

Abstract
Vascular smooth muscle (VSM) plays an important role in the regulation of vascular function. Identifying the mechanisms of VSM contraction has been a major research goal in order to determine the causes of vascular dysfunction and exaggerated vasoconstriction in vascular disease. Major discoveries over several decades have helped to better understand the mechanisms of VSM contraction. Ca2+ has been established as a major regulator of VSM contraction, and its sources, cytosolic levels, homeostatic mechanisms and subcellular distribution have been defined. Biochemical studies have also suggested that stimulation of Gq protein-coupled membrane receptors activates phospholipase C and promotes the hydrolysis of membrane phospholipids into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 stimulates initial Ca2+ release from the sarcoplasmic reticulum, and is buttressed by Ca2+ influx through voltage-dependent, receptor-operated, transient receptor potential and store-operated channels. In order to prevent large increases in cytosolic Ca2+ concentration ([Ca2+]c), Ca2+ removal mechanisms promote Ca2+ extrusion via the plasmalemmal Ca2+ pump and Na+/Ca2+ exchanger, and Ca2+ uptake by the sarcoplasmic reticulum and mitochondria, and the coordinated activities of these Ca2+ handling mechanisms help to create subplasmalemmal Ca2+ domains. Threshold increases in [Ca2+]c form a Ca2+-calmodulin complex, which activates myosin light chain (MLC) kinase, and causes MLC phosphorylation, actin–myosin interaction, and VSM contraction. Dissociations in the relationships between [Ca2+]c, MLC phosphorylation, and force have suggested additional Ca2+ sensitization mechanisms. DAG activates protein kinase C (PKC) isoforms, which directly or indirectly via mitogen-activated protein kinase phosphorylate the actin-binding proteins calponin and caldesmon and thereby enhance the myofilaments force sensitivity to Ca2+. PKC-mediated phosphorylation of PKC-potentiated phosphatase inhibitor protein-17 (CPI-17), and RhoA-mediated activation of Rho-kinase (ROCK) inhibit MLC phosphatase and in turn increase MLC phosphorylation and VSM contraction. Abnormalities in the Ca2+ handling mechanisms and PKC and ROCK activity have been associated with vascular dysfunction in multiple vascular disorders. Modulators of [Ca2+]c, PKC and ROCK activity could be useful in mitigating the increased vasoconstriction associated with vascular disease.
Keywords: blood vessels, calcium, channels, protein kinase, sarcoplasmic reticulum, signaling
Graphical Abstract
1. Introduction
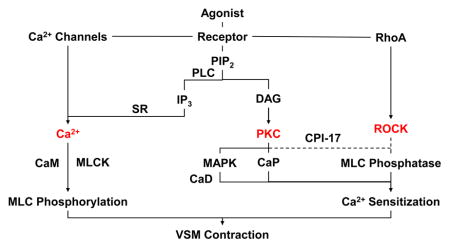
Vascular smooth muscle (VSM) is a major component of the tunica media of blood vessels, and an important regulator of vascular function. VSM contraction plays an important role in the regulation of peripheral vascular resistance and blood pressure, and vascular dysfunction, excessive vasoconstriction, and vasospasm could lead to major cardiovascular disorders such as hypertension and coronary artery disease. Over the past decades important studies and major discoveries have helped to better understand the mechanisms of VSM contraction. Under physiological conditions, agonist activation of VSM causes an initial contraction followed by a tonic contraction that can be maintained with minimal energy expenditure. Ca2+-dependent myosin light chain (MLC) phosphorylation and subsequent formation of crossbridges between actin and myosin have been recognized as a major mechanism of VSM contraction. Various sources of intracellular Ca2+ and both Ca2+ mobilization and Ca2+ removal mechanisms have been identified. VSM contraction is triggered by an increase in cytosolic free Ca2+ concentration ([Ca2+]c) due to Ca2+ release from the intracellular stores in the sarcoplasmic reticulum (SR) and Ca2+ influx from the extracellular space through plasma membrane Ca2+ channels [1, 2]. The Ca2+ concentration is several-fold higher in SR and the extracellular space than in the cytosol, and the opening of Ca2+ channels in SR or cell surface membrane causes Ca2+ mobilization into the cytosol and increases [Ca2+]c. Ca2+ then binds calmodulin (CaM) to form a Ca2+–CaM complex, which activates MLC kinase and causes MLC phosphorylation, actin–myosin interaction, and VSM contraction (Fig. 1). VSM relaxation is initiated by a decrease in [Ca2+]c due to Ca2+ uptake by SR Ca2+ pump and Ca2+ extrusion via the plasmalemmal Ca2+ pump and Na+–Ca2+ exchanger. The decrease in [Ca2+]c causes dissociation of the Ca2+–CaM complex, and the phosphorylated MLC is dephosphorylated by MLC phosphatase [1, 2]. However, dissociations in the relationships between [Ca2+]c, MLC phosphorylation and force have been observed, and Ca2+-dependent MLC phosphorylation could not explain all modalities of VSM contraction. That prompted the development of better techniques to measure [Ca2+]c and further research into its intracellular distribution and subcellular domains. Several bioluminescent and fluorescent probes have been developed for accurate measurements of [Ca2+]c, but have shown different Ca2+ sensitivities. Also, the previously thought uniformity of intracellular Ca2+ has been challenged by the discovery of uneven intracellular distribution of Ca2+ in different subcellular domains, and nanojunctions between SR, the plasma membrane and other cell organelles [3, 4]. Other mechanisms of VSM contraction have also been proposed. Activation of protein kinase C (PKC) has been suggested to increase the myofilament force sensitivity to [Ca2+]c and MLC phosphorylation, and thereby maintain VSM contraction with smaller increases in [Ca2+]c. PKC is now recognized as a family of various Ca2+-dependent and Ca2+-independent isoforms with different tissue and subcellular distribution, substrates and function. PKC translocation to the cell surface may trigger a cascade of protein kinases that ultimately interact with the contractile myofilaments and cause VSM contraction. Additional signaling pathways involving the small GTP-binding protein RhoA, RhoA-mediated increase in Rho-kinase (ROCK) activity, inhibition of MLC phosphatase and increased MLC phosphorylation and the myofilament force sensitivity to Ca2+ have also been proposed. In this review, we will discuss how the role of these Ca2+-dependent and Ca2+-sensitization pathways has evolved to better understand the mechanisms underlying the development and maintenance of VSM contraction [5–7]. We will also discuss how understanding the mechanisms of VSM contraction has helped to understand the pathogenesis of vascular disorders, and how modulators of Ca2+-dependent and Ca2+-sensitization pathways of VSM contraction could provide potential tools in the management of vascular disease.
Fig. 1.

Mechanisms of VSM contraction. A vasoconstrictor agonist (A) binding to its receptor (R) is coupled to heterotrimeric GTP-binding protein (Gq) and activates phospholipase C (PLCβ) which stimulates the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). The agonist also activates phospholipase D (PLD) which hydrolyzes phosphatidylcholine (PC) into choline and DAG. IP3 stimulates Ca2+ release from sarcoplasmic reticulum (SR). The agonist also stimulates Ca2+ influx through Ca2+ channels. Ca2+ binds calmodulin (CaM), activates MLC kinase (MLCK), causes MLC phosphorylation, and initiates VSM contraction. DAG, phosphatidylserine (PS), and Ca2+ (for cPKCs) cause activation and translocation of PKC. PKC inhibits K+ channels leading to membrane depolarization and activation of voltage-dependent Ca2+ channels. PKC phosphorylates CPI-17, which in turn inhibits MLC phosphatase and enhances the myofilament force sensitivity to Ca2+. PKC phosphorylates calponin (CaP), allowing more actin to bind myosin. PKC may activate a protein kinase cascade involving Raf, MAPK kinase (MEK), and MAPK (ERK1/2), leading to phosphorylation of the actin-binding protein caldesmon (CaD). DAG is transformed by DAG lipase into arachidonic acid (AA), and activation of phospholipase A2 (PLA2) increases the hydrolysis of phosphatidylethanolamine (PE) into AA, which in turn inhibits MLC phosphatase. Agonist-induced activation of RhoA/ROCK also inhibits MLC phosphatase and further enhances Ca2+ sensitivity of the contractile proteins. Dashed line indicates inhibition.
2. Ca2+ Mobilization Mechanisms
The role of Ca2+ in muscle function was first suggested in 1883, when Ringer observed that Ca2+ was necessary for maintaining the activity of the isolated heart [8]. Seven decades later, Heilbrunn and colleges supported the role of intracellular Ca2+ in muscle contraction [9]. The sources of intracellular Ca2+ have later been identified as Ca2+ release from intracellular Ca2+ stores and Ca2+ influx from the extracellular space. Advances in electrophysiology and voltage-clamp techniques provided evidence that the Ca2+ channel is a physiologically distinct entity that plays an important role in excitation-contraction coupling [10–12]. Further methodological advances and tight-seal single channel measurements led to the recording of Ca2+ movement through single Ca2+ channel in cardiac cells [13]. In the 1980s, the field of Ca2+ channels rapidly expanded with the discovery of multiple types of Ca2+ channels with different biophysical properties, and the molecular purification of the channels and characterization of their structure, function, activators and inhibitors.
3. Ca2+ Release from SR
Ca2+ release from the intracellular stores contributes to agonist-induced VSM contraction [1, 2]. In the absence of extracellular Ca2+, agonists often produce a transient VSM contraction [1, 2]. Also, in vascular preparations pretreated with Ca2+ channel blockers the maintained agonist-induced contraction and 45Ca2+ influx are inhibited substantially, but a smaller transient contraction can still be observed [1, 14, 15]. Also, in 45Ca2+ loaded vascular preparations and incubated in a Ca2+-free medium, agonists stimulate Ca2+ efflux [16].
Ultrastructure studies and electron probe X-ray microanalysis in smooth muscle revealed structures consistent with the SR that can accumulate Ca2+ from solutions containing micromolar Ca2+ concentrations [17]. The SR is an intracellular system of tubules or flattened cisternae [17, 18], that occupy 1.5% to 7.5% of the smooth muscle cell volume [19]. In large elastic arteries such as the rabbit aorta and main pulmonary artery, the SR occupies a larger volume, and therefore these vessels elicit a large contraction in Ca2+-free solution. In contrast, in phasic smooth muscle preparations such as the rabbit mesenteric vein and guinea pig taenia coli, the SR occupies 1.5 to 2.5% of the cell volume [20], and therefore these preparations show very small contraction in the absence of extracellular Ca2+.
Using advanced fractionation techniques, the SR has been isolated as a microsomal fraction. Isolated smooth muscle SR microsomes accumulate 45Ca2+ and release it in response to Ca2+-releasing agents such as caffeine and ryanodine. Also, Ca2+ release channels have been identified in SR vesicles planted in planar lipid bilayer [21]. Studies in smooth muscle preparations chemically permeabilized by saponin or α-toxin have avoided the loss of essential cellular components that occur during isolation and purification of SR vesicles, and thereby helped to assess the Ca2+ release mechanism under more physiological conditions [18, 22–25]. Ca2+ release from the SR can be triggered by inositol 1,4,5-trisphosphate (IP3) or by Ca2+.
3.1. IP3-Induced Ca2+ Release
Agonist–receptor interaction activates membrane-associated phospholipase C (PLC), which breaks down the plasma membrane phosphatidylinositol 4,5-bisphosphate (PIP2) into IP3 and 1,2-diacylglycerol (DAG) [26]. Because IP3 is water-soluble, it diffuses in the cytosol and stimulates Ca2+ release from SR [27–30] (Fig. 2). On the other hand, DAG is lipophilic and therefore remains in the plasma membrane where it activates PKC [6, 31]. In saponin-skinned smooth muscle cells IP3 induces large and rapid Ca2+ release. IP3 has a half maximal effective concentration (EC50) of ~1 μM which is low enough to account for the transient smooth muscle contraction [27, 29, 32]. Also, in accordance with the criteria of a second messenger, endogenous IP3-specific 5-phosphatase activity, that rapidly inactivates IP3 and converts it to inositol 1,4-bisphosphate (IP2) [26], has been identified in smooth muscle [33]. IP3 binds to IP3 receptor and activates Ca2+ release channels in SR. Heparin, through its electronegative charge, competes with IP3 and blocks IP3 receptor and IP3-induced Ca2+ release from SR [34]. Deletion of IP3 receptor in mice suppresses aortic contraction to the vasoconstrictor agonists phenylephrine, U46619, serotonin, and endothelin-1, reduces U46619-induced phosphorylation of MLC-20 and myosin phosphatase target subunit 1 (MYPT1), and attenuates the pressor response to chronic infusion of angiotensin II (AngII), supporting a role of IP3 receptor-mediated Ca2+ release in regulating VSM contraction and blood pressure [35]. IP3 also binds and activates plasmalemmal transient receptor potential-3 (TRPC3) channels and in turn promotes Ca2+ influx in airway smooth muscle cells [36].
Fig. 2.

Ca2+ mobilization and Ca2+ removal mechanisms in VSM. Agonist (A)-receptor (R) interaction causes Ca2+ release from sarcoplasmic reticulum (SR) in response to 1,4,5-inositol trisphosphate (IP3) and to Ca2+ via Ca2+-induced Ca2+ release (CICR). VSMC activation also stimulates Ca2+ influx through nonspecific Ca2+ leak, voltage-dependent Ca2+ channels (VDCC), receptor-operated channels (ROC), transient receptor potential (TRP) channels, and stretch-activated channels. Depletion of intracellular Ca2+ stores in SR causes the release of stromal interaction molecule (STIM1) which in turn stimulates Orai1 store-operated Ca2+ channels. The increased intracellular Ca2+ is taken up by SR Ca2+-ATPase (SERCA) or extruded by the plasmalemmal Ca2+-ATPase (PMCA) or Na+-Ca2+ exchanger (NCX), and the resulting excess Na+ is extruded via Na+/K+ pump and Na+/H+ exchanger. At very high and pathological increases in intracellular Ca2+, the mitochondria play a role in Ca2+ uptake and homeostasis. When Ca2+ is taken up by mitochondria, HPO42− is also taken up via HPO42−:2OH− exchange and calcium phosphate is formed. Under favorable conditions and when Ca2+ can be handled by SERCA, PMCA and NCX, mitochondrial Ca2+ is slowly released via a Ca2+ efflux pathway involving a Ca2+:2H+ or Ca2+:2Na+ antiporter. PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; DAG, diacyglycerol
3.2. Ca2+-Induced Ca2+ Release
Studies in skinned skeletal [18, 37], cardiac [38], and VSM [22] have shown that small concentrations of Ca2+ induce additional release of Ca2+ from SR. Ca2+-induced Ca2+ release (CICR) is a regenerative process that can be facilitated by Ca2+-releasing drugs such as caffeine and ryanodine [18]. A threshold 3 μM increase in Ca2+ concentration near SR. is required to trigger CICR. CICR is augmented by 3′,5′-cyclic adenosine monophosphate (cAMP) and inhibited by Mg2+ and procaine [22]. An initial IP3-induced Ca2+ release raises Ca2+ concentration near SR above the 3 μM threshold, and in turn stimulates additional Ca2+ release through CICR channels [39]. This Ca2+ release amplification mechanism is supported by the observation that Ca2+ enhances IP3-induced Ca2+ release from SR in skinned smooth muscle of guinea-pig taenia caeci [32]. Also, studies using calsequestrin-targeted Ca2+ indicator have shown that endothelin-1 (ET-1) stimulates waves of Ca2+ depletion from VSM SR. A transient elevation in SR luminal Ca2+ concentration was observed both at the site of wave initiation, just before regenerative Ca2+ release commences, and at the advancing wave front, during propagation. These observations suggest a role for SR luminal Ca2+ in the activation of IP3 receptor during agonist-induced Ca2+ waves, and that these waves are due to regenerative CICR by the IP3 receptor [40]
4. Ca2+ Influx from the Extracellular Space
Ca2+ enters VSM through non-specific Ca2+ leak and more selective channels including voltage-dependent, receptor-operated, transient receptor potential (TRP), store-operated, and stretch-activated Ca2+ channels (Fig. 2).
4.1. Ca2+ Leak
Because of the high electrochemical Ca2+ gradient across the plasma membrane, Ca2+ enters continuously into the resting VSMCs through Ca2+ leak. The Ca2+ leak pathway is lined with phosphate and carboxyl groups, partially blocked by low pH and high H+ concentration, and blocked by ~66% by cobalt or lanthanum [1]. While Ca2+ leak is thought to involve non-specific Ca2+ movement across the plasma membrane, electrophysiological studies have suggested that a divalent cation-selective channel that displays occasional spontaneous openings contributes to Ca2+ leak [41]. The Ca2+ leak channel opens at holding potentials below the threshold for activation of voltage-dependent Ca2+ channel and has a higher conductance than the adenosine triphosphate (ATP)-sensitive Ca2+ channel, a receptor-operated Ca2+ channel. In rabbit aorta under resting conditions, the 45Ca2+ leak amounts to ~14 μmole/kg/min [2]. This large Ca2+ leak does not cause VSM contraction because it is constantly balanced by Ca2+ uptake by SR and Ca2+ extrusion by the plasmalemmal Ca2+ pump. However, in conditions associated with compromised Ca2+ removal mechanisms or increased myofilament force sensitivity to Ca2+, the Ca2+ leak could cause VSM contraction.
4.2. Voltage-Dependent Ca2+ Channels
Extracellular Ca2+ is necessary for maintained contraction in most blood vessels [1]. In rabbit aorta incubated in the absence of extracellular Ca2+, contraction to membrane depolarization by high KCl solution is abolished, and norepinephrine-induced contraction is inhibited substantially. High KCl stimulates 45Ca2+ influx that is sensitive to organic Ca2+ antagonists such as dihydropyridines [14], and Ca2+ antagonist-induced blockade of 45Ca2+ influx is associated with inhibition of vascular contraction [1]. Also, the Ca2+ channel agonist Bay-K8644 stimulates Ca2+ influx and promotes vascular contraction. These observations have suggested a distinct plasma membrane Ca2+ entry pathway that is activated by membrane depolarization, and has been termed voltage-dependent Ca2+ channels (VDCCs) [42–44]. Voltage-clamp and patch-clamp studies have identified two components of voltage-activated Ca2+ current, long-lasting L-type current activated by relatively large depolarizations and inactivates relatively slowly, and transient T-type current activated by relatively small depolarizations and inactivates relatively rapidly [45]. Both L and T Ca2+ currents are blocked by cadmium, cobalt and lanthanum [46–49], but show different sensitivities to dihydropyridines. While the L current is blocked by nifedipine, nimodipine, nisoldipine and nitrendipine and augmented by Bay-K8644 and Bay-R5417, the T current is not affected by these dihydropyridines [45, 46, 48]. Also, while physiological agonists are often thought to not stimulate voltage-activated Ca2+ current [45, 46, 48], norepinephrine, acting via a non-α non-β receptor, stimulates the L-type but not T-type current in rabbit ear artery [50], and increases the open probability of VDCCs in rabbit mesenteric artery [44].
In 1990, the vascular L-type CaV1.2 channel (LTCC) was first sequenced from rabbit lungs and showed 65% amino acid sequence homology with its skeletal muscle counterpart [51]. LTCC is comprised of pore-forming α1c and auxiliary β, α2δ, and γ subunits that modulate the channel function [52]. The α1c contains the voltage sensor, gating system, and the Ca2+-permeable pore and comprises four homologous I, II, III, IV domains, each of which is composed of six transmembrane S1–S6 segments and intracellular NH2- and COOH-termini. The S5 and S6 segments of each of the homologous domains form the channel pore, two glutamate residues at the pore loop determine the Ca2+ selectivity, and the S1–S4 segments form the voltage sensor that rotates to open the channel pore [52, 53]. CaV1.2 function is prominent at more depolarized VSM membrane potentials (~ −45 to −36 mV) observed at greater intraluminal vascular pressures [54],
T-type Ca2+ channels (TTCCs) were first identified as a separate VDCC in guinea pig ventricular myocytes having a transient conductance of ~8 pS with Ba2+ as the charge carrier. T-type currents are activated at membrane potentials ~−30 mV, and dihydropyridines at nanomolar range have little effects on TTCCs. TTCCs have three isoforms; CaV3.1, CaV3.2 and CaV3.3 channels. CaV3.1 and CaV3.3 predominantly contribute to myogenic tone at lower intraluminal vascular pressures (20–40 mmHg) at which VSM membrane potential is ~ −60 to−50 mV [55]. CaV3.2 contributes to negative feedback regulation of pressure-induced vascular tone by modulating the ryanodine receptor-large conductance Ca2+ activated K+ channel (BKCa) axis [56].
Ca2+-dependent inactivation of LTCC plays a crucial feed-back role in limiting increases in [Ca2+]c, likely through binding of the Ca2+/CaM complex to the C-terminus of the pore-forming α1c subunit. Studies have also examined the biophysical properties of Ca2+ current through the three TTCC isoforms, Cav3.1, Cav3.2, and Cav3.3 using whole cell patch clamp and internal solutions containing 27 nM or l μM [Ca2+]c. Both activation and inactivation kinetics of Cav3.3 current were more rapid at l μM [Ca2+]c than 27 nM [Ca2+]c solution. In contrast, the biophysical properties of Cav3.1 and Cav3.2 isoforms were not different between the two [Ca2+]c. Overexpression of CaM1234, a calmodulin mutant that doesn’t bind Ca2+, prevented the effects of l μM [Ca2+]c on Cav3.3. Yeast two-hybrid screening and co-immunoprecipitation revealed direct interaction of CaM with the carboxyl terminus of Cav3.3. These findings have suggested that T-type Cav3.3 channel is also regulated by [Ca2+]c via interaction of Ca2+/CaM with the carboxyl terminus of Cav3.3, and represents another negative feedback mechanism restricting excessive increases in Ca2+ entry through VDCCs. [57].
4.3. Receptor-Operated Ca2+ Channels
Physiological agonists activate other Ca2+ entry pathways separate from those activated by membrane depolarization. Norepinephrine causes further contraction in rabbit aorta maximally activated by high KCl depolarizing solution. 45Ca2+ influx induced by combined stimulation with maximal concentrations of norepinephrine and KCl equals the sum of that stimulated by each one alone, suggesting that norepinephrine and KCl-induced Ca2+ influx are additive [1, 58]. 45Ca2+ influx stimulated by the Ca2+ channel agonist Bay-K8644 is additive to that induced by maximal norepinephrine concentration, but not KCl. Also, while high KCl-induced VSM contraction and Ca2+ influx are sensitive to Ca2+ channel antagonists, norepinephrine-induced VSM contraction and Ca2+ influx are refractory to organic Ca2+ antagonists [1, 58]. These observations have suggested that receptor stimulation by physiological agonists activate Ca2+ channels that are different from those activated by membrane depolarization, and have been termed receptor-operated Ca2+ channels (ROCs) [42, 43]. Electrophysiological studies provided direct evidence for ROCs and showed that ATP activates a distinct Ca2+ current in rabbit ear artery VSM [41]. The ATP-sensitive channel displays a 3:1 selectivity for Ca2+ over Na+ at near physiological ionic conditions and can be distinguished from VDCCs by its insensitivity to nifedipine or cadmium, its opening at high negative potentials, and its unitary conductance of ~5 pS in 110 mM Ca2+ or Ba2+. Also, the channel is not activated when ATP is added outside the cell-attached patch pipette, suggesting that it is directly coupled to receptor activation by ATP rather than ATP-induced generation of a freely diffusible messenger [59].
The term ROCs is not commonly used now, and is often referred to as a type of transient receptor potential (TRP) channels [60].
4.4. Transient Receptor Potential (TRP) Channels
TRP channels are a superfamily of cationic channels with 28 encoding genes. Based on their sequence homology, TRP channels have been categorized into six subfamilies; TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPA (ankyrin), and TRPML (mucolipin). Multiple TRP channels are expressed in VSM and contribute to the regulation of VSM membrane potential and contraction, and the development of myogenic tone. Additionally, certain TRP channels contribute to vascular mechanosensitivity via G-protein coupled signaling in resistance arteries. While most TRP channels are permeable to Ca2+, TRPM4 and TRPM5 are Ca2+ activated, but not permeable to Ca2+ [61].
Vasoconstrictor agonists stimulate Ca2+ entry through VDCCs activated by membrane depolarization, and non-selective cation channels, most of them members of the TRPC channels family. TRPC channels are activated following receptor occupancy (ROCs) or secondary to internal Ca2+ stores depletion that induces capacitative Ca2+ entry (store-operated cation channels or SOCs). TRPCs simultaneously induce Na+ and Ca2+ entry thus triggering cell membrane depolarization and increasing [Ca2+]c [60, 62]. With the exception of TRPC2 and TRPC7, all other TRPC isoforms are found in VSM at varying levels depending on the vessel type. TRPC1 and TRPC6 are highly expressed in VSM. TRPC4 is detected at a lower level than TRPC1 and TRPC6 in rat aorta, cerebral, mesenteric, and renal artery, and is not detected in caudal artery. TRPC3 level is higher in rat cerebral, renal, and caudal artery than in the aorta. TRPC5 shows a slight signal in rat aorta and renal artery, but is not detected in mesenteric artery [60].
4.5. Store-Operated Ca2+ Channels
During cell activation, the initial Ca2+ release from the intracellular stores is followed by maintained Ca2+ entry from the extracellular space. Depleted Ca2+ stores in sarcoplasmic/endoplasmic reticulum could act as a capacitor for “capacitative” or “store-operated” Ca2+ entry [63–65]. Studies have identified store-operated Ca2+ release-activated Ca2+ current [66, 67]. The functional significance of store-operated Ca2+ channels (SOCs) has been supported by experiments using inhibitors of the sarcoplasmic/endoplasmic reticulum Ca2+-adenosine triphosphatase (Ca2+-ATPase) (SERCA) such as cyclopiazonic acid and thapsigargin. These compounds deplete SR Ca2+ stores by inhibiting Ca2+ uptake without activating guanosine triphosphate (GTP)-binding proteins and thereby differentiate between Ca2+ entering through SOCs and ROCs. In cultured VSMCs, depletion of SR Ca2+ stores with thapsigargin activates Ca2+ influx that is independent of the generation of IP3 and resistant to the L-type VDCC blocker nicardipine [68]. Ca2+ influx induced by SERCA inhibitors is dependent on extracellular Ca2+ and sufficient to maintain vascular tone [69, 70].
Members of the canonical TRPCs such as TRPC1 and TRPC5 play a role in store-operated Ca2+ entry in VSM [71–74]. TRPC1 is linked to TRPP2 (polycystin-2) Ca2+ permeable channel [75] and TRPC5 represents another component of SOCCs [74]. Other members of the TRPC family, including TRPC3, TRPC4, and TRPC7, have been associated with store-operated Ca2+ entry in nonvascular cells [76, 77].
In mouse aortic VSMCs, depletion of Ca2+ stores triggers the release of a Ca2+ influx factor (CIF), which activates SOCCs [78]. Other studies have identified a 3-pS Ca2+-conducting channel that is activated by 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) and thapsigargin. The 3-pS channel is also activated in inside-out membrane patches from smooth muscle cells when stimulated by CIF extracted from mutant yeast cell line [79]. CIF has been partly purified in a stable form, but its molecular structure has not been well-characterized [80, 81]. A membrane-spanning protein termed stromal-interacting molecule 1 (STIM1) also plays a role in the activation of SOCCs. STIM1 serves as a sensor of Ca2+ within the stores, interacts with TRPC1 and promotes store-operated Ca2+ entry [82, 83]. Other studies have suggested that Orai1 is a pore subunit of SOCCs [84]. Studies have suggested an interaction between STIM1 and Orai1 that leads to a gain in SOCC function [85, 86]. STIM1 senses depletion of intracellular Ca2+ stores in response to physiological stimuli, and relocalizes within the sarcoplasmic/endoplasmic reticulum to plasma-membrane-apposed junctions, where it recruits and gates open plasma membrane Orai1 Ca2+ channels. Septins are cytoskeletal proteins capable of self-association, polymerization and binding to cell membranes [87]. Septin has been suggested as a potential coordinator of store-operated Ca2+ entry [88]. Septin filaments and phosphatidylinositol-4,5-bisphosphate rearrange locally at the endoplasmic reticulum-plasma membrane junction before and during formation of STIM1-Orai1 clusters, thus facilitating STIM1 targeting to these junctions and promoting stable recruitment of Orai1, and efficient STIM1-Orai1 communication and Ca2+ entry [89].
