10.1016/S1470-2045(15)00141-2. [DOI] [PubMed] [Google Scholar]
- 9.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N Engl J Med. 2015 doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen JV, Tawbi H, Margolin KA, Amravadi R, Bosenberg M, Brastianos PK, Chiang VL, de Groot J, Glitza IC, Herlyn M, Holmen SL, Jilaveanu LB, et al. Melanoma central nervous system metastases: current approaches, challenges, and opportunities. Pigment Cell Melanoma Res. 2016;29:627–42. doi: 10.1111/pcmr.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies MA, Saiag P, Robert C, Grob JJ, Flaherty KT, Arance A, Chiarion-Sileni V, Thomas L, Lesimple T, Mortier L, Moschos SJ, Hogg D, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863–73. doi: 10.1016/S1470-2045(17)30429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyle LT, Lockman PR, Adkins CE, Mohammad AS, Sechrest E, Hua E, Palmieri D, Liewehr DJ, Steinberg SM, Kloc W, Izycka-Swieszewska E, Duchnowska R, et al. Alterations in Pericyte Subpopulations Are Associated with Elevated Blood-Tumor Barrier Permeability in Experimental Brain Metastasis of Breast Cancer. Clin Cancer Res. 2016;22:5287–99. doi: 10.1158/1078-0432.CCR-15-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osswald M, Blaes J, Liao Y, Solecki G, Gommel M, Berghoff AS, Salphati L, Wallin JJ, Phillips HS, Wick W, Winkler F. Impact of Blood-Brain Barrier Integrity on Tumor Growth and Therapy Response in Brain Metastases. Clin Cancer Res. 2016;22:6078–87. doi: 10.1158/1078-0432.CCR-16-1327. [DOI] [PubMed] [Google Scholar]
- 15.Kawaguchi T, Tobai S, Nakamura K. Extravascular migration of tumor cells in the brain: an electron microscopic study. Invasion Metastasis. 1982;2:40–50. [PubMed] [Google Scholar]
- 16.Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253–7. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi Y, Nomura M, Yamagishi S, Harada S, Yamashita J, Yamamoto H. Induction of various blood-brain barrier properties in non-neural endothelial cells by close apposition to co-cultured astrocytes. Glia. 1997;19:13–26. [PubMed] [Google Scholar]
- 18.Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A. 2009;106:641–6. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–61. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 20.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lockman PR, Mittapalli RK, Taskar KS, Rudraraju V, Gril B, Bohn KA, Adkins CE, Roberts A, Thorsheim HR, Gaasch JA, Huang S, Palmieri D, et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res. 2010;16:5664–78. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, Winkler F. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16:116–22. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 23.Lorger M, Felding-Habermann B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am J Pathol. 2010;176:2958–71. doi: 10.2353/ajpath.2010.090838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avraham HK, Jiang S, Fu Y, Nakshatri H, Ovadia H, Avraham S. Angiopoietin-2 mediates blood-brain barrier impairment and colonization of triple-negative breast cancer cells in brain. J Pathol. 2014;232:369–81. doi: 10.1002/path.4304. [DOI] [PubMed] [Google Scholar]
- 25.Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, Brogi E, Brastianos PK, Hahn WC, Holsinger LJ, Massague J, Leslie CS, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol. 2014;16:876–88. doi: 10.1038/ncb3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voura EB, Ramjeesingh RA, Montgomery AM, Siu CH. Involvement of integrin alpha(v)beta(3) and cell adhesion molecule L1 in transendothelial migration of melanoma cells. Mol Biol Cell. 2001;12:2699–710. doi: 10.1091/mbc.12.9.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendes O, Kim HT, Stoica G. Expression of MMP2, MMP9 and MMP3 in breast cancer brain metastasis in a rat model. Clin Exp Metastasis. 2005;22:237–46. doi: 10.1007/s10585-005-8115-6. [DOI] [PubMed] [Google Scholar]
- 28.Denkins Y, Reiland J, Roy M, Sinnappah-Kang ND, Galjour J, Murry BP, Blust J, Aucoin R, Marchetti D. Brain metastases in melanoma: roles of neurotrophins. Neuro Oncol. 2004;6:154–65. doi: 10.1215/S115285170300067X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heyn C, Ronald JA, Ramadan SS, Snir JA, Barry AM, MacKenzie LT, Mikulis DJ, Palmieri D, Bronder JL, Steeg PS, Yoneda T, MacDonald IC, et al. In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain. Magn Reson Med. 2006;56:1001–10. doi: 10.1002/mrm.21029. [DOI] [PubMed] [Google Scholar]
