

Abstract
Acetylcholinesterase (AChE, EC 3.1.1.7) and butyrylcholinesterase (BChE, EC 3.1.1.8) are related enzymes found across the animal kingdom. The critical role of acetylcholinesterase in neurotransmission has been known for almost a century, but a physiological role for butyrylcholinesterase is just now emerging. The cholinesterases have been deliberately targeted for both therapy and toxicity, with cholinesterase inhibitors being used in the clinic for a variety of disorders and conversely for their toxic potential as pesticides and chemical weapons. Non-catalytic functions of the cholinesterases (ChEs) participate in both neurodevelopment and disease. Manipulating either the catalytic activities or the structure of these enzymes can potentially shift the balance between beneficial and adverse effect in a wide number of physiological processes.
Keywords: acetylcholinesterase, cholinergic signaling, butyrylcholinesterase, cholinesterase inhibitors, ghrelin metabolism, Alzheimer’s, organophosphates
Graphical abstract
II. INTRODUCTION
An often paraphrased statement by the 16th century Swiss physician and philosopher Paracelsus is that “dose separates poison from remedy”. Students in basic pharmacology and toxicology learn early on of the ‘therapeutic index”, a quantitative relationship between efficacy and toxicity and a direct conceptual descendent of Paracelsus’ edict. The importance of dose-response relationships in pharmacology and toxicology is difficult to overstate, as they provide chemical-specific views of drug and toxicant potency, efficacy, and selectivity. The ChEs, acetylcholinesterase (AChE, EC 3.1.1.7) and butyrylcholinesterase (BChE, EC 3.1.18) are related enzymes expressed throughout much of the animal kingdom. AChE plays a well-defined role in regulating cholinergic signaling while the physiological impact of BChE has remained unclear until very recently. Here we provide a brief overview on the biology of ChEs and consider how drug- or toxicant-induced changes in their esterase activity, or in the proteins themselves, can shift the balance between benefit and harm.
III. TWO CHOLINESTERASES
Acetylcholinesterase (AChE) and its “sister” enzyme butyrylcholinesterase (BChE) are widely expressed throughout the animal kingdom [1]. AChE and BChE share roughly 50% sequence homology and have relatively similar tertiary and quaternary structures. They both possess a catalytic triad of three amino acids (serine, glutamate and histidine) located deep inside a “gorge” in the tertiary structure [2–4]. Evidence suggests that these enzymes emerged from a carboxylesterase superfamily, with “true” AChE first emerging hundreds of millions of years ago in Platyhelminthes [5]. Higher vertebrates have one ACHE gene and one BCHE gene, while some lower species express multiple genes of one or both [1]. The cyclostomes, jawless fish including the lamprey and hagfish, only express AChE, suggesting that BChE arose later in evolution by gene duplication and divergence from AChE [1, 2, 6].
IV. ACETYLCHOLINESTERASE AND CHOLINERGIC SIGNALING
The concept of a synapse between a neuron and an innervated cell, and the receptors that mediated their interaction, was developed by Bernard, Ehrlich, Sherrington, Langley and others (see [7–9]. It was long debated whether transmission of nerve impulses to muscle cells occurs by electrical or by chemical signals until the work of Otto Loewi and Henry Dale, later recognized by their Nobel Prize in 1936 [10]. The gains in understanding of various physiological processes by these early investigators and others were aided by using natural toxins. In fact, Loewi [11] considered the primary objective of pharmacology as “revealing physiological functions by the reactions of living matter to chemical agents”. While this narrow description does not encompass the multifold aspects of modern pharmacology, the experimental use of xenobiotics has played an essential role in gaining an understanding of neurotransmission and cholinergic signaling.
Over a century ago, Dale [12] compared the effects of selected choline esters with the mushroom toxin, muscarine, and was the first to describe “muscarine-like” and “nicotine-like” actions. The relative potency of choline esters in isolated organ systems vs intact animals led him to posit that the “evanescence” of their effects could be due to rapid hydrolysis by an esterase. In Loewi’s classic studies [13], stimulating the vagus in a nerve-heart preparation in physiological solution triggered release of a substance called vagusstoff (i.e., vagus substance) that mimicked the effect of nerve stimulation when the fluid medium was transferred to a second heart with no vagal connection. Importantly, Loewi also showed that the effect of the vagal substance was (like that of acetylcholine) enhanced by eserine, a known inhibitor of ChEs [14,15]. Soon thereafter, Dale and Dudley [16] reported the isolation of acetylcholine from tissue (horse spleen), confirming its endogenous presence. These studies and others laid groundwork for an enormous amount of research on the role of acetylcholine in synaptic signaling and its regulation by AChE.
There is now a widespread consensus that AChE is the paramount or sole enzyme regulating neurotransmission in vertebrate cholinergic pathways that include brain, skeletal muscle and the autonomic nervous system. AChE serves this role in all mammals by selectively inactivating acetylcholine, within seconds or milliseconds after it is released from a presynaptic cholinergic neuron. AChE is one of the most efficient enzymes in the body, with a catalytic rate that approaches the limit of diffusion [17,18]. AChE’s function appears equally important in brain and the periphery. This view is supported by the intensely concentrated localization of this enzyme at cholinergic synapses throughout the body, and by the diversity of effects elicited by inhibiting AChE either in the brain or in the peripheral compartment.
V. Physiological role of BChE
In contrast to the long-established and well-defined role of AChE in regulating cholinergic signaling, a true physiological function for BChE remained elusive over many decades. BChE exhibits much broader substrate specificity than AChE. For example it hydrolyzes butyrylcholine and acetylcholine while AChE only hydrolyzes the latter. Also, while BChE expression in many tissues exceeds that of AChE, it exists at much lower concentrations in the brain, skeletal muscle, and peripheral nerves [19]. Although exogenous butyrylcholine has been shown to modulate intrinsic cardiac neuron activity in canines [20,21], to our knowledge no synapses in higher vertebrates use butyrylcholine as a neurotransmitter. In fact, a longstanding consensus holds that such synapses do not exist. Evidence to support that view is that, in our unpublished studies, selective inhibitors such as iso-OMPA (tetra isopropyl pyrophosphoramide) can completely inhibit BChE catalysis without eliciting obvious physiological disturbance. Not surprisingly, BChE knockout mice with no BChE expression appear perfectly healthy [22]. In particular, they show no apparent change in motor, autonomic or cognitive function. Under casual observation they are indistinguishable from wild-type mice. Moreover, there are isolated human populations who have been identified as completely lacking a functional BCHE gene, but again, by all accounts, they exhibit a normal phenotype. Their only physiologic difference from “wild-type” is an elevated risk when exposed to bioactive esters in food or ester-type muscle relaxants in the clinic [23,24].
Thus, until quite recently, BChE was considered to lack an important function apart from serving as a “backup” for AChE in regard to neurotransmission, and as a modestly protective bioscavenger of bioactive esters in the food supply. The latter could be regarded as a feature that enables humans and other species to obtain nutrients from plants, many of which could be toxic if their endogenous esters were not efficiently hydrolyzed. The enzyme’s ability to hydrolyze esters has been harnessed in surgical procedures using ester-based pharmaceuticals such as the muscle relaxant succinylcholine, which it rapidly inactivates. However, it became apparent early on that certain patients required exceptionally long recovery times after treatment with succinylcholine. The basis for this difference in clinical response is pharmacogenomic in nature, i.e., these individuals were found to express an “atypical BChE” with active site mutations leading to catalytic efficiency far below that of the native enzyme. Because of this BChE variant, they cleared succinylcholine slowly and needed assisted respiration and clinical surveillance for extended periods.
More recently, it was discovered that BChE hydrolyzes the neuropeptide gut hormone, ghrelin [25–28]. Nonetheless, because the enzyme reaction is very slow, those who first reported this finding were initially reluctant to attribute a real physiological role for that phenomenon. Our own views changed when we accidentally linked high level gene transfer of BChE in group-housed male mice to reduced stress, reduced aggression and reduced levels of plasma ghrelin [29]. We are now confident that ghrelin modulation represents an important physiological role for this enzyme and, in light of that role, there is real potential for using BChE to modulate ghrelin’s impact in many types of emotional disorders.
Ghrelin is a 28-amino acid peptide with a serine residue acylated by octanoic acid. This feature is essential for binding and activating its primary target, the G-protein-coupled growth hormone secretagogue receptor (GSHR)[30,31]. The major source of plasma ghrelin derives from endocrine cells in the stomach, which release it into the general circulation. Circulating ghrelin then feeds back on the stomach to stimulate gastric muscles that produce “hunger pangs.” It triggers afferent vagal neurons in the stomach to activate CNS regions involved in food seeking. At the same time plasma ghrelin also penetrates the blood brain barrier to stimulate GHSR and drive food cravings. In addition, brain neurons in or near the pituitary gland and hypothalamus produce and release ghrelin locally. Activating GHSR in the pituitary leads to growth hormone secretion, while activation elsewhere plays a role in many other processes including glucose homeostasis and fat storage [32,33]. Because BChE is present in both the bloodstream and the brain, its hydrolytic activity plays a role in regulating ghrelin signaling by cleaving its acyl group to form desacyl-ghrelin, which is the dominant form of the peptide in plasma and CSF [34]. Therefore, changes in blood BChE activity, e.g., in response to an anticholinesterase (anti-ChE), can shift the balance of GHSR signaling in favor of active acyl-ghrelin. However, desacyl-ghrelin also has multiple effects in a GHSR-independent manner. These relationships further highlight the physiological implications of altering ghrelin metabolism by reducing BChE activity with enzyme inhibitors or raising it with BChE gene transfer 29, 35–37].
Ghrelin serves as a stimulant for hedonic feeding, promoting food intake and fat storage [38–40]. Healthy lean individuals experience a drive for food in response to ghrelin pulses [38]. Circulating ghrelin levels are influenced by food intake, being high before a meal and low afterwards [41]. The most obvious role for BChE’s modulation of ghrelin is to regulate feeding behavior/food intake. Under ordinary conditions, circulating BChE is stable, with little change from hour to hour, day to day, or week to week. In contrast, levels of ghrelin released by the stomach or within the brain can change sharply across time. If BChE levels are too low, the drive to eat could be heightened. It has been reported that obese humans and dogs have modestly higher plasma BChE and lower plasma ghrelin than their lean counterparts [42,43]. Similar findings have been reported in mouse models of obesity [44]. By the same token, when obese humans and mice succeed in recovering their original healthy weight, plasma BChE falls and plasma ghrelin rises, often markedly. Evidence to date thus suggests that the levels of BChE activity and ghrelin activity are inversely coupled. This implies a potential for changes in plasma or tissue BChE to influence ghrelin metabolism and thereby impact the respective signaling pathways of acyl- and desacyl-ghrelin [45]. Following up these insights it seems feasible to manipulate BChE levels to impact ghrelin-driven overeating and obesity [46].
VI. Cholinesterases and pharmacological effects of inhibitors in the periphery
It is now known that cholinergic signaling plays a vital role in controlling many peripheral functions including skeletal and smooth muscle contraction, autonomic postganglionic neuron activation, parasympathetic end-organ activation (e.g., salivary, lacrimal and bronchial glands), regulation of cardiac activity, and others. Figure 1 diagrams peripheral motor and autonomic functions regulated by cholinergic signaling and illustrates relationships among cholinergic receptor activation, acetylcholinesterase activity, and functional responses.
Figure 1.
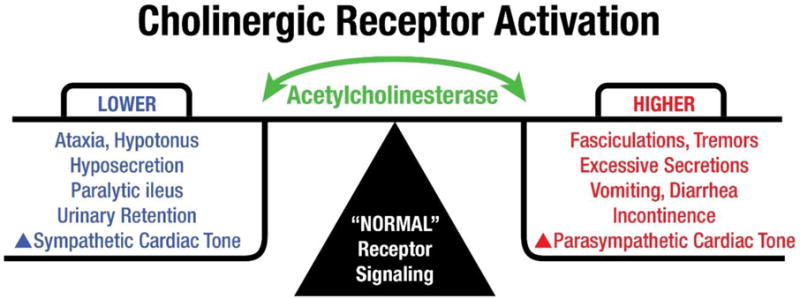
Acetylcholinesterase and cholinergic receptor activation. Under normal physiological conditions, AChE plays a dynamic role in regulating cholinergic signaling, rapidly degrading acetylcholine to terminate neurotransmission. Cholinergic motoneurons release acetylcholine to activate skeletal muscles for ambulation and fine motor control. Similarly, parasympathetic neurons stimulate smooth muscles to drive gut peristalsis, contraction of the bladder, secretion from salivary glands, slow heartbeats at the sinoatrial node. When AChE activity is suppressed, cholinergic receptor activation causes increased muscle contractions, exocrine secretions, GI disturbances, incontinence and increased parasympathetic tone leading to decreased rate and force of heartbeats. When AChE activity is high, reduction of cholinergic receptor activation, lowers force and tone in muscle and heart, and reduced exocrine secretions.