4.6. Stretch-Activated Ca2+ Channels
During “autoregulation” of blood flow, an elevation of intravascular pressure and stretch of the vascular wall can cause maintained increase in VSM tone [90]. Stretch-stimulated vascular tone is highly dependent on extracellular Ca2+ through stretch-activated Ca2+ channel [91]. Stretch-activated Ca2+ channels differ from VDCCs and ROCCs in their sensitivity to Ca2+ antagonists, being more sensitive to diltiazem but insensitive to dihydropyridines. Mechanical stretch stimulates 45Ca2+ influx in smooth muscle membranes [92]. A role of the endothelium in myogenic vascular response to stretch has also been suggested [93]. In cannulated cat cerebral arteries with intact endothelium, elevation of transmural pressure was associated with membrane depolarization, action potential generation, and reduction in internal diameter. Perfusing the vessels briefly with collagenase and elastase to disrupt the endothelium without damaging the smooth muscle cells, abolished the responses to elevation of transmural pressure, suggesting that endothelial cells serve as a transducer in the vascular autoregulatory response to pressure [93].
TRP vanilloid type 2 (TRPV2) is a Ca2+ permeable stretch-activated channel [94]. TRPV4 is also stimulated by mechanical stress, including sheer stress and cell swelling, and has been implicated in regulation of myogenic tone [52]. TRPC6 channels are activated by mechanosensation such as sheer-stress and cell swelling and promote Ca2+ entry into VSM and vasoconstriction [95]. TRPP are other mechanosensitive Ca2+-permeable channels [61]
5. Mechanisms of Ca2+ Removal
In addition to their role in Ca2+ mobilization, the smooth muscle plasma membrane and intracellular organelles play a role in maintaining Ca2+ in. The plasmalemmal Ca2+-Ca2+-ATPase (PMCA) plays a role in maintaining [Ca2+]c close to the basal levels, and the Na+–Ca2+ exchanger contributes to removal of excess cytosolic Ca2+ (Fig. 2). Also, two intracellular organelles, namely the SR and mitochondria, regulate [Ca2+]c. These organelles have pump-leak system that involves active uptake of Ca2+ from the cytosol and passive leak of Ca2+ back to the cytosol.
5.1. Plasmalemmal Ca2+-ATPase (PMCA)
Metabolic inhibition of the smooth muscle of guinea pig taenia coli using iodoacetic acid or 2,4-dinitrophenol causes a net Ca2+ uptake similar in magnitude to the passive Ca2+ leak [96, 97]. These observations have suggested that an ATP-dependent Ca2+ extrusion pump contributes to smooth muscle Ca2+ homeostasis and that inhibition of the ATP-dependent Ca2+ pump causes accumulation of Ca2+ inside the cell [98]. The smooth muscle plasmalemmal Ca2+ pump [99] shares some properties of the better studied Ca2+ pump in the squid axon and red blood cells. The Ca2+ pump has a molecular weight of 130 kDa, and is stimulated by CaM and inhibited by vanadate. Vanadate causes maximal VSM contraction, suggesting that the plasmalemmal Ca2+ pump plays a major role in the regulation of [Ca2+]j and vascular tone [100]. Also, certain agonists such as oxytocin and prostaglandins promote smooth muscle contraction in part by inhibiting the plasmalemmal Ca2+ pump [101, 102].
PMCA can be distinguished from other ATPases in the plasmalemma and endoplasmic reticulum by its insensitivity to ouabain (distinction from Na+/K+-ATPase), high sensitivity to inhibition by vanadate (more sensitive than SERCA), sensitivity to K+ (less sensitive than SERCA), and sensitivity to CaM antagonists [103]. Molecular biology studies have been successful in cloning. purification and amino acid sequencing of the plasmalemmal Ca2+ pump from several cell types including smooth muscle [104–106].
5.2. The Sodium–Calcium Exchanger
The Na+–Ca2+ exchanger (NCX) is an alternative plasma membrane pathway through which excess intracellular Ca2+ is removed to the extracellular space against a large Ca2+ gradient. NCX contributes to Ca2+ removal in many cell types including smooth muscle [107, 108]. In membrane vesicles, NCX activity copurifies with plasma membrane markers, suggesting a plasmalemmal activity. Studies have succeeded in the isolation and functional reconstitution of the plasmalemmal NCX [109, 110], and distinguished it from the mitochondrial NCX by its markedly different specificity and stoichiometry [111].
NCX is driven by the transmembrane Na+ and Ca2+ gradients and the membrane potential. The energy derived from either Na+ or Ca2+, moving down its electrochemical gradient, is balanced by an antiport movement of the coupled ion. This transport mechanism is electrogenic with 3Na+:Ca2+ stoichiometry [112, 113]. NCX plays a role in Ca2+ extrusion in VSM, but its contribution varies in different blood vessels [114, 115]. Also, depending on the membrane potential and the transmembrane Na+ and Ca2+ gradients, NCX contributes to either Ca2+ extrusion or Ca2+ influx (reverse-mode NCX). The role of NCX as a source of intracellular Ca2+ may be increased in vascular disorders such as hypertension [116].
5.3. Sarcoplasmic Reticulum Ca2+-ATPase (SERCA)
The role of SERCA in Ca2+ homeostasis has long been recognized in skeletal and cardiac muscles [117]. SERCA has a molecular weight of 100 kDa and a 2:1 stoichiometry of Ca2+ transport to ATP hydrolysis. The ability of SR to accumulate Ca2+ is markedly less in smooth muscle compared with skeletal and cardiac muscles [118]. However, smooth muscle SR microsomes show energy-dependent Ca2+ uptake. Also, Ca2+ electron probe X-ray microanalysis of saponin-permeabilized smooth muscle has demonstrated a nonmitochondrial ATP-dependent Ca2+-pump activity that is blocked by vanadate [17]. SERCA affinity for Ca2+ (Km = 0.2–0.6 μM) is sufficient to take up Ca2+ and promote muscle relaxation. Calsequestrin is a high-capacity low-affinity Ca2+-binding protein that increases SR Ca2+ storage capacity in skeletal and smooth muscle [119, 120]. Because of the limited capacity of SR to accumulate Ca2+, the mitochondria become the major Ca2+ pool during repeated and excessive Ca2+ loads [121].
Cyclopiazonic acid is a specific inhibitor of SERCA that causes slowly developing contractions in VSM, and a second application of cyclopiazonic acid causes smaller repeatable contraction that depends on the vessel type. In rat aorta, cyclopiazonic acid-induced contractions are decreased upon the second application, but are completely repeatable in the presence of PMCA inhibitor vanadate, but not the Na+/K+ pump inhibitor ouabain. The contractions are also completely repeatable in the presence of the forward mode NCX inhibitor 2′,4′-dichlorobenzamil, but not the reverse mode NCX inhibitor KBR7943. These findings indicate that cyclopiazonic acid by inducing a transient rise in [Ca2+]c causes a long-lasting stimulation of plasma membrane Ca2+ extrusion mechanisms and leading to a diminished contraction upon its second application, and thereby suggest a functional coupling between SERCA and plasma membrane Ca2+ extruders and in rat aortic VSMCs [122]. SERCA is now known to form nanojunctions with the plasma membrane and other cell organelles including lysosomes, mitochondria, and the nucleus [3, 4].
5.4. Mitochondria and Ca2+
Mitochondria occupy ~5% of smooth muscle cell volume [8], but their role in the regulation of intracellular Ca2+ has not been fully examined, and the concentration of free Ca2+ in the mitochondrial matrix space is unclear. Separate Ca2+ influx and Ca2+ efflux pathways affect Ca2+ movement across the mitochondrial membrane [111, 123]. Ca2+ influx operates as a Ca2+ uniporter driven by the large mitochondrial membrane potential (150 mV, inside negative), and Ca2+ efflux involves a Ca2+:2H+ or Ca2+:2Na+ antiporter [124, 125]. The Ca2+ efflux has lower capacity than Ca2+ influx [123]. Under physiological conditions, the major cellular cytosolic anion is phosphate (HPO42−). When Ca2+ is taken up by mitochondria, HPO42− is also taken up via: HPO42−:2OH− exchange and calcium phosphate is formed. According to Mitchell’s hypothesis of mitochondrial energy transfer [126], the primary event is the development of an electrochemical proton gradient across the mitochondrial membrane with the pH gradient greater in mitochondria than the cytoplasm. In an alkaline environment, the solubility of calcium phosphates is extremely low. Thus, the major determinants of the free Ca2+ concentration within the mitochondrial matrix space are the extra- and intramitochondrial phosphate concentration, the intramitochondrial pH, and the Km and Vmax of the efflux pathway [124]. The role of mitochondria in cellular Ca2+ homeostasis can be easily understood by considering the rate of Ca2+ uptake into mitochondria as a function of [Ca2+]c. The rate of mitochondrial Ca2+ uptake increases dramatically as [Ca2+]c rises to abnormally high levels. Since the Ca2+ efflux out of the mitochondria is saturable [123], the rate of mitochondrial Ca2+ uptake will exceed Ca2+ efflux and a net accumulation of Ca2+ by the mitochondria occurs [124]. The accumulated Ca2+ then deposits into a nonionic pool of calcium phosphate. Thus, the mitochondria function as a sink for Ca2+ during Ca2+ overload. The mitochondrial free Ca2+, however, is in equilibrium with the large nonionic calcium pool. This arrangement means that [Ca2+]c is coupled to the nonionic calcium pool in the mitochondria. Consequently, when [Ca2+]c is lower than the mitochondrial free Ca2+, the nonionic calcium pool is released to stabilize [Ca2+]c. On the other hand, when [Ca2+]c is within the normal basal level (~0.1 μM), the mitochondrial free Ca2+ will have a similar value and the plasma membrane and the SR will be largely responsible for maintaining the cellular Ca2+ homeostasis. Also, because the capacity of mitochondria, although large, is finite, it is presumed that they slowly release their stored calcium during periods of cellular quiescence when it can be handled by the plasmalemmal and SR Ca2+ pumps. The apparent Km of mitochondria for Ca2+ uptake is ~10–17 μM, which is higher than that of SR (Km ~1 μM). Thus, SR is the major Ca2+ storage site under physiological conditions, and mitochondria accumulate Ca2+ only when [Ca2+]c is abnormally high, exceeding 5 μM [17, 127]. In other words, smooth muscle mitochondria are minimally loaded with Ca2+ under physiological conditions, and the mitochondrial large Ca2+ buffering capacity plays a role mainly under pathological conditions when “Ca2+ overload” occurs and the cell viability is threatened by massive Ca2+ influx. The high Ca2+ content of mitochondria isolated from atherosclerotic blood vessels may reflect damaged smooth muscle cells, and such cells may represent the initial sites of vascular calcification [17].
6. Ca2+-Dependent Myosin Light Chain Phosphorylation
6.1. Cytosolic Free Ca2+ Concentration ([Ca2+]c)
[Ca2+]c is regulated by a balance between the Ca2+ mobilization and Ca2+ removal mechanisms. [Ca2+]c was first measured in large cells by microinjection of the cells with metallochromic dyes such as arsenazo III and antipyralzo III [128] or bioluminescent proteins such as aequorin [129, 130], or by impalement of the cell with Ca2+-sensitive microelectrodes [131]. VSMCs are very small and are not suitable for the microinjection or impalement techniques. This problem was first circumvented by administering aequorin into VSM preparations using a transient membrane permeabilization technique [132]. Thereafter, several fluorescent Ca2+ indicators including quin-2, fura-2, and indo-1 have been developed for measuring [Ca2+]c in many cell types including VSM [133–137]. The nonpolar acetoxymethyl ester of Ca2+ indicators is more lipophilic and diffuses into the cell where it is hydrolyzed by intracellular esterases into the more hydrophilic free acid that does not cross the plasma membrane and is trapped inside the cell. Regardless of the technique used, the physiological VSM [Ca2+]c is in the range between 0.1 and 1 μM.
6.2. Myosin Light Chain Phosphorylation
Ca2+-dependent MLC phosphorylation is a major determinant of smooth muscle contraction [138, 139]. The thick-filament regulation hypothesis of smooth muscle contraction predicts that Ca2+ binds CaM to form a Ca2+–CaM complex, which activates MLC kinase, and results in the phosphorylation of the 20-kDa MLC [138, 139]. The phosphorylated MLC increases the activity of actin-activated Mg2+-ATPase leading to actin–myosin interaction and smooth muscle contraction (see Fig. 1). Smooth muscle relaxation is initiated by a decrease in [Ca2+]c due to Ca2+ uptake by SR and Ca2+ extrusion by the plasmalemmal Ca2+ pump and NCX. The decrease in [Ca2+]c causes dissociation of the Ca2+–CaM complex and the phosphorylated MLC is dephosphorylated by MLC phosphatase.
6.3. Evidence for Other Mechanisms of Smooth Muscle Contraction
Agonist-induced vascular tone can not be explained only by Ca2+-dependent MLC phosphorylation. In rabbit aortic rings incubated in Ca2+-free solution, the α-adrenergic receptor agonist phenylephrine causes an initial transient contraction likely due to Ca2+ release from SR followed by a smaller but maintained contraction [2], which in the absence of extracellular Ca2+ may be due to other Ca2+ sensitization mechanisms. Simultaneous measurements of force and [Ca2+]c in VSM preparations have suggested agonist-induced increases in myofilament force sensitivity to Ca2+ [132, 140]. In rabbit inferior vena cava loaded with fura-2, norepinephrine causes an initial contraction followed by a maintained contraction in parallel with a rapid [Ca2+]c spike followed by a smaller increase in [Ca2+]c above basal levels. In contrast, membrane depolarization by high KCl causes sustained increases in contraction and [Ca2+]c. Also, for approximately the same increase in [Ca2+]c, norepinephrine causes greater contraction than that induced by high KCl. Also, when the relationship between [Ca2+]c and force was constructed by maximally stimulating inferior vena with norepinephrine or high KCl in Ca2+-free solution, then increasing extracellular Ca2+ stepwisely, the norepinephrine [Ca2+]c–force curve was enhanced and located to the left of that induced by high KCl, suggesting that norepinephrine increases the myofilament force sensitivity to Ca2+ [140].
Dissociations in the [Ca2+]c-force relationship were observed during agonist stimulation of various smooth muscle preparations including ferret aorta [141], rabbit pulmonary artery [142], and swine carotid artery [143]. Also, dissociations in the [Ca2+]c-MLC phosphorylation relationship were observed during agonist-induced activation of smooth muscle and were related to G protein-mediated change in the MLC kinase/MLC phosphatase activity ratio [144]. Agonist-induced dissociations between MLC phosphorylation and force have also been reported [145, 146], and have been explained by the “latch bridge” hypothesis, which proposes that the dephosphorylation of myosin may generate a slowly cycling cross-bridge that supports force maintenance [147]. However, Ca2+-dependent MLC phosphorylation may not be the only determinant of agonist-induced VSM contraction and other mechanisms that increase the myofilament force sensitivity to [Ca2+]c and MLC phosphorylation have been proposed.
7. Protein Kinase C
The interaction of vasoconstrictor agonists such as phenylephrine, angiotensin II (AngII), and endothelin-1 (ET-1) with their Gq protein-coupled receptors (GPCRs) activates a GTP-binding protein and PLCβ, which stimulates the hydrolysis of PIP2 into IP3 and DAG [148]. IP3 stimulates Ca2+ release from SR, while DAG activates protein kinase C (PKC). PKC is a ubiquitous enzyme found in almost all cell types including the vascular endothelium, VSM and fibroblasts. PKC is a serine/threonine kinase that phosphorylates a large number of substrates and is widely implicated in numerous physiological and pathological processes. PKC was discovered by Nishizuka and colleagues in rat brain extract [149] as a Ca2+/phospholipid protein kinase that is activated by DAG [150] and the tumor promoter phorbol ester [151]. PKC was then found to be a family of several isoforms with different subcellular localization, substrates and functions.
7.1. PKC Structure and Isoforms
The PKC molecule comprises a N-terminal regulatory domain, a hinge region and a C-terminal catalytic domain [152] (Fig. 3). The conventional PKC isoforms α, βI, βII, and γ have four conserved regions (C1, C2, C3 and C4) and five variable regions (V1, V2, V3, V4, and V5). The regulatory domain contains two conserved C1 and C2 regions. The C1 region contains cysteine-rich zinc finger-like motifs and lipid-binding sites surrounded by a band of hydrophobic residues that penetrate the lipid bilayer and anchor PKC to DAG-containing membranes. PKC also stably associates with membranes through the C2 region [153]. The C1 and possibly C2 region also bind the PKC cofactor phosphatidylserine (PS) [154, 155]. An autoinhibitory pseudosubstrate sequence immediately precedes the C1 region, and comprises a 19–36 amino acid residues that resemble the PKC substrate phosphorylation site [156]. The PKC catalytic domain contains the conserved C3 region, an ATP/Mg-binding site in a narrow hydrophobic pocket and a binding site for the phospho-acceptor sequence in the substrate [157]. The C4 region comprises the substrate-binding part of PKC [158]. The catalytic domain also contains three key phosphorylation and autophosphorylation sites in the C-terminal activation loop, turn-motif and hydrophobic-motif.
Fig. 3.

PKC structure and isoforms. PKC comprises a N-terminal regulatory domain and a C-terminal catalytic domain, connected by a V3 hinge region. The regulatory domain contains two conserved C1 and C2 regions, and the pseudosubstrate region. The catalytic or kinase activity domain contains a C3 ATP-binding site and a C4 binding site for the substrate. The catalytic domain also contains phosphorylation sites in the activation loop, turn-motif and hydrophobic-motif (The figure illustrates PKCδ phosphorylation sites, which vary in different PKCs). The PKC family is classified into conventional cPKCs α, βI, βII, and γ; novel nPKCs δ, ε, η and θ; and atypical aPKCs ζ and ι/λ isoforms. cPKCs consist of 4 conserved (C1–C4) and 5 variable regions (V1–V5) and are activated by DAG, PS and Ca2+. The C1 region binds phosphatidylserine (PS), DAG, and phorbol esters, and the C2 region contains the binding site for Ca2+. PS can also bind to the C2 region. Both cPKCs and nPKCs have twin C1 regions (C1A and C1B) and a C2 region, but the order of C1 and C2 regions is switched in nPKCs compared to cPKCs. The nPKCs have a variant form of C2 region that is insensitive to Ca2+, but still binds lipids. The aPKCs do not have a C2 region and hence not activated by Ca2+, and have a variant form of C1 that is not duplicated, but retains lipid-binding activity and sensitivity to PS. The aPKCs also have a protein–protein-interacting region Phox and Bem 1 (PB1) that controls their cellular localization. Other related kinases include PKCμ (PKD). PKC inhibitors compete with DAG at the C1 region (calphostin C), ATP at the ATP-binding site (H-7, staurosporine) or the PKC true substrate (pseudosubstrate inhibitor peptide).
PKC is a large serine/threonine kinase family that comprises ~2% of the human kinome [159] and encodes nine different genes and 10 isoforms [160]. Based on the structure of the N-terminal domain, PKC isoforms are classified into conventional cPKCs α, βI, βII, and γ; novel nPKCs δ, ε, η and θ; and atypical aPKCs ζ and ι/λ isoforms [6] (Fig. 3). The cPKCs consist of four conserved regions (C1–C4) and five variable regions (V1–V5), and are activated by Ca 2+, DAG, and PS. The N-terminal regulatory domain contains a highly homologous 60–80% C1 region among different PKC isoforms [157]. The C1 region contains the recognition site for PS, DAG, and phorbol esters. The C2 region is rich in acidic residues and contains the binding site for Ca2+ [158]. In cPKCs, the C2 region comprises 105 to 130 residue eight-stranded anti-parallel β-sandwich structures with three inter-strand Ca2+-binding loops responsible for Ca2+-dependent anionic phospholipid binding [161]. Both cPKCs and nPKCs have twin C1 regions (C1A and C1B) and a C2 region, but the ordering of the C1 and C2 regions is reversed in nPKCs compared to cPKCs [161]. Also, nPKCs have a variant C2 region that lacks the critical Ca2+-coordinating aspartic acid residues that are highly conserved in cPKCs, making it insensitive to Ca2+ [156]. The C1 region of nPKCs has a higher affinity for DAG than that of cPKCs, and functions as a lipid-binding membrane-targeting module in a Ca2+-independent manner [162]. The C2 region of PKCδ does not bind lipids, but has a protein–protein interaction domain that binds phospho-tyrosine residues flanked by the consensus sequence (Y/F)-(S/A)-(V/I)-pY-(Q/R)-X-(Y/F). PKCδ contains several tyrosine phosphorylation sites throughout its structure, including the regulatory and catalytic domains and the hinge region [163]. The aPKCs do not have a C2 region but have a variant form of C1 and are therefore activated by PS but not Ca2+ or DAG [156]. However, aPKCs do retain lipid-binding activity, and the C1 region confers DAG binding that is not duplicated, unlike the C1A–C1B tandem repeat found in cPKCs and nPKCs [164]. The aPKCs also uniquely encode the protein–protein-interacting Phox and Bem 1 (PB1) region in the N-terminal domain, which binds ZIP/p62, Par6, or MEK5 through a PB1-PB1 domain interaction that controls the localization of aPKCs [165]. PKCμ and PKCν are often considered a fourth class of PKC or members of protein kinase D (PKD) family [155, 166].
The greatest homology among PKC isoforms is in the highly conserved catalytic domain (~70%). Also, similar to other Ser/Thr kinases, PKC isoforms have a highly conserved ATP-binding site. The exception to the catalytic domain homology is the variable V5 region, consisting of 60–70 different amino acids. PKC isoforms also differ in the V3 hinge region [157]. PKCβI and βII are generated by alternative splicing from a single gene, but differ in their C-terminal 50 residues (βI) or 52 residues (βII) [158]. The amino acid in each phosphorylation site also varies in different PKCs. For example, the activation loop contains a phosphorylatable T497 in PKCα, T500 in PKCβII, T505 in PKCδ and T538 in PKCθ, and the turn motif contains a T638 in PKCα, T641 in PKCβI and PKCβII, S643 in PKCδ and S676 and S685 in PKCθ, while the hydrophobic motif contains a S657 in PKCα, S660 in PKCβII, S662 in PKCδ and S695 flanked by bulky hydrophobic residues in PKCθ [153, 167].
7.2. PKC Distribution and Translocation
PKCs are found in numerous tissues and vascular beds. PKCα, δ and ζ are expressed in most blood vessels, and other PKCs show specific distribution in certain blood vessels [168, 169] (Table 1). PKCα, β, δ and ε, but not PKCζ, are highly expressed in human VSMCs [170]. Fluorescent-tagged PKC and live imaging techniques allowed the study of PKC localization in real-time [171, 172]. In resting cells, PKCα, β and γ are localized mainly in the cytosolic fraction, and activated PKC translocates from the cytosolic to the particulate and membrane fraction [168, 173] (Fig. 4). Simple diffusion and other physico-chemical forces may drive PKC movement inside the cell, and targeting mechanisms including conformation changes, altered hydrophobicity, lipid modification, protein-protein interaction, targeting sequences, and phosphorylation allow its translocation and tight binding to different cell membranes [168, 174].
Table 1.
Distribution and scaffold proteins of PKC in representative tissues and blood vessels
| PKC | MW (kDa) |
Major Tissue Distribution |
Blood vessel | Location Inactive PKC |
Location Active PKC |
Scaffold Protein |
Ref |
|---|---|---|---|---|---|---|---|
|
cPKC α |
74–82 | Universal | Rat aorta | Cytosol | Nuclear | RACK1 | [160, 272, 446, 528–531] |
| Rat mesenteric artery | Cytosol/Membrane | Cytosol/Membrane | p32 | ||||
| Rat carotid, ferret portal vein, porcine coronary, bovine aorta | Cytosol | Plasma membrane | RACK1, AKAPs, HSP, p32 | ||||
| β | 80–82 | Adipose tissue, liver, kidney, spleen, skeletal muscle, brain | Rat aorta | Cytosol | Nuclear | RACK1, p32 | [160, 290, 529] |
| Rat carotid | Cytosol | Membrane | RACK1, AKAPs, HSP, p32, 14-3-3 | ||||
| γ | 70–82 | Adrenal gland, brain | Rat mesenteric artery | Cytosol | Cytosol | RACK1, AKAPs, HSP, 14-3-3, Importins | [160, 528, 530] |
|
nPKC δ |
76–82 | Universal | Rat aorta | Cytoskeleton/Organelles | Cytoskeleton/Organelles | RACK1, p32 | [160, 447, 530, 532] |
| Rat mesenteric artery | Membrane | Membrane | AKAPs, HSP, p32, 14-3-3 | ||||
| ε | 90–97 | Pancreas, kidney, brain | Rat mesenteric artery, porcine coronary artery | Cytosol/Membrane | Cytosol/Membrane | AKAPs, p32 | [160, 272, 528, 530, 533] |
| Ferret aorta | Cytosol | Surface membrane | RACK1, RACK2 AKAPs, HSP, p32, 14-3-3 | ||||
| η | 80 | Lung, skin, brain | NIH 3T3 fibroblasts | Cytosol/Membrane Golgi | Membrane | [160, 534, 535] | |
| θ | 79 | T cells, hematopoetic cells, skeletal muscle | cerebral microvascular endothelium | cytosol | Membrane Lipid rafts | AKAPs, HSP, p32, 14-3-3, Importins, CARMA1, Vav1 | [160, 221] |
|
aPKC ζ |
64–82 | Universal | Rat aorta, ferret aorta and portal vein | Perinuclear | Intranuclear | AKAPs, HSP, p32, 14-3-3, Importins | [160, 447, 528, 530, 533] |
| Rat mesenteric artery | Cytosol | Cytosol | |||||
| ι/λ | 70 | Kidney, testis, ovary, brain | Rabbit femoral artery and portal vein | Cytosol | Cytosol | AKAPs, HSP, 14-3-3, Importins | [160, 536, 537] |
MW, molecular weight
Fig. 4.

Activation, translocation, substrate interaction and deactivation of cPKCs. In the PKC cytosolic and inactive state, the pseudosubstrate binds the catalytic site in the C4 region, and the regulatory and catalytic domains are folded. Before it becomes catalytically competent, nascent PKC undergoes phosphorylation at three phosphorylation sites. Phosphorylation of the activation loop by phosphoinositide-dependent kinase (PDK) introduces a negative charge that properly aligns residues to form a competent catalytic domain, facilitate subsequent autophosphorylation at the turn motif and hydrophobic motif, and keep PKC in a catalytically competent and protease resistant conformation. Phosphate groups are indicated as green ovals labeled “P”. PKC activators such as PS, DAG, phorbol esters, and Ca2+ promote allosteric activation, translocation of PKC to the plasma membrane, and subsequent interaction with the substrate. Allosteric activation also induces an open conformation state, making PKC susceptible to phosphatases and proteases and allows PKC to either enter an autophosphorylation/dephosphorylation cycle, or undergo proteolytic degradation. PKC dephosphorylation terminates its kinase activity and is carried out by the PP2C member pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP) at the hydrophobic motif, which starts the process that consequently drives further dephosphorylation of PKC by PP1/PP2A protein phosphatases at the turn motif. Dephosphorylation also predisposes “naked” PKC to ubiquitination and degradation, leading to de novo synthesis and regeneration of the enzyme.
PKC binding to Ca2+ or DAG causes conformational changes and unfolding of the PKC molecule, leading to exposure of the substrate region, increased PKC hydrophobicity and binding to membrane lipids [158]. Changes in the plasma membrane lipid domains influence PKC distribution. The plasma membrane is composed of several domains of focal adhesions alternating with zones rich in caveolae, and both harbor a subset of membrane-associated proteins. PKCα exhibits binding activity in caveolae, and may not bind to non-caveolae membranes [175]. Localized [Ca2+] gradients affect the amount of PKC retained in caveolae. For instance, caveolae contain PKCα only in the presence of Ca2+, while retention of PKCε and PKCλ in caveolae is Ca2+ independent [175]. Caveolins are scaffold proteins that help PKCα and ζ localize to the caveolar microdomains where they are subsequently activated [176]. In rabbit femoral and renal arteries at rest, PKCζ is localized in punctate plasma membrane aggregates alternating with vinculin and in a perinuclear location, and such locations are conducive to regulating VSM [Ca2+]c [177]. Plasma membrane lipids are also segregated into cholesterol-rich lipid rafts and glycerophospholipid-rich non-raft regions, an arrangement that is critical for preserving the membrane architecture and for translocation of proteins. In VSMC membranes, lipid segregation is supported by annexins that target membrane sites of distinct lipid composition, and each annexin requires different [Ca2+] for its translocation to the plasma membrane, thus allowing a spatially confined graded response to external stimuli and plasmalemmal localization of PKC [178]. Several members of the annexin family function as PKC substrates and promote membrane association of PKC [179, 180] (Table 2). Annexin A1, A2, A5 and A6 (or anexxin I, II, V, and VI) display specific abilities to interact and promote membrane targeting of distinct PKCs. Also, because of the ability of annexins to create specific membrane microenvironments, they could allow PKCs to phosphorylate certain substrates and regulate their downstream effector pathways in specific subcellular locations [160]. PKC isoforms interact with specific members of the annexin family, and PKCβ, ε and α interact with annexin I, II and VI, respectively. Also, interaction between annexin V and PKCδ occurs in cells after PKCδ stimulation, but before its translocation to the membrane fraction, suggesting that PKCδ requires binding to annexin V for its translocation [181]. Whether other PKCs require annexin binding before translocation is unclear.
