

Summary
Sulfur incorporation in the biosynthesis of ergothioneine, a histidine thiol-derivative, differs from other well-characterized transsulfurations. A combination of a mononuclear non-heme iron enzyme-catalyzed oxidative C-S bond formation and a subsequent pyridoxal 5′-phosphate (PLP)-mediated C-S lyase reaction leads to the net transfer of a sulfur atom from a cysteine to a histidine. In this study, we structurally and mechanistically characterized a PLP-dependent C-S lyase Egt2, which mediates the sulfoxide C-S bond cleavage in ergothioneine biosynthesis. A cation-π interaction between substrate and enzyme accounts for Egt2’s preference of sulfoxide over thioether as a substrate. Using mutagenesis and structural biology, we captured three distinct states of the Egt2 C-S lyase reaction cycle, including a labile sulfenic intermediate captured in Egt2 crystals. Chemical trapping and high-resolution mass spectrometry were used to confirm the involvement of the sulfenic acid intermediate in Egt2 catalysis.
eTOC Blurb
Irani et al have determined the structure of Egt2, a C-S lyase at the final step in the ergothioneine biosynthesis pathways. Using X-ray crystallography and various biochemical studies, the reaction mechanism was delineated.
Introduction
Sulfur is a key component in macromolecules and small molecular metabolites (Fahey, 2001, Fontecave et al., 2003, Hand and Honek, 2005, Kessler, 2006, Lin et al., 2013, Wang et al., 2007, Wang et al., 2013). Both radical and ionic types of reactions have been employed by nature to construct C-S bonds. In the biosynthesis of biotin (Fugate and Jarrett, 2012), lipoate (Booker et al., 2007), nucleotide methylthiolation (Kessler, 2006), and thiazolidine ring formation in isopenicillin N synthase (Baldwin and Bradley, 1990), C-S bond formation commonly involves radical species. However there are also several known strategies for C-S bond construction that involve ionic species. Among them, sulfide or cysteine thiolate serve as the sulfur source, e.g., in cysteine synthetase (Rabeh and Cook, 2004) or thioether formation in lantibiotic biosynthesis (Willey and van der Donk, 2007). In many other ionic-type transsulfuration reactions, persulfide R-S-SH and thiocarboxylates R-CO-SH are the key intermediates (Johnson et al., 2005, Kessler, 2006), as found in thiamine pyrophosphate (Jurgenson et al., 2009) and molybdopterin cofactor biosynthesis (Schwarz and Mendel, 2006).
Ergothioneine is synthesized by certain species of bacteria and fungi. Its unique redox properties (E0′ = −0.06 V) (Hand and Honek, 2005) ensure its stability against the auto-oxidation that commonly causes other thiols to generate reactive oxygenated species (Saini et al., 2016). Because of its function as an antioxidant (Aruoma et al., 1999), its ability to chelate metal ions, and its biomedical potential (Yang et al., 2012), ergothioneine is used as an additive in many commercial products, such as skincare and diet supplements.
With these beneficial effects, there is an increasing interest in developing efficient ergothioneine production methods (Erdelmeier et al., 2012, Xu and Yadan, 1995). In the last few years, two different aerobic ergothioneine biosynthetic pathways were discovered: the Mycobacterium smegmatis pathway (EgtA – EgtE catalysis in Fig. S1A) (Seebeck, 2010), and the fungal Neurospora crassa pathway (Egt1 and Egt2 catalysis in Fig. S1A) (Hu et al., 2014). In both pathways, the key steps involve a non-heme iron enzyme-catalyzed oxidative C-S bond formation (EgtB/Egt1 catalysis), and a PLP-catalyzed C-S lyase (EgtE/Egt2) reaction, which result in the net transfer of a sulfur atom from a cysteine to a histidine side-chain (Cheah and Halliwell, 2012, Erdelmeier et al., 2012). When this manuscript was under revision, an anaerobic ergothioneine biosynthetic pathway in bacterium Chlorobium limicola was discovered and the sulfur transfer is mediated by a rhodonase-like enzyme (Burn et al., 2017).
The transsulfuration reaction utilized in the ergothioneine biosynthetic pathway differs from other enzymatic sulfur-transfer reactions (Hu et al., 2014, Seebeck, 2010). The non-heme iron-dependent enzyme EgtB/Egt1 catalyzes the oxidative C-S bond formation to form a sulfoxide, which is then cleaved by EgtE/Egt2 (Fig. S1A). In nature, while most C-S lyases utilize thioether as the substrate (Fahey, 2001, Hand and Honek, 2005, Jacob, 2006), the C-S lyase (EgtE/Egt2) in ergothioneine biosynthesis is among the few lyases using sulfoxides (Fig. S1B) (Bartholomeus Kuettner et al., 2002, Song et al., 2015). Furthermore, the sulfoxide C-S lyase reaction is proposed to be coupled to a reduction process. However, how these two processes are coupled remains to be addressed. Structural and biochemical characterization of the C-S lyase in the ergothioneine biosynthetic pathway would shed light on the substrate specificity and the coupling of the C-S bond cleavage with the reduction process.
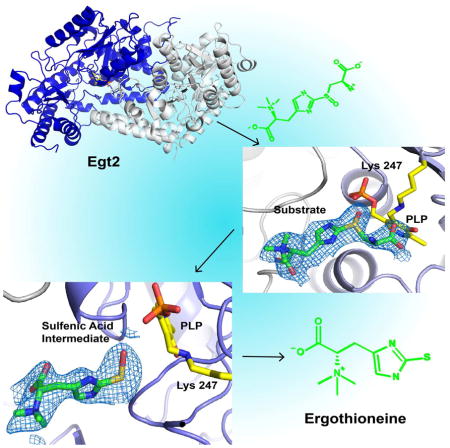
In this study, we characterized the N. crassa C-S lyase (Egt2) using a combination of biochemical and X-ray crystallographic approaches. Our Egt2 crystallographic studies reveal a novel subfamily of Type I PLP-dependent enzymes. In addition, we captured three different reaction states using X-ray crystallography and mutagenesis. The structural information explains the preference of a sulfoxide substrate for Egt2. Most importantly, the electron-density map in one of the intermediate states suggests the presence of the ergothioneine sulfenic acid, which provides evidence for the involvement of a highly reactive sulfenic intermediate in Egt2 catalysis. Further biochemical analysis allows us to formulate a mechanistic model to explain the coupling between the C-S lyase reaction and the reduction of the sulfenic intermediate to complete the catalytic cycle.
Results and Discussion
Kinetic characterization of Egt2-mediated C-S lyase activity
In ergothioneine biosynthesis, the C-S lyase reaction is coupled with a reduction process based on our recent characterization of M. smegmatis EgtE enzyme (Song et al., 2015). How is the C-S lyase reaction coupled to the reduction process remains to be addressed. The crystallization of the M. smegmatis EgtE enzyme is highly challenging. As a result, we turned our focuses to N. crassa C-S lyase Egt2 (Fig. 1 and S1B). We overexpressed and purified the N. crassa C-S lyase (Egt2) (Hu et al., 2014). The Egt2 sulfoxide substrate 4 was synthesized enzymatically using N. crassa Egt1-catalyzed oxidative coupling between cysteine and hercynine (Fig. S1C–D) (Hu et al., 2014). PLP is a cofactor that is covalently linked to the enzyme. The amount of PLP was ~ 0.83 mole PLP per mole of wild-type Egt2 monomer, according to a previously established method (Ghatge et al., 2012).
Fig. 1.

The proposed mechanistic model for Egt2 reaction using sulfoxide 4 as the substrate in the absence of reductant. The ergothioneine sulfenic acid intermediate is in brackets.
We characterized wild-type Egt2 catalysis using the same protocol as we did in our recent studies on M. smegmatis EgtE enzyme (Song et al., 2015). In the presence of dithiothreitol (DTT) as the reductant, the production of ergothioneine was detected (Fig. S1E–F) along with pyruvate and ammonia side-products. Using [13C]-labeled sulfoxide 4b as the substrate (Fig. S1G), 13C-NMR analysis of the Egt2 reaction spiked with authentic pyruvate is consistent with the production of [3-13C]-pyruvate (Fig. S2A). When pyruvate was derivatized with 4-fluorophenylhydrazine, the quantified ratio of ergothioneine and pyruvate in Egt2 reaction using 1H-NMR was ~ 1:1 (Fig. S2B–C) (Tamir and Srinivasan, 1970). The production of ammonia was quantified by a colorimetric assay using Nessler’s reagent, a solution containing potassium tetraiodomercurate(II) (K2[HgI4]), which reacts with ammonia under basic condition to give a yellow-colored species (Toney and Kirsch, 1987) (Fig. S2D). In the Egt2-catalyzed C-S lyase reaction of hercynylcysteine sulfoxide 4 in the presence of DTT, ergothioneine, pyruvate and ammonia were produced in a ratio of ~ 1:1:1. Taking advantage of pyruvate production and reaction stoichiometry, the kinetic properties of wild-type Egt2 were subsequently measured using a colorimetric assay by coupling wild-type Egt2 catalysis with the lactate dehydrogenase reaction (Song et al., 2015, Allegrini et al., 2017, Pioselli et al., 2004). When sulfoxide 4 was used as a substrate and in the presence of DTT at pH 8.0, the kinetic parameters of wild-type Egt2 were determined to be kcat = 8.7 ± 0.1 s−1 and Km = 155 ± 10 μM (Fig. S2E).