- 30.Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, Massague J. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156:1002–16. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langley RR, Fan D, Guo L, Zhang C, Lin Q, Brantley EC, McCarty JH, Fidler IJ. Generation of an immortalized astrocyte cell line from H-2Kb-tsA58 mice to study the role of astrocytes in brain metastasis. Int J Oncol. 2009;35:665–72. doi: 10.3892/ijo_00000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Placone AL, Quinones-Hinojosa A, Searson PC. The role of astrocytes in the progression of brain cancer: complicating the picture of the tumor microenvironment. Tumour Biol. 2016;37:61–9. doi: 10.1007/s13277-015-4242-0. [DOI] [PubMed] [Google Scholar]
- 33.Xing F, Kobayashi A, Okuda H, Watabe M, Pai SK, Pandey PR, Hirota S, Wilber A, Mo YY, Moore BE, Liu W, Fukuda K, et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med. 2013;5:384–96. doi: 10.1002/emmm.201201623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fitzgerald DP, Palmieri D, Hua E, Hargrave E, Herring JM, Qian Y, Vega-Valle E, Weil RJ, Stark AM, Vortmeyer AO, Steeg PS. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin Exp Metastasis. 2008;25:799–810. doi: 10.1007/s10585-008-9193-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim SJ, Kim JS, Park ES, Lee JS, Lin Q, Langley RR, Maya M, He J, Kim SW, Weihua Z, Balasubramanian K, Fan D, et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia. 2011;13:286–98. doi: 10.1593/neo.11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Q, Balasubramanian K, Fan D, Kim SJ, Guo L, Wang H, Bar-Eli M, Aldape KD, Fidler IJ. Reactive astrocytes protect melanoma cells from chemotherapy by sequestering intracellular calcium through gap junction communication channels. Neoplasia. 2010;12:748–54. doi: 10.1593/neo.10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pukrop T, Dehghani F, Chuang HN, Lohaus R, Bayanga K, Heermann S, Regen T, Van Rossum D, Klemm F, Schulz M, Siam L, Hoffmann A, et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia. 2010;58:1477–89. doi: 10.1002/glia.21022. [DOI] [PubMed] [Google Scholar]
- 38.Wang L, Cossette SM, Rarick KR, Gershan J, Dwinell MB, Harder DR, Ramchandran R. Astrocytes directly influence tumor cell invasion and metastasis in vivo. PLoS One. 2013;8:e80933. doi: 10.1371/journal.pone.0080933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.JuanYin J, Tracy K, Zhang L, Munasinghe J, Shapiro E, Koretsky A, Kelly K. Noninvasive imaging of the functional effects of anti-VEGF therapy on tumor cell extravasation and regional blood volume in an experimental brain metastasis model. Clin Exp Metastasis. 2009;26:403–14. doi: 10.1007/s10585-009-9238-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob L, Patwa R, Shah H, Xu K, Cross JR, Massague J. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493–8. doi: 10.1038/nature18268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gril B, Palmieri D, Qian Y, Anwar T, Liewehr DJ, Steinberg SM, Andreu Z, Masana D, Fernandez P, Steeg PS, Vidal-Vanaclocha F. Pazopanib inhibits the activation of PDGFRbeta-expressing astrocytes in the brain metastatic microenvironment of breast cancer cells. Am J Pathol. 2013;182:2368–79. doi: 10.1016/j.ajpath.2013.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, Li P, Li M, Wang X, Zhang C, Wang H, Ellis K, et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature. 2015;527:100–4. doi: 10.1038/nature15376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nygaard V, Prasmickaite L, Vasiliauskaite K, Clancy T, Hovig E. Melanoma brain colonization involves the emergence of a brain-adaptive phenotype. Oncoscience. 2014;1:82–94. doi: 10.18632/oncoscience.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park ES, Kim SJ, Kim SW, Yoon SL, Leem SH, Kim SB, Kim SM, Park YY, Cheong JH, Woo HG, Mills GB, Fidler IJ, et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proc Natl Acad Sci U S A. 2011;108:17456–61. doi: 10.1073/pnas.1114210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morrison BM, Lee Y, Rothstein JD. Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 2013;23:644–51. doi: 10.1016/j.tcb.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dietrich J, Han R, Yang Y, Mayer-Proschel M, Noble M. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J Biol. 2006;5:22. doi: 10.1186/jbiol50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies MA, Stemke-Hale K, Lin E, Tellez C, Deng W, Gopal YN, Woodman SE, Calderone TC, Ju Z, Lazar AJ, Prieto VG, Aldape K, et al. Integrated Molecular and Clinical Analysis of AKT Activation in Metastatic Melanoma. Clin Cancer Res. 2009;15:7538–46. doi: 10.1158/1078-0432.CCR-09-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Niessner H, Forschner A, Klumpp B, Honegger JB, Witte M, Bornemann A, Dummer R, Adam A, Bauer J, Tabatabai G, Flaherty K, Sinnberg T, et al. Targeting hyperactivation of the AKT survival pathway to overcome therapy resistance of melanoma brain metastases. Cancer medicine. 2013;2:76–85. doi: 10.1002/cam4.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen G, Chakravarti N, Aardalen K, Lazar AJ, Tetzlaff MT, Wubbenhorst B, Kim SB, Kopetz S, Ledoux AA, Gopal YNV, Pereira CG, Deng WL, et al. Molecular Profiling of Patient-Matched Brain and Extracranial Melanoma Metastases Implicates the PI3K Pathway as a Therapeutic Target. Clinical Cancer Research. 2014;20:5537–46. doi: 10.1158/1078-0432.CCR-13-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cho JH, Robinson JP, Arave RA, Burnett WJ, Kircher DA, Chen G, Davies MA, Grossmann AH, VanBrocklin MW, McMahon M, Holmen SL. AKT1 Activation Promotes Development of Melanoma Metastases. Cell Rep. 2015;13:898–905. doi: 10.1016/j.celrep.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bucheit AD, Chen G, Siroy A, Tetzlaff M, Broaddus R, Milton D, Fox P, Bassett R, Hwu P, Gershenwald JE, Lazar AJ, Davies MA. Complete loss of PTEN protein expression correlates with shorter time to brain metastasis and survival in stage IIIB/C melanoma patients with BRAFV600 mutations. Clin Cancer Res. 2014;20:5527–36. doi: 10.1158/1078-0432.CCR-14-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jilaveanu LB, Parisi F, Barr ML, Zito CR, Cruz-Munoz W, Kerbel RS, Rimm DL, Bosenberg MW, Halaban R, Kluger Y, Kluger HM. PLEKHA5 as a Biomarker and Potential Mediator of Melanoma Brain Metastasis. Clin Cancer Res. 2015;21:2138–47. doi: 10.1158/1078-0432.CCR-14-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, Van Allen EM, Lawrence MS, Horowitz PM, Cibulskis K, Ligon KL, Tabernero J, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015 doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boral D, Vishnoi M, Liu HN, Yin W, Sprouse ML, Scamardo A, Hong DS, Tan TZ, Thiery JP, Chang JC, Marchetti D. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat Commun. 2017;8:196. doi: 10.1038/s41467-017-00196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xie TX, Huang FJ, Aldape KD, Kang SH, Liu AG, Gershenwald JE, Xie KP, Sawaya R, Huang SY. Activation of Stat3 in human melanoma promotes brain metastasis. Cancer Research. 2006;66:3188–96. doi: 10.1158/0008-5472.CAN-05-2674. [DOI] [PubMed] [Google Scholar]
- 57.Cruz-Munoz W, Jaramillo ML, Man S, Xu P, Banville M, Collins C, Nantel A, Francia G, Morgan SS, Cranmer LD, O'Connor-McCourt MD, Kerbel RS. Roles for endothelin receptor B and BCL2A1 in spontaneous CNS metastasis of melanoma. Cancer Res. 2012;72:4909–19. doi: 10.1158/0008-5472.CAN-12-2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang MY, Son H, Lee YS, Lee SH. Neurons and astrocytes secrete factors that cause stem cells to differentiate into neurons and astrocytes, respectively. Mol Cell Neurosci. 2003;23:414–26. doi: 10.1016/s1044-7431(03)00068-x. [DOI] [PubMed] [Google Scholar]
- 59.Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science. 1999;283:496–7. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- 60.Garcia-Espinosa MA, Rodrigues TB, Sierra A, Benito M, Fonseca C, Gray HL, Bartnik BL, Garcia-Martin ML, Ballesteros P, Cerdan S. Cerebral glucose metabolism and the glutamine cycle as detected by in vivo and in vitro 13C NMR spectroscopy. Neurochem Int. 2004;45:297–303. doi: 10.1016/j.neuint.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 61.Chen EI, Hewel J, Krueger JS, Tiraby C, Weber MR, Kralli A, Becker K, Yates JR, 3rd, Felding-Habermann B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007;67:1472–86. doi: 10.1158/0008-5472.CAN-06-3137. [DOI] [PubMed] [Google Scholar]