Table 2.
Representative PKC substrates and the effect of their phosphorylation
| Substrate | Effect of Substrate Phosphorylation | Reference |
|---|---|---|
| Histones: H3T45 | DNA fragmentation, apoptosis | [538] |
| H3T6 | Prevents LSD1 from demethylating H3K4 during androgen receptor-dependent gene activation. Promotes cell proliferation | [539] |
| Membrane-bound proteins: MARCKS (myristoylated, alanine-rich C kinase substrate) | MARCKS binds F-actin. Functions as cross-bridge between cytoskeletal actin and plasma membrane | [182] |
| Inhibitory GTP-binding protein Gi | Facilitates the dissociation of the αi subunit from adenylyl cyclase and thereby relieves it from inhibition. | [169] |
|
Ion Channels BKCa channels |
Inhibition, leading to membrane depolarization, activation of L-type VDCCs, and increased [Ca2+]c and vascular tone, e.g. in pulmonary artery and porcine coronary artery. | [295, 296, 299, 300, 302, 303, 327] |
| Voltage-dependent K+ channel | Inhibition. Increases vascular tone | [298, 304, 305, 327] |
| KATP channels | Inhibition. Alters the channel kinetics and/or number at the cell membrane, e.g. in mesenteric artery | [309, 311, 312, 327] |
| Store-operated Ca2+ channel | Inhibition, e.g. in HEK293 cells. | [341] |
| Ion Pumps & Exchangers: PMCA | Activation. Promotes Ca2+ extrusion. Explains transient nature of agonist-induced increase in [Ca2+]c | [6] |
| α1 subunit of Na+/K+-ATPase | Inhibition. Alters membrane potential and intracellular concentrations of Na+ and K+ | [315] |
| Na+/H+ antiport exchanger | Activation. Increases cytoplasmic pH and alkalinization, leading to increased contraction | [316–318] |
| Regulatory Proteins: CPI-17 | Inhibits MLC phosphatase, increases MLC phosphorylation and enhances myofilament force sensitivity to Ca2+ and VSM contraction, e.g. in rabbit femoral artery | [322] |
| Calponin | Allows actin-myosin interaction and enhances VSM contraction | [540] |
| Raf | Initiates a cascade involving MAPK kinase (MEK) and MAPK, and phosphorylation of the actin-binding protein caldesmon which allows actin–myosin interaction and VSM contraction | [5, 339] |
| 20-kDa MLC and MLCK | Counteracts Ca2+-induced actin–myosin interaction and force development, e.g. in rabbit mesenteric artery | [343] |
| Cytoskeletal Proteins: Vinculin | Controls cell shape, and adhesion | [264] |
| Vimentin | Recycles β1-integrins to plasma membrane | [265] |
| Ribosomal Protein Kinases: S6KβII | Nucleo-cytoplasmic shuttling of S6KβII. Regulates protein synthesis and G1/S transition in the cell cycle | [266] |
| Other: Arginine-rich protein substrates | Neutralizes the acidic patch in the substrate binding site. Displaces PKC pseudosubstrate from the kinase core |
[158, 212] |
Myristoylated alanine-rich C kinase substrate (MARCKS) plays a role in PKC membrane binding. MARCKS is a major PKC substrate that binds F-actin and cross-bridges between the plasma membrane and cytoskeletal actin [182]. Phosphorylation of MARCKS by PKC has an electrostatic effect that affects its affinity to the plasma membrane and interferes with its actin cross-linking, leading to its displacement from the plasma membrane. MARCKS and CaM are co-distributed in SMCs and co-targeted simultaneously to the cell interior upon cell stimulation. PKC activation triggers the translocation of CaM which facilitates the translocation of MARCKS and its subsequent phosphorylation at multiple sites [183]. Dephosphorylation of MARCKS causes its re-association with the plasma membrane via its stably attached myristic acid membrane-targeting moiety [184].
Protein-protein interactions are crucial in signal transduction, and binding sites for arginine-rich polypeptides have been identified in the PKC molecule distal to its catalytic site and may allow targeting of PKC to precise substrates at specific cellular locations. Scaffold proteins such as receptor for activated C kinase (RACK), substrates that interacts with C kinase (STICK), receptor for inactive C kinase (RICK), and A-kinase activating proteins (AKAPs) assist in PKC translocation to the membrane [185]. RACKs and STICKs bind to active PKCs, whereas RICKs and AKAPs interact with inactive PKCs. Binding of a specific activated PKC to its RACK provides access to, and phosphorylation of, its substrates [186]. Binding of RACK increases the phosphorylation capacity of PKC several-fold independently from the substrate identity [187]. RACKs also target PKC to cytoskeletal elements [187]. The interaction of PKC and RACK is isoform specific and is largely mediated by the C2 region of cPKCs [188], and peptide fragments of this region serve as modulators of PKC activity [189]. These short peptides induce activation and translocation of the PKC isoform by mimicking the action of RACK on the isoform and, therefore, are termed ‘pseudo RACKs’ (ψRACK) [190, 191]. Disruption of the interaction between ψε RACK and the RACK-binding site is a critical rate-limiting step in translocation of PKCε [192]. Other scaffold proteins including 14-3-3, heat shock protein (HSP), importins, and even actin can tether PKC isoforms to different membranes and organelles [193–197]. Protein-protein interactions between PKC isoforms and their substrates provide further anchoring to specific subcellular sites. For PKCε, protein-protein interactions may involve a myofilament-binding site in the C2 region [198], an intra-SR calsequestrin-binding site [199], a neurocytoskeletal elements-binding site [200], an actin-binding site in the C1 region, and a Golgi-binding site [191, 197, 201]. PKCε association with Golgi membranes via its zinc finger domain can modulate Golgi function [202]. The PKC pseudosubstrate and hinge regions can facilitate its plasma membrane and cytoskeletal association [203]. Also, the V5 region can contribute to the regulation of PKCα activity by multiple mechanisms involving stabilizing the kinase through direct interaction with its N-terminal, interacting with the pseudosubstrate in the N-terminal regulatory domain, and interaction with RACK [204].
While the interaction of cPKCs at the plasma membrane has been well-studied, less is known about the activity of nPKCs and aPKCs at the plasma membrane and other membranes in the nucleus, mitochondria, endoplasmic reticulum (ER) and Golgi. For instance, c-src-dependent phosphorylation of tyrosine Y256 in PKCι, through enhanced interaction with the nuclear transporter protein importin-β, results in its translocation to the nucleus [205]. Also, the ER membrane is a major target for PKCδ recruitment. PKCε displays a similar translocation pattern to the ER following ATP binding. The localization of nPKCs in the ER membrane suggests possible role in protein synthesis and modification [206].
The allosteric model for PKC activation by lipid cofactors and the concept that membrane translocation is essential for PKC activation have been challenged. For instance, the model predicts that the cellular actions of PKC will be limited to the membranes where lipid cofactors facilitate PKC translocation. However, immunohistochemical studies have shown that the distribution of PKCα does not differ in the longitudinal and circular layers of the swine stomach under resting conditions, being predominantly localized near the plasma membrane, and stimulation with PDBu or carbachol does not alter this peripheral PKCα distribution [207]. Also, PKC is found in other cell compartments like the mitochondria, and in the soluble fraction of cells subjected to oxidative stress, a known activator of PKC [161, 208]. PKCs in the soluble fraction of VSM also phosphorylate contractile proteins located distant from the membrane lipids [161]. PKC translocation may also be dependent on cytoskeletal elements and transport along the cytoskeleton through protein-protein interactions [209–211]. Another misconception of the canonical model of PKC activation is that PKC catalytic activity is an inherent property of the enzyme that is not altered by the activation process; a model that does not explain the diverse and often opposing actions of certain PKCs [161].
7.3. PKC Phosphorylation
In the inactive PKC, both the regulatory and catalytic domains are folded together and the pseudosubstrate binds the catalytic site in the C4 region [212]. In the activated state, PKC unfolds, the pseudosubstrate dissociates from the C4 region, and PKC is ready to target its true substrate (Fig. 4). Before it becomes catalytically competent and able to respond to its allosteric activators, nascent PKCs undergo phosphorylation by a PKC kinase and autophosphorylation at three conserved Ser/Thr phosphorylation sites in the activation loop, turn motif, and hydrophobic motif of the C-terminal domain [158, 213, 214]. Phosphorylation changes PKC protein conformation and electric charge and affects its lipid affinity and binding to the plasma membrane. Phosphorylation keeps PKC in a catalytically competent and protease resistant conformation. Full activation of PKC by allosteric activators induces an open conformation that makes it susceptible to phosphatases and proteases, leading to either repeated autophosphorylation/dephosphorylation cycles, or proteolytic degradation of the PKC molecule and de novo synthesis of the enzyme [214, 215].
The first critical and rate-limiting phosphorylation of the activation loop at the conserved threonine is catalyzed by phosphoinositide-dependent kinase (PDK) [156, 216]. Mutation of phosphorylatable Thr-residues in the activation loop abolishes PKC activity [217, 218], and in the absence of PDK-1, PKC is prone to rapid degradation [219]. Phosphorylation of the activation loop introduces a negative charge that properly aligns residues to form a competent catalytic domain and facilitates the subsequent autophosphorylation of the ‘turn motif’ (which corresponds to a phosphorylation site in protein kinase A (PKA) localized at the apex of a turn), and the hydrophobic motif [220]. The hydrophobic motif is important for PKC stability, functioning as a docking-site for PDK-1 through its repeated negatively charged aspartate sequence termed PDK-1 interacting fragment [213, 219]; an interaction that allows PDK-1 to access the activation loop [221]. PDK-1 and mTOR are upstream kinases that promote PKC phosphorylation in different motifs [216, 222, 223]. Phosphorylation of the turn motif by the mTORC2 complex triggers autophosphorylation of the hydrophobic motif [224, 225]. In VSMCs, α-adrenergic receptor agonists induce translocation of the actin-binding protein calponin (CaP) from the contractile filaments to VSMC cortex, and promote CaP-dependent phosphorylation of PDK at S241, PKCα phosphorylation at the activation loop T497, and autophosphorylation at the hydrophobic motif [154]. Autophosphorylation of the turn motif contributes to relative stability of PKCδ. The aPKCs are phosphorylated at the activation loop and turn motif, and contain glutamate ‘phosphomimetic’ residues in their hydrophobic motif [213, 214, 226], while the hydrophobic motif of nPKCs contains an aspartate residue [226].
PKC phosphorylation may occur only during maturation of the newly synthesized enzyme, as with PKCα, or is dynamically regulated, as with nPKCs [227–229]. Phosphorylation of multiple sites is required for activation of mature PKCs, e.g. during H2O2-induced tyrosine phosphorylation of PKCδ [230]. Also, in cardiomyocytes, PKCδ and PKCε undergo phosphorylation of the activation loop and hydrophobic motif even in the absence of allosteric regulators [227], supporting that the regulatory pathways of PKC are isoform- and cell-specific.
The scaffold protein 14-3-3 serves as a partner of phosphorylated PKCε in mammalian cells. Phosphorylation of PKCε on Ser346 and Ser368 is required for binding to 14-3-3, and locks the enzyme in an open, active and lipid-independent conformation [164, 231]. On the other hand, direct interaction between PKCθ and 14-3-3 tau has been observed in T cells, and 14-3-3 overexpression inhibits PKCθ translocation and function [232].
Other phosphorylation patterns may be specific to certain PKC isoforms. PKCδ has tyrosine phosphorylation sites, and tyrosine-phosphorylated PKCδ is constitutively active and does not require DAG as a cofactor [208]. Tyrosine phosphorylation also underlie redox control of PKCδ activity. A Src family kinase (Lck)-driven phosphorylation of PKCδ at Tyr311 in rodents (Tyr313 in human) mediates H2O2-dependent increase in PKCδ activity [161, 208].
PKC phosphorylation has been used as a marker of its activation [233, 234]. S299-phosphorylated PKCδ is localized at both the plasma and nuclear membranes, making it the best marker of the activated enzyme [233]. However, PKCδ is phosphorylated at other sites and undergoes autophosphorylation at three sites in its V3 region (S299, S302, S304), each of which is evolutionarily conserved and unique to PKCδ. S643 is another PKCδ autophosphorylation site [235] that may not be an ideal marker of activation because it is relatively resistant to dephosphorylation and remains phosphorylated even when PKCδ releases DAG and adopts a ‘closed’ conformation [214, 233].
PKC kinase activity is terminated by dephosphorylation, when PKC is in an “open” conformation unbound by the pseudosubstrate or constitutively active [236–238]. For cPKCs and nPKCs, dephosphorylation is carried out by the PP2C member pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP) at the hydrophobic motif, which drives PKC to be further dephosphorylated by PP1/PP2A protein phosphatases at the turn motif [215, 237, 239, 240]. Phosphatases also indirectly affect PKC, e.g. dephosphorylation of the PKCθ downstream molecules CARMA1 by PP2A leads to PKCθ deactivation [241]. Dephosphorylation predisposes “naked” protein kinases to ubiquitination and degradation [242]. Partial inhibition of phosphorylation is caused by binding with HSP70, thus promoting rephosphorylation of PKCs and their subsequent reactivation [155, 243].
PKC-priming phosphorylation is also influenced by the inferred allosteric behavior caused by ATP binding. Nucleotide pocket occupation promotes a PKC conformation that is conducive to upstream kinases and protective from phosphatases. When the PKC kinase domain is compromised through mutation of the highly conserved lysine residue responsible for coordination of the α-β phosphates of ATP, it fails to be primed, but can be fully primed upon binding an ATP-competitive PKC inhibitor. Expression of inactive PKCε K437M mutant in HEK cells led to accumulation of inactive PKCε lacking phosphorylation at the priming sites, while wild-type PKCε expressed under the same conditions was constitutively phosphorylated at all priming sites. The PKC inhibitor bisindolylmaleimide induced rapid phosphorylation of the priming sites of PKCε K437M–expressing cells, but did not increase phosphorylation of wild-type PKCε, suggesting that the conformation induced by occupation of the nucleotide pocket of PKCε K437M with an inhibitor was sufficient to promote priming. Similarly, the active site PKC inhibitor Gö 6983 locks PKC in a conformation in which the priming phosphorylation sites are resistant to dephosphorylation and down-regulation by phorbol esters [244]. These findings have suggested that autophosphorylation is not critical for PKC priming, and that ATP pocket occupation is sufficient for maturation and activity of the kinase [164, 245].
7.4. PKC Activators
PKCs are activated by hormones such as epinephrine and AngII, growth factors including epidermal growth factor and insulin, and neurotransmitters like dopamine and endorphin through the hydrolysis of PIP2 and the generation of DAG [157]. High [Ca2+]c can also activate PLC and lead to PKC activation [157]. PKC isoforms respond differently to Ca2+, PS, DAG, and other phospholipids. cPKCs bind Ca2+ in a phospholipid-dependent manner, and Ca2+ may form a “bridge” holding the protein and phospholipid complex together at the membrane [246]. PS is indispensable for activation of PKC. Phosphatidylinositol and phosphatidic acid activate PKC at high Ca2+ concentrations. DAG activates PKC by reducing its Ca2+ requirement and enhancing its membrane association [247]. Lipids derived from sources other than glycerolipid hydrolysis such as cis-unsaturated free fatty acids and lysophosphatidylcholine, ceramide (a sphingomyelinase product), phosphatidylinositol 3,4,5-trisphosphate, and cholesterol sulfate can also activate PKC [248].
Phorbol 12,13-dibutyrate (PDBu), phorbol 12-myristate 13-acetate (PMA) and 12-O-tetradecanoylphorbol-13-acetate (TPA) activate PKC and stabilize its membrane association by reducing its apparent Km for Ca2+ [169]. PMA binds to PKC 1000-fold more strongly than DAG [249, 250]. PMA binding to the PKC C1B domain alone does not induce sufficient conformational change or release the pseudosubstrate from the catalytic core, but generates a hydrophobic cap covering polar groups that helps PKC to insert into membrane lipids [251].
DAG analogs and phorbol esters are not specific for a particular PKC isoform, and have other effects unrelated to PKC. For example, PMA recruits both cPKCs and nPKCs to the plasma membrane [206]. PKC activators often activate cPKCs isoforms to the greatest degree, then the nPKCs and aPKCs [252]. Also, the DAG analog 1,2-dioctanoyl-sn-glycerol (DiC8) blocks Kv, BKCa and KATP channels of mesenteric artery VSM in a PKC-independent manner. 1-oleoyl-2-acetyl-sn-glycerol (OAG) is a related compound that activates PKC without blocking K+ channels, and is a preferred over DiC8 as a pharmacological tool to study PKC [253].
Post-translational modifications affect PKC activity. Proteolysis in the hinge region activates PKCδ [254]. Oxidation, acetylation, nitration and phosphorylation also activate PKC [153]. Oxidants such as H2O2 activate PKC by oxidative modification of both the regulatory and catalytic domains [255]. The zinc-binding cysteine-rich motifs of the N-terminal regulatory domain are particularly susceptible to oxidative modification [256]. Hydroquinone, catechol, and whole cigarette smoke condensate activate PKC in Lewis lung carcinoma cells [257].
7.5. PKC Substrates
When PKC is not catalytically active, the basic autoinhibitory pseudosubstrate is protected from proteolysis by an acidic patch in the substrate-binding site. When PKC is activated, it phosphorylates arginine-rich protein substrates, which neutralize the acidic patch and displace the pseudosubstrate from the kinase core [158, 212]. The amino acid sequence near the substrate phosphorylation site assist in PKC substrate recognition. Several PKC substrates have been identified (Table 2). PKCα, β, and γ are potent histone IIIS kinases, while PKCδ, ε, and η have a poor capacity to phosphorylate histone [169]. PKC isoforms show overlapping specificities for substrates derived from modification of their pseudosubstrate regions. For example, the PKC targeting protein AKAP79 binds the catalytic core of all PKCs through a pseudosubstrate-like mechanism [258, 259]
PKC substrates include the anchoring proteins STICKs such as MARCKs, MacMARCKs, α-, β-, and γ-adducin, clone 72 (SseCKs), GTP-binding proteins and cytoskeletal proteins [185, 187, 221]. PKC causes phosphorylation of the inhibitory GTP-binding protein Gi, facilitating the dissociation of its αi subunit from adenylyl cyclase and thus relieves it from inhibition [169]. PKC phosphorylates and activates adhesion molecules such as focal adhesion kinase, paxillin, and vinculin [260–263]. PKC phosphorylation of the cytoskeletal protein vinculin affects cell shape and adhesion [264]. PKC also phosphorylates substrates involved in protein trafficking. Recycling of β1-integrins to the plasma membrane requires PKCε-mediated phosphorylation of vimentin, an intermediate filament protein upregulated upon epithelial cell transformation. Inhibition of PKC and vimentin phosphorylation causes integrins to become trapped in vesicles and attenuates directional cell motility. In vitro reconstitution assays showed that PKCε dissociates from integrin containing endocytic vesicles in a selectively phosphorylated vimentin-containing complex. Mutations of PKC-regulated sites on vimentin lead to accumulation of intracellular PKCε/integrin positive vesicles, while introduction of wild-type vimentin promotes cell motility in a PKCε-dependent manner, supporting that PKC-mediated phosphorylation of vimentin is a key process in integrin trafficking and cell motility [265]. PKC also plays a role in phosphorylation and nucleo-cytoplasmic shuttling of S6KβII, one of the forms of the ribosomal protein S6 kinase (S6K) involved in the regulation of protein synthesis and the G1/S transition in the cell cycle, and this PKC-mediated phosphorylation is induced by PMA, EGF, IGF-1, and platelet-derived growth factor (PDGF) [266]. Myosin binding protein-C slow (MyBP-C slow) is a thick filament-associated protein, that plays a role in the formation of actomyosin cross-bridges, and its activity is regulated by PKC-mediated phosphorylation at Ser-83 and Thr-84 [267]. The list of PKC substrates is growing and many of these substrates play a role in VSM contraction and growth.
7.6. PKC Inhibitors
Several PKC inhibitors have been developed (Table 3). The first generation PKC inhibitors such as H7 and staurosporine are nonspecific pan-PKC inhibitors that inhibit all PKC isoforms [268]. H7 and staurosporine bind to and compete with ATP in the catalytic domain. Because the ATP hydrophobic pocket is conserved throughout the kinome, ATP-binding PKC inhibitors have poor selectivity and intact with other ATP-binding kinases and cause severe side effects in vivo [157, 269]. Some PKC inhibitors targeting the ATP-binding site such as indolcarbazole and bisindoylmaleimide have shown selectivity, e.g. ruboxistaurin is a relatively selective PKCβ inhibitor [270, 271]. PKC inhibitors competing at the DAG/phorbol ester or the PS binding site are more specific. Calphostin C binds to the C1 domain, mimicking DAG-binding [157]. Extended exposure to phorbol esters downregulate PKCα, β and γ [272], but phorbol esters have tumor-promoting properties.
Table 3.
Representative PKC Inhibitors
| Class/Inhibitor | Chemistry | Site of Action | Selectivity | Kd or IC50 | Ref |
|---|---|---|---|---|---|
|
Isoquinolines H-7 |
1-(5-isoquinolinesulfonyl)-2-methylpiperazines | ATP-binding site | Pan-PKC | PKCβI 3.5 μM PKCζ 6 μM |
[541] |
|
Benzophenones Chelerythrine |
1,2-dimethoxy-12-methyl[1,3]benzodioxolo[5,6- c]phenanthridin-12-ium |
ATP-binding site | Pan-PKC | 0.66 μm | [260] |
|
Indolocarbazoles Gö6976 |
5,6,7,13-tetrahydro-13-methyl-5-oxo-12H- indolo[2,3-a]pyrrolo[3,4-c]carbazole-12- propanenitrile |
Catalytic domain | PKCα, βI | PKCα 2.3, βI 6.2 nM | [542, 543] |
| Gö6983 | 1H-Pyrrole-2,5-dione, 3-[1-[3- (dimethylamino)propyl]-5-methoxy-1H-indol-3- yl]-4-(1H-indol-3-yl)- |
ATP-binding site. Suppreses PKCμ auto-phosphorylation | Pan-PKC PKCα, β, γ, δ > PKCζ |
PKCα 7, β 7, γ 6, δ 10, ζ 60 nM | [544, 545] |
| Enzastaurin (LY317615) | 3-(1-methyl-1H-indol-3-yl)-4-(1-(1-(pyridin-2- ylmethyl)piperidin-4-yl)-1H-indol-3-yl)-1H- pyrrole-2,5-dione |
ATP-binding site | PKCβ > PKCα, γ, ε | PKCα 39, β 6, γ 83, ε 110 nM | [546, 547] |
| LY379196 | ATP-binding site | PKCβ | 3– 6 μM | [548] | |
| Staurosporine (CGP41251) | 9,13-Epoxy-1H,9H-diindolo[1,2,3-gh:3′,2′,1′- lm]pyrrolo[3,4-j][1,7]benzodiazonin-1-one, 2,3,10,11,12,13-hexahydro-10-methoxy-9- methyl-11-(methylamino)-, [9S- (9α,10β,11β,13α)]- |
ATP-binding site | Pan-PKC PKCα, γ, η > PKCδ, ε |
PKCα 2, γ 5, δ 20, η 4 nM | [549, 550] |
| CGP53353 | 5,6-bis[(4-Fluorophenyl)amino]-1H-isoindole- 1,3(2H)-dione |
ATP-binding site | PKCβ | PKCβI 3.8, βII 0.41 μM | [551] |
| UCN-01 | 7-hydroxystaurosporine | ATP-binding site | cPKCs | 25–50 nM | [552] |
| Sotrastaurin (AEB071) | 3-(1H-indol-3-yl)-4-(2-(4-methylpiperazin-1- yl)quinazolin-4-yl)-1H-pyrrole-2,5-dione |
ATP-binding site | Pan-PKC, especially PKCθ | PKCα 0.95, βI 0.64, δ 2.1, ε 3.2, η 1.8, θ 0.22 nM | [553, 554] |
|
Staurosporine Analogs Ruboxistaurin (LY333531) |
(9S)-9-[[(Dimethyl-d6)amino]methyl]-6,7,10,11- tetrahydro-9H,18H-5,21:12,17- Dimethenodibenzo[e, k]pyrrolo[3,4- h][1,4,13]oxadiazacyclohexadecine-18,20(19H)- dione Hydrochloride |
ATP-binding site | PKCβI, βII | PKCβI 4.7, βII 5.9 nM | [499] |
| Midostaurin (PKC412, CGP41251) | (9S,10R,11R,13R)-2,3,10,11,12,13-Hexahydro- 10-methoxy-9-methyl-11-(methylamino)-9,13- epoxy-1H,9H-diindolo[1,2,3-gh:3′,2′,1′- lm]pyrrolo[3,4-j][1,7]benzodiamzonine-1-one |
ATP-binding site | Pan-PKC | 12 nM | [555] |
| Bisindolylmaleimid e (GF 109203X, Gö 6850) | 3-(1-(3-(Dimethylamino)propyl)-1H-indol-3-yl)-4- (1H-indol-3-yl)-1H-pyrrole-2,5-dione |
ATP-binding site | Pan-PKC, especially PKCα, β, γ | PKCα 8.4, βI 18, βII 16, γ 20, δ 210, ε 132, ζ 5800 nM | [556, 557] |
| Ro 31-8220 | Carbamimidothioic acid, 3-[3-[2,5-dihydro-4-(1- methyl-1H-indol-3-yl)-2,5-dioxo-1H-pyrrol-3-yl]- 1H-indol-1-yl]propyl ester, methanesulfonate |
Catalytic domain | Pan-PKC, PKCα, βI, βII, γ, ε | PKCα 5, βI 24, βII 14, γ 27, ε 24 nM | [404, 558] |
| SCH47112 | ATP-binding site | [559] | |||
|
Dicationic, lipophilic drugs Dequalinium Cl |
Quinolinium, 1,1′-(1,10-decanediyl)bis[4-amino- 2-methyl-, chloride (1:2) |
Covalently modifies the C2-domain | Pan-PKC | 7–18 μM | [269, 560, 561] |
|
Flavonoid Myricitrin |
4H-1-Benzopyran-4-one, 3-[(6-deoxy-α-L- mannopyranosyl)oxy]-5,7-dihydroxy-2-(3,4,5- trihydroxyphenyl)- |
DAG/Phorbol ester-binding site | PKCα, ε | [562] | |
| Quercetin | 4H-1-Benzopyran-4-one, 2-(3,4- dihydroxyphenyl)-3,5,7-trihydroxy- |
Weak PKC inhibitor | [563] | ||
|
Benzothiazole Riluzole |
6-(trifluoromethoxy)benzothiazol-2-amine | ATP-binding site | PKCα | [564] | |
|
Perylenequinone Calphostin C (UCN-1028C) |
1-[3,10-dihydroxy-12-[2-(4- hydroxyphenoxy)carbonyloxypropyl]-2,6,7,11- tetramethoxy-4,9-dioxoperylen-1-yl]propan-2-yl benzoate |
DAG/phorbol ester-binding site | cPKCs, nPKCs | 50 nM | [565] |
|
Phenolic ketone Rottlerin (Mallotoxin) |
5,7-dihydroxy-2,2-dimethyl-6-(2,4,6-trihydroxy- 3-methyl-5-acetylbenzyl)-8-cinnamoyl-1,2- chromene) |
ATP-binding site | PKCδ Other nPKCs |
PKCδ 5 μM Other PKCs 30 μM |
[566] |
|
Macrolactone Bryostatin 1 (NSC 339555) |
(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23 R,25S)-25-(Acetyloxy)-1,11,21-trihydroxy-17- [(1R)-1-hydroxyethyl]-5,13-bis(2-methoxy-2- oxoethylidene)-10,10,26,26-tetramethyl-19-oxo- 18,27,28,29- tetraoxatetracyclo[21.3.1.13,7.111,15]nonacos-8- en-12-yl (2E,4E)-2,4-octadienoate |
C1 domain. DAG/phorbol ester-binding site | PKCε > PKCα, δ | [157, 269, 567] | |
|
Membrane lipids D-erythro-Sphingosine |
2-Amino-4-octadecene-1,3-diol; trans-4- Sphingenine |
Phosphatidyl-serine-binding site | 2.8 μM | [568] | |
| N, N-Dimethyl-D-erythro-sphingosine | (E,2S,3R)-2-(Dimethylamino)octadec-4-ene-1,3- dio |
Phosphatidyl-serine-binding site | 12 μM | [569] | |
|
Taxol Tamoxifen |
2-[4-[(Z)-1,2-diphenylbut-1-enyl]phenoxy]-N, N- dimethylethanamine |
Regulatory domain | cPKCs | [570] | |
|
Purine nucleoside Sangivamycin |
4-amino-5-carboxamide-7-(D- ribofuranosyl)pyrrolo[2,3-d]pyrimidine |
ATP-binding site | 10 μM | [571] | |
| Other | α-tocopherol, adriamycin, aminoacridine, apigenin, cercosporin, chlorpromazine, dexniguldipine, polymixin B, trifluoperazine, UCN-02 |
[572] | |||
| Carbonitrile | 5-vinyl-3-pyridinecarbonitriles | Catalytic domain | PKCθ | 4.7 nM | [573] |
| Pyrimidine | 2,4-Diamino-5-nitropyrimidine | Catalytic domain | PKCθ | [574] | |
| Sterols | Spheciosterol sulfate A | Catalytic domain | PKCζ | 1.59 μM | [575] |
| Spheciosterol sulfate B | Catalytic domain | PKCζ | 0.53 μM | ||
| Spheciosterol sulfate C | Catalytic domain | PKCζ | 0.11 μM | ||
|
Antisense oligonucleotides Isis3521 (CGP64128A, Aprinocarsen) |
20-mer phosphorothioate oligodeoxynucleotide | Inhibits PKCα mRNA expression | PKCα | - | [576] |
| Isis9606 | 19-mer phophorothioate oligodeoxynucleotide | Inhibits PKCα mRNA | PKCα | - | [577] |
|
Short peptides Myristoylated-pseudosubstrate peptide inhibitor |
Peptide sequence: myr-FARKGALRQ | Substrate-binding site | cPKCs | - | [273] |
| αV5-3 | Peptide sequence: QLVIAN | Site: aa 642–647 | PKCα | - | [578] |
| βIV5-3 | Peptide sequence: KLFIMN | Inhibits PKC translocation Site: aa 646–651 |
PKCβI | - | [579] |
| βIIV5-3 | Peptide sequence: QEVIRN | Inhibits PKC translocation Site: aa 645–650 |
PKCβII | - | [580] |
| βC2–4 | Peptide sequence: SLNPEWNET | Site: aa 218–226 | All cPKCs | - | [188] |
| δV1-1 (KAI-9803, Delcasertib) | Peptide sequence: SFNSYELGSL | RACK-binding site Inhibits PKC translocation Site: aa 8–17 |
PKCδ | - | [581] |
| εV1-2 (KAI-1678) | Peptide sequence: EAVSLKPT | RACK-binding site Inhibits PKC translocation Site: aa 14–21 |
PKCε | - | [582] |
| KCe-12 and KCe-16 | Substrate-binding site | PKCε | - | [583] | |
| ZIP | Peptide sequence: SIYRRGARRWRKL | ζ–pseudo substrate | PKCζ and aPKCs | - | [584] |
| γV5-3 | Peptide sequence: RLVLAS | Site: aa 659–664 | PKCγ | - | [585] |
| Other PKC Inhibitors | α-tocopherol, adriamycin, aminoacridine, apigenin, cercosporin, chlorpromazine, dexniguldipine, polymixin B, trifluoperazine, UCN-02 |
[281, 282] |
>, greater selectivity; aa, amino acid
Peptides that interfere with the intramolecular interactions within PKC have been developed [191]. For instance, myr-ψPKC, a myristoylated peptide based on the substrate motif of PKCα and PKCβ, inhibits TPA-induced PKC activation and phosphorylation of MARCKS [273]. Other peptides disrupt protein-protein interactions between the PKC regulatory domain and RACK [157]. The interaction of PKC and RACK is isoform selective and largely involves the C2 region of cPKC, and peptide fragments of this region may serve as selective cPKCs inhibitors [188]. Also, a peptide derived from the PKC binding proteins annexin I and RACKI inhibits translocation of PKCβ [187].