Overall structure of wild-type Egt2
We crystallized wild-type Egt2 from N. crassa with X-ray diffraction data collected in synchrotron beam sources at the Advanced Photon Source (APS) and the Advanced Light Source (ALS). As there was little homology with known structures, molecular replacement did not produce any reasonable solutions. The multi-wavelength anomalous dispersion (MAD) dataset for the platinum derivative of wild-type Egt2 provided a partial solution, including key secondary structural elements, which was then used as the initial phasing for selenomethionine derivative crystals. The final structure was solved with single anomalous dispersion (SAD) of the selenomethionine derivative crystals of wild-type Egt2 with a resolution of 2.3 Å (Table S1). The wild-type Egt2 crystals exhibit a P21 space group with eight molecules per asymmetric unit. The final model of N. crassa Egt2 includes residues 21 to 470, with the first 20 residues of the N-terminus and the last three residues of the C-terminus disordered (Fig. 2A). An inter-domain loop (residue 283–294) connecting the central catalytic domain and C-terminal domain is only visible in two of the eight monomers with a gap of three residues (283–285) in all molecules.
Fig. 2. Overall Egt2 structure.

A, The secondary structural elements of Egt2 are mapped along its sequence. α helices are represented by orange cylinders, β sheets by blue arrows, interconnecting loops as black lines and unstructured regions as black dashed lines. The PLP binding lysine is highlighted by red-color. The first 20 residues (residue 1–20) and the last 3 residues (residue 471–473) have not been shown since they were not visible in the crystal structure.
B, A ribbon representation of the overall structure of the Egt2 dimer. For Monomer 1 the N-terminal domain (residue 21–99) is shown in pink, the central domain (residue 100–282) in green, the C-terminal domain (residue 293–470) in blue and the inter-domain loop (residue 283–294) is shown in red. The secondary structure elements are labeled and numbered. The cofactor PLP and K247 adduct are represented as yellow sticks. Monomer 2 is shown in grey.
C, Surface representation of the active site of Egt2 in monomer 1. K247 and PLP are represented as yellow sticks. The color scheme is the same as the same as that in 2B.
D, Internal aldimine formed by K247 and PLP are shown with 2mFo-DFc map contoured to 1σ. The PLP interacting residues D221 and the Y134 are shown in sticks. The color scheme is the same as that in 2B.
E, The Egt2 cofactor PLP interaction network. Residues interacting with PLP were represented as sticks and hydrogen bonds with PLP are shown as blue dash lines. The color scheme is the same as that in 2B.
The eight molecules in each asymmetric unit form four dimers with a dimer interface of 3045 Å2, accounting for 16% of the total surface area of a monomer, as calculated by the PISA server (Krissinel and Henrick, 2007). This crystallographic observation of dimer formation is consistent with its oligomerization state in solution, as observed by size exclusion chromatography (Fig. S2F). Each wild-type Egt2 monomer has three domains: the N-terminal domain (residue 21–99), the central catalytic domain in which PLP is covalently bound (residue 100–282), and the C-terminal domain (residue 295–470). Egt2 belongs to the PLP-dependent enzyme family, an important superfamily of enzymes mediating highly diverse chemical transformations (Eliot and Kirsch, 2004). Structurally, the central catalytic domain of Egt2 exhibits a classic α/β fold with seven β strands wrapped within seven α helices (Fig. 2B), similar to the conserved folding in type I PLP-dependent enzymes (Schneider et al., 2000). By contrast, the N- and C-terminal domains of type I PLP-dependent enzymes are highly diverse and might be critical for the substrate specificity or regulation (Eliot and Kirsch, 2004). The N-terminal domain of Egt2 spans the active sites of both monomers, with an extended loop forming part of its own active site while probing the neighboring active site with a pair of long α helices (Fig. 2B). The C-terminal domain of Egt2 is the largest among type I PLP-dependent enzymes, almost as large as the catalytic domain itself. Consisting of three β-sheets and six α helices, the C-terminal domain of Egt2 spans ~60 Å and forms a lid that shelters the PLP-binding pocket and the active site from the bulky solvent (Fig. 2B). No homologs have been identified for the N- or C-terminal domains of Egt2, representing a novel subfamily in type I PLP-dependent enzymes.
Active site of Egt2
The active site of wild-type Egt2, located at the interface of the two dimers, is enclosed in a narrow, elongated tunnel of ~13 Å. The active site (volume measured ~600 Å3 using the Dogsite program) (Tepwong et al., 2012) is formed mainly by the central catalytic and C-terminal domains, along with contributions from the N-terminal and C-terminal domains of the neighboring monomer (Fig. 2C). Strong positive densities near the side chain of K247 is consistent with the formation of an internal aldimine with the PLP cofactor (Fig. 2D). The average B factor for PLP (~29 Å2) is comparable to that of the surrounding residues (~27 Å2), indicating a high occupancy of the PLP cofactor. This internal aldimine is stabilized by extensive interactions between PLP and the protein (Fig. 2E). The pyridine ring of PLP aligns parallel to the aromatic ring of Y134 via π-π stacking interactions (distance between the centers of the rings measured as 3.4 Å). The orientation in this plane is defined by a salt bridge between D221 and the protonated nitrogen in the pyridine ring, an interaction conserved among all type I PLP-dependent enzymes (Eliot and Kirsch, 2004). This orientation is further strengthened by a hydrogen bond formed between Q224 and the O3′ oxygen atom of the hydroxyl group on the pyridine ring (Fig. 2E). An extensive network of hydrophilic interactions is formed between the phosphate group of the PLP and hydrophilic sidechain of T307, T107 and N244. The position of the PLP cofactor is further anchored by a potential salt bridge between its phosphate and H246 (Fig. 2E).
Recognition of the substrate by Egt2
To understand how the substrate is recognized by wild-type Egt2, we sought to obtain the structure of wild-type Egt2•substrate 4 binary complex. PLP-dependent enzymes perform multistep catalytic reactions, in which reaction intermediates sometimes are captured by X-ray crystallography (Yan et al., 2017, Wu et al., 2012, Bhavani et al., 2008, Bisht et al., 2012). However, soaking the preformed wild-type Egt2 crystals with its substrate hercynylcysteine sulfoxide 4 showed little electron density at the active site, possibly due to the fast catalytic turnover of the wild-type Egt2 (kcat = 8.7 ± 0.1 s−1 at pH 8.0, Fig. S2E).
To capture the wild-type Egt2•substrate 4 complex formation or snapshots of the intermediates in the reaction cycle, we explored the option of using Egt2 mutants for structural determination. An inspection of the PLP-binding pocket identified eight conserved residues near the active site (D221, Q224, Y134, S47, S196, C137, T307 and H276), which could potentially attenuate the catalysis. We mutated these residues to a homolog residue in an attempt to slow down the chemical reaction rate. These mutants were then crystallized in the presence of substrate. Among them, Egt2 Y134F formed plate-shaped crystals after pre-incubation with the substrate at 4 °C and diffracted to a resolution of 2.6 Å (Table S1). The structure of Egt2 Y134F showed no significant change in the folding when compared to the wild-type Egt2. Notably, a strong positive density was observed within the catalytic pocket close to the PLP C4′ position in two of the four dimers. The electron density is consistent with the chemical structure of the sulfoxide substrate 4 (Fig. 3A). Fitting the substrate into the active site density shows that there is no covalent bond between the substrate and PLP (the substrate amino group is ~5 Å from the PLP C4′ position). Therefore, the structure captured is Egt2 Y134F• substrate 4 binary complex prior to external aldimine intermediate formation.
Fig. 3. Structure of the Egt2 Y134F •substrate 4 binary complex.

A, Sulfoxide substrate 4 in Egt2 Y134 active site shown with 2mFo-DFc map contoured to 0.75σ. The PLP cofactor is shown as yellow sticks. Monomer 2 is colored grey. Inset is the 2D chemical structure of the substrate 4.
B, Interactions between Egt2 Y134 and substrate 4. Residues interacting with the substrate are shown as sticks in pink and residues from the neighboring monomer are indicated with a prime label. Hydrogen bonds between residues are shown by black dash lines. PLP and the K247 are shown as sticks in yellow.
C, Interactions between Egt2 Y134F with tyrosine modeled at F134 position (shown as grey sticks) and its substrate 4. Residues interacting with the substrate are shown in pink and residues from the neighboring monomer are indicated with a prime label. Hydrogen bonds between tyrosine at the 134 position and the substrate are shown by black dash lines. PLP and the K247 are shown as yellow sticks.
D, Cation-π interactions between the substrate and the aromatic side chains of F48 and F134. Distances between the cation and these π system are labeled.