As noted previously, early research on cholinergic signaling was focused on the neuromuscular junction. Claude Bernard’s studies of neuromuscular blockade by the arrow poison, curare, were instrumental in developing the concept of nerve-muscle synapses [47]. Adequate control of muscle tone is an essential goal in many surgical and emergency settings. Neuromuscular blocking agents have been extensively used to facilitate tracheal intubations, perform endoscopic evaluations and maintain immobility during surgery. Curare acts as a competitive, non-depolarizing blocker of nicotinic receptors at the neuromuscular junction. A variety of non-depolarizing neuromuscular blockers such as atracurium, vecuronium and rocuronium have been developed and implemented clinically to block neuromuscular transmission. When neuromuscular blockade is no longer needed, it has to be reversed for rapid recovery of muscle tone. Cholinesterase inhibitors (e.g., neostigmine) have been used to reverse the effects of these non-depolarizing neuromuscular blocking agents. While cholinesterase inhibitors are still used for this purpose, a reversal agent that does not act by inhibiting acetylcholinesterase (sugammedex, a cyclodextrin-based scavenger) is being increasingly used to reverse the effects of rocuronium and vecuronium [48,49].
In contrast to the non-depolarizing receptor blockers, succinylcholine, a structural analog of acetylcholine, is a depolarizing blocker that remains the gold standard for neuromuscular blockade in rapid procedures such as emergency intubations [50]. Succinylcholine’s neuromuscular blockade is ordinarily reversed very quickly by the catalytic activity of plasma BChE [51]. However, as noted above, problems did arise in some individuals, owing to genetic differences in the catalytic activity of their BChE [52,53].
Obviously, a cholinesterase inhibitor would be of no use in reversing neuromuscular blockade induced by succinylcholine, and could in fact be counterproductive. But inhibiting acetylcholine breakdown by blocking AChE activity may be beneficial in disorders wherein acetylcholine signaling is impaired [54]. One clinical condition that can be improved by blocking acetylcholine hydrolysis is myasthenia gravis, a group of neuromuscular disorders involving autoimmune-mediated destruction of neuromuscular nicotinic receptors or associated proteins at the neuromuscular junction [55]. By virtue of their ability to block degradation of acetylcholine at the neuromuscular junction and enhance the activation of nicotinic receptors, cholinesterase inhibitors (e.g., pyridostigmine) have been used for decades to enhance muscular performance in human and veterinary patients [56]. However, one form of myasthenia gravis, associated with antibodies to muscle-specific kinase, does not benefit from blocking AChE and symptoms may in fact be exacerbated by cholinesterase inhibitors. Another cholinesterase inhibitor, the rapid-acting and reversible inhibitor edrophonium (Tensilon®), is often used in the Tensilon test to diagnose myasthenia gravis. Intravenous administration of edrophonium leads to a rapid (seconds) and dramatic, short-term improvement in muscle tone. Edrophonium may also be helpful in diagnosing other neuromuscular disorders, such as cervical dystonia and blepharospasm [57,58].
Cholinesterase inhibitors can be helpful in treating glaucoma, a leading cause of blindness worldwide [59]. Generally, increased intraocular pressure from aqueous humor accumulation leads to degeneration of the optic nerve and retinal ganglion cells. Strategies that reduce intraocular pressure are the only approaches proven to treat most forms of glaucoma [60]. Ocular administration of an anti-ChE can facilitate contraction of the ciliary muscle and increase the flow of aqueous humor through the trabecular meshwork to reduce intraocular pressure. Other drugs, e.g., sulfonamide carbonic anhydrase inhibitors such as acetazolamide and dorzolamide that decrease the formation of aqueous humor and prostaglandin F analogs (e.g., latanoprost, bimatoprost) that enhance aqueous humor outflow have largely supplanted cholinesterase inhibitors for these conditions [61]. Interestingly, the reversible cholinesterase inhibitor galantamine was recently shown to protect retinal ganglion cells and improve local blood flow in experimental models of glaucoma, but in a manner independent of intraocular pressure [62,63].
Gastrointestinal tone can be effectively modulated by cholinesterase inhibitors. As parasympathetic fibers release acetylcholine to contract the circular and longitudinal intestinal muscles, AChE inhibition can facilitate those actions. Postoperative ileus (delayed gastric emptying after surgery) can arise from the surgical procedure itself, anesthetic agents, opioid medications and other factors. Neostigmine can accelerate gastric emptying, but some studies suggest it contributes to adverse bowel effects, e.g., serious leaks from intestinal anastomoses [64,65]. Similarly, conditions such as functional dyspepsia, gastroparesis and colonic pseudo-obstruction may be treated with a cholinesterase inhibitor such as acotiamide 66–68]. Other prokinetic agents, e.g., metoclopramide, are used to facilitate gastric emptying and activate GI motoneurons as do cholinesterase inhibitors, but they act by increasing acetylcholine release rather than by blocking its degradation [69,70].
As noted earlier, individuals expressing BChE variants with low or absent catalytic activity are generally symptomless, and only a limited amount of research has been conducted on the pharmacological/toxicological consequences of BChE inhibition. However, the enzyme’s putative role in ghrelin signaling is likely to be a therapeutic (or toxic) target in the future. Li and colleagues [26] reported that BChE knockout mice maintained on a high-fat diet gained considerably more weight than BChE+/+ littermates. Interestingly, in that study, the mice on a path to obesity did not seem to show increased food consumption. In fact, our later studies along the same lines suggest that this apparently negative outcome may have been an artifact of the means for measuring food intake, which is easily distorted by losses hidden in bedding material. In any case, the plasma concentration of acyl-ghrelin in BChE knockouts was almost twice as high as in wildtype littermates. Other hydrolases besides BChE, including, carboxylesterases and proteases, may cleave the acyl group from acyl-ghrelin or degrade the peptide itself [26,71]. As rodents have much higher carboxylesterase blood levels than humans, such differences may influence the relative impact of BChE activity on ghrelin signaling in man compared to some test species. Nonetheless, recent findings by our research teams showed that a marked rise in plasma BChE activity after gene transfer was associated with drastically reduced acyl-ghrelin in plasma [72]. Interestingly, high-BChE mice were seen to exhibit lower levels of aggressive behavior (both spontaneous and intruder-provoked). A number of studies suggest a link between ghrelin and emotional/affective behaviors [73–78]. The role of BChE in ghrelin signaling is both intriguing and important but it remains incompletely understood [79]. Therefore, studies to evaluate the effects of BChE and its inhibitors in the complex signaling associated with ghrelin’s numerous physiological impacts appear well worthwhile. Figure 2 illustrates the widespread potential influence of BChE on ghrelin signaling and associated functions in the brain.
Figure 2.
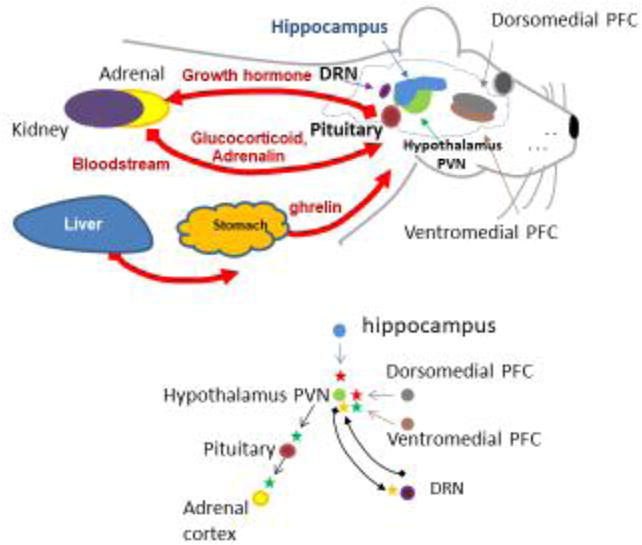
Role of BChE in ghrelin metabolism and signaling in the brain. Top panel: Main organs, brain regions, and pathways involved in fear response. Red arrows indicate blood-borne delivery. Bottom panel: Key interacting centers: parvo-ventricular nucleus of the hypothalamus (PVN); dorsomedial and ventromedial prefrontal cortex (PFC); dorsal raphé nucleus (DRN); adrenal cortex; and pituitary gland. Arrows indicate direction of interactions. Green = activation; red = inhibition; and yellow = modulation.
VII. Cholinesterases and Pharmacological Effects of Inhibition in the CNS
A number of elegant morphological and immunochemical studies led to the description of six cholinergic cell nuclei in the brain, termed Ch1-Ch6 [80–83]. Figure 3 is taken from [80], showing the cholinergic nuclei and their general projections in the mammalian brain. The diffuse innervation by these different cholinergic fibers affects many physiological functions including cognition, respiration, locomotion, and others. Moreover, some brain regions (notably striatum) contain multiple networks of cholinergic interneurons [84,85]. Thus, AChE inhibition in the CNS has the potential to influence an extremely wide array of functions, and this has been targeted in treating early-stage Alzheimer’s disease (AD).
Figure 3.
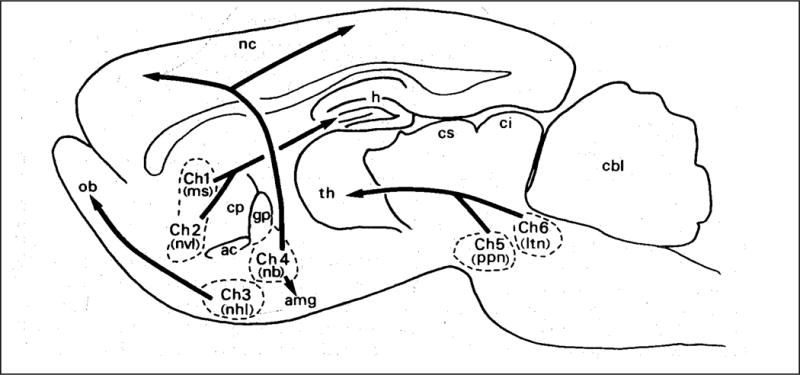
Cholinergic nuclei and projections in the rat brain. Classic schematic diagram from Mesulam et al., 1983. Ch1, medial septal nucleus; Ch2, vertical limb nucleus of the diagonal band; Ch3 Lateral part of horizontal limb nucleus of the diagonal band; Ch4, Nucleus basalis of Meynert, glogus pallidus, Substantia innominate; nucleus of the ansa lenticularis; Ch5, Nucleus pedunculopontinus; Ch6 Laterodorsal tegmental nucleus. Other structures: nc, neocortex; ob, olfactory bulb; th, thalamus; h, hippocampus; cbl, cerebellum.
AD is a devastating neurodegenerative disorder affecting an estimated 35 million people worldwide [86]. Findings of extensive loss of cells in Ch4 (nucleus basalis) and widespread reduction of cholinergic markers in Alzheimer forebrain [87–92] led to the cholinergic hypothesis of dementia (reviewed in [93,94]). This hypothesis opened the door to examine potential therapeutic benefits of cholinesterase inhibitors [95–97]. Since that time, many natural and synthetic cholinesterase inhibitors that bind selectively or show multifunctional characteristics have been evaluated to treat Alzheimer’s disease (see recent reviews [98–102].
Currently, according to Mielke and colleagues [103], drugs with US Food and Drug Administration (FDA) approval for Alzheimer therapy include the following: galantamine (Razadyn®, 4aS,6R,8aS-5,6,9,10,11,12- hexahydro- 3-methoxy- 11-methyl- 4aH [1], benzofuro[3a,3,2-ef] [2] benzazepin- 6-ol), rivastigmine (Exelon®, [3-[(1S)-1- (dimethylamino)ethyl]phenyl] N-ethyl-N-methylcarbamate) and donepezil (Aricept®, 2-((1- Benzylpiperidin-4-yl)methyl)-5,6-dimethoxy-2,3-dihydro-1H-inden-1-one). In 1993, the cholinesterase inhibitor tacrine (Cognex®, 1,2,3,4-tetrahydroacridin-9-amine) was the first drug with FDA approval to treat Alzheimer’s disease, but its use was limited by short half-life (requiring 4× daily dosing) and evidence of hepatotoxicity. A recent meta-analysis of the efficacy of cholinesterase inhibitors in Alzheimer’s disease reported that donepezil, rivastigmine and galantamine all produced relatively minimal but significant benefits including enhanced cognition and improved behavioral outcomes, while none increased the risk of serious toxicity [104]. Donepezil and galantamine are selective for CNS ChEs, but all three can elicit nausea, diarrhea and other unwanted effects [104–108]. Unfortunately, they have little if any impact on disease progression. Interestingly, Tricco and coworkers [104] reported that galantamine was associated with a decreased odds ratio for death, while pharmacovigilance databases in both the US and Canada detected an increased odds ratio for death with rivastigmine [105]. Thus, while current cholinesterase inhibitors do have minor therapeutic benefits, the search continues for more effective or multi-purpose inhibitors acting on ChEs and other macromolecular targets [109–111].