The autoinhibitory role of the PKC pseudosubstrate has been suggested from the observation that deletion of the pseudosubstrate site abrogates the inhibitory effect of the regulatory domain of PKCα on the full-length enzyme [274]. Synthetic oligopeptides based on pseudosubstrate sequence are specific PKC inhibitors because they exploit its substrate specificity and do not interfere with ATP binding [212, 258, 273] (Table 3). The synthetic peptide (19–36) inhibits PKC autophosphorylation and protein substrate phosphorylation. Replacement of Arg-27 with alanine in the peptide [Ala-27]-PKC(19–31) increases the IC50 for inhibition of substrate phosphorylation. A structure-function study of the PKC pseudosubstrate sequence R19FARK-GALRQKNV31 examined the role of specific residues using an alanine substitution scan. Arg-22 was the most important determinant in the inhibitor sequence, since its substitution by alanine led to a 600-fold increase in its IC50. Substitutions of other basic residues with Ala-19, Ala-23 and Ala-27 increased the IC50 5-, 11- and 24-fold, respectively. The importance of basic residues in determining the potency of the pseudosubstrate peptide reflects the requirement of these residues in peptide substrate phosphorylation. Gly-24, Leu-26 and Gln-28 residues are also important for pseudosubstrate inhibitor potency. The large increase in the IC50 for the [A22]PKC(19–31) peptide makes it a valuable control in studies utilizing the pseudosubstrate peptide to examine functional roles of PKC [275]. Pseudosubstrate inhibitors are also more specific because the pseudosubstrate region provides a large interface for multiple points of contact with the PKC molecule [191, 258]. However, a cell-penetrating myristoylated PKCζ pseudosubstrate inhibitor peptide (ZIP) shows affinity for all PKC isoforms and disrupts PKC translocation, suggesting that some pseudosubstrates have well-conserved residues [258]. Also, mutation of alanine in the pseudosubstrate with serine or glutamate, mimics the charge of a phosphorylated residue and in effect activates PKC [209, 274, 276].
Some compounds like β-adrenoceptor activators and antioxidants counteract the effects of PKC. In portal vein, stimulation of β-adrenoceptors activates cAMP-dependent protein kinase (PKA) and in turn opposes the effects of PKC, causes vasodilatation and inhibits store-operated Ca2+ entry [277, 278]. The PKC catalytic domain contains several reactive cysteines that can be targeted by antioxidants such as selenocompounds, vitamin E, and curcumin [256, 279, 280]. Also, α-tocopherol inhibits the expression, activity, and phosphorylation of PKCα and decreases VSM proliferation, and these effects are not mimicked, and even be opposed, by β-tocopherol [281, 282]. In animal models, hyperglycemia-induced retinal vascular dysfunction is prevented by α-tocopherol likely through inhibition of DAG-PKC [282]. Vitamin E decreases hyperglycemia-induced DAG and PKC activity and reverses the changes in the retinal and renal vessels in diabetes [283]. Glutathione inhibits PKC via a nonredox mechanism [284]. Tamoxifen is an estrogen receptor antagonist and a PKC inhibitor, and some tamoxifen analogs have high affinity and selectivity to PKC [285]. PKCZI195.17 is a novel inhibitor with high efficacy and specificity to PKCζ [286], and CGX1037 is a specific PKCδ inhibitor in platelets [287].
Post-translational modifications of PKC affect its activity. S-nitrosylation, a ubiquitous protein modification in redox-based signaling that forms S-nitrosothiol from nitric oxide (NO) on cysteine residues, decreases PKC activity and contraction in mouse aorta, and may represent a key mechanism in conditions associated with decreased vascular reactivity [288].
Transgenic animals, knockout mice and antisense techniques have been used to study the effects of PKC downregulation in vivo. PKC knockout mice have shown a critical role of PKC in the endocrine and immunological systems, and further characterization of the PKC knockout vascular phenotype should shed more light on the role of PKC in the vascular system. Also, antisense and siRNA for specific PKC isoforms can be used to study the role of PKC in various cell functions. ISSI-3521 is a phosphorothioate antisense oligonucleotide that targets the 3′-untranslated region of PKCα mRNA, and causes reduction of PKCα expression in cancer cell lines and human tumor xenograft models [269, 289].
Thus, while the ~70% homologous structure of the PKC catalytic domain poses a challenge in the development of specific inhibitors of PKC isoforms, the C2 region is less conserved among different PKCs, and pharmacological tools that target the C2 region are more selective [157]. The V5 region may also be a good target for isoform-specific PKC inhibitors. The PKC-substrate interaction can be selectively disrupted by peptide inhibitors that share the same substrate sequence. Also, protein-protein interactions can regulate the localization and activity of PKC isoforms [157].
7.7. PKC and VSM Contraction
PKC isoforms show diverse effects in different cell types including VSM. The role of PKC in vascular responses has been supported by measuring PKC mRNA expression, protein levels and activity, and by testing the effects of PKC inhibitors and the vascular changes in PKC knockout mice and transgenic rats [290]. PKC can affect VSM contraction through regulation of ion channels, pumps and [Ca2+]c, Ca2+ sensitization of the contractile proteins, and activation of Ca2+ independent contraction pathways. PKC translocation to the cell surface could also trigger a cascade of protein kinases that ultimately interact with the contractile myofilaments and cause VSM contraction. In some instances, PKC inhibits VSM contraction.
7.8. PKC, Ion Channels, and [Ca2+]c
PKC modulates the activity of plasmalemmal K+ and Ca2+ channels, and in turn [Ca2+]c. K+ channels play a role in the regulation of the resting membrane potential, and inactivation of K+ channels causes membrane depolarization, elevation of [Ca2+]c and VSM contraction [291]. Membrane depolarization activates Ca2+ entry through L-type VDCCs and could facilitate Ca2+ release from IP3- and ryanodine-sensitive Ca2+ stores [292, 293]. Large conductance Ca2+-activated K+ channels (BKCa) are the predominant K+ channels in VSMCs [291, 294]. PKC activation by PDBu inhibits BKCa and increases vascular tone [295–298] in pulmonary [296], coronary [299], cerebral [300], and uterine vessels [301]. PKC activators inhibit BKCa by phosphorylation of the channel protein and decreasing its sensitivity to activation by cGMP-dependent protein kinase [302, 303].
Voltage-gated K+ channels (Kv) can be modulated by vasoconstrictors such as arginine-vasopressin, ET-1 and AngII through activation of PKC. In rat mesenteric artery VSMCs, vasopressin regulates Kv7.4 and Kv7.5 subunits of Kv7 channels via activation of PKC. PKCα-dependent phosphorylation of the K+ channel proteins on serine residues is sufficient to reduce Kv7 channel activity, and the extent of PKC-mediated Kv7.4 and Kv7.5 phosphorylation and K+ current suppression depends on the subunit composition of the channel proteins [304]. Also, thromboxane A2 induces pulmonary vasoconstriction by a mechanism involving PKCζ and inhibition of Kv [305]. In rabbit coronary arterial VSMCs, ET-1 and AngII inhibit Kv currents by activating PKCε, and inhibit KIR channel by activating PKCα [306, 307].
Vasoconstrictor agonists also inhibit KATP through PKC signaling [291, 308]. Phorbol esters inhibit KATP currents in mesenteric arteries [309]. In human embryonic kidney cells (HEK293) AngII and PDBu induce PKC-mediated inhibition of KATP channel through the formation of channel complexes comprising four Kir6.1 and their associated SUR2B subunits [310]. PKC-mediated phosphorylation of KATP alters the channel properties, kinetics and/or number at the cell membrane [311, 312]. Protein trafficking studies have suggested that PKC initiates internalization and inactivation of KATP channel complex [313].
7.9. PKC, Ion Pumps, Co-transporters, and [Ca2+]c
PMCA and SERCA are important Ca2+ removal mechanisms in VSM. PKC activates PMCA or SERCA, leading to Ca2+ extrusion and re-uptake and decreased VSM [Ca2+]c. In isolated cardiac SR preparations, PKC activates SERCA [314]. Also, PKC-mediated inhibition of the α1 subunit of Na+/K+-ATPase affects the intracellular concentrations of Na+ and K+ and in turn membrane potential [315]. PKC activation by phorbol esters and DAG analogs phosphorylates and activates the Na+/H+ antiport exchanger and thereby increases the cytoplasmic pH, leading to alkalinization which generally increases vascular contraction [316–319].
7.10. PKC and Ca2+-Sensitization of Contractile Proteins
PKC activation increases the myofilament force sensitivity to [Ca2+]c, thereby maintaining VSM contraction with smaller increases in [Ca2+]c. In pulmonary arteries, PKC inhibitors attenuate ET-1 induced constriction and [Ca2+]c and the vasoconstrictor responses associated with store-operated Ca2+ entry, suggesting that PKC contributes to both Ca2+ influx and Ca2+ sensitization [320]. The nPKC isoforms play an important role in mediating VSM contraction through a Ca2+ sensitizing pathway, and inhibition of nPKCs attenuates norepinephrine-induced VSM contraction [321]. PKC phosphorylates CPI-17, which in turn inhibits MLC phosphatase, increases MLC phosphorylation, and enhances VSM contraction [322]. PKC also inhibits MLC-phosphatase through phosphorylation of myosin phosphatase target subunit 1 (MYPT1) [323]. PKC may contribute to VSM contraction in a MLC phosphorylation-independent manner. In rat middle cerebral artery, PKC activation by PDBu is associated with sustained vasoconstriction that is much larger than that expected with the same level of MLC phosphorylation achieved by 5-HT [323]. Also, activation of PKCα causes phosphorylation of the actin-binding protein CaP, and thereby reverses its inhibition of actin-activated myosin ATPase, and enhances VSM contraction. Interestingly, CaP activates PKC in vitro in the absence of lipid cofactors, and knockdown of CaP inhibits PKC-dependent contraction in ferret arterial VSM [5, 324].
PKC may also be involved in stretch-induced vascular myogenic response. In rat cerebral artery VSMCs, DAG, PMA and cell swelling activate a cation current through stretch-activated TRP channels, and these effects are blocked by PKC inhibitors. It has been suggested that myogenic tone involves mechanotransduction through stimulation of PLC, hydrolysis of phosphoinositides, DAG production, and PKC activation, which increase cation channel activity, and the associated depolarization activates L-type VDCCs, leading to increased [Ca2+]c and vasoconstriction [277, 325]. PKCδ, PKCθ and PKCμ participate in VSM mechanotransduction in response to mechanical and cyclic stretch [263, 326].
AngII, ET-1, serotonin, norepinephrine, and neuropeptide Y activate PKC-dependent pathways and cause VSMC membrane depolarization and contraction [308, 327, 328]. Of note, AngII activates multiple PKC isoforms in VSM [329] and PKC increases VSM contraction via other pathways involving downregulation of atrial natriuretic peptide (ANP) receptor and decreased ANP-induced inhibition of contraction [330]. PKC affects not only arterial but also venous contraction. In deoxycorticosterone salt-sensitive rat model of hypertension ET-1 increases venomotor tone, suggesting a role of the venous system in regulation of blood pressure likely through increases in venous return and cardiac output [331]. The PKC inhibitor chelerythrine attenuates ET-1-induced contraction in both the aorta and vena cava. However, in the aorta, ET-1-induced contraction is dependent on PLC activation and IP3-mediated Ca2+ release, while in the vena cava ET-1 induced contraction is unaffected by the IP3 receptor antagonist 2-APB. Also, only the vena cava contracts in response to the DAG analog OAG, highlighting the differences in the venous and arterial pathways of contraction [331]. Endothelium-derived NO also regulates VSM tone by activating guanylate cyclase, increasing cGMP and promoting vasodilation, and PKC inhibits NO-mediated vasodilation by inhibiting guanylate cyclase, leading to increased vasoconstriction [332].
7.11. PKC and Cytoskeletal Proteins
Studies in cerebral microvessels have shown that PKC mediates myogenic constriction through dynamic reorganization of the cytoskeleton and increased actin polymerization [333]. Also, both in the presence and absence of Ca2+, PKC promotes cerebral vasoconstriction by increasing the phosphorylation of paxillin and HSP27, reducing G-actin content, and promoting actin cytoskeleton reorganization [323]. In rat middle cerebral arteries, PDBu-induced constriction is more sensitive to disruption of actin cytoskeleton compared to inhibition of cross-bridge cycling, supporting the pivotal contribution of PKC-mediated cytoskeletal actin polymerization to force generation in cerebral arteries [323].
PKC modulates certain genes that code for structural proteins such as fibronectin and type IV collagen by changing the binding of nuclear transcription factors to the promoter regions on responsive genes [268]. PKC affects the expression of the regulator of G-protein signaling 2 (RGS2), which affects vascular tone. In cultured VSMCs, adrenotensin increases RGS2 expression, while the PKC inhibitor chelerythrine reduces RGS2 expression, suggesting that adrenotensin increases RGS2 expression via a PKC-mediated pathway [334].
7.12. PKC-Dependent Signaling Cascades
PKC activation may trigger a cascade of protein kinases that ultimately stimulate VSM contraction. PKC affects Akt signaling [260, 335]. Also, mitogen-activated protein kinases (MAPK) such as extracellular signal-regulated kinase (ERK), p38 and JNK are common downstream effectors of PKC [336, 337]. MAPK is a Ser/Thr kinase that is activated by its dual phosphorylation at Thr and Tyr residues. PKC, MAPK, and c-Raf-1 have been implicated in VSM growth. In cultured VSMCs, MAPK is mainly cytosolic, and translocates to the nucleus during activation by mitogens [338]. Tyrosine kinase and MAPK activities have also been identified in differentiated contractile VSM. During VSM activation, MAPK transiently translocates to the plasma membrane, then undergoes redistribution to the cytoskeleton during maintained VSM contraction [339]. DAG promotes translocation of cytosolic PKCε to the plasma membrane, where it is fully activated. Activated PKCε then stimulates the translocation of cytosolic MAPK kinase (MEK) and MAPK to the plasmalemma, where they form a kinase complex. PKC phosphorylates and activates MEK, which in turn phosphorylates MAPK at both Thr and Tyr residues. Tyrosine phosphorylation targets MAPK to the cytoskeleton, where it phosphorylates the actin-binding protein caldesmon (CaD) and reverses its inhibition of Mg2+-ATPase activity, thus increasing actin-myosin interaction and VSM contraction [5, 339]. Also, in aortic VSM, phenylephrine promotes CaP-dependent PKC autophosphorylation and activation, followed by a delayed ERK activation, CaD phosphorylation and VSM contraction [154]. These PKC-dependent pathways function in parallel with the spike in [Ca2+]c and MLC phosphorylation to enhance VSM contraction [154]. Biochemical studies have shown that PDBu does not directly change the phospho-content of CaP or CaD [323], supporting that other kinases downstream of PKC cause phosphorylation of CaP or CaD [154]. In, esophageal smooth muscle, PKC-dependent resting tone and contraction are associated with ERK and HSP27-linked p38 MAPK phosphorylation [340], supporting that MAPK serves as a link in the signal transduction cascade between membrane-bound PKC and smooth muscle contraction.
7.13. PKC and Vasodilation
PKC affects Ca2+ channel permeability in VSM. In VSMCs, GPCR agonists affect ROCs, TRPCs, and SOCs activity via activation of PKC. While low levels of DAG activate TRPC6 via a PKC-independent mechanism, high levels of DAG inhibit TRPC6 SOCs activity in a PKC- and Ca2+-dependent manner [277, 341]. In mesenteric artery and ear artery VSMCs, DAG inhibits TRPCs through a PKC-dependent pathway, and such mechanism may limit ROC activity at high agonist concentrations [342].
The 20-kDa MLC and MLC kinase also serve as substrates for PKC, and their phosphorylation counteracts Ca2+-induced actin-myosin interaction and VSM contraction [343]. In human VSMCs, PKC activation stimulates secretion of C-type natriuretic peptide (CNP), a known endogenous vasodilator [344, 345]. PMA increases CNP expression via PKCα- and PKCδ-mediated pathways, and PDGF increases CNP in SMCs via a PKCδ-dependent pathway [346]. PKC also regulates the activity of endothelial NO synthase, and in turn NO production and vasodilation [321].
8. Rho Kinase
GPCR agonists, particularly those coupling to Ga12/13 proteins, can also activate the small G-protein RhoA. In its active GTP-bound form, RhoA activates Rho-associated coiled-coil protein kinase or Rho-kinase (ROCK), which then phosphorylates and inhibits MLC phosphatase, increases MLC phosphorylation and promotes VSM contraction. The ROCK-mediated enhancement of VSM contraction often occurs in the absence of substantial increases in [Ca2+]c and is therefore considered a Ca2+ sensitization mechanism [347–350].
8.1. ROCK Structure and Isoforms
ROCK was initially described in the mid 1990s as a member of AGC family of protein kinases [351, 352], and a major target of the small GTPase RhoA. ROCKs are Ser/Thr kinases with a molecular mass of ~160 kDa. Two ROCK isoforms encoded by two different genes have been identified: ROCK-1 (ROCK-I, ROKα) and ROCK-2 (ROCK-II, ROKβ) [351, 353–356]. Human ROCK-1 and ROCK-2 genes are located on chromosome 18 (18q11.1) and chromosome 2 (2p24), respectively. ROCK structure comprises a kinase domain located at the N-terminus of the protein (amino acids 1–420), a coiled-coil region containing the Rho-binding domain, and a pleckstrin-homology domain with a cysteine-rich domain at the C-terminus that helps in ROCK binding to the plasma membrane (Fig. 5). ROCK-1 and ROCK-2 are highly homologous, with 65% overall amino acid sequence identity; 92% in the kinase domain and 58% in the Rho-binding domain [355].
Fig. 5.

Structure and activation of ROCKs. (A) ROCK amino acid sequence comprises a kinase domain located at the N-terminus, a coiled-coil region containing the Rho-binding domain, and a pleckstrin-homology domain (PHD) with a cysteine-rich domain (CRD). ROCK-1 and ROCK-2 are highly homologous with an overall amino acid sequence identity of 65%. (B) In the inactive form, the C-terminus region of ROCK is folded over the N-terminus region, allowing the autoinhibitory region to block the kinase site. Binding of activated GTP-bound RhoA causes unfolding and activation of ROCK, and thereby exposes the kinase domain and allows phosphorylation of the true substrate.
8.2. Tissue Expression of ROCK
ROCK-1 and ROCK-2 are ubiquitously expressed. Both ROCK-1 and ROCK-2 are expressed in VSM and the heart [357], and ROCK-2 mRNA is highly expressed in the brain and skeletal muscle [355, 358]. ROCK-1 and ROCK-2 expression is up-regulated by AngII via angiotensin type 1 receptor and by interleukin-1β [359], possibly through PKC- and nuclear factor κB-dependent pathways. Chronic administration of AngII in mice causes up-regulation of ROCK in the coronary artery [359].
8.3. Subcellular Distribution of ROCK
Similar to other GTPases, RhoA cycles between an inactive guanosine diphosphate (GDP)-bound form and an active GTP-bound form. In unstimulated cells, RhoA resides mainly in the cytosol bound to GDP, but upon receptor stimulation it undergoes translocation to the plasma membrane where GDP–GTP exchange takes place [350]. RhoA translocation to the plasma membrane is facilitated by the hydrophobic geranylgeranyl tail that is attached to its C-terminal during post-translational modification of the protein. ROCKs are also essentially distributed in the cytosol and are partially translocated to the plasma membrane during activation by RhoA [351, 354]. The mechanisms responsible for translocation of ROCKs could be similar to those involved in the translocation of PKC.
8.4. Regulation of ROCK Activity
The C-terminal of ROCK contains an autoinhibitory region [360], including the pleckstrin-homology domain and Rho-binding domain, which binds to the N-terminal kinase domain and inhibits ROCK activity [361] (Fig. 5). During ROCK activation, the binding of RhoA disrupts the interaction between the C-terminal autoinhibitory region and kinase domain and yields an active kinase. Truncated forms of ROCK lacking the C-terminal region that contains the Rho-binding domain and pleckstrin-homology domain are constitutively active. However, when the C-terminal region of ROCK is expressed in cells, it acts as dominant negative [362], suggesting that the C-terminal region is a negative regulatory region responsible for autoinhibition of kinase activity in resting cells [363]. Binding of active GTP-bound form of RhoA to Rho-binding domain increases the phosphotransferase activity of ROCK 1.5- to 2-fold [364]. Arachidonic acid is a metabolic by-product of the agonist–receptor interaction produced from the hydrolysis of membrane phospholipids by phospholipase A2 or through the transformation of DAG to arachidonic acid by DAG lipase. At micromolar concentrations, arachidonic acid inhibits MLC phosphatase and Ca2+-dependent MLC phosphorylation [365]. Arachidonic acid and sphingosine phosphorylcholine interact with the regulatory region and pleckstrin-homology domain of ROCK, disrupt their inhibitory action on the catalytic domain, and increase ROCK activity 5- to 6-fold independently of RhoA [363, 364, 366].
Dimerization affects ROCK activity and affinity for ATP [367]. ROCK2 harbors a C-terminal extension within the kinase domain that contains a hydrophobic cluster of phenylalanine and tyrosine residues surrounding a key threonine residue. The hydrophobic motif at Thr405 of ROCK is essential for substrate phosphorylation and kinase domain dimerization. Mechanistically, both ROCK2 activity and dimerization are dependent upon the interaction between Thr405 of the hydrophobic motif and Asp39 of the N-terminal extension. The observation that ROCK2 hydrophobic motif requires association with the N-terminal extension for kinase activity provides the rationale for the development of small-molecule inhibitors designed to block ROCK activation by selectively interfering with hydrophobic motif-mediated activation and dimer formation [368].
Other Rho proteins exert negative control on ROCK activity. The small G-protein RhoE binds to the N-terminal region of ROCK1 containing the kinase domain, thus preventing RhoA from binding to Rho-binding domain and inhibiting ROCK [369]. Other small G proteins such as Gem and Rad bind and inhibit ROCK through an unclear mechanism [370].
8.5. ROCK Substrates
Activated RhoA interacts with the Rho-binding domain of ROCK and induces a conformational change that allows the interaction of the Ser/Thr kinase with its substrates [371]. The consensus sequence of ROCK phosphorylation site is RXXS/T or RXS/T [372–375]. ROCKs require basic amino acids such as Arg (R) close to its phosphorylation site. Several ROCK substrates have been identified, and the functional consequence of their ROCK-mediated phosphorylation is often related to actin filament formation and organization, and cytoskeleton rearrangements [376, 377]. Proteomics analysis has identified more than 100 proteins as potential substrates of ROCK [378].
ROCK targets include the myosin phosphatase target subunit-1 (MYPT-1) [379], CPI-17 [380], the 20-kDa MLC [372], and CaP [381], which are known effectors of VSM contraction. MYPT-1 is a major effector of ROCK-mediated Ca2+ sensitization of smooth muscle contraction. Cardiac troponin is another ROCK substrate, but ROCK-mediated phosphorylation of troponin reduces contraction in cardiac myocytes [382].
Phosphatase and tensin homologue (PTEN) is another ROCK substrate [383]. PTEN dephosphorylates the phosphatidylinositol 3-kinase (PI3-kinase)/Akt pathway involved in the regulation of mRNA transcription, protein synthesis, and cell growth and survival. Phosphorylation of PTEN by ROCK stimulates its phosphatase activity, and ROCK inhibitors reduce ROCK-mediated PTEN phosphorylation and enhance Akt signaling in endothelial cells [384]. Activated ROCK also phosphorylates the insulin receptor substrate-1 (IRS-1) in VSMCs, leading to inhibition of insulin-induced IRS-1 tyrosine phosphorylation and PI3-kinase activation [385]. The interaction between ROCK and IRS-1 is increased, and insulin signaling is markedly decreased in VSMCs from hypertensive rats [385].