Substrate recognition is dictated by the interaction between the substrate 4 and the Egt2 Y134F N- and C-terminal domains. The substrate cysteine moiety forms extensive interactions with the enzyme (Fig. 3B). In addition to forming a salt bridge with R438, the cysteine carboxylate group forms hydrogen bonds with Q224 and S196 and its amino group forms a hydrogen bond with the side chain of S47 (Fig. 3B). The substrate sulfoxide group is sandwiched between the benzyl rings of F134 and F48. The aromatic side chain of F134 is in the same position in the Egt2 Y134F as Y134 in Egt2 wild-type, indicating no significant change in the conformation of this residue. This position of Y134 places its hydroxyl group at distances favorable for hydrogen bonding interactions to the amino group of the substrate imidazole ring and the sulfoxide group (3.0 Å and 3.5 Å, respectively) (Fig. 3C). Unlike the cysteine moiety, the substrate histidine moiety that is located near the exit of the active site cavity is only loosely associated with the enzyme through bonding with the imidazole ring of H276. There is also a cation-π interaction formed between the trimethylamino group of the substrate and the aromatic ring of F304 at a distance of ~ 4.3 Å (Fig. 3B).
Rationale for the unusual substrate specificity of Egt2
The structure of the Egt2 Y134F•substrate 4 binary complex reveals the structural rationale for its unique substrate specificity, which prefers a sulfoxide over thioethers recognized in other C-S lyases. In our recent characterization of EgtE (an Egt2 homolog from M. smegmatis ergothioneine biosynthesis), the kcat/Km for the sulfoxide substrate is 52-fold greater than that for the thioether substrate (Song et al., 2015). The complex structure of Egt2 Y134F with the substrate 4 provides a plausible explanation for the Egt2 substrate specificity, highlighting the cation-π interaction between the sulfoxide of the substrate and the phenylalanine residues in the active site (Fig. 3D). Cation-π interactions are formed as a result of the electrostatic, dispersive and induction forces between a positively charged group and a π system (Dougherty, 2013). The cation-π interaction is involved in maintaining the protein structure (Gallivan and Dougherty, 1999) and enables the molecular recognition (Zacharias and Dougherty, 2002). The oxidation of the thioether groups, e.g., methionine, significantly strengthens their interactions with aromatic groups by up to 0.5 to 1.5 kcal/mol, corresponding to a binding preference of five- to sixteen-fold (Lewis et al., 2016). The structure of the Egt2 Y134F •substrate 4 binary complex has a prominent cation-π stacking interaction between the sulfoxide group of substrate 4 and the F48 of the Egt2 Y134F at a distance of ~ 4.9 Å (within the range of a cation-π interaction (Gallivan and Dougherty, 2000)) and could have an off-centered cation-π interaction (Marshall et al., 2009) with F134 at a distance of ~ 4.4 Å (Fig. 3D). To provide a comparison to the sulfoxide substrate, we also obtained the complex structure of Egt2 Y134F with thioether substrate at a resolution of 2.55 Å (Fig. S3A). 50 fold more thioether compound compared to the sulfoxide was required in the crystallization setup to detect electron density at the active site. While the structure reveals that the thioether substrate assumes the same binding mode as the sulfoxide substrate, the aminoacrylate of the thioether substrate moiety shows much less density, indicating weaker interaction and/or higher flexibility. The observation in these structures is consistent with our analysis that stronger cation-π interactions in sulfoxide substrate structure contributes to the preference for a sulfoxide over a thioether as substrate for Egt2.
Capture of the potential ergothioneine sulfenic acid intermediate
To understand the function of Y134, we characterized kinetic activity of Egt2 Y134F. Despite of a reduction relative to wild-type Egt2, Egt2 Y134F still maintains some enzymatic activity (Km of 743.2 ± 52.5 μM and kcat of 3.6 ± 0.1 s-1 at pH 7.0) (Fig. S3B) and actually shows increased activity with pH (Fig. S3C). Therefore, we explored the option of the C-S bond cleavage in situ in Egt2 crystals by increasing the pH condition of mother liquor, which will promote the chemical reaction but may be slow enough in the crystalline form to be captured by X-ray crystallography. We transferred the pre-formed crystal of the Egt2 Y134F-substrate obtained at pH 7.0 to a cryo buffer with higher pH prior to flash freezing in liquid nitrogen. When the crystals were transferred to a cryo buffer with a final pH 8.0, the structure showed a strong positive electron density observed in the active sites in two of the four Egt2 Y134F dimers (Fig. S3D–E). The large volume of the density eliminates the possibility of some salt from mother liquor or cryo buffer. Modeling the substrate 4 into these electron densities fits well mostly with overall densities. However, a strong negative density was observed at the position of the C-S bond subject to cleavage by Egt2 Y134F (Fig. S3F). After rounds of iterative manual building, the density is most consistent with the structure of ergothioneine sulfenic acid (Fig. 4A, Fig. 6 compound 10). The most significant difference between this structure with the substrate-bound Egt2 Y134F at pH 7.0 is the loss of aminoacrylate moiety density. In the complex structure of substrate with Egt2 Y134F (Fig. 3B), an extensive interaction was observed for the aminoacrylate moiety of the substrate with the enzyme. With the increasing pH, the only residue density near R438 is consistent with a formate ion from the crystallization buffer forming a salt bridge interaction (Fig. 4B). The sulfur atom shows no connecting electron density to the region that would be occupied by the aminoacrylate moiety. This observation suggests that the chemical reaction occurs and the intermediate captured is a sulfenic acid in Egt2 catalysis.
Fig. 4. Structure of the Egt2 Y134 in complex with a sulfenic acid intermediate.

A, The ergothioneine sulfenic acid intermediate bound at the active site is shown with 2mFo-DFc map contoured to 0.8σ. The PLP is shown as yellow sticks. A formate ion is shown in white. Inset is the 2D chemical structure of the sulfenic acid intermediate 10.
B, The interaction network of the ergothioneine sulfenic acid intermediate at the active site of Egt2 Y134F. Residues interacting with the substrate are shown as pink sticks. The formate ions are shown as grey sticks. Hydrogen bonds are shown by black dashed lines. PLP and the K247 are shown as yellow sticks. A water molecule hydrogen bonded to PLP and the sulfenic acid are shown as a red sphere.
C – D, Egt2 reaction in the presence of 1,3-cyclohexanedione shows an appearance of a new peak at 6.86 ppm (C) while the control reaction without trapping reagent in D shows two signals at 6.65 ppm and 6.87, which correspond to ergothioneine 5 and ergothioneine-2-sulfinic acid 9 respectively.
E, The 2mFo-DFc map of the geminial diamine intermediate contoured to 1σ. Inset is the 2D chemical structure of the geminial diamine intermediate 15.
Fig. 6.

The proposed Egt2 catalytic mechanism showing the involvement of the ergothioneine sulfenic acid, which could form a covalent adduct with C156 (Pathway I) or get released directly into solution (Pathway II). In both pathways, cellular thiol reduction system can reduce the intermediate to give ergothioneine as the final product. The ergothioneine sulfenic acid intermediate is in brackets.
Compared with the structure of the Egt2 Y134F•substrate 4 binary complex, C-S bond cleavage allows the sulfenic group to move 1.6 Å closer to PLP, forming a triad among the sulfenic acid and the oxygen atoms of the phosphate group of PLP (Fig. 4B). A hydrogen bond network is formed among the PLP phosphate group, the sulfenic acid, the T307 hydroxyl group, and a solvent molecule (Fig. 4B). The sulfenic acid is further stabilized by the cation-π interaction between the partial positive charge on the sulfur atom and the aromatic ring of F48 at a distance of ~ 5 Å (Fig. 4B). The imidazole ring of the ergothioneine sulfenic acid intermediate is sandwiched between the side chains of R75 and H276. The N-trimethylamine is stabilized by cation-π interaction with F304 from the neighboring monomer.
Confirmation of sulfenic acid intermediate using chemical trapping
In our previous characterization of M. smegmatis EgtE enzyme in solution, the involvement of a sulfenic acid intermediate in EgtE-catalysis was observed (Song et al., 2015). Sulfenic acid compounds are widely acknowledged as important transient intermediates in organic and bioorganic sulfur chemistry (Allison, 1976). Unlike sulfinic or sulfonic acid, sulfenic acid is reactive and labile. Hence, it is unlikely to directly visualize a sulfenic acid moiety during a chemical reaction pathway unless it is stabilized by other factors such as a steric, electronic, or hydrogen bonding environment (Gupta and Carroll, 2014). For example, the crystal structures of small molecular sulfenic acids such as thiophenetriptycene-8-sulfenic acid, (4-tert-butyl-2,6-bis[(2,2″,6,6″-tetramethyl-m-terphenyl-2′-yl)methyl]phenyl) sulfenic acid (Bmt-SOH), and 4,6-demethoxyl triazine-2 sulfenic acid have been obtained due to their chemical scaffolds, which restricts further reaction (Claiborne et al., 1999). In the biological system, protein sulfenic acid is an important post-translational modification as part of a signaling pathway in response to an oxidative environment (Devarie-Baez et al., 2016). In the protein environment, the sulfenic acid is a precursor to a disulfide bond (Poole et al., 2004) or cyclic sulfenyl-amide (as found in tyrosine phosphatase) (Salmeen et al., 2003). In extremely rare cases, for example, methionine sulfoxide reductase (Ranaivoson et al., 2008), phosphatase cdc25B (Buhrman et al., 2005), peptidyl prolyl isomerase (Pin1) (Chen et al., 2015), and redox-sensitive transcription factors (SarZ) (Poor et al., 2009), a free cysteine sulfenic acid is observed due to its stabilization by a hydrogen bonding network.