Non-catalytic Roles of CNS Cholinesterases
In addition to serving as drug targets for inhibitors that increase synaptic acetylcholine levels, both AChE and BChE have been proposed to play non-catalytic roles in neurobiology. A number of studies suggest that both cholinesterases have non-catalytic roles in neurodevelopment, possibly playing a “morphogenic” role in vertebrate systems [112–116]. Some of the most striking findings in support of a morphogenic role for the cholinesterases includes the spatiotemporal expression patterns of AChE in the thalamocortical and geniculocortical projections during neonatal rat brain development [113,117]. These neurons are not cholinergic nor do they receive cholinergic innervation in adulthood, but during a discrete window of development (peaking during week 2 of postnatal development correlating with the timing of respective fiber ingrowth into the cortex) they exhibit intense AChE staining. Interestingly, inhibition of the transiently expressed AChE activity had no effect on subsequent patterning of innervation [118,119]. On the other hand, Yang and colleagues [120] reported that chlorpyrifos and chlorpyrifos oxon could disrupt axonal growth in cultured sensory neurons expressing AChE at low concentrations not inhibiting the enzyme, but similar exposures had no such effect in AChE −/− neurons. Transfection of the neurons with cDNA coding for AChE restored sensitivity. Using chicken neurodevelopmental models, Layer and coworkers [121] provided evidence suggesting both BChE and AChE play morphogenic roles in vertebrate neurodevelopment. Of p articular interest was the “foreshadowing” of AChE-positive areas cholinergic fibers by BChE expression during neural tube development [112,121,122]. It was proposed that these enzymes both regulate neurite extension by inactivating acetylcholine, at least partially independent from cholinergic synaptogenesis. The ChEs may play non-catalytic roles in the adult CNS as well. For example, Graybiel and Ragsdale [123] reported marked, differential staining of AChE and BChE in the primate visual pathway. BChE showed distinct labeling of parvocellular layers of the lateral geniculate and area 17 of the visual cortex, independent of AChE labeling. The authors suggested that the differential distribution of both cholinesterases could be an indicator that BChE played some role in signaling within this pathway.
Substantial evidence points to non-catalytic roles for AChE and BChE in the development and progression of Alzheimer’s disease. Alzheimer’s disease greatly enhances the rate of irreversible neuronal cell death in the aging brain. Its characteristic lesions are accumulations of amyloid protein and neurofibrillary tangles that replace functional synaptic structures. These lesions typically incorporate deposits of excess AChE and BChE, in patterns that have no clear relation to functional neuronal circuitry. This feature has raised a key question: Are the enzyme deposits compensatory reactions to amyloid toxicity, or are they actually damaging stressed neurons? Adding to complexity, in addition to modulating synaptic acetylcholine levels, AChE appears to have non-cholinergic functions that involve protein– protein interactions rather than enzymatic catalysis [124–128]. Thus, studies on neuronal development suggest that AChE promotes neurite outgrowth, possibly through adhesive interactions with the extracellular matrix [128].
In AD research, consistent local accumulations of AChE in senile plaques led early on to the hypothesis that this enzyme binds beta-amyloid (A-beta) and promotes its deposition. Indeed, in vitro models have shown that AChE accelerates the formation of insoluble fibrils of A-beta and AChE proteins, which are more cytotoxic than A-beta alone. Crystal structure data suggest a potential locus of interaction between amyloid and the AChE protein [129]. This locus is near the enzyme’s peripheral site, located on the external surface near the entrance to the catalytic gorge. Consistent with this structural inference, peripheral site-directed AChE ligands like propidium iodide and certain monoclonal antibodies have been found to prevent AChE from enhancing the formation of amyloid fibrils in vitro [130]. These observations are intriguing, but their relevance to the pathogenesis of Alzheimer’s disease has been largely speculative. Now we need a realistic appraisal of AChE’s influence on amyloid deposition in the intact brain.
In vitro studies with neuronal tissue culture have shown that AChE interacts with A-beta to promote its deposition and aggregation, which lead to neuronal death [130]. To test that the hypothesis in vivo, Tg2576 mice were engineered in one of our labs to express human AChE, and then crossed with a line of mice that express human amyloid precursor protein [126]. These subjects reliably developed plaques at 9 months. The resulting F1 hybrids expressed both of these human transgenes in brain, and by 6 months of age their cerebral cortex showed authentic plaques that stained readily by immunochemistry for A-beta 1–40 and 1–42. Those early onset plaques also stained positively for other components found in the brain lesions of human patients as hallmarks of AD pathology. These included Cd11b, GFAP, and again, AChE. Overall, plaque onset in the hybrid mice began 30%–50% sooner than in either of the two parental lines. Their plaque burdens increased with age, growing markedly larger and more numerous in these doubly transgenic animals at 9 and 12 months. Quantitative immunoassay via ELISA also confirmed an increase of total amyloid content in brain at 9–12 months. These histological and biochemical results bear a striking resemblance to human AD pathology, and they strongly support a conclusion that AChE plays a role in initiating and promoting the pathogenesis of Alzheimer’s disease.
Taken together, several findings show increased plaque formation when A-beta is expressed against a background of high AChE levels. The mechanism for this interaction remains unclear, but a plausible sequence of pathological events can be inferred from prior in vitro work. Direct measurements of protein aggregation in culture show that AChE can act as a nucleating factor to promote conversion of soluble amyloid peptide into insoluble amyloid fibrils [130]. Structural analysis by X-ray crystallography, competition assays, and computational docking with AChE ligands all point to the enzyme’s peripheral site as a probable locus of protein interaction with A-beta [131,132]. Therefore, the most effective compound to block this interaction would not necessarily be a “classical inhibitor” that binds in AChE’s active site within the gorge. In fact it seems likely that ligands directed to more superficial regions may suppress the aggregation of beta amyloid monomers and, in turn, reduce subsequent neuronal toxicity.
Among the anti-ChEs currently approved for Alzheimer therapy, tacrine, galantamine, and rivastigmine all bind primarily at the central site of AChE [133]. Interestingly, donepezil can orient from the catalytic triad region to the peripheral anionic site via aromatic stacking with aromatic acid residues [134]. This unique feature adds interest to the debate as to whether donepezil slows disease progression, as it has appeared to do in recent clinical trials. With advanced molecular modeling technology it should be possible to discover drugs that more effectively disrupt interactions between AChE and A-beta, ideally with minimal inhibition of AChE in the periphery, especially skeletal muscle. Hybrid transgenic mice with early plaque deposition in a background of high AChE and APP may represent an especially good model for testing such agents to determine whether they can help in slowing the progression of AD.
BChE’s role in AD is much more controversial. This enzyme is primarily generated in the liver for release into the systemic circulation, where, as noted earlier, it serves to inactivate various bio-active ester compounds from plant-based foods that otherwise might be toxic. But a second locus of BChE production is the brain, where it contributes to inactivation of acetylcholine and thereby participates in regulating key neural pathways. Like AChE, BChE also concentrates in amyloid plaques associated with dying neurons in AD. These features have convinced many neurologists and neurobiologists that BChE is a significant contributor to AD pathogenesis, or even a major driver. That view led to clinical trials of selective BChE inhibitors like cymserine, with the expectation of enhancing acetylcholine levels at brain synapses and restoring the neurochemistry for cholinergic transmission and cognition. In fact, this concept did lead to improved cognitive function and better short term memory, but only temporarily. Unfortunately, none of the tested BChE-selective inhibitors were able to slow, much less reverse, the inexorable process of neuronal death and irreversible cognitive loss. In other words, these drugs can indeed be useful as palliative treatments, primarily in the early stages of AD, but they are not disease modifiers. As the brain pathology inevitably progresses to severe neuronal loss, their efficacy declines accordingly.
As for the issue of BChE’s mechanistic role in AD, if any, full clarity remains elusive despite intensive and ongoing research. Some investigators have recently concluded that BChE is an actual driver of AD pathology [135–137] but other equally well-known scientists have published data suggesting the direct opposite. The former view is largely based on the ubiquitous presence of BChE protein in plaque lesions, and the growing abundance of this protein as lesions progress. However, one can make an argument that BChE’s abundance in plaque lesions is instead a “bystander effect”. Specifically, BChE may be attracted to plaques as amyloid increases and its impact may be entirely neutral or even one that mitigates amyloid-induced damage to neuronal circuitry. Support for that concept came from Diamant and coworkers [130] who showed that native BChE attenuates spontaneous formation of amyloid fibrils in vitro, while C-terminal mutated “K-variant BChE” (strongly associated with increased risk of early onset AD) is deficient in that respect.
In one of our labs, studies have begun with so- called “5×FAD mice” prone to early-onset AD, hoping to shed fresh light on BChE’s role in AD plaque lesions, neuronal death, and cognitive impairment. It is far too early to draw any conclusions from the preliminary data. Nonetheless we have been able to demonstrate an essentially permanent (i.e., lifetime) elevation of brain BChE levels after direct intracerebral injection of AAV8 viral vector encoding mRNA for this enzyme. In addition, using a novel “PHP gene transfer vector” that readily crosses the blood-brain barrier [138], we found it possible to generate very high and essentially permanent BChE expression across virtually all regions of the cerebral cortex, cerebellum, and basal ganglia [139]. This capability should facilitate studies aimed at clarifying BChE’s roles in the central nervous system.
VIII. Toxicological Uses of Cholinesterase Inhibitors
While ChE inhibitors can and have been used to treat a variety of illnesses, the other side of therapy is toxicity. A remarkable number of microorganisms, plants, fungi and animals have developed anti-ChEs [140–142]. As the ChEs are widespread across the animal kingdom and play an important role in neuronal signaling in all higher species of animals, widespread expression of ChE inhibitors by these many organisms has likely evolved as a defense against predation.
The toxic potential of ChE inhibitors has been recognized for over 170 years. Robert Christison, a leading toxicologist of the time reported the first results of toxicological testing of the “trial by ordeal” bean [143]. Indigenous people in the Calabar region of West Africa were known to use an “ordeal poison” to determine whether a person was innocent or guilty of a crime, usually “witchcraft.” As we noted earlier herein, extracts of the calabar bean (Physostigma venenosum) contain eserine (physostigmine), subsequently found to be a potent inhibitor of ChEs. The deadly potency of the extract was noted by European missionaries as early as 1840. It has been speculated that this toxicant actually had inherent properties that led to its effective “judicial” use (see review [144]).
Since discovery of eserine’s toxicity, carbamate structure and anti-ChE action, many other carbamate cholinesterase inhibitors have been synthesized and characterized [145]. A substantial number were introduced as pesticides in the 1960s and some of these continue extensive use throughout the world [146]. Before widespread introduction of the carbamates, starting in the 1940s, many organophosphorus cholinesterase inhibitors were developed as insecticides [147]. Carbamate and organophosphorus cholinesterase inhibitors both covalently modify the active site serine residue in AChE and BChE, thereby inhibiting all choline ester hydrolysis. Cleavage of carbamoyl moieties by an esterase, while far slower than hydrolysis of the acyl group in a choline ester, is still rapid compared to the painfully prolonged reactivation of esterase inhibited by an organophosphorus agent [148].
Organophosphorus cholinesterase inhibitors did not emerge from a natural source: unlike the carbamates, they are all synthetic entities. Tetraethylpyrophosphate was the first organophosphorus inhibitor, synthesized and reported in the 1850s. Further development and characterization of organophosphorus anti-ChEs was not initiated until the mid-1930s in pre-WWII Germany, under the direction of Dr. Gerhard Schrader [149]. Schrader was searching for synthetic insecticides to address the high cost and low availability of common natural insecticides in use at that time. Tabun (N-dimethyl phosphoramidocyanidate), one of Schrader’s most potent inhibitors, subsequently came under German military control to use as a chemical warfare agent [150]. Other organophosphorus compounds to come from that laboratory included parathion (O,O’-diethyl-O-p-nitrophenyl phosphorothioate) and its oxygen analog (O,O’-diethyl-O-p-nitrophenyl phosphate), paraoxon [147]. Since that time, many organophosphorus anti-ChEs have been synthesized. Most were developed for commercial use as pesticides and, in some cases, as drugs in veterinary and human therapeutics (e.g., diisopropylfluorophosphate for glaucoma, trichlorfon as an anthelminth).
Carbamate and OP insecticides are still among the most widely used pesticides in the world [151]. In 2012, the OP insecticides chlorpyrifos and acephate were ranked 14th and 22nd overall in estimated amounts of agricultural pesticides used in the US. The carbamate insecticide carbaryl and the OP insecticides acephate and malathion were until recently among the most common pesticides for home use. Acephate was also widely used in the industrial sector. Over the last two decades, a number of OP insecticides have been withdrawn from the market, banned, or use-restricted in the US, but others remain a large part of total pesticide use. In 2012, OP insecticides made up roughly 38% of all insecticides used in the US [151]. As of now, multiple regulatory actions in the US have curtailed the use of such pesticides, but certain populations may still be exposed to cholinesterase-inhibiting levels [152].
Worldwide concern for anti-ChE pesticide exposure remains: more sinister is the threat of OP nerve agent intoxication. While virtually all developed countries have joined agreements to eliminate stockpiles of these extremely toxic compounds, their relative ease of synthesis makes them a continuing concern. As recently as April of 2017, the Organization for Prohibition of Chemical Weapons confirmed that the “nerve gas” sarin had been used on residents of Khan Sheikoun, Syria. Thus the potential use of OP nerve agents by rogue nations and/or terrorist groups is a remaining global problem.