Because the kinase domains of ROCK-1 and ROCK-2 are nearly identical, it has been thought that they share the same substrates. The N-terminal region that precedes the kinase domain determines the substrate specificity of the ROCK isoforms [369]. ROCK-1, but not ROCK-2, binds to and phosphorylates RhoE [386], while elongation initiation factor-1-α-1 is a ROCK-2 substrate [387].
8.6. ROCK and VSM Function
A large body of evidence suggests important functions of ROCK in VSMCs. A major role of ROCKs is in the organization of actin cytoskeleton, a process involved in multiple cell functions including proliferation, apoptosis, contraction, migration and adhesion [388–390]. ROCK plays a role in phosphorylation and inhibition of MLC phosphatase [391], activation of LIM-kinase 2 and formation of stress fibers, focal adhesions and membrane blebs [392], phosphorylation of ezrin/radixin/moesin [374] and phosphorylation of adducin and regulation of cell motility [375]. RhoA-mediated ROCK activation phosphorylates MYPT-1, the regulatory subunit of MLC phosphatase, and thereby inhibits MLC phosphatase, causes Ca2+ sensitization of the contractile proteins, and enhances VSM contraction. While both ROCK-1 and ROCK-2 have been implicated in microfilament bundle assembly and smooth muscle contraction, ROCK-2, but not ROCK-1, binds to and is sensitive to phosphatidylinositol 3,4,5-P3 [393]. Also, some studies suggest that ROCK-2 is the major isoform regulating VSMC contraction through direct binding to MLC phosphatase [394].
ROCK also regulates cell migration, proliferation, apoptosis/survival, gene transcription, and differentiation. ROCK-1-deficient mice have open eyelids at birth [395]. ROCK-2-deficient mice show placental dysfunction and intrauterine fetal growth restriction [396, 397]. The vascular phenotype of ROCK-1 and ROCK-2 knockout mice needs to be further analyzed.
8.7. ROCK Inhibitors
ROCK activation involves RhoA translocation to the plasma membrane, RhoA binding to ROCK, and ATP-dependent phosphorylation of various substrates. Disruption of prenylation by 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins) or protein prenyltransferase inhibitors prevents the membrane translocation and activation of RhoA and impairs its ability to activate ROCK.
Some ROCK inhibitors compete with ATP for binding at the kinase domain, or “active site” of the enzyme. Commonly known ROCK inhibitors include Y-27632 and fasudil. Y-27632 is a synthetic pyridine derivative that inhibits ROCK by competing with ATP for the kinase active site. Y-27632 has ~200- and ~2000-fold higher affinity for ROCK than the structurally similar kinases PKC and MLC kinase, respectively. Y-27632 lowers blood pressure in animal models of experimental hypertension, supporting a link between VSM ROCK and increased blood pressure [397]. Experiments with Y-27632 have also suggested that ROCK activity might be enhanced in the cerebral circulation during chronic hypertension [398–401]. Despite the utility of Y-27632 in numerous studies and its promise as an antihypertensive agent [397, 402, 403], its specificity and safety profile have not been fully verified. Y-27632 inhibits PKC-related kinase (PRK2) with a similar potency to that of ROCK-2 [404]. At 10 μM concentration Y-27632 inhibits PKC-dependent vasoconstriction of the aorta and superior mesenteric artery [405], suggesting that ROCK activation occurs downstream of PKC, or that Y-27632 causes nonselective inhibition of PKC.
Fasudil is an isoquinoline derivative that inhibits ROCK by competing with ATP for the kinase active site. Fasudil has been used to assess the role of ROCK in vascular function in small-scale clinical studies [406–408]. The good safety profile of fasudil contributed to its approval for treatment of cerebral vasospasm following subarachnoid hemorrhage in Japan. Fasudil is a prodrug, and after its oral administration, it is metabolized to the more selective ROCK inhibitor hydroxyfasudil. In porcine model of coronary vasospasm, hyroxyfasudil potently inhibited vasospasm and hypercontraction [409]. Of note, the active site of ROCK is similar to that of protein kinase A (PKA), the target of cAMP that mediates vasodilation and inhibits platelet aggregation, and therefore fasudil inhibits ROCK and PKA with equal potency and is prone to cause clinical side-effects. Although hydroxyfasudil is 15-fold more selective for ROCK than for PKA, the possibility that fasudil could exert unwanted vascular effects if its metabolism is compromised should be considered. Studies have identified amino acid sequence differences between ROCK and PKA, which could be used to further improve the selectivity of ROCK inhibitors [410].
Other ROCK inhibitors include PT-262 which inhibits RhoA-ROCK-MLC pathway by interacting with the ATP-binding site of ROCK protein [411]. DJ4 is a multi-kinase inhibitor of ROCK and MLC kinase in an ATP competitive manner [412]. FSD-C10 and K-115 showed superior neuroprotective effects and lower cytotoxicity compared to fasudil [413, 414]. L-F001 also showed inhibitory effect on ROCK activity [415].
9. VSM Dysfunction and Vascular Disease
Identification of the mechanisms of VSM contraction has helped to understand the mechanisms of vascular disease and to develop new tools for the management of vascular disorders. Increased Ca2+ permeability of plasma membrane channels and [Ca2+]c have been demonstrated in VSMCs isolated from animal models of hypertension and coronary vasospasm [137, 416, 417], and Ca2+ channel blockers could be useful in these conditions. Ca2+ antagonist-insensitive forms of hypertension and coronary vasospasm require other treatment modalities that target other pathways such as PKC and ROCK
PKC plays a role in the pathogenesis of vascular restenosis, coronary artery disease, cerebral vasospasm, hypertension, and vascular complications of diabetes [6, 418, 419]. PKC contributes to vascular restenosis following vascular bypass and angioplasty procedures by promoting thrombosis and inflammation and subsequent VSMC migration and proliferation [420]. PKCα, β, δ and θ are expressed in platelets, and cPKCs promote while nPKCs inhibit platelet aggregation and thrombus formation [421]. Knocking out PKCδ or PKCθ potentiates murine platelet aggregation, and the PKCδ inhibitor rottlerin potentiates human platelet aggregation [422]. PKCα, β and ζ also potentiate the expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), a key step in leucocyte recruitment and VSMC migration [423–425]. PKCα, β and δ further affect VSMC migration by promoting actin polymerization and cell adhesion [426–428]. PKCα is involved in VSMCs migration induced by ROS [429], and PKCε promotes VSMC migration by upregulating matrix metalloproteinases MMP-2 and MMP-9 [420, 430, 431]. PKC also contributes to VSMC proliferation. PKCβ mediates synergistic proliferative effect of PDGF and high glucose in human coronary VSMCs [432], and the PKCβ inhibitor LY-379196 attenuates DNA synthesis and cell growth [260]. In rat models of aortic balloon injury, the PKCε activator ψεRACK promotes neointimal development, while the PKCε inhibitor εV1-2 reduces luminal narrowing, neointimal proliferation, VSMC ERK phosphorylation, and PDGF-induced VSMC proliferation/migration [433]. Repeated stimulation with phorbol myristate acetate (PMA) downregulates PKCε and inhibits VSMC proliferation [434].
PKCδ contributes to coronary artery disease through decreased ATP generation and increased ROS formation, apoptosis and necrosis [157, 435, 436]. On the other hand, PKCε protects mitochondrial functions and proteasomal activity, activates aldehyde dehydronage 2 (ALDH2) and reduces aldehyde load [157, 189, 437, 438]. A combination of a PKCδ inhibitor and PKCε activator could be useful for organ preservation, and prevention of ischemia-reperfusion injury and graft coronary artery disease in cardiac transplantation [439].
PKCδ has a deleterious effect in cerebral reperfusion by promoting neutrophil migration into ischemic tissue. In a model of transient middle cerebral artery occlusion, PKCδ-null mice showed decreased neutrophil migration into ischemic tissues and a 70% reduction in stroke size compared with wildtype. Transplantation of bone marrow from PKCδ-null mice into wildtype mice reduced infarct size, while bone marrow transplantation from wildtype donors increased infarction size and worsened neurological scores in PKCδ-null mice [440, 441]. Inhibition of PKCδ also improves microvascular pathology and function in transient focal ischemia and reduces ischemic damage in normotensive and hypertensive animals. Therefore, PKCδ could be a therapeutic target for the preservation of cerebrovascular function following stroke, and its inhibition may reduce stroke risk in hypertensive patients [442]. PKCζ mRNA is also induced in the cerebral cortex after focal brain ischemia, and inhibiting PKCζ prevents N-methyl-D-aspartate (NMDA)-induced excito-toxic neuronal cell death [440, 443]. In contrast, PKCε mediates ischemic tolerance in response to cerebral ischemic stress. Systemic delivery of PKCε activator ψεRACK immediately prior to stroke confers neuroprotection against a subsequent cerebral ischemic event, and decreases microvascular cerebral blood flow thus further contributing to cerebral protection [444].
PKC plays a role in hypertension, and mutations of PKC may influence the individual susceptibility to vascular hyper-reactivity and hypertension. A consistent association has been found between the single nucleotide polymorphism (SNP) rs9922316 in PKCβ gene (PRKCB) and inter-individual variation in the constriction responses of dorsal hand vein to the α2-adrenergic receptor agonist dexmedetomidine [445]. PKCα plays a role in Ca2+-dependent contraction of VSM [446], and overexpression of PKCα has been implicated in the pathogenesis of hypertension [447]. In normotensive rats, PKCα is localized mainly in the cytosol of VSMCs, and is hyperactivated and concentrated at VSM plasma membrane in hypertensive animals [447]. Also, PKCδ expression is increased in VSM from spontaneously hypertensive rats, and increases VSM contraction by decreasing BKCa channel conductance, as the PKC inhibitor chelerythrine restores K+ channel activity [298].
Sleep apnea could cause systemic and pulmonary hypertension [448, 449]. Chronic hypoxia attenuates store-operated Ca2+ entry in pulmonary arteries from Sprague-Dawley rats, while augmenting this response in Wistar rats. PKC inhibition restored store-operated Ca2+ entry in Sprague-Dawley arteries, and had no effect in Wistar arteries, suggesting rat strain difference in the role of PKC and store-operated Ca2+ entry in the pulmonary arteries following chronic hypoxia [450]. Rat models of sleep apnea produced by exposure to eucapnic intermittent hypoxia show increased circulating ET-1 levels and ET-1-dependent systemic hypertension that is likely mediated by PKCδ-dependent VSM Ca2+ sensitization in systemic arteries [449, 451]. On the other hand, intermittent hypoxia mediates a PKCβ-dependent increase in the pulmonary arterial reactivity to multiple vasoconstrictors including ET-1 [452]. In fawnhooded rat model of pulmonary hypertension, PKC inhibits BKCa, resulting in indirect activation of VDCCs and pulmonary vasoconstriction [453]. PKC-mediated Ca2+ sensitization is also demonstrated by an increase in phosphorylated CPI-17 in pulmonary arteries from newborn swine exposed to hypoxia [454]. PKCε may have divergent effects in pulmonary hypertension. PKCε null mice show decreased acute hypoxic pulmonary vasoconstriction, increased Kv3.1b channel expression and membrane hyperpolarization [455]. However, PKCε null mice exposed to chronic hypoxia show a greater pulmonary arterial pressure compared to wild-type mice, and the increase in pressure is reversed by inhaled NO, suggesting that PKCε mediates hypoxic downregulation of NO synthase [456].
Hypertension in pregnancy and preeclampsia are major complications of pregnancy, and placental ischemia/hypoxia could be an initiating event. Hypoxia during pregnancy attenuates the effects of sex steroids, and enhance PKC activity and vascular tone in uterine arteries of pregnant sheep [457]. Increased BKCa channel activity inhibits PKC-mediated contraction in ovine uterine arteries during pregnancy, and gestational hypoxia upregulates PKC and inhibits BKCa [458]. Hypoxia also inhibits KIR channels via PKC-dependent mechanism, and contributes to the maladaptation of uterine vascular hemodynamics in preeclampsia [327]. In cultured rat cardiomyocytes, IgG obtained from preeclamptic women enhances angiotensin type 1 receptor-mediated response, which is ameliorated with the PKC inhibitor calphostin C, supporting a role of PKC in preeclampsia [459].
PKC plays a role in diabetes-related vascular pathology by promoting cell growth and proliferation, cell permeability, oxidative stress, and phospholipase A2 activity; inhibition of K+ channels and Na+/K+-ATPase; increasing vascular reactivity, extracellular matrix and remodeling; and increasing vascular inflammation and pro-inflammatory cytokines [247, 460, 461]. In diabetes, PKC is activated by advanced glycation end (AGE) products and polyol pathway flux [270, 293, 462]. Activated PKC increases endothelial cell permeability and angiogenic factors, which could contribute to the loss of capillary pericytes, and increased retinal capillary permeability, ischemia, and neovascularization associated with diabetes [463–467]. High glucose stimulates DAG production and activates PKC in bovine aortic endothelial cells and VSMCs [468], and PKC activation alters the expression of vascular endothelial growth factor (VEGF), PDGF, and transforming growth factor-β [467, 469], and in turn affect extracellular matrix proteins and vascular remodeling [470]. PKC is also activated by ROS generated by different oxidases and the mitochondrial electron transport chain, and following AGE:RAGE (AGE receptor) interactions [471], and PKC in turn activates NADPH oxidases and further increases ROS [293, 472, 473]. High glucose via PKC activation and oxidative stress also reduces VSMC Kv current resulting in VSMC depolarization, activation of L-VDCCs and increased vasoconstriction [474–477]. PKC promotes Ca2+ sensitization in VSM myofilaments and vascular reactivity in diabetes [293]. Diabetic patients have reduced nocturnal dip in blood pressure and increased vascular complications, partly due to lack of diurnal PKC inhibition [478, 479]. Unsaturated fatty acids and their coenzyme A esters work synergistically with DAG to activate PKC [268], while long-chain omega-3 polyunsaturates from fish oil limit PKC activity. Fish oil-rich diets reduce blood pressure [480] and vasoconstriction to AngII and norepinephrine in humans [481], likely through decreased DAG production and PKC activity, and treatment of rat VSMCs with eicosapentaenoic acid prevents vasopressin-induced increase in DAG production [482]. In VSMCs, high glucose activates PKCα, β, δ, and ε, but not PKCζ [467, 483, 484]. PKCβ and δ are the dominant PKCs in diabetic vasculopathy. In rat VSMCs, high glucose increases PKCβ and δ in the membrane fraction, and p38 MAPK phosphorylation [270, 484]. PKCβ is implicated in insulin resistance, and transgenic mice overexpressing PKCβII exhibit decreased insulin-induced Akt activation in vascular cells [270, 485]. PKC inhibits insulin’s anti-atherosclerotic mechanisms by inhibiting the PI3K/Akt pathway at the insulin receptor substrate (IRS) level [486], but accentuates insulin’s pro-atherosclerotic mechanisms via the ERK1/2 signaling pathway [270, 487]. PKCβ activation by hyperglycemia may mediate the diabetic microvascular complications of retinopathy, nephropathy, and neuropathy. Hyperglycemia-induced activation of PKCβ causes abnormal signaling, cytokine activation, vascular alterations, cell cycle and transcriptional factor dysregulation, and abnormal angiogenesis [157, 270]. PKCβ mediates diabetic retinopathy by affecting VEGF expression through the mRNA-stabilizing human embryonic lethal abnormal vision protein HuR in the retina [488, 489]. In arteries of streptozotocin-induced diabetic mice and human coronary artery VSMCs, high glucose increases the expression of PKCβ which in turn increases vascular contraction by reducing the expression of the BKβI channel subunit and BKCa channel activity, and PKCβ inhibition restores BKCa-mediated vasodilation in diabetic mice [293, 490]. The nPKCs also contribute to insulin resistance through phosphorylation and inhibition of IRS1 [491, 492]. PKCδ plays a role in islet cell function and insulin response, and changes in PKCδ expression/activity in mice correlate with insulin resistance and glucose intolerance. Also, mice with global or liver-specific downregulation of PKCδ show increased hepatic insulin signaling and improved glucose tolerance with aging. Conversely, mice with liver-specific overexpression of PKCδ develop hepatic insulin resistance and decreased insulin signaling [493]. Diabetes-induced PKCδ activation also decreases responsiveness to PDGF leading to pericyte apoptosis, acellular capillaries, and retinopathy [494]. PKCδ may also be involved in poor collateral vessel formation, as the ischemic adductor muscles of diabetic PRKCD knockout mice show increased blood flow and capillary density compared with diabetic PRKCD+/+ mice. The poor angiogenesis response in ischemic diabetic muscles could be caused by PKCδ-induced expression of Src homology-2 domain-containing phosphatase-1 (SHP-1), which contributes to VEGF and PDGF unresponsiveness [467]. PKCδ also inhibits K+ current in aortic VSMCs, and PKCδ gene silencing by siRNAs restores VSMCs K + current and endothelium-dependent vasodilatation in aorta of streptozotocin-induced diabetic rats [293, 495]. Endothelium-independent vasoconstriction mediated by prostaglandin E2 EP1-/EP3-receptor activation is enhanced in mesenteric arteries of diabetic rats and is highly sensitive to PKCδ inhibition [293, 496]. Indolylmaleimide and its derivatives are nonselective PKC inhibitors that reduced diabetic nephropathy, cardiomyopathy and neuropathy in clinical trials [293, 497], but their lack of specificity raises safety concerns. Ruboxistaurin (LY333531) is an oral PKCβII inhibitor that is well-tolerated in diabetic retinopathy, nephropathy and neuropathy [297, 498, 499]. Ruboxistaurin decreases vessel permeability and diabetic macular edema, improves retinal condition, and prevents reduction of visual acuity in diabetic patients [270, 489]. Some antidiabetic drugs have inhibitory effects on PKC. In cultured human endothelial cells, combined use of metformin and liraglutide (a glucagon like peptide-1) inhibits high glucose-induced PKCβII translocation and phosphorylation, oxidative stress through inhibition of PKC-NADPH oxidase, p47phox translocation and NADPH oxidase activation, and high glucose-induced production of DAG and phosphorylation of AMP-activated protein kinase [500].
ROCK plays a role in metabolic and neurological disorders, cancer, and systemic and pulmonary hypertension [501–503]. Animal models of hypertension show increased RhoA/ROCK activity [504], and the ROCK inhibitors Y-27632 and fasudil normalize arterial pressure in experimental hypertension [397]. Chronic inhibition of ROCK suppresses vascular media hypertrophy and perivascular fibrosis in coronary arteries of spontaneously hypertensive rats [505], and in a rat model of hypertension induced by chronic inhibition of NO synthesis, where RhoA/ROCK activity is increased [506]. In hypertensive rats, inhibition of angiotensin type 1 receptor decreases RhoA/ROCK activity, suggesting that ROCK activation is likely due to increased AngII activity [506]. Long-term infusion of AngII increases RhoA/ROCK activity, media thickness and perivascular fibrosis in coronary arteries, and ROCK inhibitors inhibit AngII-induced coronary hypertrophy and fibrosis [507], and reduce production of superoxide anion [507], and monocyte chemoattractant protein-1 and PAI-1 [508, 509]. Hypertension is associated with increased mechanical strain on the vessel wall which in turn stimulates VSMC proliferation [510], and ROCK inhibition inhibits stretch-induced activation of MAPK and VSMC growth [511, 512]. Pulmonary hypertension involves sustained vasoconstriction and structural remodeling of pulmonary arteries leading to narrowing of the pulmonary microvessels and increased pulmonary vascular resistance. Reduced endothelium-derived NO in pulmonary arteries has been implicated in pulmonary hypertension [513], and ROCK mediates hypoxia-induced decrease in eNOS expression in human pulmonary endothelial cells [514]. RhoA/ROCK also contributes to both vasoconstriction and vascular remodeling in pulmonary hypertension [515, 516]. Chronic hypoxia in rats is associated with 2-fold increase in ROCK expression and enhanced ROCK-dependent Ca2+ sensitization in small pulmonary arteries [517]. The ROCK inhibitor Y-27632 attenuates hypoxia-induced pulmonary vasoconstriction, hypertension and vascular remodeling [518]. Inhibition of ROCK by oral or inhaled fasudil also improves monocrotaline-induced pulmonary hypertension in rats by inhibiting VSMC proliferation and macrophage infiltration, and improving endothelium-dependent pulmonary artery relaxation [516, 519, 520].
10. Summary
In the past decades, great advances have been made in our understanding of the mechanisms of VSM contraction and their role in the pathogenesis of vascular disease. [Ca2+]c is a major determinant of VSM contraction, and is controlled by Ca2+ channels and Ca2+ pumps in the plasma membrane and intracellular organelles. The balance between Ca2+ mobilizing and Ca2+ removal mechanisms maintains resting [Ca2+]c constant. Vasoconstrictor agonists and pathological states such as hypertension disrupt the balance between Ca2+ mobilizing and Ca2+ removal mechanisms leading to increased [Ca2+]c, Ca2+-dependent MLC phosphorylation, and vasoconstriction. PKC is also a major regulator of VSM function. PKC is a family of conventional, novel and atypical isoforms with different Ca2+ and phospholipid dependency, cellular localization, and substrates. PKC is mainly cytosolic, and upon VSMC activation it undergoes phosphorylation, maturation and translocation to the plasma membrane, nucleus, endoplasmic reticulum, and other cell organelles; a process facilitated by scaffold proteins. Activated PKC phosphorylates different substrates including ion channels, pumps and nuclear proteins. PKC phosphorylates CPI-17 leading to inhibition of MLC phosphatase, increased MLC phosphorylation and enhanced VSM contraction. PKC also activates a cascade of protein kinases leading to phosphorylation of the actin-binding proteins CaP and CaD, increased actin-myosin interaction, and VSM contraction. Agonists also increase RhoA/ROCK activity leading to inhibition of MLC phosphatase and enhancement of the myofilament force sensitivity to Ca2+. Knockout animals lacking a specific channel subunit, PKC isoform, or ROCK have supported their role in the regulation of vascular function. Of note, cyclic nucleotides such as cAMP and cGMP activate PKA and PKG, respectively, which in turn affect Ca2+ handling mechanisms and [Ca2+]c, smooth muscle membrane potential, and the sensitivity of the contractile machinery to Ca2+, and thereby cause opposing effects and decrease smooth muscle contraction [521].
Increases in [Ca2+]c, PKC and ROCK activity play a role in the exaggerated vasoconstriction, VSM growth and proliferation, and vascular remodeling associated with coronary artery disease, hypertension, and diabetic vasculopathy. The subcellular location of PKC may determine the state of VSM activity, and could be useful in the diagnosis/prognosis of hypertension [6]. Ca2+ channel blockers have been used in treatment of hypertension and coronary vasospasm. PKC may represent an alternative target for treatment of vascular disease. The first generation of PKC inhibitors such as staurosporine and chelerythrine have shown mixed results in experimental and clinical trials [157], but isoform-specific PKC inhibitors show some promise. PKC inhibitors could be useful in Ca2+ antagonist-resistant forms of hypertension [6]. Inhibitors of PKCβ and δ improve glucose tolerance and reduce fat accumulation, hepatosteatosis, and foam cell formation in obesity and hyperlipidemia-induced atherosclerosis [493, 522, 523]. A PKCβ inhibitor or PKCε activator may reduce vascular damage secondary to endothelial dysfunction and VSMC proliferation in patients with atherosclerosis [522, 524–526]. PKCε activators may be useful in coronary artery disease, and the PKCε activator acadesine reduced the 2 year mortality in patients with postoperative acute myocardial infarction after coronary bypass grafting [157]. Ruboxistaurin is a PKCβII inhibitor that has been tested in diabetic retinopathy, nephropathy and neuropathy [297, 498, 499]. PKC siRNA could target specific PKC isoforms in vascular disease. PKCδ gene silencing with the short hairpin RNAs (shRNAs)-plasmid delivery system administered intravenously normalizes vascular function and blood pressure in spontaneously hypertensive rats [298]. Also, PKCδ siRNA attenuates the proinflammatory effect of human C-reactive protein in diabetic rats [527]. Target-delivery of PKC pseudosubstrate inhibitory peptides may be useful in localized vascular disease. PKC inhibitors could be coated onto stents and directly released at effective concentrations in vasospastic areas. PKCβII and PKCδ inhibitors coated stents or balloons showed efficacy in experimental trials [260]. ROCK inhibitors such as fasudil have shown some promise in pulmonary hypertension and cerebral vasospasm, but more specific inhibitors are needed for clinical use in humans.
Acknowledgments
This work was supported by grants from National Heart, Lung, and Blood Institute (HL-65998).