- 62.Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L, Wang Y, He J, Bollu LR, Gao G, Su F, Briggs J, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015;75:554–65. doi: 10.1158/0008-5472.CAN-14-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 64.Fedorenko IV, Paraiso KH, Smalley KS. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem Pharmacol. 2011;82:201–9. doi: 10.1016/j.bcp.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arozarena I, Sanchez-Laorden B, Packer L, Hidalgo-Carcedo C, Hayward R, Viros A, Sahai E, Marais R. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45–57. doi: 10.1016/j.ccr.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 66.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR, Ribas A, Palma MD, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Research. 2011;71:2750–60. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, Meyerson M, Garraway LA. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, Wolchok JD, de Stanchina E, et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell. 2012;22:668–82. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jakob JA, Bassett RL, Ng CS, Curry JL, Joseph RW, Alvarado GC, Rohlfs ML, Richard J, Gershenwald JE, Kim KB, Lazar AJ, Hwu P, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118:4014–23. doi: 10.1002/cncr.26724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haueis S, Kranzlin P, Mangana J, Cheng PF, Urosevic-Maiwald M, Braun R, Levesque M, Dummer R, Goldinger SM. Does the distribution of pattern of brain metastases during BRAF inhibitor therapy reflect phenotype switching. Melanoma Res. 2017 doi: 10.1097/CMR.0000000000000338. [DOI] [PubMed] [Google Scholar]
- 73.Seifert H, Hirata E, Gore M, Khabra K, Messiou C, Larkin J, Sahai E. Extrinsic factors can mediate resistance to BRAF inhibition in central nervous system melanoma metastases. Pigment Cell Melanoma Res. 2016;29:92–100. doi: 10.1111/pcmr.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rusthoven CG, Doebele RC. Management of Brain Metastases in ALK-Positive Non-Small-Cell Lung Cancer. J Clin Oncol. 2016;34:2814–9. doi: 10.1200/JCO.2016.67.2410. [DOI] [PubMed] [Google Scholar]
- 75.Proto C, Imbimbo M, Gallucci R, Brissa A, Signorelli D, Vitali M, Macerelli M, Corrao G, Ganzinelli M, Greco FG, Garassino MC, Lo Russo G. Epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of central nervous system metastases from non-small cell lung cancer: the present and the future. Transl Lung Cancer Res. 2016;5:563–78. doi: 10.21037/tlcr.2016.10.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mittapalli RK, Vaidhyanathan S, Sane R, Elmquist WF. Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on the brain distribution of a novel BRAF inhibitor: vemurafenib (PLX4032) J Pharmacol Exp Ther. 2012;342:33–40. doi: 10.1124/jpet.112.192195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mittapalli RK, Vaidhyanathan S, Dudek AZ, Elmquist WF. Mechanisms limiting distribution of the threonine-protein kinase B-RaF(V600E) inhibitor dabrafenib to the brain: implications for the treatment of melanoma brain metastases. J Pharmacol Exp Ther. 2013;344:655–64. doi: 10.1124/jpet.112.201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Azer MW, Menzies AM, Haydu LE, Kefford RF, Long GV. Patterns of response and progression in patients with BRAF-mutant melanoma metastatic to the brain who were treated with dabrafenib. Cancer. 2014;120:530–6. doi: 10.1002/cncr.28445. [DOI] [PubMed] [Google Scholar]
- 79.Gerstner ER, Fine RL. Increased permeability of the blood-brain barrier to chemotherapy in metastatic brain tumors: establishing a treatment paradigm. J Clin Oncol. 2007;25:2306–12. doi: 10.1200/JCO.2006.10.0677. [DOI] [PubMed] [Google Scholar]
- 80.Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, Sznol M, Tsiouris AJ, Cohen J, Vortmeyer A, Jilaveanu L, Yu J, Hegde U, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016;17:976–83. doi: 10.1016/S1470-2045(16)30053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sakji-Dupre L, Le Rhun E, Templier C, Desmedt E, Blanchet B, Mortier L. Cerebrospinal fluid concentrations of vemurafenib in patients treated for brain metastatic BRAF-V600 mutated melanoma. Melanoma Res. 2015;25:302–5. doi: 10.1097/CMR.0000000000000162. [DOI] [PubMed] [Google Scholar]