The visualization of a potential ergothioneine sulfenic acid intermediate captured in our Egt2 Y134F structure is an important piece of evidence in understanding the mechanism of this C-S lyase (Song et al., 2015). To corroborate our structural observation of the existence of sulfenic acid in the wild-type Egt2 reaction, we conducted a chemical sulfenic acid intermediate trapping experiment in solution. Herein, we used 1,3-cyclohexanedione, which reacts selectively with sulfenic acid groups to give a thioether derivative, as a chemical probe for detecting the sulfenic acid intermediate of an enzymatic reaction (Song et al., 2015, Gupta and Carroll, 2014). A wild-type Egt2 reaction containing the sulfoxide 4 as the substrate with excess 1,3-cyclohexanedione in the absence of reductant was conducted, as was a control reaction. 1H NMR analysis of these reactions showed that, in the presence of 1,3-cyclohexanedione, a signal at 6.86 ppm was observed (Fig. 4C). However, in the control reaction without 1,3-cyclohexanedione, two signals (6.65 ppm and 6.87 ppm) were present (Fig. 4D). The difference in NMR signals between these two reaction conditions suggested that Egt2 may release the reactive sulfenic acid intermediate into solution, which can then be trapped by 1,3-cyclohexanedione. The proposed sulfenic acid-1,3-cyclohexanedione adduct was isolated using a cellulose chromatography (Song et al., 2015). Further 1H NMR and high-resolution mass spectrometry characterizations confirmed the structural assignment of the ergothioneine sulfenic acid-1,3-cyclohexanedione adduct 8 (Fig. S4A–B). Therefore, chemical trapping results in solution support the crystal structure suggesting the presence of a sulfenic acid intermediate as part of the Egt2 C-S lyase reaction.
Capture of the aminoacrylate germinal diamine intermediate
Besides the sulfenic acid intermediate, careful inspection of the active sites of all eight molecules within each asymmetric unit of Egt2 Y134F structure at pH 8.0 identified positive density next to the PLP C4′ position in one of the molecules (Fig. S4C). The density is most consistent with the aminoacrylate geminal diamine intermediate (Fig. 4E, Fig. 6 compound 15), which is formed immediately prior to the release of pyruvate and the regeneration of the PLP intra-aldimine bond for the next cycle. The aminoacrylate is stabilized by a salt bridge interaction between its carboxylate group and R75 from the adjacent monomer (Fig. 4E). Although this intermediate (Fig. 6 compound 15) is presumed to be produced concurrently with the sulfenic acid intermediate, we did not observe density for the sulfenic acid intermediate in the active site containing the aminoacrylate. Superimposition of the active site containing the two intermediates shows an over-crowded active site. It is highly possible that the sulfenic acid intermediate might have been released from the active site already.
To understand the role of Y134 in Egt2-mediated reaction, we conducted an alignment of wild-type Egt2 and seventeen of its closest homologs from a Basic Local Alignment Search Tool (BLAST) search using ClutsalOmega (McWilliam et al., 2013), which revealed that the residue Y134 was strictly conserved (Fig. S4D). A comprehensive survey of the literature highlights that an active site tyrosine residue at a position comparable to wild-type Egt2 has a dual role both in substrate binding and subsequent catalysis. Several members of the type I PLP-dependent enzyme family have an active site tyrosine that plays a key role in PLP binding, e.g., aspartate aminotransferase (Toney and Kirsch, 1987), aminolevulinate synthase (Tan et al., 1998), cystathionine β-lyase (Clausen et al., 1996), and cystalysin (Cellini et al., 2005). The extensive interaction formed by Y134 with sulfoxide substrate as well as PLP binding highlights its importance for substrate recognition for Egt2. In other cases, an active site tyrosine is reported to act as a general acid/base to facilitate catalysis (e.g., alanine racemase) (Toney, 2005). A structure-based Dali search (Holm and Rosenström, 2010) identified the cysteine/cystine C-S lyase (PDB ID: 1ELQ) and cysteine desulfurase (PDB ID: 4Q76) as the closest Egt2 structural homologs. The superimposition of these three structures reveals that the Y134 in Egt2 is replaced by a histidine in cysteine/cystine C-S lyase and cysteine desulfurase (Fig. S4E). The histidine imidazole ring stacks with the PLP pyridine ring and functions as a base to deprotonate the substrate amine group and to initiate the internal aldimine into the external aldimine intermediate transition (Clausen et al., 2000, Roret et al., 2014). Consistently, the reduced activity of Y134F fits its role as a general acid/base candidate. The mutation of Y134 did reduce, but not abolish the catalytic activity, suggesting that Y134 is not the sole acid/base in the enzymatic catalysis. In the Egt2 Y134F structure in complex with the substrate, the nitrogen of the K247 residue that forms an internal aldimine with the PLP is positioned at a distance of ~ 4.3Å. The formation of the external aldimine would release K247, which may play the role of the active site acid/base (Fig. S4F).
Coupling of the sulfenic acid intermediate reduction
The presence of sulfenic acid intermediate in the catalytic cycle determined by X-ray crystallography and a chemical trapping experiment immediately raises the next key question regarding wild-type Egt2 catalysis: how is C-S lyase activity coupled to the reduction of this sulfenic acid intermediate? There are two possible strategies in which ergothioneine sulfenic acid produced at the active site could be reduced to ergothioneine. It could be reduced by a thiol group of the protein itself or released directly into solution and reduced by reductants in the cell.
To test if sulfenic acid can form a covalent adduct with a thiol in Egt2, we conducted a single-turnover experiment in the absence of DTT for wild-type Egt2. Equal molar ratio of protein and substrate were incubated together and then subject to proteolysis by trypsin. The digested peptides were then analyzed by ESI-LC/MS/MS, which shows that residue C156 is covalently modified with a sulfenic acid adduct after the single turnover (Fig. 5A–B). When the labeled wild-type protein was treated with DTT, the C156 containing peptide was regenerated (Fig. S5A). Using Egt2 C156A as control (Fig. S5B), the MS/MS analysis of the C156A from a single-turnover reaction indicates that the corresponding peptide is not modified (Fig. S5C–D). When we examined the wild-type Egt2 crystal structure, we noticed that C156 is located next to the exit of the wild-type Egt2 active site (Fig. 5C). All the evidence supports the modification at C156 position by forming an adduct with sulfenic acid intermediate in wild-type Egt2 under non-reducing condition.
Fig. 5. Reactions of sulfenic acid intermediate.

A, MS/MS spectrum of a tryptic peptide of the wild-type Egt2 (residue 156–173) before single turnover reaction. The parent ion has the signal with m/z 1003.998.
B, MS/MS spectrum of a tryptic peptide of the wild-type Egt2 (residue 154–173) after the single turnover reaction. The non-specific cleavage of trypsin at S154 could be due to the presence of modification at C156. The parent ion has the signal with m/z 827.0616.
C, Surface representation of the active site of Egt2 Y134 with the sulfenic acid intermediate shown in sticks and the C156 lying at the exit of the substrate binding pocket (colored in orange). Color scheme is identical to that in Fig. 2B and having C156 in orange.
In the presence of DTT as the reductant, wild-type Egt2 produced ergothioneine as the sole product, as shown by our 1H-NMR assay (Fig. S6A). However, in the absence of the reductant DTT, chemical characterization of the reaction by wild-type Egt2 reveals the existence of ergothioneine 5 and ergothioneine-2-sulfinic acid 9 (Fig. 4D and Fig. S6B–C) as products. This result can be explained by the disproportionation of the sulfenic acid intermediate 10 to a thiosulfinate 11 (Fig. 1). The subsequent hydrolysis of the thiosulfinate produces ergothioneine 5 and ergothioneine-2-sulfinic acid 9 in a 1:1 ratio. In addition, under acidic condition, ergothioneine-2-sulfinic acid can undergo elimination of sulfur dioxide resulting in hercynine (Loksha et al., 2004). The production of both 5 and 9 in this case suggests that, in the absence of reductants, wild-type Egt2 can also release the sulfenic acid intermediate directly to the solution.
Based on the above results, we proposed that two non-exclusive mechanisms for Egt2 catalysis. After its formation, ergothioneine sulfenic acid can be reduced by C156 to form a disulfide bond, which can then be reduced by a thiol reduction system inside the cell to release ergothioneine and restore C156 for the next cycle. Alternatively, ergothioneine sulfenic acid may be directly released to the solution and reduced by intracellular reductants to produce ergothioneine. In vivo, such a reduction system can be provided by other intra-cellular thiols (e.g., glutathione) or the thioredoxin/thioredoxin reductase system.