List of Abbreviations
- AGE
Advanced Glycation End products
- ALDH2
aldehyde dehydrogenase 2
- AngII
angiotensin II
- BKCa
large conductance Ca2+-activated K+ channel
- Ca2+
calcium
- [Ca2+]c
cytosolic free Ca2+ concentration
- CaD
caldesmon
- CaM
calmodulin
- cAMP
cyclic adenosine monophosphate
- CaP
calponin
- cGMP
cyclic guanosine monophosphate
- CICR
Ca2+-induced Ca2+ release
- CNP
C-type natriuretic peptide
- CPI-17
PKC-potentiated phosphatase inhibitor protein-17
- DAG
diacyglycerol
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- ET-1
endothelin-1
- HSP
heat shock protein
- ICAM-1
intercellular adhesion molecule-1
- IP3
inositol 1,4,5-trisphosphate
- IRS1
insulin receptor substrate 1
- Kv
voltage-gated K+ channel
- LTCC
L-Type CaV1.2 channel
- MARCKS
myristoylated alanine-rich C kinase substrate
- MLC
myosin light chain
- PDBu
phorbol 12,13-dibutyrate
- PDGF
platelet-derived growth factor
- PDK
phosphoinositide-dependent kinase
- PKA
cAMP-dependent protein kinase
- PKC
protein kinase C
- PKG
cGMP-dependent protein kinase
- PMA
phorbol 12-myristate 13-acetate
- PMCA
plasmalemmal Ca2+-ATPase
- PLC
phospholipase C
- PS
phosphatidylserine
- RAGE
AGE receptor
- ROC
receptor-operated Ca2+ channel
- ROCK
Rho-kinase
- ROS
reactive oxygen species
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+-ATPase
- SOC
store-operated Ca2+ channel
- SR
sarcoplasmic reticulum
- TRP
transient receptor potential channel
- TTCC
T-type CaV3.1/3.2/3.3
- VCAM-1
vascular cell adhesion molecule-1
- VEGF
vascular endothelial growth factor
- VDCC
voltage-dependent Ca2+ channel
- VSM
vascular smooth muscle
Footnotes
CONFLICT OF INTEREST
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Khalil R, Lodge N, Saida K, van Breemen C. Mechanism of calcium activation in vascular smooth muscle. J Hypertens Suppl. 1987;5:S5–15. doi: 10.1097/00004872-198712004-00003. [DOI] [PubMed] [Google Scholar]
- 2.Khalil RA, van Breemen C. Sustained contraction of vascular smooth muscle: calcium influx or C-kinase activation? The Journal of pharmacology and experimental therapeutics. 1988;244:537–42. [PubMed] [Google Scholar]
- 3.van Breemen C, Fameli N, Evans AM. Pan-junctional sarcoplasmic reticulum in vascular smooth muscle: nanospace Ca2+ transport for site- and function-specific Ca2+ signalling. The Journal of physiology. 2013;591:2043–54. doi: 10.1113/jphysiol.2012.246348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans AM. Nanojunctions of the Sarcoplasmic Reticulum Deliver Site- and Function-Specific Calcium Signaling in Vascular Smooth Muscles. Adv Pharmacol. 2017;78:1–47. doi: 10.1016/bs.apha.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 5.Kim HR, Appel S, Vetterkind S, Gangopadhyay SS, Morgan KG. Smooth muscle signalling pathways in health and disease. J Cell Mol Med. 2008;12:2165–80. doi: 10.1111/j.1582-4934.2008.00552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salamanca DA, Khalil RA. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochemical pharmacology. 2005;70:1537–47. doi: 10.1016/j.bcp.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Somlyo AP, Somlyo AV. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. The Journal of physiology. 2000;522(Pt 2):177–85. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ringer S. A further Contribution regarding the influence of the different Constituents of the Blood on the Contraction of the Heart. The Journal of physiology. 1883;4:29–423. doi: 10.1113/jphysiol.1883.sp000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heilbrunn LV, Wiercinski FJ. The action of various cations on muscle protoplasm. J Cell Comp Physiol. 1947;29:15–32. doi: 10.1002/jcp.1030290103. [DOI] [PubMed] [Google Scholar]
- 10.Reuter H. On the effect of adrenaline on the cellular Ca-metabolism in the guinea pig atrium. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol. 1965;251:401–12. [PubMed] [Google Scholar]
- 11.Beeler GW, Jr, Reuter H. Membrane calcium current in ventricular myocardial fibres. The Journal of physiology. 1970;207:191–209. doi: 10.1113/jphysiol.1970.sp009056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beeler GW, Jr, Reuter H. The relation between membrane potential, membrane currents and activation of contraction in ventricular myocardial fibres. The Journal of physiology. 1970;207:211–29. doi: 10.1113/jphysiol.1970.sp009057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reuter H, Stevens CF, Tsien RW, Yellen G. Properties of single calcium channels in cardiac cell culture. Nature. 1982;297:501–4. doi: 10.1038/297501a0. [DOI] [PubMed] [Google Scholar]
- 14.Cauvin C, Loutzenhiser R, Van Breemen C. Mechanisms of calcium antagonist-induced vasodilation. Annu Rev Pharmacol Toxicol. 1983;23:373–96. doi: 10.1146/annurev.pa.23.040183.002105. [DOI] [PubMed] [Google Scholar]
- 15.Deth R, van Breemen C. Agonist induced release of intracellular Ca2+ in the rabbit aorta. J Membr Biol. 1977;30:363–80. doi: 10.1007/BF01869677. [DOI] [PubMed] [Google Scholar]
- 16.Hwang KS, van Breemen C. Ryanodine modulation of 45Ca efflux and tension in rabbit aortic smooth muscle. Pflugers Archiv: European journal of physiology. 1987;408:343–50. doi: 10.1007/BF00581127. [DOI] [PubMed] [Google Scholar]
- 17.Somlyo AP, Somlyo AV. The sarcoplasmic reticulum: then and now. Novartis Found Symp. 2002;246:258–68. discussion 68–71, 72–6. [PubMed] [Google Scholar]
- 18.Endo M. Calcium release from the sarcoplasmic reticulum. Physiological reviews. 1977;57:71–108. doi: 10.1152/physrev.1977.57.1.71. [DOI] [PubMed] [Google Scholar]
- 19.Devine CE, Somlyo AV, Somlyo AP. Sarcoplasmic reticulum and mitochondria as cation accumulation sites in smooth muscle. Philos Trans R Soc Lond B Biol Sci. 1973;265:17–23. doi: 10.1098/rstb.1973.0005. [DOI] [PubMed] [Google Scholar]
- 20.Bond M, Kitazawa T, Somlyo AP, Somlyo AV. Release and recycling of calcium by the sarcoplasmic reticulum in guinea-pig portal vein smooth muscle. The Journal of physiology. 1984;355:677–95. doi: 10.1113/jphysiol.1984.sp015445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehrlich BE, Watras J. Inositol 1,4,5-trisphosphate activates a channel from smooth muscle sarcoplasmic reticulum. Nature. 1988;336:583–6. doi: 10.1038/336583a0. [DOI] [PubMed] [Google Scholar]
- 22.Saida K, Van Breemen C. Cyclic AMP modulation of adrenoreceptor-mediated arterial smooth muscle contraction. J Gen Physiol. 1984;84:307–18. doi: 10.1085/jgp.84.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cassidy P, Hoar PE, Kerrick WG. Irreversible thiophosphorylation and activation of tension in functionally skinned rabbit ileum strips by [35S]ATP gamma S. The Journal of biological chemistry. 1979;254:11148–53. [PubMed] [Google Scholar]
- 24.Nishimura J, Kolber M, van Breemen C. Norepinephrine and GTP-gamma-S increase myofilament Ca2+ sensitivity in alpha-toxin permeabilized arterial smooth muscle. Biochemical and biophysical research communications. 1988;157:677–83. doi: 10.1016/s0006-291x(88)80303-6. [DOI] [PubMed] [Google Scholar]
- 25.Kitazawa T, Kobayashi S, Horiuti K, Somlyo AV, Somlyo AP. Receptor-coupled, permeabilized smooth muscle. Role of the phosphatidylinositol cascade, G-proteins, and modulation of the contractile response to Ca2+ The Journal of biological chemistry. 1989;264:5339–42. [PubMed] [Google Scholar]
- 26.Berridge MJ, Irvine RF. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature. 1984;312:315–21. doi: 10.1038/312315a0. [DOI] [PubMed] [Google Scholar]
- 27.Suematsu E, Hirata M, Hashimoto T, Kuriyama H. Inositol 1,4,5-trisphosphate releases Ca2+ from intracellular store sites in skinned single cells of porcine coronary artery. Biochemical and biophysical research communications. 1984;120:481–5. doi: 10.1016/0006-291x(84)91279-8. [DOI] [PubMed] [Google Scholar]
- 28.Somlyo AV, Bond M, Somlyo AP, Scarpa A. Inositol trisphosphate-induced calcium release and contraction in vascular smooth muscle. Proc Natl Acad Sci U S A. 1985;82:5231–5. doi: 10.1073/pnas.82.15.5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto H, van Breemen C. Inositol-1,4,5-trisphosphate releases calcium from skinned cultured smooth muscle cells. Biochemical and biophysical research communications. 1985;130:270–4. doi: 10.1016/0006-291x(85)90412-7. [DOI] [PubMed] [Google Scholar]
- 30.Saida K, van Breemen C. GTP requirement for inositol-1,4,5-trisphosphate-induced Ca2+ release from sarcoplasmic reticulum in smooth muscle. Biochemical and biophysical research communications. 1987;144:1313–6. doi: 10.1016/0006-291x(87)91453-7. [DOI] [PubMed] [Google Scholar]
- 31.Griendling KK, Rittenhouse SE, Brock TA, Ekstein LS, Gimbrone MA, Jr, Alexander RW. Sustained diacylglycerol formation from inositol phospholipids in angiotensin II-stimulated vascular smooth muscle cells. The Journal of biological chemistry. 1986;261:5901–6. [PubMed] [Google Scholar]
- 32.Iino M. Calcium dependent inositol trisphosphate-induced calcium release in the guinea-pig taenia caeci. Biochemical and biophysical research communications. 1987;142:47–52. doi: 10.1016/0006-291x(87)90449-9. [DOI] [PubMed] [Google Scholar]
- 33.Walker JW, Somlyo AV, Goldman YE, Somlyo AP, Trentham DR. Kinetics of smooth and skeletal muscle activation by laser pulse photolysis of caged inositol 1,4,5-trisphosphate. Nature. 1987;327:249–52. doi: 10.1038/327249a0. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi S, Somlyo AV, Somlyo AP. Heparin inhibits the inositol 1,4,5-trisphosphate-dependent, but not the independent, calcium release induced by guanine nucleotide in vascular smooth muscle. Biochemical and biophysical research communications. 1988;153:625–31. doi: 10.1016/s0006-291x(88)81141-0. [DOI] [PubMed] [Google Scholar]
- 35.Lin Q, Zhao G, Fang X, Peng X, Tang H, Wang H, et al. IP3 receptors regulate vascular smooth muscle contractility and hypertension. JCI Insight. 2016;1:e89402. doi: 10.1172/jci.insight.89402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song T, Hao Q, Zheng YM, Liu QH, Wang YX. Inositol 1,4,5-trisphosphate activates TRPC3 channels to cause extracellular Ca2+ influx in airway smooth muscle cells. American journal of physiology Lung cellular and molecular physiology. 2015;309:L1455–66. doi: 10.1152/ajplung.00148.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ford LE, Podolsky RJ. Regenerative calcium release within muscle cells. Science. 1970;167:58–9. doi: 10.1126/science.167.3914.58. [DOI] [PubMed] [Google Scholar]
- 38.Fabiato A, Fabiato F. Excitation-contraction coupling of isolated cardiac fibers with disrupted or closed sarcolemmas. Calcium-dependent cyclic and tonic contractions. Circulation research. 1972;31:293–307. doi: 10.1161/01.res.31.3.293. [DOI] [PubMed] [Google Scholar]
- 39.van Breemen C, Saida K. Cellular mechanisms regulating [Ca2+]i smooth muscle. Annu Rev Physiol. 1989;51:315–29. doi: 10.1146/annurev.ph.51.030189.001531. [DOI] [PubMed] [Google Scholar]
- 40.Esfandiarei M, Fameli N, Choi YY, Tehrani AY, Hoskins JG, van Breemen C. Waves of calcium depletion in the sarcoplasmic reticulum of vascular smooth muscle cells: an inside view of spatiotemporal Ca2+ regulation. PloS one. 2013;8:e55333. doi: 10.1371/journal.pone.0055333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benham CD, Tsien RW. A novel receptor-operated Ca2+-permeable channel activated by ATP in smooth muscle. Nature. 1987;328:275–8. doi: 10.1038/328275a0. [DOI] [PubMed] [Google Scholar]
- 42.Bolton TB. Mechanisms of action of transmitters and other substances on smooth muscle. Physiological reviews. 1979;59:606–718. doi: 10.1152/physrev.1979.59.3.606. [DOI] [PubMed] [Google Scholar]
- 43.Van Breemen C, Aaronson P, Loutzenhiser R. Sodium-calcium interactions in mammalian smooth muscle. Pharmacological reviews. 1978;30:167–208. [PubMed] [Google Scholar]
- 44.Nelson MT, Standen NB, Brayden JE, Worley JF., 3rd Noradrenaline contracts arteries by activating voltage-dependent calcium channels. Nature. 1988;336:382–5. doi: 10.1038/336382a0. [DOI] [PubMed] [Google Scholar]
- 45.Benham CD, Hess P, Tsien RW. Two types of calcium channels in single smooth muscle cells from rabbit ear artery studied with whole-cell and single-channel recordings. Circulation research. 1987;61:I10–6. [PubMed] [Google Scholar]
- 46.Yatani A, Seidel CL, Allen J, Brown AM. Whole-cell and single-channel calcium currents of isolated smooth muscle cells from saphenous vein. Circulation research. 1987;60:523–33. doi: 10.1161/01.res.60.4.523. [DOI] [PubMed] [Google Scholar]
- 47.Sturek M, Hermsmeyer K. Calcium and sodium channels in spontaneously contracting vascular muscle cells. Science. 1986;233:475–8. doi: 10.1126/science.2425434. [DOI] [PubMed] [Google Scholar]
- 48.Loirand G, Pacaud P, Mironneau C, Mironneau J. Evidence for two distinct calcium channels in rat vascular smooth muscle cells in short-term primary culture. Pflugers Archiv: European journal of physiology. 1986;407:566–8. doi: 10.1007/BF00657519. [DOI] [PubMed] [Google Scholar]
- 49.Bean BP, Sturek M, Puga A, Hermsmeyer K. Calcium channels in muscle cells isolated from rat mesenteric arteries: modulation by dihydropyridine drugs. Circulation research. 1986;59:229–35. doi: 10.1161/01.res.59.2.229. [DOI] [PubMed] [Google Scholar]
- 50.Benham CD, Tsien RW. Noradrenaline modulation of calcium channels in single smooth muscle cells from rabbit ear artery. The Journal of physiology. 1988;404:767–84. doi: 10.1113/jphysiol.1988.sp017318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Biel M, Ruth P, Bosse E, Hullin R, Stuhmer W, Flockerzi V, et al. Primary structure and functional expression of a high voltage activated calcium channel from rabbit lung. FEBS Lett. 1990;269:409–12. doi: 10.1016/0014-5793(90)81205-3. [DOI] [PubMed] [Google Scholar]
- 52.Ghosh D, Syed AU, Prada MP, Nystoriak MA, Santana LF, Nieves-Cintron M, et al. Calcium Channels in Vascular Smooth Muscle. Adv Pharmacol. 2017;78:49–87. doi: 10.1016/bs.apha.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bezanilla F. How membrane proteins sense voltage. Nat Rev Mol Cell Biol. 2008;9:323–32. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- 54.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. The Journal of physiology. 1998;508(Pt 1):199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.VanBavel E, Sorop O, Andreasen D, Pfaffendorf M, Jensen BL. Role of T-type calcium channels in myogenic tone of skeletal muscle resistance arteries. American journal of physiology Heart and circulatory physiology. 2002;283:H2239–43. doi: 10.1152/ajpheart.00531.2002. [DOI] [PubMed] [Google Scholar]
- 56.Harraz OF, Visser F, Brett SE, Goldman D, Zechariah A, Hashad AM, et al. CaV1.2/CaV3.x channels mediate divergent vasomotor responses in human cerebral arteries. J Gen Physiol. 2015;145:405–18. doi: 10.1085/jgp.201511361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee N, Jeong S, Kim KC, Kim JA, Park JY, Kang HW, et al. Ca2+ Regulation of Cav3. 3 T-type Ca2+ Channel Is Mediated by Calmodulin. Mol Pharmacol. 2017;92:347–57. doi: 10.1124/mol.117.108530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meisheri KD, Hwang O, van Breemen C. Evidence for two separated Ca2+ pathways in smooth muscle plasmalemma. J Membr Biol. 1981;59:19–25. doi: 10.1007/BF01870817. [DOI] [PubMed] [Google Scholar]
- 59.Reuter H, Sigel E. Ionic channels: modulation by G proteins and by phosphorylation. Curr Opin Neurobiol. 1991;1:27–31. doi: 10.1016/0959-4388(91)90007-t. [DOI] [PubMed] [Google Scholar]
- 60.Martinsen A, Dessy C, Morel N. Regulation of calcium channels in smooth muscle: new insights into the role of myosin light chain kinase. Channels (Austin) 2014;8:402–13. doi: 10.4161/19336950.2014.950537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiological reviews. 2015;95:645–90. doi: 10.1152/physrev.00026.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Albert AP. Gating mechanisms of canonical transient receptor potential channel proteins: role of phosphoinositols and diacylglycerol. Adv Exp Med Biol. 2011;704:391–411. doi: 10.1007/978-94-007-0265-3_22. [DOI] [PubMed] [Google Scholar]
- 63.Putney JW, Jr, Broad LM, Braun FJ, Lievremont JP, Bird GS. Mechanisms of capacitative calcium entry. J Cell Sci. 2001;114:2223–9. doi: 10.1242/jcs.114.12.2223. [DOI] [PubMed] [Google Scholar]
- 64.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiological reviews. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 65.Leung FP, Yung LM, Yao X, Laher I, Huang Y. Store-operated calcium entry in vascular smooth muscle. British journal of pharmacology. 2008;153:846–57. doi: 10.1038/sj.bjp.0707455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parekh AB. Functional consequences of activating store-operated CRAC channels. Cell Calcium. 2007;42:111–21. doi: 10.1016/j.ceca.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 67.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–6. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 68.Xuan YT, Wang OL, Whorton AR. Thapsigargin stimulates Ca2+ entry in vascular smooth muscle cells: nicardipine-sensitive and -insensitive pathways. The American journal of physiology. 1992;262:C1258–65. doi: 10.1152/ajpcell.1992.262.5.C1258. [DOI] [PubMed] [Google Scholar]
- 69.Xuan YT, Glass PS. Propofol regulation of calcium entry pathways in cultured A10 and rat aortic smooth muscle cells. British journal of pharmacology. 1996;117:5–12. doi: 10.1111/j.1476-5381.1996.tb15147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tosun M, Paul RJ, Rapoport RM. Coupling of store-operated Ca++ entry to contraction in rat aorta. The Journal of pharmacology and experimental therapeutics. 1998;285:759–66. [PubMed] [Google Scholar]
- 71.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, et al. Upregulated TRP and enhanced capacitative Ca(2+) entry in human pulmonary artery myocytes during proliferation. American journal of physiology Heart and circulatory physiology. 2001;280:H746–55. doi: 10.1152/ajpheart.2001.280.2.H746. [DOI] [PubMed] [Google Scholar]
- 72.Xu SZ, Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca(2+) channels in native vascular smooth muscle cells. Circulation research. 2001;88:84–7. doi: 10.1161/01.res.88.1.84. [DOI] [PubMed] [Google Scholar]
- 73.Bergdahl A, Gomez MF, Wihlborg AK, Erlinge D, Eyjolfson A, Xu SZ, et al. Plasticity of TRPC expression in arterial smooth muscle: correlation with store-operated Ca2+ entry. American journal of physiology Cell physiology. 2005;288:C872–80. doi: 10.1152/ajpcell.00334.2004. [DOI] [PubMed] [Google Scholar]
- 74.Xu SZ, Boulay G, Flemming R, Beech DJ. E3-targeted anti-TRPC5 antibody inhibits store-operated calcium entry in freshly isolated pial arterioles. American journal of physiology Heart and circulatory physiology. 2006;291:H2653–9. doi: 10.1152/ajpheart.00495.2006. [DOI] [PubMed] [Google Scholar]
- 75.Giamarchi A, Padilla F, Coste B, Raoux M, Crest M, Honore E, et al. The versatile nature of the calcium-permeable cation channel TRPP2. EMBO Rep. 2006;7:787–93. doi: 10.1038/sj.embor.7400745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. The Journal of biological chemistry. 2003;278:39014–9. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- 77.Zagranichnaya TK, Wu X, Villereal ML. Endogenous TRPC1, TRPC3, and TRPC7 proteins combine to form native store-operated channels in HEK-293 cells. The Journal of biological chemistry. 2005;280:29559–69. doi: 10.1074/jbc.M505842200. [DOI] [PubMed] [Google Scholar]
- 78.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol. 2004;6:113–20. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 79.Trepakova ES, Gericke M, Hirakawa Y, Weisbrod RM, Cohen RA, Bolotina VM. Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. The Journal of biological chemistry. 2001;276:7782–90. doi: 10.1074/jbc.M010104200. [DOI] [PubMed] [Google Scholar]
- 80.Kim HY, Thomas D, Hanley MR. Chromatographic resolution of an intracellular calcium influx factor from thapsigargin-activated Jurkat cells. Evidence for multiple activities influencing calcium elevation in Xenopus oocytes. The Journal of biological chemistry. 1995;270:9706–8. doi: 10.1074/jbc.270.17.9706. [DOI] [PubMed] [Google Scholar]
- 81.Bolotina VM, Csutora P. CIF and other mysteries of the store-operated Ca2+-entry pathway. Trends Biochem Sci. 2005;30:378–87. doi: 10.1016/j.tibs.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 82.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lopez JJ, Jardin I, Bobe R, Pariente JA, Enouf J, Salido GM, et al. STIM1 regulates acidic Ca2+ store refilling by interaction with SERCA3 in human platelets. Biochemical pharmacology. 2008;75:2157–64. doi: 10.1016/j.bcp.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 84.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–3. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 85.Navarro-Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. The Journal of physiology. 2008;586:5383–401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. The Journal of biological chemistry. 2006;281:20661–5. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 87.Valadares NF, d’ Muniz Pereira H, Ulian Araujo AP, Garratt RC. Septin structure and filament assembly. Biophys Rev. 2017;9:481–500. doi: 10.1007/s12551-017-0320-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lopez JJ, Albarran L, Gomez LJ, Smani T, Salido GM, Rosado JA. Molecular modulators of store-operated calcium entry. Biochim Biophys Acta. 2016;1863:2037–43. doi: 10.1016/j.bbamcr.2016.04.024. [DOI] [PubMed] [Google Scholar]
- 89.Sharma S, Quintana A, Findlay GM, Mettlen M, Baust B, Jain M, et al. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature. 2013;499:238–42. doi: 10.1038/nature12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. The Journal of physiology. 1902;28:220–31. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bohr DF, Webb RC. Vascular smooth muscle function and its changes in hypertension. Am J Med. 1984;77:3–16. doi: 10.1016/s0002-9343(84)80032-7. [DOI] [PubMed] [Google Scholar]
- 92.Kirber MT, Walsh JV, Jr, Singer JJ. Stretch-activated ion channels in smooth muscle: a mechanism for the initiation of stretch-induced contraction. Pflugers Archiv: European journal of physiology. 1988;412:339–45. doi: 10.1007/BF01907549. [DOI] [PubMed] [Google Scholar]
- 93.Harder DR. Pressure-induced myogenic activation of cat cerebral arteries is dependent on intact endothelium. Circulation research. 1987;60:102–7. doi: 10.1161/01.res.60.1.102. [DOI] [PubMed] [Google Scholar]
- 94.Aguettaz E, Bois P, Cognard C, Sebille S. Stretch-activated TRPV2 channels: Role in mediating cardiopathies. Prog Biophys Mol Biol. 2017;130:273–80. doi: 10.1016/j.pbiomolbio.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 95.Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circulation research. 2002;90:248–50. doi: 10.1161/hh0302.105662. [DOI] [PubMed] [Google Scholar]
- 96.Van Breemen C, Daniel EE. The influence of high potassium depolarization and acetylcholine on calcium exchange in the rat uterus. J Gen Physiol. 1966;49:1299–317. doi: 10.1085/jgp.0491299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Somlyo AP, Somlyo AV. Vascular smooth muscle. II. Pharmacology of normal and hypotensive vessels. Pharmacological reviews. 1970;22:249–353. [PubMed] [Google Scholar]
- 98.Casteels R, Van Breemen C. Active and passive Ca2+ fluxes across cell membranes of the guinea-pig taenia coli. Pflugers Archiv: European journal of physiology. 1975;359:197–207. doi: 10.1007/BF00587379. [DOI] [PubMed] [Google Scholar]
- 99.Raeymaekers L, Wuytack F, Casteels R. Subcellular fractionation of pig stomach smooth muscle. A study of the distribution of the (Ca2+ + Mg2+)-ATPase activity in plasmalemma and endoplasmic reticulum. Biochim Biophys Acta. 1985;815:441–54. doi: 10.1016/0005-2736(85)90372-4. [DOI] [PubMed] [Google Scholar]
- 100.Rapp JP. Aortic responses to vanadate: independence from (Na, K)-ATPase and comparison of Dahl salt-sensitive and salt-resistant rats. Hypertension. 1981;3:I168–72. doi: 10.1161/01.hyp.3.3_pt_2.i168. [DOI] [PubMed] [Google Scholar]
- 101.Popescu LM, Nutu O, Panoiu C. Oxytocin contracts the human uterus at term by inhibiting the myometrial Ca2+-extrusion pump. Biosci Rep. 1985;5:21–8. doi: 10.1007/BF01117437. [DOI] [PubMed] [Google Scholar]
- 102.Eggermont JA, Vrolix M, Raeymaekers L, Wuytack F, Casteels R. Ca2+-transport ATPases of vascular smooth muscle. Circulation research. 1988;62:266–78. doi: 10.1161/01.res.62.2.266. [DOI] [PubMed] [Google Scholar]
- 103.Popescu LM, Ignat P. Calmodulin-dependent Ca2+-pump ATPase of human smooth muscle sarcolemma. Cell Calcium. 1983;4:219–35. doi: 10.1016/0143-4160(83)90001-5. [DOI] [PubMed] [Google Scholar]
- 104.Heim R, Iwata T, Zvaritch E, Adamo HP, Rutishauser B, Strehler EE, et al. Expression, purification, and properties of the plasma membrane Ca2+ pump and of its N-terminally truncated 105-kDa fragment. The Journal of biological chemistry. 1992;267:24476–84. [PubMed] [Google Scholar]
- 105.Kumar R, Haugen JD, Penniston JT. Molecular cloning of a plasma membrane calcium pump from human osteoblasts. J Bone Miner Res. 1993;8:505–13. doi: 10.1002/jbmr.5650080415. [DOI] [PubMed] [Google Scholar]
- 106.De Jaegere S, Wuytack F, Eggermont JA, Verboomen H, Casteels R. Molecular cloning and sequencing of the plasma-membrane Ca2+ pump of pig smooth muscle. Biochem J. 1990;271:655–60. doi: 10.1042/bj2710655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grover AK, Kwan CY, Rangachari PK, Daniel EE. Na-Ca exchange in a smooth muscle plasma membrane-enriched fraction. The American journal of physiology. 1983;244:C158–65. doi: 10.1152/ajpcell.1983.244.3.C158. [DOI] [PubMed] [Google Scholar]
- 108.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiological reviews. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 109.Barzilai A, Spanier R, Rahamimoff H. Isolation, purification, and reconstitution of the Na+ gradient-dependent Ca2+ transporter (Na+-Ca2+ exchanger) from brain synaptic plasma membranes. Proc Natl Acad Sci U S A. 1984;81:6521–5. doi: 10.1073/pnas.81.20.6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Matlib MA, Reeves JP. Solubilization and reconstitution of the sarcolemmal Na+-Ca2+ exchange system of vascular smooth muscle. Biochim Biophys Acta. 1987;904:145–8. doi: 10.1016/0005-2736(87)90096-4. [DOI] [PubMed] [Google Scholar]
- 111.Carafoli E, Crompton M. The regulation of intracellular calcium by mitochondria. Annals of the New York Academy of Sciences. 1978;307:269–84. doi: 10.1111/j.1749-6632.1978.tb41957.x. [DOI] [PubMed] [Google Scholar]
- 112.Reeves JP, Sutko JL. Sodium-calcium exchange activity generates a current in cardiac membrane vesicles. Science. 1980;208:1461–4. doi: 10.1126/science.7384788. [DOI] [PubMed] [Google Scholar]
- 113.Reeves JP, Hale CC. The stoichiometry of the cardiac sodium-calcium exchange system. The Journal of biological chemistry. 1984;259:7733–9. [PubMed] [Google Scholar]
- 114.Mulvany MJ, Aalkjaer C, Petersen TT. Intracellular sodium, membrane potential, and contractility of rat mesenteric small arteries. Circulation research. 1984;54:740–9. doi: 10.1161/01.res.54.6.740. [DOI] [PubMed] [Google Scholar]
- 115.Ashida T, Blaustein MP. Regulation of cell calcium and contractility in mammalian arterial smooth muscle: the role of sodium-calcium exchange. The Journal of physiology. 1987;392:617–35. doi: 10.1113/jphysiol.1987.sp016800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Blaustein MP, Hamlyn JM. Sodium transport inhibition, cell calcium, and hypertension. The natriuretic hormone/Na+-Ca2+ exchange/hypertension hypothesis. Am J Med. 1984;77:45–59. doi: 10.1016/s0002-9343(84)80037-6. [DOI] [PubMed] [Google Scholar]
- 117.Inesi G. Mechanism of calcium transport. Annu Rev Physiol. 1985;47:573–601. doi: 10.1146/annurev.ph.47.030185.003041. [DOI] [PubMed] [Google Scholar]