- 82.Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, Puzanov I, Hauschild A, Robert C, Algazi A, Mortier L, Tawbi H, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087–95. doi: 10.1016/S1470-2045(12)70431-X. [DOI] [PubMed] [Google Scholar]
- 83.Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao SL, Rizos H, Sucker A, Scolyer RA, Gutzmer R, Gogas H, Kefford RF, et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. European Journal of Cancer. 2015;51:2792–9. doi: 10.1016/j.ejca.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Niessner H, Schmitz J, Tabatabai G, Schmid AM, Calaminus C, Sinnberg T, Weide B, Eigentler TK, Garbe C, Schittek B, Quintanilla-Fend L, Bender B, et al. PI3K Pathway Inhibition Achieves Potent Antitumor Activity in Melanoma Brain Metastases In Vitro and In Vivo. Clin Cancer Res. 2016;22:5818–28. doi: 10.1158/1078-0432.CCR-16-0064. [DOI] [PubMed] [Google Scholar]
- 85.Kodack DP, Askoxylakis V, Ferraro GB, Sheng Q, Badeaux M, Goel S, Qi X, Shankaraiah R, Cao ZA, Ramjiawan RR, Bezwada D, Patel B, et al. The brain microenvironment mediates resistance in luminal breast cancer to PI3K inhibition through HER3 activation. Science translational medicine. 2017;9 doi: 10.1126/scitranslmed.aal4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gilbertson RJ, Rich JN. Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–6. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 87.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–8. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mulcahy Levy JM, Zahedi S, Griesinger AM, Morin A, Davies KD, Aisner DL, Kleinschmidt-DeMasters BK, Fitzwalter BE, Goodall ML, Thorburn J, Amani V, Donson AM, et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife. 2017;6 doi: 10.7554/eLife.19671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kaverina N, Borovjagin AV, Kadagidze Z, Baryshnikov A, Baryshnikova M, Malin D, Ghosh D, Shah N, Welch DR, Gabikian P, Karseladze A, Cobbs C, et al. Astrocytes promote progression of breast cancer metastases to the brain via a KISS1-mediated autophagy. Autophagy. 2017:1–19. doi: 10.1080/15548627.2017.1360466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berghoff AS, Fuchs E, Ricken G, Mlecnik B, Bindea G, Spanberger T, Hackl M, Widhalm G, Dieckmann K, Prayer D, Bilocq A, Heinzl H, et al. Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall survival time in patients with brain metastases. Oncoimmunology. 2016;5:e1057388. doi: 10.1080/2162402X.2015.1057388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ogiya R, Niikura N, Kumaki N, Bianchini G, Kitano S, Iwamoto T, Hayashi N, Yokoyama K, Oshitanai R, Terao M, Morioka T, Tsuda B, et al. Comparison of tumor-infiltrating lymphocytes between primary and metastatic tumors in breast cancer patients. Cancer Sci. 2016;107:1730–5. doi: 10.1111/cas.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Abou-Ghazal M, Yang DS, Qiao W, Reina-Ortiz C, Wei J, Kong LY, Fuller GN, Hiraoka N, Priebe W, Sawaya R, Heimberger AB. The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin Cancer Res. 2008;14:8228–35. doi: 10.1158/1078-0432.CCR-08-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zuazo M, Gato-Canas M, Llorente N, Ibanez-Vea M, Arasanz H, Kochan G, Escors D. Molecular mechanisms of programmed cell death-1 dependent T cell suppression: relevance for immunotherapy. Ann Transl Med. 2017;5:385. doi: 10.21037/atm.2017.06.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. 2017 doi: 10.1016/j.semcancer.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 95.Lo B, Abdel-Motal UM. Lessons from CTLA-4 deficiency and checkpoint inhibition. Curr Opin Immunol. 2017;49:14–9. doi: 10.1016/j.coi.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 96.Schartz NE, Farges C, Madelaine I, Bruzzoni H, Calvo F, Hoos A, Lebbe C. Complete regression of a previously untreated melanoma brain metastasis with ipilimumab. Melanoma Res. 2010;20:247–50. doi: 10.1097/CMR.0b013e3283364a37. [DOI] [PubMed] [Google Scholar]