Mechanism of Egt2-mediated C-S lyase reaction
Based on these biochemical and structural data, we proposed a mechanistic model for Egt2-catalysis (Fig. 6). Similar to other PLP-containing enzymes, the first step is the formation of external aldimine intermediate 12 between the PLP cofactor and the sulfoxide substrate 4, resulting in a Schiff base followed by deprotonation of the Cys α-carbon producing the quinonoid intermediate 13. The subsequent C-S bond cleavage produces the ergothioneine sulfenic acid 10 and the PLP-based aminoacrylate intermediate 14, which undergoes the amine exchange with K247 at the active site forming intermediate 15, leading to the formation of pyruvate and ammonia as the side-products.
When the ergothioneine sulfenic acid is released, the C156 at the exit of the substrate binding pocket then traps 10 forming a disulfide bond in species 17, which can be further reduced by a reductant such as DTT or a thiol-reduction system (e.g., the thioredoxin/thioredoxin reductase pair), releasing ergothioneine 5 as the final product and regenerating C156 for the next cycle (Pathway I, Fig. 6). Alternatively, the sulfenic acid intermediate 10 might be released from the active site to the solution directly, which can be then reduced by a thiol reduction system (e.g., glutathione or the thioredoxin/thioredoxin reductase pair) to give ergothioneine as a final product (Pathway II, Fig. 6).
As a summary, we determined the crystal structure of Egt2, the C-S lyase in the last step of ergothioneine biosynthesis pathway. Using a combination of biochemical and crystallographic studies, we have provided mechanistic evidence of how C-S lyase activity is coupled to a reduction process for Egt2. In particular, we have gained structural insight for the unique substrate recognition and the sulfenic acid intermediate has been successfully captured in C-S lyases.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the lead contact, Dr. Yan Jessie Zhang (jzhang@cm.utexas.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strain E.coli BL21 (DE3) were used for protein purification.
Cloning, protein expression and purification
The N. Crassa Egt2 protein was overexpressed and purified as described in E.coli BL21 (DE3) (Hu et al., 2014), with some modifications. Egt2 protein eluted from the Strep-Tactin resin was reconstituted with PLP in 25 mM HEPES buffer at pH 7.5 containing 50 mM NaCl and 10 mM β-mercaptoethanol (BME) with overnight incubation at 4 °C. Before it was used for crystallization studies, Egt2 protein was further purified using size exclusion chromatography to near homogeneity, as visualized on SDS-PAGE. The purified protein was concentrated to ~ 8 mg/ml and stored at −80 °C until kinetic analysis or crystallographic set up.
The Egt2 selenomethionine derivative was obtained by overexpression in minimal medium supplemented with an amino acid mixture. Egt2 overexpression was induced by anhydrotetracycline at a final concentration of 500 ng/L when the OD600 of the cell culture reached 0.5. Approximately 30 minutes prior to the induction of protein expression, the amino acid mix (per liter of culture: 100 mg of lysine, threonine, phenylalanine, 50 mg of leucine, isoleucine, valine and 60 mg of selenomethionine) was added to the culture medium. Cells were harvested 6 hours after induction and selenomethionine-labeled Egt2 was purified following the same procedure as the wild-type Egt2 protein described above.
The Egt2 Y134F construct was prepared using site-directed mutagenesis following recommended procedures (Stratagene). The mutant was overexpressed, purified, and reconstituted by PLP following the same procedure used for the wild-type enzyme. There have been no experimental models or cell lines used in this study.
METHOD DETAILS
All reagents were purchased from Sigma-Aldrich unless otherwise specified. Kinetic measurements were conducted in triplicates.
PLP Content Determination
The PLP cofactor in Egt2 and its mutants was released to the solution after treatment using 0.2 N of NaOH and the UV visible spectrum was then recorded. Protein concentration was determined from amino acid analysis. The PLP content was then calculated using ε390 = 6600 M−1cm−1 (Ghatge et al., 2012). For Egt2 wild-type and Y134F, the PLP content per monomer was ~0.83 and ~0.63, respectively.
Preparation of Egt2 Sulfoxide Substrate
Sulfoxide substrate 4 was prepared through Egt1 enzymatic reaction as reported previously (Hu et al., 2014). In particular, Egt1 was used for the enzymatic coupling between hercynine and cysteine in the presence of ascorbate and an air saturated buffer at pH 8.0. The Egt1 was subsequently removed by ultrafiltration and the desired product was purified by ion-exchange chromatography.
1H NMR (5003MHz, D2O) of sulfoxide substrate 4: δ 3.10–3.19 (m, 13H), δ 3.48 (dd, J3=33.9, 15.1 Hz, 1H), δ 3.65 (dd, J3=33.9, 8.83Hz, 1H), δ 3.77 (dd, J3=33.9, 11.9, 1H), δ 7.12 (s, 1 H)
High resolution ESI-MS of 4: The calculated [M-H]− in the negative mode for compound 4 was m/z 333.1233, and found m/z 333.1246.
Quantification of Pyruvate in Egt2 Reaction
The amount of pyruvate from Egt2 reaction was calculated based on the amount of dinitrophenylhydrazine derivative determined by 1H NMR spectra. Egt2 reaction containing 1 mM substrate, 1 mM DTT, and 2 μM Egt2 in 50 mM KPi buffer, pH 8.0, was incubated at 28 °C for 1 hr. After the Egt2 reaction was completed, 4-fluorophenylhydrazine was added to the reaction mixture to 2 mM final concentration and incubated at 50 °C for 3 hr. 4-Fluorophenylhydrazine derivatizes pyruvate to a hydrazine adduct 7, which converts the solvent-exchangeable methyl group on pyruvate to non-exchangeable one. The reaction was then lyophilized and characterized by 1H NMR.
However, the chemical shift for the methyl group of the pyruvate adduct is 1.88 (s, 3H) while that of ergothioneine imidazole hydrogen was 6.63 ppm, direct quantification between these two signals would be inaccurate since they are located on two sides of a large water signal. To solve the issue, ethyl viologen was chosen as an internal standard, since its signals are present at both low field and high field: δ 1.54 (t, 7.23Hz, 6H), δ 8.37 (d, 6.53Hz, 4H), and δ 8.98 (d, 6.53Hz, 4H). The ratio between ergothioneine and pyruvate calculated using ethyl viologen as an internal standard was determined to be ~1:1.
Determination of Ammonium in Egt2 Reaction
Besides pyruvate and ergothioneine, ammonium is another proposed product of the Egt2 catalysis. To determine the ratio between ergothioneine and ammonium, we need to prepare ammonium free substrate first. To achieve that, 7 μL of 91 mM sulfoxide substrate was adjusted to pH 13.0 using NaOH and lyophilized. The dry substrate was re-dissolved into 100 μL of H2O and lyophilized. This process was repeated three times to remove ammonium. The final powder was dissolved in 100 μL H2O and neutralized with HCl. The substrate was aliquoted into two portions: one as the control and the other for Egt2 reaction.
A 1-mL reaction containing 5.03μM Egt2, 0.323mM substrate, 0.53mM DTT in 503mM KPi buffer, pH38.0 was incubated at 283°C for one hour. The control reaction containing the same component as the above reaction except that denatured Egt2 was used. Then, 100 μL of Nessler’s reagent was added to the reaction mixture and A462 was measured to be 0.7333 ± 0.06 and the ammonium concentration derived from the standard curve was 0.29 mM while the concentration of ergothioneine was 0.32 mM. This result suggested that the ratio between ergothioneine and ammonium produced from Egt2 reaction was found to be approximately 1: 1.
Characterization of Ergothioneine from Egt2 Reaction
The reaction containing 1 μM Egt2 protein, 2 mM sulfoxide substrate in 10 ml of 50 mM KPi buffer pH 8.0 was incubated at 28 °C for 1 hr. After the reaction was completed, the protein was removed by ultrafiltration. Ergothioneine was purified through DOWEX50 cation exchange resin and lyophilized. The purified product was then characterized by 1H NMR and HRMS.
1H NMR (5003MHz, D2O) of ergothioneine 5: δ 3.05 (t, J = 12.5 Hz, 1H), δ 3.12 (dd, J = 3.4, 12.5 Hz, 1H), δ 3.14 (s, 9H), δ 3.75 (dd, J3=33.6, 10.2 Hz, 1H), δ 6.66 (s, 1 H).
High resolution ESI-MS of 5: The calculated [M-H]− in negative mode for ergothioneine was m/z 230.0963, and found m/z 230.0974.
Crystallization of Egt2 and co-crystallization of Egt2 Y134F with the substrate
The Egt2 crystallization conditions were screened using the sparse matrix on a Phoenix Robotic system (Art Robinson). After identification of initial hits, systematic optimization led to crystals with a plate morphology in 24% PEG3350, 0.2 M sodium formate, and 50 mM Tris-HCl at pH 8.0. Wild-type Egt2 and selenomethionine derivative crystals were cryo-protected in a solution containing 30% glycerol, 27.5% PEG 3350, 0.2 M sodium formate, and 50 mM Tris-HCl at pH 8.0 before vitrification in liquid nitrogen. Platinum derivative crystals used in phasing were obtained by soaking wild-type Egt2 crystals in mother liquor containing 4 mM potassium chloroplatinate (K2PtCl4) at 4°C for 2 hrs prior to crystal harvesting.