- 118.Borle AB. Control, Modulation, and regulation of cell calcium. Rev Physiol Biochem Pharmacol. 1981;90:13–153. doi: 10.1007/BFb0034078. [DOI] [PubMed] [Google Scholar]
- 119.MacLennan DH, Wong PT. Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1971;68:1231–5. doi: 10.1073/pnas.68.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wuytack F, Raeymaekers L, Verbist J, Jones LR, Casteels R. Smooth-muscle endoplasmic reticulum contains a cardiac-like form of calsequestrin. Biochim Biophys Acta. 1987;899:151–8. doi: 10.1016/0005-2736(87)90395-6. [DOI] [PubMed] [Google Scholar]
- 121.Waisman DM, Gimble JM, Goodman DB, Rasmussen H. Studies of the Ca2+ transport mechanism of human erythrocyte inside-out plasma membrane vesicles. II. Stimulation of the Ca2+ pump by phosphate. The Journal of biological chemistry. 1981;256:415–9. [PubMed] [Google Scholar]
- 122.Zhang WB, Kwan CY. Pharmacological evidence that potentiation of plasmalemmal Ca(2+)-extrusion is functionally coupled to inhibition of SR Ca(2+)-ATPases in vascular smooth muscle cells. Naunyn Schmiedebergs Arch Pharmacol. 2016;389:447–55. doi: 10.1007/s00210-016-1209-7. [DOI] [PubMed] [Google Scholar]
- 123.Puskin JS, Gunter TE, Gunter KK, Russell PR. Evidence for more than one Ca2+ transport mechanism in mitochondria. Biochemistry. 1976;15:3834–42. doi: 10.1021/bi00662a029. [DOI] [PubMed] [Google Scholar]
- 124.Rasmussen H, Barrett PQ. Calcium messenger system: an integrated view. Physiological reviews. 1984;64:938–84. doi: 10.1152/physrev.1984.64.3.938. [DOI] [PubMed] [Google Scholar]
- 125.Carafoli E. Intracellular calcium homeostasis. Annu Rev Biochem. 1987;56:395–433. doi: 10.1146/annurev.bi.56.070187.002143. [DOI] [PubMed] [Google Scholar]
- 126.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–8. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 127.Yamamoto H, van Breemen C. Ca2+ compartments in saponin-skinned cultured vascular smooth muscle cells. J Gen Physiol. 1986;87:369–89. doi: 10.1085/jgp.87.3.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dubyak GR, Scarpa A. Sarcoplasmic Ca2+ transients during the contractile cycle of single barnacle muscle fibres: measurements with arsenazo III-injected fibres. J Muscle Res Cell Motil. 1982;3:87–112. doi: 10.1007/BF00711882. [DOI] [PubMed] [Google Scholar]
- 129.Ashley CC. Calcium ion regulation in barnacle muscle fibers and its relation to force development. Annals of the New York Academy of Sciences. 1978;307:308–29. doi: 10.1111/j.1749-6632.1978.tb41959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Blinks JR, Wier WG, Hess P, Prendergast FG. Measurement of Ca2+ concentrations in living cells. Prog Biophys Mol Biol. 1982;40:1–114. doi: 10.1016/0079-6107(82)90011-6. [DOI] [PubMed] [Google Scholar]
- 131.Gasser R, Frey M, Fleckenstein-Grun G. Free calcium in rat papillary muscle at contraction assessed with Ca-selective microelectrodes. Angiology. 1989;40:736–42. doi: 10.1177/000331978904000809. [DOI] [PubMed] [Google Scholar]
- 132.Morgan JP, Morgan KG. Stimulus-specific patterns of intracellular calcium levels in smooth muscle of ferret portal vein. The Journal of physiology. 1984;351:155–67. doi: 10.1113/jphysiol.1984.sp015239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tsien RY. Intracellular measurements of ion activities. Annu Rev Biophys Bioeng. 1983;12:91–116. doi: 10.1146/annurev.bb.12.060183.000515. [DOI] [PubMed] [Google Scholar]
- 134.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. The Journal of biological chemistry. 1985;260:3440–50. [PubMed] [Google Scholar]
- 135.Kobayashi S, Kanaide H, Nakamura M. Cytosolic-free calcium transients in cultured vascular smooth muscle cells: microfluorometric measurements. Science. 1985;229:553–6. doi: 10.1126/science.3927484. [DOI] [PubMed] [Google Scholar]
- 136.Himpens B, Casteels R. Measurement by Quin2 of changes of the intracellular calcium concentration in strips of the rabbit ear artery and of the guinea-pig ileum. Pflugers Archiv: European journal of physiology. 1987;408:32–7. doi: 10.1007/BF00581837. [DOI] [PubMed] [Google Scholar]
- 137.Sugiyama T, Yoshizumi M, Takaku F, Urabe H, Tsukakoshi M, Kasuya T, et al. The elevation of the cytoplasmic calcium ions in vascular smooth muscle cells in SHR--measurement of the free calcium ions in single living cells by lasermicrofluorospectrometry. Biochemical and biophysical research communications. 1986;141:340–5. doi: 10.1016/s0006-291x(86)80374-6. [DOI] [PubMed] [Google Scholar]
- 138.Ikebe M, Hartshorne DJ. The role of myosin phosphorylation in the contraction-relaxation cycle of smooth muscle. Experientia. 1985;41:1006–10. doi: 10.1007/BF01952122. [DOI] [PubMed] [Google Scholar]
- 139.Kamm KE, Stull JT. Regulation of smooth muscle contractile elements by second messengers. Annu Rev Physiol. 1989;51:299–313. doi: 10.1146/annurev.ph.51.030189.001503. [DOI] [PubMed] [Google Scholar]
- 140.Khalil RA, van Breemen C. Intracellular free calcium concentration/force relationship in rabbit inferior vena cava activated by norepinephrine and high K+ Pflugers Archiv: European journal of physiology. 1990;416:727–34. doi: 10.1007/BF00370622. [DOI] [PubMed] [Google Scholar]
- 141.DeFeo TT, Morgan KG. Calcium-force relationships as detected with aequorin in two different vascular smooth muscles of the ferret. The Journal of physiology. 1985;369:269–82. doi: 10.1113/jphysiol.1985.sp015900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Himpens B, Matthijs G, Somlyo AV, Butler TM, Somlyo AP. Cytoplasmic free calcium, myosin light chain phosphorylation, and force in phasic and tonic smooth muscle. J Gen Physiol. 1988;92:713–29. doi: 10.1085/jgp.92.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rembold CM, Murphy RA. Myoplasmic [Ca2+] determines myosin phosphorylation in agonist-stimulated swine arterial smooth muscle. Circulation research. 1988;63:593–603. doi: 10.1161/01.res.63.3.593. [DOI] [PubMed] [Google Scholar]
- 144.Kitazawa T, Gaylinn BD, Denney GH, Somlyo AP. G-protein-mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. The Journal of biological chemistry. 1991;266:1708–15. [PubMed] [Google Scholar]
- 145.Rembold CM. Modulation of the [Ca2+] sensitivity of myosin phosphorylation in intact swine arterial smooth muscle. The Journal of physiology. 1990;429:77–94. doi: 10.1113/jphysiol.1990.sp018245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Suematsu E, Resnick M, Morgan KG. Change of Ca2+ requirement for myosin phosphorylation by prostaglandin F2 alpha. The American journal of physiology. 1991;261:C253–8. doi: 10.1152/ajpcell.1991.261.2.C253. [DOI] [PubMed] [Google Scholar]
- 147.Hai CM, Murphy RA. Ca2+, crossbridge phosphorylation, and contraction. Annu Rev Physiol. 1989;51:285–98. doi: 10.1146/annurev.ph.51.030189.001441. [DOI] [PubMed] [Google Scholar]
- 148.Berridge MJ. Inositol trisphosphate and diacylglycerol as second messengers. Biochem J. 1984;220:345–60. doi: 10.1042/bj2200345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Takai Y, Kishimoto A, Inoue M, Nishizuka Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. I. Purification and characterization of an active enzyme from bovine cerebellum. The Journal of biological chemistry. 1977;252:7603–9. [PubMed] [Google Scholar]
- 150.Takai Y, Kishimoto A, Kikkawa U, Mori T, Nishizuka Y. Unsaturated diacylglycerol as a possible messenger for the activation of calcium-activated, phospholipid-dependent protein kinase system. Biochemical and biophysical research communications. 1979;91:1218–24. doi: 10.1016/0006-291x(79)91197-5. [DOI] [PubMed] [Google Scholar]
- 151.Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. The Journal of biological chemistry. 1982;257:7847–51. [PubMed] [Google Scholar]
- 152.Kishimoto A, Mikawa K, Hashimoto K, Yasuda I, Tanaka S, Tominaga M, et al. Limited proteolysis of protein kinase C subspecies by calcium-dependent neutral protease (calpain) The Journal of biological chemistry. 1989;264:4088–92. [PubMed] [Google Scholar]
- 153.Steinberg SF. Structural basis of protein kinase C isoform function. Physiological reviews. 2008;88:1341–78. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kim HR, Gallant C, Morgan KG. Regulation of PKC autophosphorylation by calponin in contractile vascular smooth muscle tissue. BioMed research international. 2013;2013:358643. doi: 10.1155/2013/358643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Poli A, Mongiorgi S, Cocco L, Follo MY. Protein kinase C involvement in cell cycle modulation. Biochem Soc Trans. 2014;42:1471–6. doi: 10.1042/BST20140128. [DOI] [PubMed] [Google Scholar]
- 156.Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev. 2001;101:2353–64. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 157.Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov. 2012;11:937–57. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Newton AC. Protein kinase C: structure, function, and regulation. The Journal of biological chemistry. 1995;270:28495–8. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 159.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332(Pt 2):281–92. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hoque M, Rentero C, Cairns R, Tebar F, Enrich C, Grewal T. Annexins - scaffolds modulating PKC localization and signaling. Cell Signal. 2014;26:1213–25. doi: 10.1016/j.cellsig.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 161.Steinberg SF. Mechanisms for redox-regulation of protein kinase C. Front Pharmacol. 2015;6:128. doi: 10.3389/fphar.2015.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. The Journal of biological chemistry. 2007;282:826–30. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 163.Benes CH, Wu N, Elia AE, Dharia T, Cantley LC, Soltoff SP. The C2 domain of PKCdelta is a phosphotyrosine binding domain. Cell. 2005;121:271–80. doi: 10.1016/j.cell.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 164.Linch M, Riou P, Claus J, Cameron AJ, de Naurois J, Larijani B, et al. Functional implications of assigned, assumed and assembled PKC structures. Biochem Soc Trans. 2014;42:35–41. doi: 10.1042/BST20130192. [DOI] [PubMed] [Google Scholar]
- 165.Hirano Y, Yoshinaga S, Ogura K, Yokochi M, Noda Y, Sumimoto H, et al. Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5. The Journal of biological chemistry. 2004;279:31883–90. doi: 10.1074/jbc.M403092200. [DOI] [PubMed] [Google Scholar]
- 166.Rozengurt E. Protein kinase D signaling: multiple biological functions in health and disease. Physiology (Bethesda) 2011;26:23–33. doi: 10.1152/physiol.00037.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Xu ZB, Chaudhary D, Olland S, Wolfrom S, Czerwinski R, Malakian K, et al. Catalytic domain crystal structure of protein kinase C-theta (PKCtheta) The Journal of biological chemistry. 2004;279:50401–9. doi: 10.1074/jbc.M409216200. [DOI] [PubMed] [Google Scholar]
- 168.Khalil RA. Protein Kinase C Inhibitors as Modulators of Vascular Function and their Application in Vascular Disease. Pharmaceuticals. 2013;6:407–39. doi: 10.3390/ph6030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kanashiro CA, Khalil RA. Signal transduction by protein kinase C in mammalian cells. Clinical and experimental pharmacology & physiology. 1998;25:974–85. doi: 10.1111/j.1440-1681.1998.tb02170.x. [DOI] [PubMed] [Google Scholar]
- 170.Grange JJ, Baca-Regen LM, Nollendorfs AJ, Persidsky Y, Sudan DL, Baxter BT. Protein kinase C isoforms in human aortic smooth muscle cells. Journal of vascular surgery. 1998;27:919–26. doi: 10.1016/s0741-5214(98)70273-3. discussion 26–7. [DOI] [PubMed] [Google Scholar]
- 171.Sakai N, Shirai Y, Saito N. Analysis of PKC targeting mechanism using PKC fused with fluorescent proteins. Nihon Yakurigaku Zasshi. 2003;121:421–34. doi: 10.1254/fpj.121.421. [DOI] [PubMed] [Google Scholar]
- 172.Khalil RA, Morgan KG. Imaging of protein kinase C distribution and translocation in living vascular smooth muscle cells. Circulation research. 1991;69:1626–31. doi: 10.1161/01.res.69.6.1626. [DOI] [PubMed] [Google Scholar]
- 173.Kraft AS, Anderson WB. Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature. 1983;301:621–3. doi: 10.1038/301621a0. [DOI] [PubMed] [Google Scholar]
- 174.Saito K, Ito E, Takakuwa Y, Tamura M, Kinjo M. In situ observation of mobility and anchoring of PKCbetaI in plasma membrane. FEBS Lett. 2003;541:126–31. doi: 10.1016/s0014-5793(03)00324-7. [DOI] [PubMed] [Google Scholar]
- 175.Mineo C, Ying YS, Chapline C, Jaken S, Anderson RG. Targeting of protein kinase Calpha to caveolae. J Cell Biol. 1998;141:601–10. doi: 10.1083/jcb.141.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Oka N, Yamamoto M, Schwencke C, Kawabe J, Ebina T, Ohno S, et al. Caveolin interaction with protein kinase C. Isoenzyme-dependent regulation of kinase activity by the caveolin scaffolding domain peptide. The Journal of biological chemistry. 1997;272:33416–21. doi: 10.1074/jbc.272.52.33416. [DOI] [PubMed] [Google Scholar]
- 177.Ratz PH, Miner AS. Role of protein kinase Czeta and calcium entry in KCl-induced vascular smooth muscle calcium sensitization and feedback control of cellular calcium levels. The Journal of pharmacology and experimental therapeutics. 2009;328:399–408. doi: 10.1124/jpet.108.142422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Draeger A, Wray S, Babiychuk EB. Domain architecture of the smooth-muscle plasma membrane: regulation by annexins. Biochem J. 2005;387:309–14. doi: 10.1042/BJ20041363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dubois T, Oudinet JP, Mira JP, Russo-Marie F. Annexins and protein kinases C. Biochim Biophys Acta. 1996;1313:290–4. doi: 10.1016/0167-4889(96)00102-4. [DOI] [PubMed] [Google Scholar]
- 180.Xu TR, Rumsby MG. Phorbol ester-induced translocation of PKC epsilon to the nucleus in fibroblasts: identification of nuclear PKC epsilon-associating proteins. FEBS Lett. 2004;570:20–4. doi: 10.1016/j.febslet.2004.05.080. [DOI] [PubMed] [Google Scholar]
- 181.Kheifets V, Bright R, Inagaki K, Schechtman D, Mochly-Rosen D. Protein kinase C delta (deltaPKC)-annexin V interaction: a required step in deltaPKC translocation and function. The Journal of biological chemistry. 2006;281:23218–26. doi: 10.1074/jbc.M602075200. [DOI] [PubMed] [Google Scholar]
- 182.Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature. 1992;356:618–22. doi: 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- 183.Gallant C, You JY, Sasaki Y, Grabarek Z, Morgan KG. MARCKS is a major PKC-dependent regulator of calmodulin targeting in smooth muscle. J Cell Sci. 2005;118:3595–605. doi: 10.1242/jcs.02493. [DOI] [PubMed] [Google Scholar]
- 184.Thelen M, Rosen A, Nairn AC, Aderem A. Regulation by phosphorylation of reversible association of a myristoylated protein kinase C substrate with the plasma membrane. Nature. 1991;351:320–2. doi: 10.1038/351320a0. [DOI] [PubMed] [Google Scholar]
- 185.Ron D, Kazanietz MG. New insights into the regulation of protein kinase C and novel phorbol ester receptors. FASEB J. 1999;13:1658–76. [PubMed] [Google Scholar]
- 186.Qvit N, Mochly-Rosen D. Highly Specific Modulators of Protein Kinase C Localization: Applications to Heart Failure. Drug Discov Today Dis Mech. 2010;7:e87–e93. doi: 10.1016/j.ddmec.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ron D, Mochly-Rosen D. Agonists and antagonists of protein kinase C function, derived from its binding proteins. The Journal of biological chemistry. 1994;269:21395–8. [PubMed] [Google Scholar]
- 188.Ron D, Luo J, Mochly-Rosen D. C2 region-derived peptides inhibit translocation and function of beta protein kinase C in vivo. The Journal of biological chemistry. 1995;270:24180–7. doi: 10.1074/jbc.270.41.24180. [DOI] [PubMed] [Google Scholar]
- 189.Mochly-Rosen D, Kauvar LM. Pharmacological regulation of network kinetics by protein kinase C localization. Semin Immunol. 2000;12:55–61. doi: 10.1006/smim.2000.0207. [DOI] [PubMed] [Google Scholar]
- 190.Dorn GW, 2nd, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ, et al. Sustained in vivo cardiac protection by a rationally designed peptide that causes epsilon protein kinase C translocation. Proc Natl Acad Sci U S A. 1999;96:12798–803. doi: 10.1073/pnas.96.22.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Churchill EN, Qvit N, Mochly-Rosen D. Rationally designed peptide regulators of protein kinase C. Trends Endocrinol Metab. 2009;20:25–33. doi: 10.1016/j.tem.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Schechtman D, Craske ML, Kheifets V, Meyer T, Schechtman J, Mochly-Rosen D. A critical intramolecular interaction for protein kinase Cepsilon translocation. The Journal of biological chemistry. 2004;279:15831–40. doi: 10.1074/jbc.M310696200. [DOI] [PubMed] [Google Scholar]
- 193.Welch EJ, Jones BW, Scott JD. Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol Interv. 2010;10:86–97. doi: 10.1124/mi.10.2.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Toker A, Ellis CA, Sellers LA, Aitken A. Protein kinase C inhibitor proteins. Purification from sheep brain and sequence similarity to lipocortins and 14-3-3 protein. Eur J Biochem. 1990;191:421–9. doi: 10.1111/j.1432-1033.1990.tb19138.x. [DOI] [PubMed] [Google Scholar]
- 195.Lum MA, Balaburski GM, Murphy ME, Black AR, Black JD. Heat shock proteins regulate activation-induced proteasomal degradation of the mature phosphorylated form of protein kinase C. The Journal of biological chemistry. 2013;288:27112–27. doi: 10.1074/jbc.M112.437095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Adwan TS, Ohm AM, Jones DN, Humphries MJ, Reyland ME. Regulated binding of importin-alpha to protein kinase Cdelta in response to apoptotic signals facilitates nuclear import. The Journal of biological chemistry. 2011;286:35716–24. doi: 10.1074/jbc.M111.255950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Prekeris R, Mayhew MW, Cooper JB, Terrian DM. Identification and localization of an actin-binding motif that is unique to the epsilon isoform of protein kinase C and participates in the regulation of synaptic function. J Cell Biol. 1996;132:77–90. doi: 10.1083/jcb.132.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Huang X, Walker JW. Myofilament anchoring of protein kinase C-epsilon in cardiac myocytes. J Cell Sci. 2004;117:1971–8. doi: 10.1242/jcs.01044. [DOI] [PubMed] [Google Scholar]
- 199.Rodriguez MM, Chen CH, Smith BL, Mochly-Rosen D. Characterization of the binding and phosphorylation of cardiac calsequestrin by epsilon protein kinase C. FEBS Lett. 1999;454:240–6. doi: 10.1016/s0014-5793(99)00697-3. [DOI] [PubMed] [Google Scholar]
- 200.Zeidman R, Lofgren B, Pahlman S, Larsson C. PKCepsilon, via its regulatory domain and independently of its catalytic domain, induces neurite-like processes in neuroblastoma cells. J Cell Biol. 1999;145:713–26. doi: 10.1083/jcb.145.4.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Csukai M, Chen CH, De Matteis MA, Mochly-Rosen D. The coatomer protein beta’-COP, a selective binding protein (RACK) for protein kinase Cepsilon. The Journal of biological chemistry. 1997;272:29200–6. doi: 10.1074/jbc.272.46.29200. [DOI] [PubMed] [Google Scholar]
- 202.Lehel C, Olah Z, Jakab G, Anderson WB. Protein kinase C epsilon is localized to the Golgi via its zinc-finger domain and modulates Golgi function. Proc Natl Acad Sci U S A. 1995;92:1406–10. doi: 10.1073/pnas.92.5.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lehel C, Olah Z, Jakab G, Szallasi Z, Petrovics G, Harta G, et al. Protein kinase C epsilon subcellular localization domains and proteolytic degradation sites. A model for protein kinase C conformational changes. The Journal of biological chemistry. 1995;270:19651–8. doi: 10.1074/jbc.270.33.19651. [DOI] [PubMed] [Google Scholar]
- 204.Yang Y, Igumenova TI. The C-terminal V5 domain of Protein Kinase Calpha is intrinsically disordered, with propensity to associate with a membrane mimetic. PloS one. 2013;8:e65699. doi: 10.1371/journal.pone.0065699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.White WO, Seibenhener ML, Wooten MW. Phosphorylation of tyrosine 256 facilitates nuclear import of atypical protein kinase C. J Cell Biochem. 2002;85:42–53. [PubMed] [Google Scholar]
- 206.Hui X, Reither G, Kaestner L, Lipp P. Targeted activation of conventional and novel protein kinases C through differential translocation patterns. Mol Cell Biol. 2014;34:2370–81. doi: 10.1128/MCB.00040-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhang Y, Hermanson ME, Eddinger TJ. Tonic and phasic smooth muscle contraction is not regulated by the PKCalpha - CPI-17 pathway in swine stomach antrum and fundus. PloS one. 2013;8:e74608. doi: 10.1371/journal.pone.0074608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Konishi H, Yamauchi E, Taniguchi H, Yamamoto T, Matsuzaki H, Takemura Y, et al. Phosphorylation sites of protein kinase C delta in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc Natl Acad Sci U S A. 2001;98:6587–92. doi: 10.1073/pnas.111158798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kheifets V, Mochly-Rosen D. Insight into intra- and inter-molecular interactions of PKC: design of specific modulators of kinase function. Pharmacol Res. 2007;55:467–76. doi: 10.1016/j.phrs.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Schmalz D, Kalkbrenner F, Hucho F, Buchner K. Transport of protein kinase C alpha into the nucleus requires intact cytoskeleton while the transport of a protein containing a canonical nuclear localization signal does not. J Cell Sci. 1996;109(Pt 9):2401–6. doi: 10.1242/jcs.109.9.2401. [DOI] [PubMed] [Google Scholar]
- 211.Dykes AC, Fultz ME, Norton ML, Wright GL. Microtubule-dependent PKC-alpha localization in A7r5 smooth muscle cells. American journal of physiology Cell physiology. 2003;285:C76–87. doi: 10.1152/ajpcell.00515.2002. [DOI] [PubMed] [Google Scholar]
- 212.House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–8. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 213.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–71. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. EMBO J. 2000;19:496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Newton AC. Protein kinase C: poised to signal. American journal of physiology Endocrinology and metabolism. 2010;298:E395–402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–5. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- 217.Liu Y, Graham C, Li A, Fisher RJ, Shaw S. Phosphorylation of the protein kinase C-theta activation loop and hydrophobic motif regulates its kinase activity, but only activation loop phosphorylation is critical to in vivo nuclear-factor-kappaB induction. Biochem J. 2002;361:255–65. doi: 10.1042/bj3610255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Cazaubon S, Bornancin F, Parker PJ. Threonine-497 is a critical site for permissive activation of protein kinase C alpha. Biochem J. 1994;301(Pt 2):443–8. doi: 10.1042/bj3010443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Balendran A, Hare GR, Kieloch A, Williams MR, Alessi DR. Further evidence that 3-phosphoinositide-dependent protein kinase-1 (PDK1) is required for the stability and phosphorylation of protein kinase C (PKC) isoforms. FEBS Lett. 2000;484:217–23. doi: 10.1016/s0014-5793(00)02162-1. [DOI] [PubMed] [Google Scholar]
- 220.Behn-Krappa A, Newton AC. The hydrophobic phosphorylation motif of conventional protein kinase C is regulated by autophosphorylation. Curr Biol. 1999;9:728–37. doi: 10.1016/s0960-9822(99)80332-7. [DOI] [PubMed] [Google Scholar]
- 221.Hage-Sleiman R, Hamze AB, Reslan L, Kobeissy H, Dbaibo G. The Novel PKCtheta from Benchtop to Clinic. J Immunol Res. 2015;2015:348798. doi: 10.1155/2015/348798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1) Curr Biol. 1998;8:1366–75. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- 223.Jacinto E, Lorberg A. TOR regulation of AGC kinases in yeast and mammals. Biochem J. 2008;410:19–37. doi: 10.1042/BJ20071518. [DOI] [PubMed] [Google Scholar]
- 224.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–31. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 226.Cameron AJ, De Rycker M, Calleja V, Alcor D, Kjaer S, Kostelecky B, et al. Protein kinases, from B to C. Biochem Soc Trans. 2007;35:1013–7. doi: 10.1042/BST0351013. [DOI] [PubMed] [Google Scholar]
- 227.Rybin VO, Sabri A, Short J, Braz JC, Molkentin JD, Steinberg SF. Cross-regulation of novel protein kinase C (PKC) isoform function in cardiomyocytes. Role of PKC epsilon in activation loop phosphorylations and PKC delta in hydrophobic motif phosphorylations. The Journal of biological chemistry. 2003;278:14555–64. doi: 10.1074/jbc.M212644200. [DOI] [PubMed] [Google Scholar]
- 228.Rybin VO, Guo J, Sabri A, Elouardighi H, Schaefer E, Steinberg SF. Stimulus-specific differences in protein kinase C delta localization and activation mechanisms in cardiomyocytes. The Journal of biological chemistry. 2004;279:19350–61. doi: 10.1074/jbc.M311096200. [DOI] [PubMed] [Google Scholar]
- 229.Cenni V, Doppler H, Sonnenburg ED, Maraldi N, Newton AC, Toker A. Regulation of novel protein kinase C epsilon by phosphorylation. Biochem J. 2002;363:537–45. doi: 10.1042/0264-6021:3630537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, et al. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci U S A. 1997;94:11233–7. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Saurin AT, Durgan J, Cameron AJ, Faisal A, Marber MS, Parker PJ. The regulated assembly of a PKCepsilon complex controls the completion of cytokinesis. Nat Cell Biol. 2008;10:891–901. doi: 10.1038/ncb1749. [DOI] [PubMed] [Google Scholar]
- 232.Meller N, Liu YC, Collins TL, Bonnefoy-Berard N, Baier G, Isakov N, et al. Direct interaction between protein kinase C theta (PKC theta) and 14-3-3 tau in T cells: 14-3-3 overexpression results in inhibition of PKC theta translocation and function. Mol Cell Biol. 1996;16:5782–91. doi: 10.1128/mcb.16.10.5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Durgan J, Michael N, Totty N, Parker PJ. Novel phosphorylation site markers of protein kinase C delta activation. FEBS Lett. 2007;581:3377–81. doi: 10.1016/j.febslet.2007.06.035. [DOI] [PubMed] [Google Scholar]
- 234.Ng T, Squire A, Hansra G, Bornancin F, Prevostel C, Hanby A, et al. Imaging protein kinase Calpha activation in cells. Science. 1999;283:2085–9. doi: 10.1126/science.283.5410.2085. [DOI] [PubMed] [Google Scholar]
- 235.Li W, Zhang J, Bottaro DP, Pierce JH. Identification of serine 643 of protein kinase C-delta as an important autophosphorylation site for its enzymatic activity. The Journal of biological chemistry. 1997;272:24550–5. doi: 10.1074/jbc.272.39.24550. [DOI] [PubMed] [Google Scholar]
- 236.Dutil EM, Keranen LM, DePaoli-Roach AA, Newton AC. In vivo regulation of protein kinase C by trans-phosphorylation followed by autophosphorylation. The Journal of biological chemistry. 1994;269:29359–62. [PubMed] [Google Scholar]
- 237.Gao T, Brognard J, Newton AC. The phosphatase PHLPP controls the cellular levels of protein kinase C. The Journal of biological chemistry. 2008;283:6300–11. doi: 10.1074/jbc.M707319200. [DOI] [PubMed] [Google Scholar]
- 238.Lee HW, Smith L, Pettit GR, Bingham Smith J. Dephosphorylation of activated protein kinase C contributes to downregulation by bryostatin. The American journal of physiology. 1996;271:C304–11. doi: 10.1152/ajpcell.1996.271.1.C304. [DOI] [PubMed] [Google Scholar]
- 239.Ahn JH, McAvoy T, Rakhilin SV, Nishi A, Greengard P, Nairn AC. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc Natl Acad Sci U S A. 2007;104:2979–84. doi: 10.1073/pnas.0611532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sontag E, Sontag JM, Garcia A. Protein phosphatase 2A is a critical regulator of protein kinase C zeta signaling targeted by SV40 small t to promote cell growth and NF-kappaB activation. EMBO J. 1997;16:5662–71. doi: 10.1093/emboj/16.18.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Eitelhuber AC, Warth S, Schimmack G, Duwel M, Hadian K, Demski K, et al. Dephosphorylation of Carma1 by PP2A negatively regulates T-cell activation. EMBO J. 2011;30:594–605. doi: 10.1038/emboj.2010.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- 243.Gao T, Newton AC. Invariant Leu preceding turn motif phosphorylation site controls the interaction of protein kinase C with Hsp70. The Journal of biological chemistry. 2006;281:32461–8. doi: 10.1074/jbc.M604076200. [DOI] [PubMed] [Google Scholar]