A seemingly paradoxical pharmacological use of more readily reversible cholinesterase inhibitors is to block the long-term inactivation of AChE by “irreversible” inhibitors, the OP nerve agents [153]. In essence, individuals at risk of exposure to an OP nerve agent can be protected by pre-exposure to pyridostigmine or another carbamate anti-ChE, transiently blocking OP access to the active site of the enzyme. If the agent with short-lived inhibition is given before exposure to the irreversible inhibitor, the latter cannot bind to the enzyme, which will then spontaneously reactivate. In fact this scenario has shown protection against nerve agent intoxication in several animal models [153–158]. The logic behind this prophylactic approach is based on the concept that cholinergic synapses have “spare” enzyme, so a certain level of AChE inhibition can be tolerated without disrupting the dynamics of cholinergic signaling. This clever prophylactic strategy was implemented in the first Persian Gulf War. Soldiers were given varying doses of pyridostigmine with concern for possible exposure to chemical weapons. For some time, a cloud of suspicion has hovered over this application however because of the potential role of pyridostigmine in the unexplained “Gulf War Illnesses” following the first Persian Gulf War [159]. Those disorders may well have had other contributing causes however, e.g., post-traumatic stress. Even if pyridostigmine had no role in these illnesses, delivering a dose of drug to shield enough enzyme molecules without excessive inhibition, in a diverse group of people with high variability and exposure to environmental stressors, was a risky approach. But one thing is certain: if one is to use cholinesterase inhibitors for therapy or prophylaxis, there will always be a delicate balance between beneficial and toxic inhibition of AChE. Too little drug will be ineffective, too much will be dangerous.
IX. Increasing ChEs in vivo
Elevating cholinesterase levels in the body may be beneficial to protect against chemicals that elicit toxicity by inhibiting AChE, or that are inactivated primarily by ChEs. The main incentives for enhancing ChE activity are to overcome or prevent the toxicity of anti-ChEs caused by 1) accidental or intentional exposure to anti-ChE pesticides in agricultural settings, 2) ingestion of foods carrying residual pesticides, or 3) intentional exposure to nerve agents in chemical warfare or terrorism. All these scenarios are possible in present day conditions. Some are especially likely in developing countries and regions that lack active oversight and strict environmental regulations. Increasing the levels of BChE in the circulation has been evaluated as a protective strategy in two very different settings.
First, administering large amounts of purified human BChE has been shown to protect experimental animals against lethal exposures to various OP nerve agents. BChE can sequester or scavenge OP molecules in a stoichiometric manner, inactivating the toxicant molecules before they can inhibit AChE in brain and peripheral tissues to disrupt cholinergic signaling [160]. In fact, both BChE and AChE have been evaluated as bioscavengers for OP nerve agents. In sufficient quantities, administration of these stoichiometric binding proteins can minimize AChE inhibition in target tissues and protect against lethality from OP toxicants in multiple experimental models, including primates.
There are obstacles in the prophylactic use of ChEs as OP bioscavengers including a need for large amounts of purified human protein, due in part to their rapid clearance from the circulation. For example, intramuscular (im) administration of human BChE in mice (13 mg/kg, about 0.3 mg) leads to peak blood BChE activities at about 10–12 h, but only ~ 25% of peak remains at 70 h [161]. Both im and intraperitoneal (ip) administration of 0.1–3 mg of human BChE in mice caused marked elevation of circulating BChE enzyme activity, peaking 12–24 h later, but returning to near baseline by 120 h [162]. Doctor and Saxena [163] and Saxena and coworkers [164] reported on the pharmacokinetics of human BChE in mice and guinea pigs given im and ip administration as well as long-term stability of the lyophilized enzyme (at least 2 years), and complete, sign-free survival in guinea pigs subsequently given an LD50 dosage of the nerve agents VX and soman. Higher im dosages of human BChE (up to 60 mg/kg), led to peak levels of enzyme around 24 h that remained substantial for at least four days [165]. The same investigators reported no overt physiological or behavioral signs after high-dose BChE administration, and no changes in serum chemistry or tissue histopathology. Intravenous administration of 30 mg/kg human BChE in rhesus monkeys led to marked elevation of blood BChE activity that returned to within about 25% of peak levels by 100 h [166]. These and other recent studies illustrate the potential for exogenously administered human BChE to elevate circulating enzyme activity and protect against toxicity from potent OP nerve agents, with little evidence of adverse reaction to high doses of the enzyme.
As noted earlier, while exogenously administered BChE can protect against OP toxicity, its efficacy is limited by clearance of the protein from the circulation within a few days. A number of investigations have attempted to increase the circulation time of ChEs by chemical modification, gene transfer, and “nanoformulation.” Studies using AChE as a bioscavenger [167–171] led to the conclusion that the degree of AChE sialyation has a direct influence on the enzyme’s circulatory residence time. Chemical modification of recombinant BChE by polysialylation led to a 6-fold increase in mean residence time after iv dosing in mice, and such enzyme proved to be a bioscavenger offering protection against several OP nerve agents [172,173]. Modifying proteins with polyethylene glycol (PEG, i.e., PEGylation) has been used for decades to reduce their clearance in vivo [174–177]. Cohen and coworkers [171] reported that PEGylation of rhesus and human AChE led to large increases in mean residence times of recombinant rhesus and human AChE given to rhesus monkeys. PEGylation also doubled the mean circulatory residence time of recombinant tetrameric BChE from 18.3 to 36.2 h [178]. An almost 10-fold increase in circulatory time was gained by PEGylating recombinant monomeric BChE [179]. Thus, modifying ChEs by PEG generally leads to increased circulation times, but there are reports of antibody development and more rapid clearance of enzyme activity with subsequent doses of PEGylated proteins [179,180].
Some studies have evaluated gene transfer as an approach to increase and prolong BChE activity for protection against OP toxicants. Chilukuri and coworkers [179,181] showed that BChE−/− mice treated (iv but not ip) with recombinant adenoviruses encoding rHu-BChE showed elevated blood BChE levels (about 200-fold higher than wild-type controls) peaking about 5 days after treatment, but returning to baseline by 10 days post-inoculation. Antibodies to the native protein were detected in the serum. In a similar approach, Parikh and coworkers [182] reported up to 3,400- fold increase in plasma BChE activity and transient protection against high (5-30-fold LD50) doses of the OP anti-ChEs echothiophate and VX (O-ethyl-S-2-N,N-diisopropylaminoethyl methylphosphonothiolate). Again BChE activity returned to baseline by day 10, however.
Nanoformulations of BChE that might be useful as bioscavengers have been reported. Gaydess and coworkers [183] described a polyionic complex made of BChE with a poly-L-lysine and poly(ethylene glycol) copolymer, with an estimated diameter of about 15 nm. Fluorescence-labeled BChE-copolymer complexes injected into mice showed a small amount of BChE entered the brain. In 2015, Pope and colleagues [184] reported on a series of BChE-copolymer complexes synthesized following the general approach of Gaydess et al., [183]. A subset of these complexes was spherical, with a median diameter of about 35 nm [185]. In vitro sensitivity to the OP anti-ChE paraoxon, resistance to proteases and heat-inactivation, and in vitro “bioscavenging” activity against paraoxon were all equivalent to native BChE [184,185]. Recombinant dimeric BChE has also been conjugated with CdSe/CdZnS quantum dots [186]. These conjugates retained partial enzyme activity and showed similar sensitivity to paraoxon. Sokolov and colleagues [187] reported that human BChE conjugated with gold nanoparticles had a diameter about 15 nm and showed an interesting increase of BChE activity. In another recent study, equine serum BChE was coated with a zwitterionic polymer gel [188], leading to nanoparticles of 15–30 nm diameter that resisted inactivation by heat and trypsin. They also showed about a 3-fold increase in circulating time versus the free enzyme, and importantly no immune sensitization with repeated dosing. Rahhal and colleagues [189] reported an interesting formulation of equine BChE, laminating a film of protein onto a mold to produce 1 μm BChE microparticles. After purification, the preparation was administered by insufflation, with BChE retention times similar to free enzyme following oro-tracheal administration (48–72 h). Optimization of such formulations holds promise for further development of bioscavengers for OP and possibly other toxic chemicals.
It has long been known that native BChE is a major factor in the inactivation of cocaine, an ester-type drug of abuse [190]. But recently some researchers conceived the idea that BChE mutations could improve that function to a point that would favorably impact cocaine overdose. Rapid progress was made in different laboratories approaching this goal, assisted by computer-based models of cocaine docking in the enzyme’s active site, which led to successful predictions to improve drug binding and hydrolysis [29]. At present, near-optimal BChE versions that enhance the rate of cocaine inactivation by more than a thousand-fold have been generated [191]. Efficacy with exogenous administration of such cocaine-hydrolyzing BChE variants can also suffer because they are more rapidly cleared from the circulation than native BChE. However, site-directed mutagenesis introducing disulfide bonds between two mutant BChE subunits led to an approximate doubling of circulatory half-life in rats [192]. In our hands, mice and rats treated with a cocaine hydrolase showed no reaction whatsoever to doses of cocaine that would ordinarily have been lethal within 1–2 minutes. In contrast, they merely continued normal cage-side activities, eating and grooming, just like vehicle-controls [72].
The theoretical importance of a highly effective cocaine hydrolase is that treatment-seeking addicts who have been “clean” for a while would be less likely to relapse after, in a weak moment, “one last hit” delivered a greatly reduced drug reward. For that effect to be maintained for periods of months to years the most promising approach would be one that doesn’t require former users to make repeated decisions to remain in treatment on a daily or weekly basis. We consider that only a durable gene transfer can meet that requirement. In fact, our recent studies with such agents indicate that very high levels of BChE can be generated by gene transfer with no overt disturbances. Furthermore, these levels were sustained for more than a year in experimental animals ranging from mice to Rhesus monkeys (S. Brimijoin and M. Carroll, unpublished data).
Several studies on the safety of BChE as an OP bioscavenger or cocaine hydrolase have reported no overt signs of toxicity, even with massive increases in circulating BChE activity. While the severity of either nerve agent intoxication or cocaine abuse may warrant taking the risk for possible side effects of large increases in circulating BChE levels for extended periods, the emerging role of BChE in the metabolism of ghrelin could be of concern. Long-term studies should be conducted to evaluate more subtle physiological changes that may accompany prolonged BChE elevation. Of special note, altered affective behaviors in mice with high levels of genetically-modified BChE were disconcerting at first, but they led to highly positive effects such as reductions in aggression and stress-induced fear [72,193,194].
Brain AChE and BChE both concentrate in senile plaques associated with amyloid protein. While a systemic administration of BChE would not be expected to lead to increased activity in the CNS, gene transfer approaches may in fact lead to increased expression in the brain. If BChE has some role in the aggregation of amyloid and plaque generation, the potential for its enhancement by increasing BChE expression would be of great concern. However, as noted earlier, at Mayo, extensive but unpublished studies on mice carrying genes for premature formation of brain plaques of amyloid and tau proteins with neurofibrillary tangles showed no increase in lesions when brain BChE levels were raised many fold.
X. Conclusion
In summary, AChE and BChE are related enzymes that evolved throughout the animal kingdom. Complex organisms have developed chemical signaling pathways relying primarily on AChE as a key regulator. On the other hand, a physiological role for BChE has previously been obscure. In fact, there was no real consensus that this enzyme had any role beyond the detoxification of bioactive esters in plant foods and ester-type medications, but its status is now rapidly changing. A number of clinical disorders have been therapeutically addressed by use of cholinesterase inhibitors. In general, any condition that involves reduced activation of acetylcholine receptors in cholinergic synapses may be partially or fully relieved by application of a cholinesterase inhibitor. Such conditions include neuromuscular disorders, autonomic dysfunction, and Alzheimer’s disease. All share a deficiency of cholinergic receptor activation that can be restored by inhibiting AChE. Of course, excessive AChE inhibition in these patients can lead from therapy to non-selective toxicity. Abnormally low levels of BChE can lead to inefficient clearance of esters in the food supply and slowed inactivation of ester-based drugs, potentially leading to toxicity. Moreover, recent studies point to a likely role for BChE in regulating levels and activity of the peptide hormone ghrelin, with altered ghrelin signaling a potential consequence of xenobiotics that affect BChE activity.
It seems fair to say, the two cholinesterase enzymes, AChE and BChE, have catalytic properties with important impacts on neuromuscular, cognitive and emotional functions. In addition they show adhesive and other properties that contribute to protein-protein interactions, with potential roles in disorders like Alzheimer’s disease. For this reason, manipulating either the protein levels of AChE or BChE or their enzymatic activities may influence the toxic aggregation of proteins in this and other neurodegenerative diseases, with positive (therapeutic) or negative (toxic) outcomes. While each of these two ChEs has been studied for decades, remarkable new information continues to emerge regarding their pleiotropic relationships with key signaling pathways and functions in the nervous system. It has become clear that changes in either catalytic or structural properties of these ChEs are likely to shift the delicate balance between poison and remedy in regard to numerous crucially important physiological functions. And, with the rising tide of discovery regarding the range and power of these roles we can look forward to harnessing this knowledge to avoid toxicity while reaping a harvest of advances in treatments for a wide range of medical conditions.