- 97.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Weber JS, Amin A, Minor D, Siegel J, Berman D, O'Day SJ. Safety and clinical activity of ipilimumab in melanoma patients with brain metastases: retrospective analysis of data from a phase 2 trial. Melanoma Res. 2011;21:530–4. doi: 10.1097/CMR.0b013e32834d3d88. [DOI] [PubMed] [Google Scholar]
- 99.Silk AW, Bassetti MF, West BT, Tsien CI, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013;2:899–906. doi: 10.1002/cam4.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grimaldi AM, Simeone E, Giannarelli D, Muto P, Falivene S, Borzillo V, Giugliano FM, Sandomenico F, Petrillo A, Curvietto M, Esposito A, Paone M, et al. Abscopal effects of radiotherapy on advanced melanoma patients who progressed after ipilimumab immunotherapy. Oncoimmunology. 2014;3:e28780. doi: 10.4161/onci.28780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Margolin K, Ernstoff MS, Hamid O, Lawrence D, McDermott D, Puzanov I, Wolchok JD, Clark JI, Sznol M, Logan TF, Richards J, Michener T, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13:459–65. doi: 10.1016/S1470-2045(12)70090-6. [DOI] [PubMed] [Google Scholar]
- 102.Kluger HM, Zito CR, Barr ML, Baine MK, Chiang VL, Sznol M, Rimm DL, Chen L, Jilaveanu LB. Characterization of PD-L1 Expression and Associated T-cell Infiltrates in Metastatic Melanoma Samples from Variable Anatomic Sites. Clin Cancer Res. 2015;21:3052–60. doi: 10.1158/1078-0432.CCR-14-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Anderson ES, Postow MA, Wolchok JD, Young RJ, Ballangrud A, Chan TA, Yamada Y, Beal K. Melanoma brain metastases treated with stereotactic radiosurgery and concurrent pembrolizumab display marked regression; efficacy and safety of combined treatment. J Immunother Cancer. 2017;5:76. doi: 10.1186/s40425-017-0282-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tawbi H, Forsyth P, Algazi A, Hamid O, Hodi FS, Moschos S, Khushalani NI, Gonzalez R, Lao CD, Postow M, Atkins MB, Ernstoff MS, et al. Efficacy and safety of nivolumab (NIVO) plus ipilimumab (IPI) in patients with melanoma (MEL) metastatic to the brain: Results of the phase II study CheckMate 204. Journal of Clinical Oncology. 2017;35 [Google Scholar]
- 105.Long GV, Atkinson V, Menzies A, Lo S, Guminski A, Brown MP, Gonzalez MM, Diamante K, Sandhu S, Scolyer R, Emmett L, McArthur G. A randomized phase II study of nivolumab or nivolumab combined with ipilimumab in patients (pts) with melanoma brain metastases (mets): The Anti-PD1 Brain Collaboration (ABC) Journal of Clinical Oncology. 2017;35 [Google Scholar]
- 106.Prasad V. Immunotherapy: Tisagenlecleucel - the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol. 2017 doi: 10.1038/nrclinonc.2017.156. [DOI] [PubMed] [Google Scholar]
- 107.FDA Approves Second CAR T-cell Therapy. Cancer Discov. 2017 doi: 10.1158/2159-8290.CD-NB2017-155. [DOI] [PubMed] [Google Scholar]
- 108.Abate-Daga D, Davila ML. CAR models: next-generation CAR modifications for enhanced T-cell function. Mol Ther Oncolytics. 2016;3:16014. doi: 10.1038/mto.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257:56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US, Klebanoff CA, Hughes MS, et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J Clin Oncol. 2016;34:2389–97. doi: 10.1200/JCO.2016.66.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, Sherry RM. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010;16:4892–8. doi: 10.1158/1078-0432.CCR-10-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lonser RR, Song DK, Klapper J, Hagan M, Auh S, Kerr PB, Citrin DE, Heiss JD, Camphausen K, Rosenberg SA. Surgical management of melanoma brain metastases in patients treated with immunotherapy. J Neurosurg. 2011;115:30–6. doi: 10.3171/2011.3.JNS091107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, Kurien A, Priceman SJ, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016;375:2561–9. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sloot S, Chen YA, Zhao X, Weber JL, Benedict JJ, Mule JJ, Smalley KS, Weber JS, Zager JS, Forsyth PA, Sondak VK, Gibney GT. Improved survival of patients with melanoma brain metastases in the era of targeted BRAF and immune checkpoint therapies. Cancer. 2017 doi: 10.1002/cncr.30946. [DOI] [PMC free article] [PubMed] [Google Scholar]