The co-crystallization of Egt2 and the substrate setup included a Egt2 Y134F variant incubated with 100 μM substrate hercynylcysteine sulfoxide for five minutes before crystallization in a similar condition, with 50 mM HEPES at pH 7.0. When transferred to higher pH, the crystals obtained at pH 7.0 was transferred to a cryo buffer containing mother liquor made with buffer at pH 8.0 and 30% glycerol. The pH was then adjusted for the final solution. The pHs in the results and discussion section are pHs measured for the final solutions rather than the initial stock buffer pH values.
Data collection and Processing
The multiple anomalous dispersion (MAD) data for the platinum derivative and the single anomalous dispersion (SAD) data for the selenomethionine derivative were collected at the Advanced Light Source (ALS), Berkeley beamline BL 5.0.3. The datasets for the Egt2 Y134F co-crystallized with the substrates were collected at Advanced Photon Source (APS), Chicago, BL 23–ID-B. The diffraction data were integrated and scaled using HKL2000 (Otwinowski and Minor, 1997). The statistics for data collection for all crystallographic data sets were summarized in Table S1.
Structure determination
Multiple anomalous dispersion (MAD) datasets from platinum derivative crystals provided a partial solution which was used for phasing selenomethionine SAD data in Phenix Autosol (Terwilliger et al., 2009). The structure was refined using Phenix Refine excluding ~2000 diffraction spots (1.2%–1.5% of the data) as the Rfree flag (Brünger, 1992). The structures of ligands were prepared in jLigand (Lebedev et al., 2012). Iterative cycles of optimization were carried out to improve the quality of the model using the refinement program Phenix. Refine (Echols et al., 2012) followed by manual rebuilding in COOT (Emsley et al., 2010). The final structures have ~ 96% of the residues with Ramachandran (Ramachandran et al., 1963) favored phi/psi angles with the exception of G225 as outlier, which is located in a loop interacting with the cofactor and shows excellent electron density. The Molprobity scores exceeds 98 percentile of other structures at the same resolution. The statistics of refinement for all reported structures are summarized in Table S1.
Egt2 activity measurement
The steady-state kinetic parameters were determined using a coupled assay as developed in characterizing the M. smegmatis EgtE enzyme recently (Song et al., 2015). Detailed information about the chemical characterization of substrates and product can be found in Experimental Procedures. To measure Egt2 activity, a typical 1-mL reaction contained 0.13 mM NADH, 1 mM DTT, 22.5 U/mL lactate dehydrogenase (2000× of Egt2 activity in the assay), 10 nM wild-type Egt2, and variable concentrations of sulfoxide 4 (0.05 – 3 mM) in the assay buffer (25 mM NaOAc, 25 mM MES, 25 mM glycine, and 75 mM Tris) adjusted to the desired pH with HCl or NaOH. The consumption of NADH at 340 nm was monitored with a Carry 100 Bio UV-Vis spectrophotometer at 25°C and the graph was fitted using GraphPad Prism. For Y134F and C156A mutant kinetic studies, a similar experimental set up was carried out.
Chemical trapping and isolation of sulfenic acid intermediate in Egt2 catalysis
A chemical trapping experiment using 1,3-cyclohexanedione was performed to trap the sulfenic acid intermediate in the Egt2 reaction following a reported procedure (Song et al., 2015). A 5-mL reaction contained 3.5 mM sulfoxide substrate, 165 mM 1,3-cyclohexanedione (50× of substrate concentration), and 4 μM of Egt2 in 100 mM potassium phosphate buffer pH 8.0. The reaction was stirred at 28 °C for 1 hr. The reaction was lyophilized and purified using cellulose resin packed with iPrOH: ACN = 4.5: 2.5 (1 × 20 cm). The column was washed with 18 × 5.0 mL of iPrOH: ACN = 4.5: 2.5. The adduct was eluted with iPrOH: ACN: 0.1 M NH4HCO3 = 4.5: 2.5: 3. The adduct was lyophilized and characterized by 1H NMR and high-resolution mass spectrometry.
Egt2 protein modification analysis
In a 1-mL assay, a single-turnover reaction containing 489 μM wild-type or C156A Egt2 and 489 μM sulfoxide substrate in 50 mM KPi, pH 8.0, was incubated at 28 °C for 15 min. The wild-type reaction was divided into two portions, and one of them was treated with DTT at 10 mM final concentration. Both wild-type and C156A Egt2 protein before the reaction were also analyzed by tandem mass spectrometry as the control.
The protein was denatured in 6 M Urea with 50 mM Tris-HCl pH 8.0 and incubated at 60 °C for 60 min. Next, 500 μL of 50 mM NH4HCO3 was added and Sequencing Grade Modified trypsin (Promega US) was added to the mixture in a 1:100 (w/w) ratio. The protein was digested overnight at 37 °C and purified using C-18 spin columns (Thermo Scientific, Rockford, IL). A 6550i Funnel Quadrupole Time-of-Flight (Q-TOF) mass spectrometer coupled to a 1200 series HPLC-Chip Cube interface equipped with a Polaris-HR-Chip-3C18 LC chip (Agilent Corp, Santa Clara, CA) was used for peptide sequencing. The total peptide digestion mixture was separated by a reversed phase nanoLC, followed by MS/MS analysis with collision-induced dissociation (CID) as the fragmentation method. Peptide sequencing and identification of modifications was achieved using the proteomics software program PEAKS Studio (Waterloo, ON, Canada). Targeted peptides were identified by comparing the experimental MS/MS product ions to expected values predicted by Protein Prospector from the University of California, San Francisco and manual interpretation.
Isolation and Characterization of Egt2 Product in the Absence of Reductant
A 10-mL reaction containing 1 μM Egt2 protein, 2 mM sulfoxide substrate 4, 2 mM DTT, and 50 mM KPi buffer pH 8.0 was incubated at 28 °C for 1 hr. Egt2 protein was removed by ultrafiltration and the product was purified following reported procedure using HPLC (Song et al., 2015). After purification, the product was characterized by 1H NMR and HRMS.
1H-NMR (5003MHz, D2O) of ergothineine-2-sulfinic acid 9: δ 1.83 (dt, J3=36.3, 12.8, 2H), δ 2.35 (t, J3=36.3, 4H), δ 2.98 (dd, J3= 6.3, 12.8, 2H), δ 3.02 (dd, J3=33.9, 11.63Hz, 1H), δ 3.09 (s, 9H), δ 3.73 (dd, J3=34.4, 10.83Hz, 1H), δ 3.74 (dd, J3=33.9, 11.73Hz, 1H), δ 6.68 (s, 1 H)
High resolution ESI-MS of 9: The calculated [M-H]− in negative mode for compound 9 was m/z 260.0711, and found m/z 260.0720.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details on the statistical tests employed are mentioned in figure legends.
DATA AND SOFTWARE AVAILABILITY
Diffraction data was processed using HKL2000 (Otwinowski and Minor, 1997) ( http://www.hkl-xray.com/), Phenix (Echols et al., 2012) (https://www.phenix-online.org/) and Coot (Emsley et al.,2010) (https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/) was used for solving the structure and refinement runs. Graphpad prism (https://www.graphpad.com/) was used for fitting kinetic data. Further details on the software used is present in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial Strains | ||
| E. coli BL21(DE3) | NEB | Catalog # C25271 |
| Critical Commercial Assays | ||
| Nextal Tubes PEGs Suite | Qiagen | Catalog # 130704 |
| Deposited Data | ||
| Structure of Egt2 WT | http://www.rcsb.org/ | Deposited in Protein Data Bank as 5UTS |
| Structure of Egt2 Y134F with substrate 4 | http://www.rcsb.org/ | Deposited in Protein Data Bank as 5V1X |
| Structure of Egt2 Y134F with sulfenic acid and amino acrylate intermediates | http://www.rcsb.org/ | Deposited in Protein Data Bank as 5V12 |
| Software and Algorithms | ||
| HKL2000 | (Otwinowski and Minor, 1997) | http://www.hkl-xray.com/ |
| Phenix | (Echols et al., 2012) | https://www.phenix-online.org/ |
| Coot | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| GraphPad Prism | https://www.graphpad.com/ | |
The structural factors and coordinates of Egt2 and its Y134F mutation variant in complex with substrate or reaction intermediates have been deposited in the Protein Data Bank with accession codes, 5UTS, 5V12 and 5V1X. Further details are mentioned in the key resources table. All residue numbers used in this manuscript are based on the sequence information from the reported Egt2 gene sequence (accession number in Uniprot: A7UX13).
Supplementary Material
Significance.