Acknowledgments
This work was primarily supported by grants from the Defense Threat Reduction Agency (HDTRA1-13-1-0042, CP), National Institute for Environmental Health Sciences (ES008739, CP) and a Translational Avant-Garde Award from the National Institute on Drug Abuse (DA42492, SB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pezzementi L, Chatonnet A. Evolution of cholinesterases in the animal kingdom. Chem Biol Interact. 2010;187:27–33. doi: 10.1016/j.cbi.2010.03.043. [DOI] [PubMed] [Google Scholar]
- 2.Chatonnet A, Lockridge O. Comparison of butyrylcholinesterase and acetylcholinesterase. Biochem J. 1989;260:625–34. doi: 10.1042/bj2600625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science. 1991;253:872–9. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 4.Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J Biol Chem. 2003;278:41141–7. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- 5.Bentley GN, Jones AK, Agnew A. Expression and comparative functional characterisation of recombinant acetylcholinesterase from three species of Schistosoma. Mol Biochem Parasitol. 2005;141:119–23. doi: 10.1016/j.molbiopara.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 6.Pezzementi L, Reinheimer EJ, Pezzementi ML. Acetylcholinesterase from the skeletal muscle of the lamprey Petromyzon marinus exists in globular and asymmetric forms. J Neurochem. 1987;48:1753–60. doi: 10.1111/j.1471-4159.1987.tb05733.x. [DOI] [PubMed] [Google Scholar]
- 7.Maehle A-H. “Receptive substances”: John Newport Langley (1852–1925) and his path to a receptor theory of drug action. Med Hist. 2004;48:153–174. doi: 10.1017/s0025727300000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burke RE. Sir Charles Sherrington’s the integrative action of the nervous system: a centenary appreciation. Brain. 2007;130:887–894. doi: 10.1093/brain/awm022. [DOI] [PubMed] [Google Scholar]
- 9.Regnier C. Claude Bernard (1813–1878) and experimental medicine. Medicographia. 2013;35:474–484. [Google Scholar]
- 10.Karczmar A. Exploring the Vertebrate Central Cholinergic Nervous System. Springer; New York, NY: 2007. Cholinergic cell pathways; pp. 33–79. [Google Scholar]
- 11.Loewi O. Über humorale Übertragbarkeit der Herznervenwirkung. I. Mitteilung. Pflügers Arch Ges Physiol. 1921;189:239–242. [Google Scholar]
- 12.Dale HH. The action of certain esters and ethers of choline and their relation to muscarine. J Pharmacol Exp Ther. 1914;6:147–190. [Google Scholar]
- 13.Loewi O. An autobiographic sketch. Perspect Biol Med. 1960;4:3–25. [Google Scholar]
- 14.Loewi O, Navratil E. Über humorale Übertragbarkeit der Herznervenwirkung. X. Mitteilung. Über das Schicksal des Vagusstoffes. Pflügers Arch Ges Physiol. 1926a;214:678–688. [Google Scholar]
- 15.Loewi O, Navratil E. Über humorale Übertragbarkeit der Herznervenwirkung. XI. Über den Mechanismus der Vaguswirkung von Physostigmin und Ergotamine. Pflügers Arch Ges Physiol. 1926b;214:678–688. [Google Scholar]
- 16.Dale HH, Dudley HW. The presence of histamine and acetylcholine in the spleen of the ox and the horse. J Physiol. 1929;68:97–123. doi: 10.1113/jphysiol.1929.sp002598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bazelyansky M, Robey E, Kirsch JF. Fractional diffusion-limited component of reactions catalyzed by acetylcholinesterase. Biochemistry. 1986;25:125–30. doi: 10.1021/bi00349a019. [DOI] [PubMed] [Google Scholar]
- 18.Silman I, Sussman JL. Acetylcholinesterase: how is structure related to function? Chem Biol Interact. 2008;175:3–10. doi: 10.1016/j.cbi.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 19.Lockridge O. Review of human butyrylcholinesterase structure, function, genetic variants, history of use in the clinic, and potential therapeutic uses. Pharmacol Ther. 2015;148:34–46. doi: 10.1016/j.pharmthera.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 20.Darvesh S, MacDonald SE, Losier AM, Martin E, Hopkins DA, Armour JA. Cholinesterases in cardiac ganglia and modulation of canine intrinsic cardiac neuronal activity. J Auton Nerv Syst. 1998;71:75–84. doi: 10.1016/s0165-1838(98)00064-2. [DOI] [PubMed] [Google Scholar]
- 21.Darvesh S, Arora RC, Martin E, Magee D, Hopkins DA, Armour JA. Cholinesterase inhibitors modify the activity of intrinsic cardiac neurons. Exp Neurol. 2004;188:461–70. doi: 10.1016/j.expneurol.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Duysen EG, Li B, Lockridge O. The butyrylcholinesterase knockout mouse a research tool in the study of drug sensitivity, bio-distribution, obesity and Alzheimer’s disease. Expert Opin Drug Metab Toxicol. 2009;5:523–8. doi: 10.1517/17425250902915555. [DOI] [PubMed] [Google Scholar]
- 23.Manoharan I, Boopathy R, Darvesh S, Lockridge O. A medical health report on individuals with silent butyrylcholinesterase in the Vysya community of India. Clin Chim Acta. 2007;378:128–135. doi: 10.1016/j.cca.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Lockridge O, Norgren RB, Jr, Johnson RC, Blake TA. Naturally occurring genetic variants of human acetylcholinesterase and butyrylcholinesterase and their potential impact on the risk of toxicity from cholinesterase inhibitors. Chem Res Toxicol. 2016;29:1381–92. doi: 10.1021/acs.chemrestox.6b00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Vriese C, Gregoire F, Lema-Kisoka R, Waelbroeck M, Robberecht P, Delporte C. Ghrelin degradation by serum and tissue homogenates: identification of the cleavage sites. Endocrinology. 2004;145:4997–5005. doi: 10.1210/en.2004-0569. [DOI] [PubMed] [Google Scholar]
- 26.Li B, Duysen EG, Carlson M, Lockridge O. The butyrylcholinesterase knockout mouse as a model for human butyrylcholinesterase deficiency. J Pharmacol Exp Ther. 2008;324:1146–54. doi: 10.1124/jpet.107.133330. [DOI] [PubMed] [Google Scholar]
- 27.Tham E, Liu J, Innis S, Thompson D, Gaylinn BD, Bogarin R, Haim A, Thorner MO, Chanoine JP. Acylated ghrelin concentrations are markedly decreased during pregnancy in mothers with and without gestational diabetes: relationship with cholinesterase. Amer J Physiol Endocrinol Metab. 2009;296:E1093–E1100. doi: 10.1152/ajpendo.90866.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schopfer LM, Lockridge O, Brimijoin S. Pure human butyrylcholinesterase hydrolyzes octanoyl ghrelin to desacyl ghrelin. Gen Comp Endocrinol. 2015;224:61–68. doi: 10.1016/j.ygcen.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 29.Brimijoin S, Chen VP, Pang YP, Geng L, Gao Y. Physiological roles for butyrylcholinesterase: a BChE-ghrelin axis. Chem Biol Interact. 2016;259(Part B):271–275. doi: 10.1016/j.cbi.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–60. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 31.Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, Matsukura S, Kangawa K, Nakazato M. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology. 2000;141:4255–4261. doi: 10.1210/endo.141.11.7757. [DOI] [PubMed] [Google Scholar]
- 32.Smith RG, Jiang H, Sun Y. Developments in ghrelin biology and potential clinical relevance. Trends Endocrinol Metab. 2005;16:436–42. doi: 10.1016/j.tem.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Yin Y, Li Y, Zhang W. The growth hormone secretagogue receptor: its intracellular signaling and regulation. Int J Mol Sci. 2014;15:4837–55. doi: 10.3390/ijms15034837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toshinai K, Mondal MS, Nakazato M, Date Y, Murakami N, Kojima M, Kangawa K, Matsukura S. Upregulation of ghrelin expression in the stomach upon fasting, insulin-induced hypoglycemia, and leptin administration. Biochem Biophys Res Commun. 2001;281:1220–5. doi: 10.1006/bbrc.2001.4518. [DOI] [PubMed] [Google Scholar]
- 35.Chung H, Seo S, Moon M, Park S. Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3 beta and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen-glucose deprivation-induced apoptosis in primary rat cortical neuronal cells. J Endocrinol. 2008;198:511–21. doi: 10.1677/JOE-08-0160. [DOI] [PubMed] [Google Scholar]
- 36.Hwang S, Moon M, Kim S, Hwang L, Ahn KJ, Park S. Neuroprotective effect of ghrelin is associated with decreased expression of prostate apoptosis response-4. Endocr J. 2009;56:609–17. doi: 10.1507/endocrj.k09e-072. [DOI] [PubMed] [Google Scholar]
- 37.Ku JM, Andrews ZB, Barsby T, Reichenbach A, Lemus MB, Drummond GR, Sleeman MW, Spencer SJ, Sobey CG, Miller AA. Ghrelin-related peptides exert protective effects in the cerebral circulation of male mice through a nonclassical ghrelin receptor(s) Endocrinology. 2015;156:280–90. doi: 10.1210/en.2014-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, Batterham RL, Taheri S, Stanley SA, Ghatei MA, Bloom SR. Ghrelin causes hyperphagia and obesity in rats. Diabetes. 2001;50:2540–7. doi: 10.2337/diabetes.50.11.2540. [DOI] [PubMed] [Google Scholar]
- 39.Neary NM, Small CJ, Wren AM, Lee JL, Druce MR, Palmieri C, Frost GS, Ghatei MA, Coombes RC, Bloom SR. Ghrelin increases energy intake in cancer patients with impaired appetite: acute, randomized, placebo-controlled trial. J Clin Endocrinol Metab. 2004;89:2832–6. doi: 10.1210/jc.2003-031768. [DOI] [PubMed] [Google Scholar]
- 40.Kouno T, Akiyama N, Ito T, Okuda T, Nanchi I, Notoya M, Oka S, Yukioka H. Ghrelin O-acyltransferase knockout mice show resistance to obesity when fed high-sucrose diet. J Endocrinol. 2016;228:115–25. doi: 10.1530/JOE-15-0330. [DOI] [PubMed] [Google Scholar]
- 41.Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50:1714–9. doi: 10.2337/diabetes.50.8.1714. [DOI] [PubMed] [Google Scholar]
- 42.Tschop M, Weyer C, Tataranni A, Devanarayan V, Ravussin E, Heiman ML. Circulating ghrelin levels are decreased in human obesity. Diabetes. 2001;50:707–709. doi: 10.2337/diabetes.50.4.707. [DOI] [PubMed] [Google Scholar]
- 43.Tvarijonaviciute A, Tecles F, Ceron JJ. Relationship between serum butyrylcholinesterase and obesity in dogs: a preliminary report. Vet J. 2010;186:197–200. doi: 10.1016/j.tvjl.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 44.Alcantara VM, Oliveira LC, Rea RR, Suplicy HL. Butyrylcholinesterase and obesity in individuals with the CHE2 C5+ and CHE2 C5− phenotypes. Int J Obesity. 2003;27:1557–1564. doi: 10.1038/sj.ijo.0802464. [DOI] [PubMed] [Google Scholar]
- 45.Chen VP, Gao Y, Geng L, Brimijoin S. Butyrylcholinesterase regulates central ghrelin signaling and has an impact on food intake and glucose homeostasis. Int J Obes (Lond) 2017a;41:1413–1419. doi: 10.1038/ijo.2017.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen VP, Gao Y, Geng L, Brimijoin S. Butyrylcholinesterase gene transfer in obese mice prevents postdieting body weight rebound by suppressing ghrelin signaling. Proc Natl Acad Sci U S A. 2017b;114:10960–10965. doi: 10.1073/pnas.1706517114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernard C. Analyse physiologique des proprieties des systemes musculaire et nerveux au moyen du curare. Comptes Rendus Acad Sci. 1856;43:825–829. [Google Scholar]
- 48.Schepens T, Cammu G. Neuromuscular blockade: what was, is and will be. Acta Anaesthesiol Belg. 2014;65:151–9. [PubMed] [Google Scholar]
- 49.Hristovska AM, Duch P, Allingstrup M, Afshari A. Efficacy and safety of sugammadex versus neostigmine in reversing neuromuscular blockade in adults. Cochrane Database Systemat Rev. 2017 doi: 10.1002/14651858.CD012763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tran DTT, Newton EK, Mount VAH, Lee JS, Mansour C, Wells GA, Perry JJ. Rocuronium vs. succinylcholine for rapid sequence intubation: a Cochrane systematic review. Anaesthesia. 2017;72:765–777. doi: 10.1111/anae.13903. [DOI] [PubMed] [Google Scholar]
- 51.Lockridge O, Masson P. Pesticides and susceptible populations: people with butyrylcholinesterase genetic variants may be at risk. Neurotoxicology. 2000;21:113–26. [PubMed] [Google Scholar]
- 52.Bretlau C, Sørensen MK, Vedersoe AL, Rasmussen LS, Gätke MR. Response to succinylcholine in patients carrying the K-variant of the butyrylcholinesterase gene. Anesth Analg. 2013;116:596–601. doi: 10.1213/ANE.0b013e318280a3f3. [DOI] [PubMed] [Google Scholar]
- 53.Wichmann S, Færk G, Bundgaard JR, Gätke MR. Patients with prolonged effect of succinylcholine or mivacurium had novel mutations in the butyrylcholinesterase gene. Pharmacogenet Genomics. 2016;26:351–6. doi: 10.1097/FPC.0000000000000221. [DOI] [PubMed] [Google Scholar]
- 54.Taylor P. Anticholinesterase agents. In: Brunton LL, Chabner BA, Knollmann BC, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 12th. McGraw Hill; New York, NY: 2011. pp. 239–254. [Google Scholar]
- 55.Guptill JT, Soni M, Meriggioli MN. Current treatment, emerging translational therapies, and new therapeutic targets for autoimmune myasthenia gravis. Neurotherapeutics. 2016;13:118–31. doi: 10.1007/s13311-015-0398-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilhus NE. Myasthenia gravis. N Engl J Med. 2016;375:2570–2581. doi: 10.1056/NEJMra1602678. [DOI] [PubMed] [Google Scholar]
- 57.Matsumoto S, Murakami N, Koizumi H, Takahashi M, Izumi Y, Kaji R. Edrophonium challenge test for blepharospasm. Front Neurosci. 2016;10:226. doi: 10.3389/fnins.2016.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsumoto S, Murakami N, Koizumi H, Takahashi M, Izumi Y, Kaji R. Evaluation of the edrophonium challenge test for cervical dystonia. Intern Med. 2017;56:2415–2421. doi: 10.2169/internalmedicine.8555-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311:1901–11. doi: 10.1001/jama.2014.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goel M, Picciani RG, Lee RK, Bhattacharya SK. Aqueous humor dynamics: a review. Open Ophthalmol J. 2010;4:52–9. doi: 10.2174/1874364101004010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doucette LP, Walter MA. Prostaglandins in the eye: Function, expression, and roles in glaucoma. Ophthalmic Genet. 2017;38:108–116. doi: 10.3109/13816810.2016.1164193. [DOI] [PubMed] [Google Scholar]
- 62.Almasieh M, MacIntyre JN, Pouliot M, Casanova C, Vaucher E, Kelly ME, Di Polo A. Acetylcholinesterase inhibition promotes retinal vasoprotection and increases ocular blood flow in experimental glaucoma. Invest Ophthalmol Vis Sci. 2013;54:3171–83. doi: 10.1167/iovs.12-11481. [DOI] [PubMed] [Google Scholar]
- 63.Almasieh M, Zhou Y, Kelly ME, Casanova C, Di Polo A. Structural and functional neuroprotection in glaucoma: role of galantamine-mediated activation of muscarinic acetylcholine receptors. Cell Death Dis. 2010;1:e27. doi: 10.1038/cddis.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ogilvy AJ, Smith G. The gastrointestinal tract after anaesthesia. Eur J Anaesthesiol Suppl. 1995;10:35–42. [PubMed] [Google Scholar]
- 65.Hirst GR, Karandikar SS, Brown G, Slowey H, Beynon J. Colonic anastomotic disruption in the immediate postoperative period. Int J Colorectal Dis. 2004;19:281–2. doi: 10.1007/s00384-003-0564-2. [DOI] [PubMed] [Google Scholar]
- 66.Altan E, Masaoka T, Farré R, Tack J. Acotiamide, a novel gastroprokinetic for the treatment of patients with functional dyspepsia: postprandial distress syndrome. Expert Rev Gastroenterol Hepatol. 2012;6:533–44. doi: 10.1586/egh.12.34. [DOI] [PubMed] [Google Scholar]
- 67.Parthasarathy G, Ravi K, Camilleri M, Andrews C, Szarka LA, Low PA, Zinsmeister AR, Bharucha AE. Effect of neostigmine on gastroduodenal motility in patients with suspected gastrointestinal motility disorders. Neurogastroenterol Motil. 2015;27:1736–46. doi: 10.1111/nmo.12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Camilleri M. Functional Dyspepsia and Gastroparesis. Dig Dis. 2016;34:491–9. doi: 10.1159/000445226. [DOI] [PubMed] [Google Scholar]
- 69.Rao AS, Camilleri M. Review article: metoclopramide and tardive dyskinesia. Aliment Pharmacol Ther. 2010;31:11–9. doi: 10.1111/j.1365-2036.2009.04189.x. [DOI] [PubMed] [Google Scholar]
- 70.Bielefeldt K. From harmful treatment to secondary gain: adverse event reporting in dyspepsia and gastroparesis. Dig Dis Sci. 2017;62:2999–3013. doi: 10.1007/s10620-017-4633-8. [DOI] [PubMed] [Google Scholar]
- 71.Ni H, Walia P, Chanoine JP. Ontogeny of acylated ghrelin degradation in the rat. Peptides. 2010;31:301–6. doi: 10.1016/j.peptides.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 72.Chen VP, Gao Y, Geng L, Parks RJ, Pang YP, Brimijoin S. Plasma butyrylcholinesterase regulates ghrelin to control aggression. Proc Natl Acad Sci U S A. 2015;112:2251–6. doi: 10.1073/pnas.1421536112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carlini VP, Monzón ME, Varas MM, Cragnolini AB, Schiöth HB, Scimonelli TN, de Barioglio SR. Ghrelin increases anxiety-like behavior and memory retention in rats. Biochem Biophys Res Commun. 2002;299:739–43. doi: 10.1016/s0006-291x(02)02740-7. [DOI] [PubMed] [Google Scholar]
- 74.Seoane LM, Al-Massadi O, Lage M, Dieguez C, Casanueva FF. Ghrelin: from a GH-secretagogue to the regulation of food intake, sleep and anxiety. Pediatr Endocrinol Rev. 2004;1(Suppl 3):432–7. [PubMed] [Google Scholar]
- 75.Spencer SJ, Xu L, Clarke MA, Lemus M, Reichenbach A, Geenen B, Kozicz T, Andrews ZB. Ghrelin regulates the hypothalamic-pituitary-adrenal axis and restricts anxiety after acute stress. Biol Psychiatry. 2012;72:457–65. doi: 10.1016/j.biopsych.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 76.Jensen M, Ratner C, Rudenko O, Christiansen SH, Skov LJ, Hundahl C, Woldbye DP, Holst B. Anxiolytic-like effects of increased ghrelin receptor signaling in the amygdala. Int J Neuropsychopharmacol. 2016;19:1–12. doi: 10.1093/ijnp/pyv123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stark R, Santos VV, Geenen B, Cabral A, Dinan T, Bayliss JA, Lockie SH, Reichenbach A, Lemus MB, Perello M, Spencer SJ, Kozicz T, Andrews ZB. Des-acyl ghrelin and ghrelin O-acyltransferase regulate hypothalamic-pituitary-adrenal axis activation and anxiety in response to acute stress. Endocrinology. 2016;157:3946–3957. doi: 10.1210/en.2016-1306. [DOI] [PubMed] [Google Scholar]
- 78.Huang HJ, Zhu XC, Han QQ, Wang YL, Yue N, Wang J, Yu R, Li B, Wu GC, Liu Q, Yu J. Ghrelin alleviates anxiety- and depression-like behaviors induced by chronic unpredictable mild stress in rodents. Behav Brain Res. 2017;326:33–43. doi: 10.1016/j.bbr.2017.02.040. [DOI] [PubMed] [Google Scholar]
- 79.Brimijoin S, Tye S. Favorable impact on stress-related behaviors by modulating plasma butyrylcholinesterase. Cell Mol Neurobiol. 2017 doi: 10.1007/s10571-017-0523-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mesulam MM, Mufson EJ, Wainer BH, Levey AI. Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6) Neuroscience. 1983;10:1185–201. doi: 10.1016/0306-4522(83)90108-2. [DOI] [PubMed] [Google Scholar]
- 81.Butcher LL, Woolf NJ. Central cholinergic systems: synopsis of anatomy and overview of physiology and pathology. In: Scheibel AB, Wechsler AF, editors. The Biological Substrates of Alzheimer’s Disease. Academic Press; New York, NY: 1986. pp. 73–86. [Google Scholar]
- 82.Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- 83.Lucas-Meunier E, Fossier P, Baux G, Amar M. Cholinergic modulation of the cortical neuronal network. Pflugers Arch. 2003;446:17–29. doi: 10.1007/s00424-002-0999-2. [DOI] [PubMed] [Google Scholar]
- 84.Zhou FM, Wilson CJ, Dani JA. Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol. 2002;53:590–605. doi: 10.1002/neu.10150. [DOI] [PubMed] [Google Scholar]
- 85.Gonzales KK, Smith Y. Cholinergic interneurons in the dorsal and ventral striatum: anatomical and functional considerations in normal and diseased conditions. Ann N Y Acad Sci. 2015;1349:1–45. doi: 10.1111/nyas.12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.World Health Organization. The epidemiology and impact of dementia: current state and future trends. 2015 http://www.who.int/mental_health/neurology/dementia/dementia_thematicbrief_epidemiology.pdf.
- 87.Divac I. Magnocellular nuclei of the basal forebrain project to neocortex, brain stem, and olfactory bulb. Review of some functional correlates. Brain Res. 1975;93:385–98. doi: 10.1016/0006-8993(75)90178-x. [DOI] [PubMed] [Google Scholar]
- 88.Kievit J, Kuypers HG. Subcortical afferents to the frontal lobe in the rhesus monkey studied by means of retrograde horseradish peroxidase transport. Brain Res. 1975;85:261–6. doi: 10.1016/0006-8993(75)90079-7. [DOI] [PubMed] [Google Scholar]
- 89.Davies P, Maloney AJF. Selective loss of central cholinergic neurons in Alzheimer’s disease. The Lancet. 1976;308:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- 90.Drachman DA. Memory dementia and cholinergic system. In: Katzman R, Terry RD, Bick KL, editors. Azheimer’s Disease: Senile Dementia and Related Disorders. Raven Press; San Diego, CA: 1978. pp. 141–171. [Google Scholar]
- 91.Perry EK, Tomlinson BE, Blessed G, Bergman K, Bigson PH, Perry RH. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Brit Med J. 1978;2:1457–1459. doi: 10.1136/bmj.2.6150.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bowen DM, Spillane JA, Curzon G, Meier-Ruge W, White P, Goodhardt MJ, Iwangoff P, Davison AN. Accelerated ageing or selective neuronal loss as an important cause of dementia? Lancet. 1979;313:11–14. doi: 10.1016/s0140-6736(79)90454-9. [DOI] [PubMed] [Google Scholar]
- 93.Bartus RT, Dean RL, 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–14. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 94.Contestabile A. The history of the cholinergic hypothesis. Behav Brain Res. 2011;221:334–40. doi: 10.1016/j.bbr.2009.12.044. [DOI] [PubMed] [Google Scholar]
- 95.Comfort A. Cholinesterase inhibition in treatment of Alzheimer’s dementia. Lancet. 1978;311:659–60. doi: 10.1016/s0140-6736(78)91161-3. [DOI] [PubMed] [Google Scholar]
- 96.Smith CM, Swash M. Physostigmine in Alzheimer’s disease. Lancet. 1979;313:42. doi: 10.1016/s0140-6736(79)90479-3. [DOI] [PubMed] [Google Scholar]
- 97.Giacobini E. Modulation of brain acetylcholine levels with cholinesterase inhibitors as a treatment of Alzheimer disease. Keio J Med. 1987;36:381–91. doi: 10.2302/kjm.36.381. [DOI] [PubMed] [Google Scholar]
- 98.Jones M, Wang J, Harmon S, Kling B, Heilmann J, Gilmer JF. Novel selective butyrylcholinesterase inhibitors incorporating antioxidant functionalities as potential bimodal therapeutics for Alzheimer’s disease. Molecules. 2016;21:440. doi: 10.3390/molecules21040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ismaili L, Refouvelet B, Benchekroun M, Brogi S, Brindisi M, Gemma S, Campiani G, Filipic S, Agbaba D, Esteban G, Unzeta M, Nikolic K, Butini S, Marco-Contelles J. Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog Neurobiol. 2017;151:4–34. doi: 10.1016/j.pneurobio.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 100.Liu Su J, Guo H, Chen K, Yang L, Chen MQ. Research advances and detection methodologies for microbe-derived acetylcholinesterase inhibitors: a systemic review. Molecules. 2017;22 doi: 10.3390/molecules22010176. pii: E176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li Q, Yang H, Chen Y, Sun H. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur J Med Chem. 2017;132:294–309. doi: 10.1016/j.ejmech.2017.03.062. [DOI] [PubMed] [Google Scholar]
- 102.Shah AA, Dar TA, Dar PA, Ganie SA, Kamal MA. A current perspective on the inhibition of cholinesterase by natural and synthetic inhibitors. Curr Drug Metab. 2017;18:96–111. doi: 10.2174/1389200218666161123122734. [DOI] [PubMed] [Google Scholar]
- 103.Mielke MM, Leoutsakos JM, Corcoran CD, Green RC, Norton MC, Welsh-Bohmer KA, Tschanz JT, Lyketsos CG. Effects of Food and Drug Administration-approved medications for Alzheimer’s disease on clinical progression. Alzheimers Dement. 2012;8:180–7. doi: 10.1016/j.jalz.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tricco AC, Ashoor HM, Soobiah C, Rios P, Veroniki AA, Hamid JS, Ivory JD, Khan PA, Yazdi F, Ghassemi M, Blondal E, Ho JM, Ng CH, Hemmelgarn B, Majumdar SR, Perrier L, Straus SE. Comparative effectiveness and safety of cognitive enhancers for treating Alzheimer’s disease: systematic review and network metaanalysis. J Am Geriatr Soc. 2017 doi: 10.1111/jgs.15069. [DOI] [PubMed] [Google Scholar]