The sulfur incorporation strategy in the ergothioneine biosynthetic pathway differs from that of other sulfur-containing natural products. In this paper, we performed structural and kinetic characterization of Egt2, the enzyme mediating carbon sulfoxide bond cleavage in the last step of the biosynthetic pathways. Its structure reveals a labile sulfenic acid bound to protein at the active site, representing the first reported sulfenic intermediate-protein complex captured by X-ray crystallography. With snapshots of three distinct intermediate stages of reaction cycle and analytical tests, we were able to formulate a mechanistic model to explain the coupling between the C-S lyase reaction and the reduction of the sulfenic intermediate in this sulfoxide lyase reaction.
Highlights.
Structure of a C-S lyase in ergothionine synthesis pathway determined de novo.
Cation-π interactions was found to be critical for substrate specificity.
Sulfenic acid intermediate was captioned at the active site in X-ray structure.
Multiple crystal structures elucidate reaction mechanism.
Acknowledgments
We thank Dr. Kevin Chandler and Dr. Yi Pu for their help on the mass spectrometry studies. This work is supported in part by grants from the National Institutes of Health (R01 GM093903 to P.L.; P41 GM104603 to C.E.C.; and R01 GM104896 to Y.J.Z), the National Science Foundation (CHE-1309148 to P.L.) and the Welch Foundation (F-1778 to Y.J.Z). Crystallographic data collection was conducted at Advanced Light Sources (Beamline 5.0.3) and Advanced Photon Sources (BL23-ID-B), Department of Energy (DOE) National User Facility.
Footnotes
Author contributions—S. I., K. K., and W. Y. conducted crystallography experiments. N. N., S. W., and A. D. performed biochemical studies. Y. T. and N. L. performed mass spectrometry experiments. S.I. and N.N. contributed to the manuscript writing. C. E. C., P. L., and Y. J. Z. contributed to experimental designs, results interpretations, and manuscript preparation.
Declaration of Interests-
The authors declare competing financial interests. A patent (WO 2014100752 A1) on ergothioneine production through metabolic engineering has been submitted.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allegrini A, Astegno A, La Verde V, Dominici P. Characterization of C-S lyase from Lactobacillus delbrueckii subsp bulgaricus ATCC BAA-365 and its potential role in food flavour applications. J Biochem. 2017;161:349–360. doi: 10.1093/jb/mvw079. [DOI] [PubMed] [Google Scholar]
- Allison WS. Formation and reactions of sulfenic acids in proteins. Acc Chem Res. 1976;9:293–299. [Google Scholar]
- Aruoma OI, Spencer JPE, Mahmood N. Protection Against Oxidative Damage and Cell Death by the Natural Antioxidant Ergothioneine. Food Chem Toxicol. 1999;37:1043–1053. doi: 10.1016/s0278-6915(99)00098-8. [DOI] [PubMed] [Google Scholar]
- Baldwin JE, Bradley M. Isopenicillin N synthase: mechanistic studies. Chem Rev. 1990;90:1079–1088. [Google Scholar]
- Bartholomeus Kuettner E, Hilgenfeld R, Weiss MS. Purification, characterization, and crystallization of alliinase from garlic. Arch Biochem Biophys. 2002;402:192–200. doi: 10.1016/S0003-9861(02)00088-7. [DOI] [PubMed] [Google Scholar]
- Bhavani BS, Rajaram V, Bisht S, Kaul P, Prakash V, Murthy MR, Appaji Rao N, Savithri HS. Importance of tyrosine residues of Bacillus stearothermophilus serine hydroxymethyltransferase in cofactor binding and L-allo-Thr cleavage. FEBS J. 2008;275:4606–19. doi: 10.1111/j.1742-4658.2008.06603.x. [DOI] [PubMed] [Google Scholar]
- Bisht S, Rajaram V, Bharath SR, Kalyani JN, Khan F, Rao AN, Savithri HS, Murthy MR. Crystal structure of Escherichia coli diaminopropionate ammonia-lyase reveals mechanism of enzyme activation and catalysis. J Biol Chem. 2012;287:20369–81. doi: 10.1074/jbc.M112.351809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker SJ, Cicchillo RM, Grove TL. Self-sacrifice in radical S-adenosylmethionine proteins. Curr Opin Chem Biol. 2007;11:543–552. doi: 10.1016/j.cbpa.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–5. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Buhrman G, Parker B, Sohn J, Johannes Rudolph A, Mattos C. Structural Mechanism of Oxidative Regulation of the Phosphatase Cdc25B via an Intramolecular Disulfide Bond. Biochemistry. 2005;44:7602–7602. doi: 10.1021/bi047449f. [DOI] [PubMed] [Google Scholar]
- Burn R, Misson L, Meury M, Seebeck FP. Angewandte. Chemie International Edition; 2017. Anaerobic origin of ergothioneine. [DOI] [PubMed] [Google Scholar]
- Cellini B, Bertoldi M, Montioli R, Borri Voltattorni C. Probing the Role of Tyr 64 of Treponema denticola Cystalysin by Site-Directed Mutagenesis and Kinetic Studies. Biochemistry. 2005;44:13970–13980. doi: 10.1021/bi051433n. [DOI] [PubMed] [Google Scholar]
- Cheah IK, Halliwell B. Ergothioneine; antioxidant potential, physiological function and role in disease. Biochim Biophys Acta. 2012;1822:784–793. doi: 10.1016/j.bbadis.2011.09.017. [DOI] [PubMed] [Google Scholar]
- Chen CH, Li W, Sultana R, You MH, Kondo A, Shahpasand K, Kim BM, Luo ML, Nechama M, Lin YM, Yao Y, Lee TH, Zhou XZ, Swomley AM, Allan Butterfield D, Zhang Y, Lu KP. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol Dis. 2015;76:13–23. doi: 10.1016/j.nbd.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, Charrier V, Parsonage D. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–16. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- Clausen T, Huber R, Laber B, Pohlenz HD, Messerschmidt A. Crystal Structure of the Pyridoxal-5′-phosphate Dependent Cystathionine β-lyase fromEscherichia coliat 1.83 Å. J Mol Biol. 1996;262:202–224. doi: 10.1006/jmbi.1996.0508. [DOI] [PubMed] [Google Scholar]
- Clausen T, Kaiser JT, Steegborn C, Huber R, Kessler D. Crystal structure of the cystine C-S lyase from Synechocystis: stabilization of cysteine persulfide for FeS cluster biosynthesis. Proc Natl Acad Sci. 2000;97:3856–61. doi: 10.1073/pnas.97.8.3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarie-Baez NO, Silva Lopez EI, Furdui CM. Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radic Res. 2016;50:172–194. doi: 10.3109/10715762.2015.1090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty DA. The cation-pi interaction. Acc Chem Res. 2013;46:885–893. doi: 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echols N, Grosse-Kunstleve RW, Afonine PV, Bunkóczi G, Chen VB, Headd JJ, Mccoy AJ, Moriarty NW, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Adams PD. Graphical tools for macromolecular crystallography in PHENIX. J Appl Crystallogr. 2012;45:581–586. doi: 10.1107/S0021889812017293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliot AC, Kirsch JF. Pyridoxal Phosphate Enzymes: Mechanistic, Structural, and Evolutionary Considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdelmeier I, Daunay S, Lebel R, Farescour L, Yadan JC. Cysteine as a sustainable sulfur reagent for the protecting-group-free synthesis of sulfur-containing amino acids: biomimetic synthesis of L-ergothioneine in water. Green Chem. 2012;14:2256–2265. [Google Scholar]
- Fahey RC. Novel Thiols of Prokaryotes. Annu Rev Microbiol. 2001;55:333–356. doi: 10.1146/annurev.micro.55.1.333. [DOI] [PubMed] [Google Scholar]
- Fontecave M, Ollagnier-De-Choudens S, Mulliez E. Biological radical sulfur insertion reactions. Chem Rev. 2003;103:2149–2166. doi: 10.1021/cr020427j. [DOI] [PubMed] [Google Scholar]
- Fugate CJ, Jarrett JT. Biotin synthase: Insights into radical-mediated carbon–sulfur bond formation. Biochim Biophys Acta. 2012;1824:1213–1222. doi: 10.1016/j.bbapap.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. Proc Natl Acad Sci. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallivan JP, Dougherty DA. A Computational Study of Cation–π Interactions vs Salt Bridges in Aqueous Media:3 Implications for Protein Engineering. Journal of the American Chemical Society. 2000;122:870–874. [Google Scholar]
- Ghatge MS, Contestabile R, Di Salvo ML, Desai JV, Gandhi AK, Camara CM, Florio R, González IN, Parroni A, Schirch V, Safo MK. Pyridoxal 5′-Phosphate Is a Slow Tight Binding Inhibitor of E. coli Pyridoxal Kinase. PLoS ONE. 2012;7:e41680. doi: 10.1371/journal.pone.0041680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Carroll KS. Sulfenic acid chemistry, detection and cellular lifetime. Biochim Biophys Acta. 2014;1840:847–875. doi: 10.1016/j.bbagen.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand CE, Honek JF. Biological chemistry of naturally occurring thiols of microbial and marine origin. J Nat Prod. 2005;68:293–308. doi: 10.1021/np049685x. [DOI] [PubMed] [Google Scholar]
- Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Song H, Her AS, Bak DW, Naowarojna N, Elliott SJ, Qin L, Chen X, Liu P. Bioinformatic and biochemical characterizations of C-S bond formation and cleavage enzymes in the fungus neurospora crassa ergothioneine biosynthetic pathway. Org Lett. 2014;16:5382–5385. doi: 10.1021/ol502596z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob C. A scent of therapy: pharmacological implications of natural products containing redox-active sulfur atoms. Nat Prod Rep. 2006;23:851–863. doi: 10.1039/b609523m. [DOI] [PubMed] [Google Scholar]
- Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, functions, and formation of biological iron-sulfur clusters. Annu Rev Microbiol. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- Jurgenson CT, Begley TP, Ealick SE. The Structural and Biochemical Foundations of Thiamin Biosynthesis. Annu Rev Biochem. 2009;78:569–603. doi: 10.1146/annurev.biochem.78.072407.102340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol Rev. 2006:30. doi: 10.1111/j.1574-6976.2006.00036.x. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr D Biol Crystallogr. 2012;68:431–40. doi: 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AK, Dunleavy KM, Senkow TL, Her C, Horn BT, Jersett MA, Mahling R, Mccarthy MR, Perell GT, Valley CC, Karim CB, Gao J, Pomerantz WC, Thomas DD, Cembran A, Hinderliter A, Sachs JN. Oxidation increases the strength of the methionine-aromatic interaction. Nat Chem Biol. 2016;12:860–866. doi: 10.1038/nchembio.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CI, Mccarty RM, Liu HW. The biosynthesis of nitrogen-, sulfur-, and high-carbon chain-containing sugars. Chem Soc Rev. 2013;42:4377–4407. doi: 10.1039/c2cs35438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loksha YM, Ahmed A, El-Badawi MA, Nielsen C, Pedersen EB. Synthesis of 2-hydroxymethyl-1H-imidazoles from 1, 3-dihydroimidazole-2-thiones. Synthesis. 2004;2004:116–120. [Google Scholar]
- Marshall MS, Steele RP, Thanthiriwatte KS, Sherrill CD. Potential Energy Curves for Cation–π Interactions: Off-Axis Configurations Are Also Attractive. The Journal of Physical Chemistry A. 2009;113:13628–13632. doi: 10.1021/jp906086x. [DOI] [PubMed] [Google Scholar]
- Mcwilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pioselli B, Bettati S, Demidkina TV, Zakomirdina LN, Phillips RS, Mozzarelli A. Tyrosine phenol-lyase and tryptophan indole-lyase encapsulated in wet nanoporous silica gels: Selective stabilization of tertiary conformations. Protein Science: A Publication of the Protein Society. 2004;13:913–924. doi: 10.1110/ps.03492904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- Poor CB, Chen PR, Duguid E, Rice PA, He C. Crystal structures of the reduced, sulfenic acid, and mixed disulfide forms of SarZ, a redox active global regulator in Staphylococcus aureus. J Biol Chem. 2009;284:23517–24. doi: 10.1074/jbc.M109.015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabeh WM, Cook PF. Structure and mechanism of O-acetylserine sulfhydrylase. J Biol Chem. 2004;279:26803–6. doi: 10.1074/jbc.R400001200. [DOI] [PubMed] [Google Scholar]
- Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963;7:95–9. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- Ranaivoson FM, Antoine M, Kauffmann B, Boschi-Muller S, Aubry A, Branlant G, Favier F. A Structural Analysis of the Catalytic Mechanism of Methionine Sulfoxide Reductase A from Neisseria meningitidis. J Mol Biol. 2008;377:268–280. doi: 10.1016/j.jmb.2008.01.021. [DOI] [PubMed] [Google Scholar]
- Roret T, Pégeot H, Couturier J, Mulliert G, Rouhier N, Didierjean C. X-ray structures of Nfs2, the plastidial cysteine desulfurase from Arabidopsis thaliana. Acta Crystallogr Sect F Struct Biol Commun. 2014;70:1180–1185. doi: 10.1107/S2053230X14017026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini V, Cumming BM, Guidry L, Lamprecht DA, Adamson JH, Reddy VP, Chinta KC, Mazorodze JH, Glasgow JN, Richard-Greenblatt M, Gomez-Velasco A, Bach H, Av-Gay Y, Eoh H, Rhee K, Steyn AJ. Ergothioneine Maintains Redox and Bioenergetic Homeostasis Essential for Drug Susceptibility and Virulence of Mycobacterium tuberculosis. Cell Rep. 2016;14:572–585. doi: 10.1016/j.celrep.2015.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- Schneider G, Käck H, Lindqvist Y. The manifold of vitamin B6 dependent enzymes. Structure. 2000;8:R1–R6. doi: 10.1016/s0969-2126(00)00085-x. [DOI] [PubMed] [Google Scholar]
- Schwarz G, Mendel RR. Molybdenum cofactor biosynthesis and molybdenum enzymes. annu Rev Plant Biol. 2006;57:623–647. doi: 10.1146/annurev.arplant.57.032905.105437. [DOI] [PubMed] [Google Scholar]
- Seebeck FP. In Vitro Reconstitution of Mycobacterial Ergothioneine Biosynthesis. J Am Chem Soc. 2010;132:6632–6633. doi: 10.1021/ja101721e. [DOI] [PubMed] [Google Scholar]
- Song H, Hu W, Naowarojna N, Her AS, Wang S, Desai R, Qin L, Chen X, Liu P. Mechanistic studies of a novel C-S lyase in ergothioneine biosynthesis: the involvement of a sulfenic acid intermediate. Sci Rep. 2015;5:11870. doi: 10.1038/srep11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamir H, Srinivasan PR. Studies of the mechanism of anthranilate synthase reaction. Proc Natl Acad Sci USA. 1970;66:547–551. doi: 10.1073/pnas.66.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan D, Barber MJ, Ferreira GC. The role of tyrosine 121 in cofactor binding of 5-aminolevulinate synthase. Protein Sci. 1998;7:1208–1213. doi: 10.1002/pro.5560070516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepwong P, Giri A, Ohshima T. Effect of mycelial morphology on ergothioneine production during liquid fermentation of Lentinula edodes. Mycoscience. 2012;53:102–112. [Google Scholar]
- Terwilliger TC, Adams PD, Read RJ, Mccoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, Hung LW. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr D Biol Crystallogr. 2009;65:582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toney MD. Reaction specificity in pyridoxal phosphate enzymes. Arch Biochem Biophys. 2005;433:279–287. doi: 10.1016/j.abb.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Toney MD, Kirsch JF. Tyrosine 70 increases the coenzyme affinity of aspartate aminotransferase. A site-directed mutagenesis study. J Biol Chem. 1987;262:12403–5. [PubMed] [Google Scholar]
- Wang L, Chen S, Xu T, Taghizadeh K, Wishnok JS, Zhou X, You D, Deng Z, Dedon PC. Phosphorothioation of DNA in bacteria by dnd genes. Nat Chem Biol. 2007;3:709–710. doi: 10.1038/nchembio.2007.39. [DOI] [PubMed] [Google Scholar]
- Wang Q, Song F, Xiao X, Huang P, Li L, Monte A, Abdel-Mageed WM, Wang J, Guo H, He W, Xie F, Dai H, Liu M, Chen C, Xu H, Liu M, Piggott AM, Liu X, Capon RJ, Zhang L. Abyssomicins from the South China Sea Deep-Sea Sediment Verrucosispora sp.: Natural Thioether Michael Addition Adducts as Antitubercular Prodrugs. Angew Chemie Int Ed. 2013;52:1231–1234. doi: 10.1002/anie.201208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willey JM, Van Der Donk WA. Lantibiotics: Peptides of Diverse Structure and Function. Annu Rev Microbiol. 2007;61:477–501. doi: 10.1146/annurev.micro.61.080706.093501. [DOI] [PubMed] [Google Scholar]
- Wu HM, Kuan YC, Chu CH, Hsu WH, Wang WC. Crystal structures of lysine-preferred racemases, the non-antibiotic selectable markers for transgenic plants. PLoS One. 2012;7:e48301. doi: 10.1371/journal.pone.0048301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Yadan JC. Synthesis of L-(+)-Ergothioneine. J Org Chem. 1995;60:6296–6301. [Google Scholar]
- Yan W, Stone E, Zhang YJ. Structural Snapshots of an Engineered Cystathionine-gamma-lyase Reveal the Critical Role of Electrostatic Interactions in the Active Site. Biochemistry. 2017;56:876–885. doi: 10.1021/acs.biochem.6b01172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang NC, Lin HC, Wu JH, Ou HC, Chai YC, Tseng CY, Liao JW, Song TY. Ergothioneine protects against neuronal injury induced by beta-amyloid in mice. Food Chem Toxicol. 2012;50:3902–3911. doi: 10.1016/j.fct.2012.08.021. [DOI] [PubMed] [Google Scholar]
- Zacharias N, Dougherty DA. Cation-pi interactions in ligand recognition and catalysis. Trends Pharmacol Sci. 2002;23:281–287. doi: 10.1016/s0165-6147(02)02027-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.