- 105.Ali TB, Schleret TR, Reilly BM, Chen WY, Abagyan R. Adverse effects of cholinesterase inhibitors in dementia, according to the pharmacovigilance databases of the United-States and Canada. PLoS One. 2015;10(12):e0144337. doi: 10.1371/journal.pone.0144337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mohammad D, Chan P, Bradley J, Lanctôt K, Herrmann N. Acetylcholinesterase inhibitors for treating dementia symptoms – a safety evaluation. Expert Opin Drug Saf. 2017;16:1009–1019. doi: 10.1080/14740338.2017.1351540. [DOI] [PubMed] [Google Scholar]
- 107.Pourmand A, Shay C, Redha W, Aalam A, Mazer-Amirshahi M. Cholinergic symptoms and QTc prolongation following donepezil overdose. Am J Emerg Med. 2017;35:1386.e1–1386.e3. doi: 10.1016/j.ajem.2017.06.044. [DOI] [PubMed] [Google Scholar]
- 108.Suzuki Y, Kamijo Y, Yoshizawa T, Fujita Y, Usui K, Kishino T. Acute cholinergic syndrome in a patient with mild Alzheimer’s type dementia who had applied a large number of rivastigmine transdermal patches on her body. Clin Toxicol (Phila) 2017;55:1008–1010. doi: 10.1080/15563650.2017.1329536. [DOI] [PubMed] [Google Scholar]
- 109.Das S, Basu S. Multi-targeting strategies for Alzheimer’s disease therapeutics: pros and cons. Curr Top Med Chem. 2017;17:3017–3061. doi: 10.2174/1568026617666170707130652. [DOI] [PubMed] [Google Scholar]
- 110.Lan JS, Ding Y, Liu Y, Kang P, Hou JW, Zhang XY, Xie SS, Zhang T. Design, synthesis and biological evaluation of novel coumarin-N-benzyl pyridinium hybrids as multi-target agents for the treatment of Alzheimer’s disease. Eur J Med Chem. 2017;139:48–59. doi: 10.1016/j.ejmech.2017.07.055. [DOI] [PubMed] [Google Scholar]
- 111.Ramsay RR, Tipton KF. Assessment of enzyme inhibition: a review with examples from the development of monoamine oxidase and cholinesterase inhibitory drugs. Molecules. 2017;22 doi: 10.3390/molecules22071192. pii: E1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Layer PG, Sporns O. Spatiotemporal relationship of embryonic cholinesterases with cell proliferation in chicken brain and eye. Proc Natl Acad Sci U S A. 1987;84:284–8. doi: 10.1073/pnas.84.1.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Robertson RT. A morphogenic role for transiently expressed acetylcholinesterase in developing thalamocortical systems? Neurosci Lett. 1987;75:259–64. doi: 10.1016/0304-3940(87)90531-3. [DOI] [PubMed] [Google Scholar]
- 114.Layer PG, Willbold E. Cholinesterases in avian neurogenesis. Int Rev Cytol. 1994;151:139–81. doi: 10.1016/s0074-7696(08)62632-7. [DOI] [PubMed] [Google Scholar]
- 115.Layer PG. Cholinesterases preceding major tracts in vertebrate neurogenesis. Bioessays. 1990;12:415–20. doi: 10.1002/bies.950120904. [DOI] [PubMed] [Google Scholar]
- 116.Brimijoin S, Koenigsberger C. Cholinesterases in neural development: new findings and toxicologic implications. Env Health Persp. 1999;107:59–64. doi: 10.1289/ehp.99107s159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Robertson RT, Mostamand F, Kageyama GH, Gallardo KA, Yu J. Primary auditory cortex in the rat: transient expression of acetylcholinesterase activity in developing geniculocortical projections. Brain Res Dev Brain Res. 1991;58:81–95. doi: 10.1016/0165-3806(91)90240-j. [DOI] [PubMed] [Google Scholar]
- 118.Ling JJ, Yu J, Robertson RT. Sustained inhibition of acetylcholinesterase activity does not disrupt early geniculocortical ingrowth to developing rat visual cortex. Brain Res Dev Brain Res. 1995;86:354–8. doi: 10.1016/0165-3806(95)00035-c. [DOI] [PubMed] [Google Scholar]
- 119.Kimm EJ, Perez CE, Yu CC, Yu J, Robertson RT. Reduction of transiently expressed acetylcholinesterase activity in developing thalamocortical projections does not affect the mature pattern of basal forebrain projections to visual cortex. Brain Res Dev Brain Res. 1995;85:283–7. doi: 10.1016/0165-3806(95)00006-y. [DOI] [PubMed] [Google Scholar]
- 120.Yang D, Howard A, Bruun D, Ajua-Alemanj M, Pickart C, Lein PJ. Chlorpyrifos and chlorpyrifos-oxon inhibit axonal growth by interfering with the morphogenic activity of acetylcholinesterase. Toxicol Appl Pharmacol. 2008;228:32–41. doi: 10.1016/j.taap.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Layer PG, Alber R, Sporns O. Quantitative development and molecular forms of acetyl-and butyrylcholinesterase during morphogenesis and synaptogenesis of chick brain and retina. J Neurochem. 1987;49:175–82. doi: 10.1111/j.1471-4159.1987.tb03411.x. [DOI] [PubMed] [Google Scholar]
- 122.Vollmer G, Layer PG. Cholinesterases and cell proliferation in “nonstratified” and “stratified” cell aggregates from chicken retina and tectum. Cell Tissue Res. 1987;250:481–7. doi: 10.1007/BF00218938. [DOI] [PubMed] [Google Scholar]
- 123.Graybiel AM, Ragsdale CW., Jr Pseudocholinesterase staining in the primary visual pathway of the macaque monkey. Nature. 1982;299:439–42. doi: 10.1038/299439a0. [DOI] [PubMed] [Google Scholar]
- 124.Sharma KV, Brimijoin S, Koenigsberger C, Bigbee J. Direct evidence for an adhesive function in the noncholinergic role of acetylcholinesterase in neurite outgrowth. J Neurosci Res. 2001;63:165–175. doi: 10.1002/1097-4547(20010115)63:2<165::AID-JNR1008>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 125.Rees T, Brimijoin S. The role of acetylcholinesterase in the pathogenesis of Alzheimer disease. Drugs of Today. 2003;39:75–83. doi: 10.1358/dot.2003.39.1.740206. [DOI] [PubMed] [Google Scholar]
- 126.Rees T, Hammond PI, Soreq H, Younkin S, Brimijoin S. Acetylcholinesterase promotes beta-amyloid plaques in cerebral cortex. Neurobiol Aging. 2003;24:777–787. doi: 10.1016/s0197-4580(02)00230-0. [DOI] [PubMed] [Google Scholar]
- 127.Hu W, Gray NW, Brimijoin S. Amyloid-beta increases acetylcholinesterase expression in neuroblastoma cells by reducing enzyme degradation. J Neurochem. 2003;86:470–478. doi: 10.1046/j.1471-4159.2003.01855.x. [DOI] [PubMed] [Google Scholar]
- 128.Rees T, Berson A, Sklan EH, Younkin L, Younkin S, Brimijoin S, Soreq H. Memory deficits correlating with acetylcholinesterase splice shift and amyloid burden in doubly transgenic mice. Curr Alzheimer Res. 2005;2:291–300. doi: 10.2174/1567205054367847. [DOI] [PubMed] [Google Scholar]
- 129.Morgan C, Colombres M, Tuliio Nunez M, Inestrosa NC. Structure and function of amyloid in Alzheimer’s disease. Prog Neurobiol. 2004;74:323–349. doi: 10.1016/j.pneurobio.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 130.Diamant S, Podoly E, Friedler A, Ligumsky H, Livnah O, Soreq H. Butyrylcholinesterase attenuates amyloid fibril formation in vitro. Proc Natl Acad Sci U S A. 2006;103:8628–33. doi: 10.1073/pnas.0602922103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dickerson TJ, Beuscher AE, 4th, Rogers CJ, Hixon MS, Yamamoto N, Xu Y, Olson AJ, Janda KD. Discovery of acetylcholinesterase peripheral anionic site ligands through computational refinement of a directed library. Biochemistry. 2005;44:14845–53. doi: 10.1021/bi051613x. [DOI] [PubMed] [Google Scholar]
- 132.De Ferrari GV, Canales MA, Shin I, Weiner LM, Silman I, Inestrosa NC. A structural motif of acetylcholinesterase that promotes amyloid beta-peptide fibril formation. Biochemistry. 2001;40:10447–57. doi: 10.1021/bi0101392. [DOI] [PubMed] [Google Scholar]
- 133.Nordberg A, Svensson AL. Cholinesterase inhibitors in the treatment of Alzheimer’s disease: a comparison of tolerability and pharmacology. Drug Safety. 1998;19:465–480. doi: 10.2165/00002018-199819060-00004. [DOI] [PubMed] [Google Scholar]
- 134.Kryger G, Silman I, Sussman JL. Structure of acetylcholinesterase complexed with E2020 (Aricept): implications for the design of new anti-Alzheimer drugs. Structure. 1999;7:297–307. doi: 10.1016/s0969-2126(99)80040-9. [DOI] [PubMed] [Google Scholar]
- 135.Reid GA, Darvesh S. Butyrylcholinesterase-knockout reduces brain deposition of fibrillar β-amyloid in an Alzheimer mouse model. Neuroscience. 2015;298:424–35. doi: 10.1016/j.neuroscience.2015.04.039. [DOI] [PubMed] [Google Scholar]
- 136.Darvesh S, Reid GA. Reduced fibrillar β-amyloid in subcortical structures in a butyrylcholinesterase-knockout Alzheimer disease mouse model. Chem Biol Interact. 2016;259:307–312. doi: 10.1016/j.cbi.2016.04.022. [DOI] [PubMed] [Google Scholar]
- 137.Darreh-Shori T, Modiri N, Blennow K, Baza S, Kamil C, Ahmed H, Andreasen N, Nordberg A. The apolipoprotein E ε4 allele plays pathological roles in AD through high protein expression and interaction with butyrylcholinesterase. Neurobiol Aging. 2011;32:1236–48. doi: 10.1016/j.neurobiolaging.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 138.Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, Wu WL, Yang B, Huber N, Pasca SP, Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gao Y, Geng L, Chen VP, Brimijoin S. Therapeutic delivery of butyrylcholinesterase by brain-wide viral gene transfer to mice. Molecules. 2017;22 doi: 10.3390/molecules22071145. (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Konrath EL, Passos Cdos S, Klein LC, Jr, Henriques AT. Alkaloids as a source of potential anticholinesterase inhibitors for the treatment of Alzheimer’s disease. J Pharm Pharmacol. 2013;65:1701–25. doi: 10.1111/jphp.12090. [DOI] [PubMed] [Google Scholar]
- 141.Pinho BR, Ferreres F, Valentão P, Andrade PB. Nature as a source of metabolites with cholinesterase-inhibitory activity: an approach to Alzheimer’s disease treatment. J Pharm Pharmacol. 2013;65:1681–700. doi: 10.1111/jphp.12081. [DOI] [PubMed] [Google Scholar]
- 142.Orhan IE. Nature: a substantial source of auspicious substances with acetylcholinesterase inhibitory action. Curr Neuropharmacol. 2013;11:379–87. doi: 10.2174/1570159X11311040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Christison R. On the properties of the ordeal bean of Old Calabar, Western Africa. Edinb Med J. 1855;1:193–204. [PMC free article] [PubMed] [Google Scholar]
- 144.Proudfoot A. The early toxicology of physostigmine: a tale of beans, great men and egos. Toxicol Rev. 2006;25:99–138. doi: 10.2165/00139709-200625020-00004. [DOI] [PubMed] [Google Scholar]
- 145.Kuhr RJ, Dorough HW. Carbamate Insecticides: Chemistry, Biochemistry and Toxicology. CRC Press; Boca Raton, FL: 1976. [Google Scholar]
- 146.Casida JE, Durkin KA. Pesticide chemical research in toxicology: lessons from nature. Chem Res Toxicol. 2017;30:94–104. doi: 10.1021/acs.chemrestox.6b00303. [DOI] [PubMed] [Google Scholar]
- 147.Murphy SD. Pesticides. In: Doull J, Klaassen CD, Amdur MO, editors. Casarett and Doull’s Toxicology. 2nd. Macmillan Publishing Co; New York, NY: 1980. pp. 357–408. [Google Scholar]
- 148.Fukuto TR. Mechanism of action of organophosphorus and carbamate insecticides. Environ Health Perspect. 1990;87:245–54. doi: 10.1289/ehp.9087245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schmaltz F. Neurosciences and research on chemical weapons of mass destruction in Nazi Germany. J Hist Neurosci. 2006;15:186–209. doi: 10.1080/09647040600658229. [DOI] [PubMed] [Google Scholar]
- 150.Tucker JB. War of Nerves: Chemical Warfare from World War I to Al-Queda. Pantheon Books; New York, NY: 2006. p. 479. [Google Scholar]
- 151.Atwood D, Paisley-Jones C. Pesticide industry sales and usage: 2008–2012 market estimates. US Environmental Protection Agency; Washington, DC: 2017. [Google Scholar]
- 152.Quandt SA, Pope CN, Chen H, Summers P, Arcury TA. Longitudinal assessment of blood cholinesterase activities over 2 consecutive years among Latino nonfarmworkers and pesticide-exposed farmworkers in North Carolina. J Occup Environ Med. 2015;57:851–7. doi: 10.1097/JOM.0000000000000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dirnhuber P, French MC, Green DM, Leadbeater L, Stratton JA. The protection of primates against soman poisoning by pretreatment with pyridostigmine. J Pharm Pharmacol. 1979;31:295–299. doi: 10.1111/j.2042-7158.1979.tb13504.x. [DOI] [PubMed] [Google Scholar]
- 154.Berry WK, Davies DR. The use of carbamates and atropine in the protection of animals against poisoning by 1,2,2-trimethylpropyl methylphosphonofluoridate. Biochem Pharmacol. 1970;19:927–34. doi: 10.1016/0006-2952(70)90256-x. [DOI] [PubMed] [Google Scholar]
- 155.Leadbeater L, Inns RH, Rylands JM. Treatment of poisoning by soman. Fund Appl Toxicol. 1985;5:S225–S231. doi: 10.1016/0272-0590(85)90132-0. [DOI] [PubMed] [Google Scholar]
- 156.Miller SA, Blick DW, Kerenyi SZ, Murphy MR. Efficacy of physostigmine as a pretreatment for organophosphate poisoning. Pharmacol Biochem Behav. 1993;44:343–7. doi: 10.1016/0091-3057(93)90472-6. [DOI] [PubMed] [Google Scholar]
- 157.Lavon O, Eisenkraft A, Blanca M, Raveh L, Ramaty E, Krivoy A, Atsmon J, Grauer E, Brandeis R. Is rivastigmine safe as pretreatment against nerve agents poisoning? A pharmacological, physiological and cognitive assessment in healthy young adult volunteers. Neurotoxicology. 2015;49:36–44. doi: 10.1016/j.neuro.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 158.Myhrer T, Aas P. Pretreatment and prophylaxis against nerve agent poisoning: Are undesirable behavioral side effects unavoidable? Neurosci Biobehav Rev. 2016;71:657–670. doi: 10.1016/j.neubiorev.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 159.Research Advisory Committee on Gulf War Veterans’ Illnesses. Gulf War illness and the health of Gulf War veterans: scientific findings and recommendations. U.S. Government Printing Office; Washington, DC: 2008. [Google Scholar]
- 160.Doctor BP, Raveh L, Wolfe AD, Maxwell DM, Ashani Y. Enzymes as pretreatment drugs for organophosphate toxicity. Neurosci Biobehav Rev. 1991;15:123–8. doi: 10.1016/s0149-7634(05)80103-4. [DOI] [PubMed] [Google Scholar]
- 161.Raveh L, Grunwald J, Marcus D, Papier Y, Cohen E, Ashani Y. Human butyrylcholinesterase as a general prophylactic antidote for nerve agent toxicity. In vitro and in vivo quantitative characterization. Biochem Pharmacol. 1993;45:2465–74. doi: 10.1016/0006-2952(93)90228-o. [DOI] [PubMed] [Google Scholar]
- 162.Saxena A, Sun W, Luo C, Doctor BP. Human serum butyrylcholinesterase: in vitro and in vivo stability, pharmacokinetics, and safety in mice. Chem Biol Interact. 2005;157–158:199–203. doi: 10.1016/j.cbi.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 163.Doctor BP, Saxena A. Bioscavengers for the protection of humans against organophosphate toxicity. Chem Biol Interact. 2005;157–158:167–71. doi: 10.1016/j.cbi.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 164.Saxena A, Sun W, Luo C, Myers TM, Koplovitz I, Lenz DE, Doctor BP. Bioscavenger for protection from toxicity of organophosphorus compounds. J Mol Neurosci. 2006;30:145–8. doi: 10.1385/jmn:30:1:145. [DOI] [PubMed] [Google Scholar]
- 165.Saxena A, Sun W, Fedorko JM, Koplovitz I, Doctor BP. Prophylaxis with human serum butyrylcholinesterase protects guinea pigs exposed to multiple lethal doses of soman or VX. Biochem Pharmacol. 2011;81:164–9. doi: 10.1016/j.bcp.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 166.Myers TM, Sun W, Naik RS, Clark MG, Doctor BP, Saxena A. Characterization of human serum butyrylcholinesterase in rhesus monkeys: behavioral and physiological effects. Neurotoxicol Teratol. 2012;34:323–30. doi: 10.1016/j.ntt.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 167.Kronman C, Velan B, Marcus D, Ordentlich A, Reuveny S, Shafferman A. Involvement of oligomerization, N-glycosylation and sialylation in the clearance of cholinesterases from the circulation. Biochem J. 1995;311:959–967. doi: 10.1042/bj3110959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chitlaru T, Kronman C, Zeevi M, Kam M, Harel A, Ordentlich A, Velan B, Shafferman A. Modulation of circulatory residence of recombinant acetylcholinesterase through biochemical or genetic manipulation of sialylation levels. Biochem J. 1998;336:647–58. doi: 10.1042/bj3360647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kronman C, Chitlaru T, Elhanany E, Velan B, Shafferman A. Hierarchy of post-translational modifications involved in the circulatory longevity of glycoproteins. Demonstration of concerted contributions of glycan sialylation and subunit assembly to the pharmacokinetic behavior of bovine acetylcholinesterase. J Biol Chem. 2000;275:29488–29502. doi: 10.1074/jbc.M004298200. [DOI] [PubMed] [Google Scholar]
- 170.Chitlaru T, Kronman C, Velan B, Shafferman A. Effect of human acetylcholinesterase subunit assembly on its circulatory residence. Biochem J. 2001;354:613–625. doi: 10.1042/0264-6021:3540613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Cohen O, Kronman C, Velan B, Shafferman A. Amino acid domains control the circulatory residence time of primate acetylcholinesterases in rhesus macaques (Macaca mulatta) Biochem J. 2004;378:117–28. doi: 10.1042/BJ20031305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ilyushin DG, Smirnov IV, Belogurov AA, Jr, Dyachenko IA, Zharmukhamedova TIu, Novozhilova TI, Bychikhin EA, Serebryakova MV, Kharybin ON, Murashev AN, Anikienko KA, Nikolaev EN, Ponomarenko NA, Genkin DD, Blackburn GM, Masson P, Gabibov AG. Chemical polysialylation of human recombinant butyrylcholinesterase delivers a long-acting bioscavenger for nerve agents in vivo. Proc Natl Acad Sci USA. 2013;110:1243–8. doi: 10.1073/pnas.1211118110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Terekhov SS, Smirnov IV, Shamborant OG, Bobik TV, Ilyushin DG, Murashev AN, Dyachenko IA, Palikov VA, Knorre VD, Belogurov AA, Ponomarenko NA, Kuzina ES, Genkin DD, Masson P, Gabibov AG. Chemical polysialylation and in vivo tetramerization improve pharmacokinetic characteristics of recombinant human butyrylcholinesterase-based bioscavengers. Acta Naturae. 2015;7:136–41. [PMC free article] [PubMed] [Google Scholar]
- 174.Mumtaz S, Bachhawat BK. Conjugation of proteins and enzymes with hydrophilic polymers and their applications. Indian J Biochem Biophys. 1991;28:346–51. [PubMed] [Google Scholar]
- 175.Sharifi J, Khawli LA, Hornick JL, Epstein AL. Improving monoclonal antibody pharmacokinetics via chemical modification. Q J Nucl Med. 1998;42:242–9. [PubMed] [Google Scholar]
- 176.Harris JM, Martin NE, Modi M. Pegylation: a novel process for modifying pharmacokinetics. Clin Pharmacokinet. 2001;40:539–51. doi: 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- 177.Bhadra D, Bhadra S, Jain P, Jain NK. Pegnology: a review of PEG-ylated systems. Pharmazie. 2002;57:5–29. [PubMed] [Google Scholar]
- 178.Chilukuri N, Parikh K, Sun W, Naik R, Tipparaju P, Doctor BP, Saxena A. Polyethylene glycosylation prolongs the circulatory stability of recombinant human butyrylcholinesterase. Chem Biol Interact. 2005;157–158:115–21. doi: 10.1016/j.cbi.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 179.Chilukuri N, Duysen EG, Parikh K, Sun W, Doctor BP, Lockridge O, Saxena A. Adenovirus-mediated gene transfer of human butyrylcholinesterase results in persistent high-level transgene expression in vivo. Chem Biol Interact. 2008;175:327–31. doi: 10.1016/j.cbi.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 180.Sun W, Luo C, Tipparaju P, Doctor BP, Saxena A. Effect of polyethylene glycol conjugation on the circulatory stability of plasma-derived human butyrylcholinesterase in mice. Chem Biol Interact. 2013;203:172–6. doi: 10.1016/j.cbi.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 181.Chilukuri N, Duysen EG, Parikh K, diTargiani R, Doctor BP, Lockridge O, Saxena A. Adenovirus-transduced human butyrylcholinesterase in mouse blood functions as a bioscavenger of chemical warfare nerve agents. Mol Pharmacol. 2009;76:612–7. doi: 10.1124/mol.109.055665. [DOI] [PubMed] [Google Scholar]
- 182.Parikh K, Duysen EG, Snow B, Jensen NS, Manne V, Lockridge O, Chilukuri N. Gene-delivered butyrylcholinesterase is prophylactic against the toxicity of chemical warfare nerve agents and organophosphorus compounds. J Pharmacol Exp Ther. 2011;337:92–101. doi: 10.1124/jpet.110.175646. [DOI] [PubMed] [Google Scholar]
- 183.Gaydess A, Duysen E, Li Y, Gilman V, Kabanov A, Lockridge O, Bronich T. Visualization of exogenous delivery of nanoformulated butyrylcholinesterase to the central nervous system. Chem Biol Interact. 2010;187:295–8. doi: 10.1016/j.cbi.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pope C, Uchea C, Flynn N, Poindexter K, Geng L, Brimijoin WS, Hartson S, Ranjan A, Ramsey JD, Liu J. In vitro characterization of cationic copolymer-complexed recombinant human butyrylcholinesterase. Biochem Pharmacol. 2015;98:531–9. doi: 10.1016/j.bcp.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 185.Hester K, Liu J, Flynn N, Sultatos LG, Geng L, Brimijoin S, Ramsey JD, Hartson S, Ranjan A, Pope C. Polyionic complexes of butyrylcholinesterase and poly-l-lysine-g-poly(ethylene glycol): Comparative kinetics of catalysis and inhibition and in vitro inactivation by proteases and heat. Chem Biol Interact. 2017;275:86–94. doi: 10.1016/j.cbi.2017.07.019. [DOI] [PubMed] [Google Scholar]
- 186.Waiskopf N, Shweky I, Lieberman I, Banin U, Soreq H. Quantum dot labeling of butyrylcholinesterase maintains substrate and inhibitor interactions and cell adherence features. ACS Chem Neurosci. 2011;2:141–50. doi: 10.1021/cn1000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sokolov OI, Selivanov NY, Bogatyrev VA, Selivanova OG, Velikorodnaya YI, Pocheptsov AY, Filatov BN, Shchyogolev SY, Dykman LA. Synthesis and study on activity in vitro of the high purity human butyrylcholinesterase conjugated with gold nanoparticles. Dokl Biochem Biophys. 2016;468:232–4. doi: 10.1134/S1607672916030212. [DOI] [PubMed] [Google Scholar]
- 188.Zhang P, Jain P, Tsao C, Sinclair A, Sun F, Hung HC, Bai T, Wu K, Jiang S. Butyrylcholinesterase nanocapsule as a long circulating bioscavenger with reduced immune response. J Control Release. 2016;230:73–8. doi: 10.1016/j.jconrel.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 189.Rahhal TB, Fromen CA, Wilson EM, Kai MP, Shen TW, Luft JC, DeSimone JM. Pulmonary delivery of butyrylcholinesterase as a model protein to the lung. Mol Pharmacol. 2016;13:1626–35. doi: 10.1021/acs.molpharmaceut.6b00066. [DOI] [PubMed] [Google Scholar]
- 190.Gatley SJ. Activities of the enantiomers of cocaine and some related compounds as substrates and inhibitors of plasma butyrylcholinesterase. Biochem Pharmacol. 1991;41:1249–54. doi: 10.1016/0006-2952(91)90665-r. [DOI] [PubMed] [Google Scholar]
- 191.Chen X, Huang X, Geng L, Xue L, Hou S, Zheng X, Brimijoin S, Zheng F, Zhan CG. Kinetic characterization of a cocaine hydrolase engineered from mouse butyrylcholinesterase. Biochem J. 2015;466:243–251. doi: 10.1042/BJ20141266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fang L, Hou S, Xue L, Zheng F, Zhan CG. Amino-acid mutations to extend the biological half-life of a therapeutically valuable mutant of human butyrylcholinesterase. Chem Biol Interact. 2014;214:18–25. doi: 10.1016/j.cbi.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Meyer RM, Burgos-Robles A, Liu E, Correia SS, Goosens KA. A ghrelin-growth hormone axis drives stress-induced vulnerability to enhanced fear. Mol Psychiatry. 2014;19:1284–94. doi: 10.1038/mp.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Harmatz ES, Stone L, Lim SH, Lee G, McGrath A, Gisabella B, Peng X, Kosoy E, Yao J, Liu E, Machado NJ, Weiner VS, Slocum W, Cunha RA, Goosens KA. Central ghrelin resistance permits the overconsolidation of fear memory. Biol Psychiatry. 2016;81:1003–1013. doi: 10.1016/j.biopsych.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]