- 244.Gould CM, Antal CE, Reyes G, Kunkel MT, Adams RA, Ziyar A, et al. Active site inhibitors protect protein kinase C from dephosphorylation and stabilize its mature form. The Journal of biological chemistry. 2011;286:28922–30. doi: 10.1074/jbc.M111.272526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Cameron AJ, Escribano C, Saurin AT, Kostelecky B, Parker PJ. PKC maturation is promoted by nucleotide pocket occupation independently of intrinsic kinase activity. Nat Struct Mol Biol. 2009;16:624–30. doi: 10.1038/nsmb.1606. [DOI] [PubMed] [Google Scholar]
- 246.Bazzi MD, Nelsestuen GL. Protein kinase C interaction with calcium: a phospholipid-dependent process. Biochemistry. 1990;29:7624–30. doi: 10.1021/bi00485a012. [DOI] [PubMed] [Google Scholar]
- 247.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–14. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 248.Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–96. [PubMed] [Google Scholar]
- 249.Dries DR, Newton AC. Kinetic analysis of the interaction of the C1 domain of protein kinase C with lipid membranes by stopped-flow spectroscopy. The Journal of biological chemistry. 2008;283:7885–93. doi: 10.1074/jbc.M709943200. [DOI] [PubMed] [Google Scholar]
- 250.Sanchez-Bautista S, Corbalan-Garcia S, Perez-Lara A, Gomez-Fernandez JC. A comparison of the membrane binding properties of C1B domains of PKCgamma, PKCdelta, and PKCepsilon. Biophys J. 2009;96:3638–47. doi: 10.1016/j.bpj.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Zhang G, Kazanietz MG, Blumberg PM, Hurley JH. Crystal structure of the cys2 activator-binding domain of protein kinase C delta in complex with phorbol ester. Cell. 1995;81:917–24. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 252.Mukherjee A, Roy S, Saha B, Mukherjee D. Spatio-Temporal Regulation of PKC Isoforms Imparts Signaling Specificity. Front Immunol. 2016;7:45. doi: 10.3389/fimmu.2016.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Rainbow R, Parker A, Davies N. Protein kinase C-independent inhibition of arterial smooth muscle K(+) channels by a diacylglycerol analogue. British journal of pharmacology. 2011;163:845–56. doi: 10.1111/j.1476-5381.2011.01268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Persaud SD, Hoang V, Huang J, Basu A. Involvement of proteolytic activation of PKCdelta in cisplatin-induced apoptosis in human small cell lung cancer H69 cells. Int J Oncol. 2005;27:149–54. [PubMed] [Google Scholar]
- 255.Gopalakrishna R, Anderson WB. Ca2+- and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc Natl Acad Sci U S A. 1989;86:6758–62. doi: 10.1073/pnas.86.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349–61. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 257.Gopalakrishna R, Chen ZH, Gundimeda U. Tobacco smoke tumor promoters, catechol and hydroquinone, induce oxidative regulation of protein kinase C and influence invasion and metastasis of lung carcinoma cells. Proc Natl Acad Sci U S A. 1994;91:12233–7. doi: 10.1073/pnas.91.25.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Bogard AS, Tavalin SJ. Protein Kinase C (PKC)zeta Pseudosubstrate Inhibitor Peptide Promiscuously Binds PKC Family Isoforms and Disrupts Conventional PKC Targeting and Translocation. Mol Pharmacol. 2015;88:728–35. doi: 10.1124/mol.115.099457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Faux MC, Rollins EN, Edwards AS, Langeberg LK, Newton AC, Scott JD. Mechanism of A-kinase-anchoring protein 79 (AKAP79) and protein kinase C interaction. Biochem J. 1999;343(Pt 2):443–52. [PMC free article] [PubMed] [Google Scholar]
- 260.Ding RQ, Tsao J, Chai H, Mochly-Rosen D, Zhou W. Therapeutic potential for protein kinase C inhibitor in vascular restenosis. J Cardiovasc Pharmacol Ther. 2011;16:160–7. doi: 10.1177/1074248410382106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Lewis JM, Cheresh DA, Schwartz MA. Protein kinase C regulates alpha v beta 5-dependent cytoskeletal associations and focal adhesion kinase phosphorylation. J Cell Biol. 1996;134:1323–32. doi: 10.1083/jcb.134.5.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kappert K, Furundzija V, Fritzsche J, Margeta C, Kruger J, Meyborg H, et al. Integrin cleavage regulates bidirectional signalling in vascular smooth muscle cells. Thromb Haemost. 2010;103:556–63. doi: 10.1160/TH09-07-0478. [DOI] [PubMed] [Google Scholar]
- 263.Li C, Wernig F, Leitges M, Hu Y, Xu Q. Mechanical stress-activated PKCdelta regulates smooth muscle cell migration. FASEB J. 2003;17:2106–8. doi: 10.1096/fj.03-0150fje. [DOI] [PubMed] [Google Scholar]
- 264.Perez-Moreno M, Avila A, Islas S, Sanchez S, Gonzalez-Mariscal L. Vinculin but not alpha-actinin is a target of PKC phosphorylation during junctional assembly induced by calcium. J Cell Sci. 1998;111(Pt 23):3563–71. doi: 10.1242/jcs.111.23.3563. [DOI] [PubMed] [Google Scholar]
- 265.Ivaska J, Vuoriluoto K, Huovinen T, Izawa I, Inagaki M, Parker PJ. PKCepsilon-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 2005;24:3834–45. doi: 10.1038/sj.emboj.7600847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Valovka T, Verdier F, Cramer R, Zhyvoloup A, Fenton T, Rebholz H, et al. Protein kinase C phosphorylates ribosomal protein S6 kinase betaII and regulates its subcellular localization. Mol Cell Biol. 2003;23:852–63. doi: 10.1128/MCB.23.3.852-863.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Ackermann MA, Kontrogianni-Konstantopoulos A. Myosin binding protein-C slow is a novel substrate for protein kinase A (PKA) and C (PKC) in skeletal muscle. J Proteome Res. 2011;10:4547–55. doi: 10.1021/pr200355w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Clarke M, Dodson PM. PKC inhibition and diabetic microvascular complications. Best Pract Res Clin Endocrinol Metab. 2007;21:573–86. doi: 10.1016/j.beem.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 269.Roffey J, Rosse C, Linch M, Hibbert A, McDonald NQ, Parker PJ. Protein kinase C intervention: the state of play. Curr Opin Cell Biol. 2009;21:268–79. doi: 10.1016/j.ceb.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 270.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circulation research. 2010;106:1319–31. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. The Journal of clinical investigation. 1997;100:115–26. doi: 10.1172/JCI119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kanashiro CA, Altirkawi KA, Khalil RA. Preconditioning of coronary artery against vasoconstriction by endothelin-1 and prostaglandin F2alpha during repeated downregulation of epsilon-protein kinase C. Journal of cardiovascular pharmacology. 2000;35:491–501. doi: 10.1097/00005344-200003000-00021. [DOI] [PubMed] [Google Scholar]
- 273.Eichholtz T, de Bont DB, de Widt J, Liskamp RM, Ploegh HL. A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. The Journal of biological chemistry. 1993;268:1982–6. [PubMed] [Google Scholar]
- 274.Parissenti AM, Kirwan AF, Kim SA, Colantonio CM, Schimmer BP. Inhibitory properties of the regulatory domains of human protein kinase Calpha and mouse protein kinase Cepsilon. The Journal of biological chemistry. 1998;273:8940–5. doi: 10.1074/jbc.273.15.8940. [DOI] [PubMed] [Google Scholar]
- 275.House C, Kemp BE. Protein kinase C pseudosubstrate prototope: structure-function relationships. Cell Signal. 1990;2:187–90. doi: 10.1016/0898-6568(90)90022-3. [DOI] [PubMed] [Google Scholar]
- 276.Pears CJ, Kour G, House C, Kemp BE, Parker PJ. Mutagenesis of the pseudosubstrate site of protein kinase C leads to activation. Eur J Biochem. 1990;194:89–94. doi: 10.1111/j.1432-1033.1990.tb19431.x. [DOI] [PubMed] [Google Scholar]
- 277.Albert AP, Large WA. Signal transduction pathways and gating mechanisms of native TRP-like cation channels in vascular myocytes. The Journal of physiology. 2006;570:45–51. doi: 10.1113/jphysiol.2005.096875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Liu M, Large WA, Albert AP. Stimulation of beta-adrenoceptors inhibits store-operated channel currents via a cAMP-dependent protein kinase mechanism in rabbit portal vein myocytes. The Journal of physiology. 2005;562:395–406. doi: 10.1113/jphysiol.2004.077602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Boscoboinik D, Szewczyk A, Hensey C, Azzi A. Inhibition of cell proliferation by alpha-tocopherol. Role of protein kinase C The Journal of biological chemistry. 1991;266:6188–94. [PubMed] [Google Scholar]
- 280.Liu JY, Lin SJ, Lin JK. Inhibitory effects of curcumin on protein kinase C activity induced by 12-O-tetradecanoyl-phorbol-13-acetate in NIH 3T3 cells. Carcinogenesis. 1993;14:857–61. doi: 10.1093/carcin/14.5.857. [DOI] [PubMed] [Google Scholar]
- 281.Clement S, Tasinato A, Boscoboinik D, Azzi A. The effect of alpha-tocopherol on the synthesis, phosphorylation and activity of protein kinase C in smooth muscle cells after phorbol 12-myristate 13-acetate down-regulation. Eur J Biochem. 1997;246:745–9. doi: 10.1111/j.1432-1033.1997.t01-2-00745.x. [DOI] [PubMed] [Google Scholar]
- 282.Engin KN. Alpha-tocopherol: looking beyond an antioxidant. Mol Vis. 2009;15:855–60. [PMC free article] [PubMed] [Google Scholar]
- 283.Bursell SE, King GL. Can protein kinase C inhibition and vitamin E prevent the development of diabetic vascular complications? Diabetes Res Clin Pract. 1999;45:169–82. doi: 10.1016/s0168-8227(99)00047-9. [DOI] [PubMed] [Google Scholar]
- 284.Ward NE, Pierce DS, Chung SE, Gravitt KR, O’Brian CA. Irreversible inactivation of protein kinase C by glutathione. The Journal of biological chemistry. 1998;273:12558–66. doi: 10.1074/jbc.273.20.12558. [DOI] [PubMed] [Google Scholar]
- 285.Newman AH. A Novel PKC Inhibitor Shows Promise for Amphetamine Use Disorders. Neuropsychopharmacology. 2017;42:1929–30. doi: 10.1038/npp.2017.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wu J, Liu S, Fan Z, Zhang L, Tian Y, Yang R. A novel and selective inhibitor of PKC zeta potently inhibits human breast cancer metastasis in vitro and in mice. Tumour Biol. 2016;37:8391–401. doi: 10.1007/s13277-015-4744-9. [DOI] [PubMed] [Google Scholar]
- 287.Bhavanasi D, Kostyak JC, Swindle J, Kilpatrick LE, Kunapuli SP. CGX1037 is a novel PKC isoform delta selective inhibitor in platelets. Platelets. 2015;26:2–9. doi: 10.3109/09537104.2013.868877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Choi H, Tostes RC, Webb RC. S-nitrosylation Inhibits protein kinase C-mediated contraction in mouse aorta. Journal of cardiovascular pharmacology. 2011;57:65–71. doi: 10.1097/FJC.0b013e3181fef9cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Song HF, Tang ZM, Yuan SJ, Zhu BZ, Liu XW. Antisense candidates against protein kinase C-alpha designed based on phylogenesis and simulant structure of mRNA. Acta Pharmacol Sin. 2003;24:269–76. [PubMed] [Google Scholar]
- 290.Mehta KD. Emerging role of protein kinase C in energy homeostasis: A brief overview. World J Diabetes. 2014;5:385–92. doi: 10.4239/wjd.v5.i3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. The American journal of physiology. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 292.Nauli SM, Williams JM, Akopov SE, Zhang L, Pearce WJ. Developmental changes in ryanodine- and IP(3)-sensitive Ca(2+) pools in ovine basilar artery. American journal of physiology Cell physiology. 2001;281:C1785–96. doi: 10.1152/ajpcell.2001.281.6.C1785. [DOI] [PubMed] [Google Scholar]
- 293.Kizub IV, Klymenko KI, Soloviev AI. Protein kinase C in enhanced vascular tone in diabetes mellitus. Int J Cardiol. 2014;174:230–42. doi: 10.1016/j.ijcard.2014.04.117. [DOI] [PubMed] [Google Scholar]
- 294.Ghatta S, Nimmagadda D, Xu X, O’Rourke ST. Large-conductance, calcium-activated potassium channels: structural and functional implications. Pharmacology & therapeutics. 2006;110:103–16. doi: 10.1016/j.pharmthera.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 295.Taguchi K, Kaneko K, Kubo T. Protein kinase C modulates Ca2+-activated K+ channels in cultured rat mesenteric artery smooth muscle cells. Biol Pharm Bull. 2000;23:1450–4. doi: 10.1248/bpb.23.1450. [DOI] [PubMed] [Google Scholar]
- 296.Barman SA, Zhu S, White RE. Protein kinase C inhibits BKCa channel activity in pulmonary arterial smooth muscle. American journal of physiology Lung cellular and molecular physiology. 2004;286:L149–55. doi: 10.1152/ajplung.00207.2003. [DOI] [PubMed] [Google Scholar]
- 297.Kizub IV, Pavlova OO, Ivanova IV, Soloviev AI. Protein kinase C-dependent inhibition of BK(Ca) current in rat aorta smooth muscle cells following gamma-irradiation. Int J Radiat Biol. 2010;86:291–9. doi: 10.3109/09553000903564042. [DOI] [PubMed] [Google Scholar]
- 298.Novokhatska T, Tishkin S, Dosenko V, Boldyriev A, Ivanova I, Strielkov I, et al. Correction of vascular hypercontractility in spontaneously hypertensive rats using shRNAs-induced delta protein kinase C gene silencing. European journal of pharmacology. 2013;718:401–7. doi: 10.1016/j.ejphar.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 299.Minami K, Fukuzawa K, Nakaya Y. Protein kinase C inhibits the Ca(2+)-activated K+ channel of cultured porcine coronary artery smooth muscle cells. Biochemical and biophysical research communications. 1993;190:263–9. doi: 10.1006/bbrc.1993.1040. [DOI] [PubMed] [Google Scholar]
- 300.Lange A, Gebremedhin D, Narayanan J, Harder D. 20-Hydroxyeicosatetraenoic acid-induced vasoconstriction and inhibition of potassium current in cerebral vascular smooth muscle is dependent on activation of protein kinase C. The Journal of biological chemistry. 1997;272:27345–52. doi: 10.1074/jbc.272.43.27345. [DOI] [PubMed] [Google Scholar]
- 301.Hu XQ, Xiao D, Zhu R, Huang X, Yang S, Wilson S, et al. Pregnancy upregulates large-conductance Ca(2+)-activated K(+) channel activity and attenuates myogenic tone in uterine arteries. Hypertension. 2011;58:1132–9. doi: 10.1161/HYPERTENSIONAHA.111.179952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Crozatier B. Central role of PKCs in vascular smooth muscle cell ion channel regulation. J Mol Cell Cardiol. 2006;41:952–5. doi: 10.1016/j.yjmcc.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 303.Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 2006;21:69–78. doi: 10.1152/physiol.00040.2005. [DOI] [PubMed] [Google Scholar]
- 304.Brueggemann LI, Mackie AR, Cribbs LL, Freda J, Tripathi A, Majetschak M, et al. Differential protein kinase C-dependent modulation of Kv7.4 and Kv7. 5 subunits of vascular Kv7 channels. The Journal of biological chemistry. 2014;289:2099–111. doi: 10.1074/jbc.M113.527820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase Czeta. Circulation research. 2003;93:656–63. doi: 10.1161/01.RES.0000095245.97945.FE. [DOI] [PubMed] [Google Scholar]
- 306.Park WS, Han J, Kim N, Youm JB, Joo H, Kim HK, et al. Endothelin-1 inhibits inward rectifier K+ channels in rabbit coronary arterial smooth muscle cells through protein kinase C. Journal of cardiovascular pharmacology. 2005;46:681–9. doi: 10.1097/01.fjc.0000182846.08357.ed. [DOI] [PubMed] [Google Scholar]
- 307.Park WS, Kim N, Youm JB, Warda M, Ko JH, Kim SJ, et al. Angiotensin II inhibits inward rectifier K+ channels in rabbit coronary arterial smooth muscle cells through protein kinase Calpha. Biochemical and biophysical research communications. 2006;341:728–35. doi: 10.1016/j.bbrc.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 308.Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiological reviews. 1997;77:1165–232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- 309.Bonev AD, Nelson MT. Vasoconstrictors inhibit ATP-sensitive K+ channels in arterial smooth muscle through protein kinase C. J Gen Physiol. 1996;108:315–23. doi: 10.1085/jgp.108.4.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Thorneloe KS, Maruyama Y, Malcolm AT, Light PE, Walsh MP, Cole WC. Protein kinase C modulation of recombinant ATP-sensitive K(+) channels composed of Kir6.1 and/or Kir6. 2 expressed with SUR2B. The Journal of physiology. 2002;541:65–80. doi: 10.1113/jphysiol.2002.018101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Levitan IB. Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu Rev Physiol. 1994;56:193–212. doi: 10.1146/annurev.ph.56.030194.001205. [DOI] [PubMed] [Google Scholar]
- 312.Light P. Regulation of ATP-sensitive potassium channels by phosphorylation. Biochim Biophys Acta. 1996;1286:65–73. doi: 10.1016/0304-4157(96)00004-4. [DOI] [PubMed] [Google Scholar]
- 313.Manna PT, Smith AJ, Taneja TK, Howell GJ, Lippiat JD, Sivaprasadarao A. Constitutive endocytic recycling and protein kinase C-mediated lysosomal degradation control K(ATP) channel surface density. The Journal of biological chemistry. 2010;285:5963–73. doi: 10.1074/jbc.M109.066902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Limas CJ. Phosphorylation of cardiac sarcoplasmic reticulum by a calcium-activated, phospholipid-dependent protein kinase. Biochemical and biophysical research communications. 1980;96:1378–83. doi: 10.1016/0006-291x(80)90103-5. [DOI] [PubMed] [Google Scholar]
- 315.Bertorello AM, Aperia A, Walaas SI, Nairn AC, Greengard P. Phosphorylation of the catalytic subunit of Na+, K(+)-ATPase inhibits the activity of the enzyme. Proc Natl Acad Sci U S A. 1991;88:11359–62. doi: 10.1073/pnas.88.24.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Austin C, Wray S. Interactions between Ca(2+) and H(+) and functional consequences in vascular smooth muscle. Circulation research. 2000;86:355–63. doi: 10.1161/01.res.86.3.355. [DOI] [PubMed] [Google Scholar]
- 317.Wray S, Smith RD. Mechanisms of action of pH-induced effects on vascular smooth muscle. Mol Cell Biochem. 2004;263:163–72. doi: 10.1023/B:MCBI.0000041858.78005.d2. [DOI] [PubMed] [Google Scholar]
- 318.Aviv A. Cytosolic Ca2+, Na+/H+ antiport, protein kinase C trio in essential hypertension. American journal of hypertension. 1994;7:205–12. doi: 10.1093/ajh/7.2.205. [DOI] [PubMed] [Google Scholar]
- 319.Rosoff PM, Stein LF, Cantley LC. Phorbol esters induce differentiation in a pre-B-lymphocyte cell line by enhancing Na+/H+ exchange. The Journal of biological chemistry. 1984;259:7056–60. [PubMed] [Google Scholar]
- 320.Jernigan NL, Resta TC. Calcium homeostasis and sensitization in pulmonary arterial smooth muscle. Microcirculation. 2014;21:259–71. doi: 10.1111/micc.12096. [DOI] [PubMed] [Google Scholar]
- 321.Wang Y, Zhou H, Wu B, Zhou Q, Cui D, Wang L. Protein Kinase C Isoforms Distinctly Regulate Propofol-induced Endothelium-dependent and Endothelium-independent Vasodilation. Journal of cardiovascular pharmacology. 2015;66:276–84. doi: 10.1097/FJC.0000000000000275. [DOI] [PubMed] [Google Scholar]
- 322.Woodsome TP, Eto M, Everett A, Brautigan DL, Kitazawa T. Expression of CPI-17 and myosin phosphatase correlates with Ca(2+) sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. The Journal of physiology. 2001;535:553–64. doi: 10.1111/j.1469-7793.2001.t01-1-00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.El-Yazbi AF, Abd-Elrahman KS, Moreno-Dominguez A. PKC-mediated cerebral vasoconstriction: Role of myosin light chain phosphorylation versus actin cytoskeleton reorganization. Biochemical pharmacology. 2015;95:263–78. doi: 10.1016/j.bcp.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 324.Je HD, Gangopadhyay SS, Ashworth TD, Morgan KG. Calponin is required for agonist-induced signal transduction--evidence from an antisense approach in ferret smooth muscle. The Journal of physiology. 2001;537:567–77. doi: 10.1111/j.1469-7793.2001.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Slish DF, Welsh DG, Brayden JE. Diacylglycerol and protein kinase C activate cation channels involved in myogenic tone. American journal of physiology Heart and circulatory physiology. 2002;283:H2196–201. doi: 10.1152/ajpheart.00605.2002. [DOI] [PubMed] [Google Scholar]
- 326.Yang YC, Wang XD, Huang K, Wang L, Jiang ZL, Qi YX. Temporal phosphoproteomics to investigate the mechanotransduction of vascular smooth muscle cells in response to cyclic stretch. J Biomech. 2014;47:3622–9. doi: 10.1016/j.jbiomech.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 327.Zhu R, Xiao D, Zhang L. Potassium channels and uterine vascular adaptation to pregnancy and chronic hypoxia. Current vascular pharmacology. 2013;11:737–47. doi: 10.2174/1570161111311050011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Cole WC, Malcolm T, Walsh MP, Light PE. Inhibition by protein kinase C of the K(NDP) subtype of vascular smooth muscle ATP-sensitive potassium channel. Circulation research. 2000;87:112–7. doi: 10.1161/01.res.87.2.112. [DOI] [PubMed] [Google Scholar]
- 329.Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts Hypertension. 1997;29:366–73. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 330.Jiao Y, Yang Q. Downregulation of natriuretic peptide clearance receptor mRNA in vascular smooth muscle cells by angiotensin II. Fundam Clin Pharmacol. 2015;29:260–8. doi: 10.1111/fcp.12111. [DOI] [PubMed] [Google Scholar]
- 331.Tykocki NR, Wu B, Jackson WF, Watts SW. Divergent signaling mechanisms for venous versus arterial contraction as revealed by endothelin-1. Journal of vascular surgery. 2014;62:721–33. doi: 10.1016/j.jvs.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Johnson JA, Barman SA. Protein kinase C modulation of cyclic GMP in rat neonatal pulmonary vascular smooth muscle. Lung. 2004;182:79–89. doi: 10.1007/s00408-003-1046-6. [DOI] [PubMed] [Google Scholar]
- 333.Moreno-Dominguez A, El-Yazbi AF, Zhu HL, Colinas O, Zhong XZ, Walsh EJ, et al. Cytoskeletal reorganization evoked by Rho-associated kinase- and protein kinase C-catalyzed phosphorylation of cofilin and heat shock protein 27, respectively, contributes to myogenic constriction of rat cerebral arteries. The Journal of biological chemistry. 2014;289:20939–52. doi: 10.1074/jbc.M114.553743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Mao Y, Su J, Lei L, Meng L, Qi Y, Huo Y, et al. Adrenomedullin and adrenotensin increase the transcription of regulator of Gprotein signaling 2 in vascular smooth muscle cells via the cAMPdependent and PKC pathways. Mol Med Rep. 2013;9:323–7. doi: 10.3892/mmr.2013.1751. [DOI] [PubMed] [Google Scholar]
- 335.Radhakrishnan Y, Maile LA, Ling Y, Graves LM, Clemmons DR. Insulin-like growth factor-I stimulates Shc-dependent phosphatidylinositol 3-kinase activation via Grb2-associated p85 in vascular smooth muscle cells. The Journal of biological chemistry. 2008;283:16320–31. doi: 10.1074/jbc.M801687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Yamaguchi H, Igarashi M, Hirata A, Sugae N, Tsuchiya H, Jimbu Y, et al. Altered PDGF-BB-induced p38 MAP kinase activation in diabetic vascular smooth muscle cells: roles of protein kinase C-delta. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:2095–101. doi: 10.1161/01.ATV.0000144009.35400.65. [DOI] [PubMed] [Google Scholar]
- 337.Ginnan R, Singer HA. PKC-delta-dependent pathways contribute to PDGF-stimulated ERK1/2 activation in vascular smooth muscle. American journal of physiology Cell physiology. 2005;288:C1193–201. doi: 10.1152/ajpcell.00499.2004. [DOI] [PubMed] [Google Scholar]
- 338.Mii S, Khalil RA, Morgan KG, Ware JA, Kent KC. Mitogen-activated protein kinase and proliferation of human vascular smooth muscle cells. The American journal of physiology. 1996;270:H142–50. doi: 10.1152/ajpheart.1996.270.1.H142. [DOI] [PubMed] [Google Scholar]
- 339.Khalil RA, Menice CB, Wang CL, Morgan KG. Phosphotyrosine-dependent targeting of mitogen-activated protein kinase in differentiated contractile vascular cells. Circulation research. 1995;76:1101–8. doi: 10.1161/01.res.76.6.1101. [DOI] [PubMed] [Google Scholar]
- 340.Cao W, Sohn UD, Bitar KN, Behar J, Biancani P, Harnett KM. MAPK mediates PKC-dependent contraction of cat esophageal and lower esophageal sphincter circular smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2003;285:G86–95. doi: 10.1152/ajpgi.00156.2002. [DOI] [PubMed] [Google Scholar]
- 341.Shi J, Mori E, Mori Y, Mori M, Li J, Ito Y, et al. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. The Journal of physiology. 2004;561:415–32. doi: 10.1113/jphysiol.2004.075051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Large WA, Saleh SN, Albert AP. Role of phosphoinositol 4,5-bisphosphate and diacylglycerol in regulating native TRPC channel proteins in vascular smooth muscle. Cell Calcium. 2009;45:574–82. doi: 10.1016/j.ceca.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 343.Inagaki M, Yokokura H, Itoh T, Kanmura Y, Kuriyama H, Hidaka H. Purified rabbit brain protein kinase C relaxes skinned vascular smooth muscle and phosphorylates myosin light chain. Arch Biochem Biophys. 1987;254:136–41. doi: 10.1016/0003-9861(87)90089-0. [DOI] [PubMed] [Google Scholar]
- 344.Ahluwalia A, MacAllister RJ, Hobbs AJ. Vascular actions of natriuretic peptides. Cyclic GMP-dependent and -independent mechanisms. Basic Res Cardiol. 2004;99:83–9. doi: 10.1007/s00395-004-0459-6. [DOI] [PubMed] [Google Scholar]
- 345.Mendonca MC, Doi SQ, Glerum S, Sellitti DF. Increase of C-type natriuretic peptide expression by serum and platelet-derived growth factor-BB in human aortic smooth muscle cells is dependent on protein kinase C activation. Endocrinology. 2006;147:4169–78. doi: 10.1210/en.2006-0239. [DOI] [PubMed] [Google Scholar]
- 346.Mendonca MC, Koles N, Sellitti DF. Protein kinase C-delta (PKC-delta) and PKC-alpha mediate Ca(2+)-dependent increases in CNP mRNA in human vascular cells. Vascular pharmacology. 2012;57:98–104. doi: 10.1016/j.vph.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 347.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiological reviews. 2003;83:1325–58. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 348.Hilgers RH, Webb RC. Molecular aspects of arterial smooth muscle contraction: focus on Rho. Experimental biology and medicine. 2005;230:829–35. doi: 10.1177/153537020523001107. [DOI] [PubMed] [Google Scholar]
- 349.Budzyn K, Sobey CG. Vascular rho kinases and their potential therapeutic applications. Curr Opin Drug Discov Devel. 2007;10:590–6. [PubMed] [Google Scholar]
- 350.Gong MC, Fujihara H, Somlyo AV, Somlyo AP. Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. The Journal of biological chemistry. 1997;272:10704–9. doi: 10.1074/jbc.272.16.10704. [DOI] [PubMed] [Google Scholar]
- 351.Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15:2208–16. [PMC free article] [PubMed] [Google Scholar]
- 352.Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996;15:1885–93. [PMC free article] [PubMed] [Google Scholar]
- 353.Loirand G, Guerin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circulation research. 2006;98:322–34. doi: 10.1161/01.RES.0000201960.04223.3c. [DOI] [PubMed] [Google Scholar]
- 354.Leung T, Manser E, Tan L, Lim L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. The Journal of biological chemistry. 1995;270:29051–4. doi: 10.1074/jbc.270.49.29051. [DOI] [PubMed] [Google Scholar]
- 355.Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392:189–93. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- 356.Szasz T, Webb RC. Rho-Mancing to Sensitize Calcium Signaling for Contraction in the Vasculature: Role of Rho Kinase. Adv Pharmacol. 2017;78:303–22. doi: 10.1016/bs.apha.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 357.Wibberley A, Chen Z, Hu E, Hieble JP, Westfall TD. Expression and functional role of Rho-kinase in rat urinary bladder smooth muscle. British journal of pharmacology. 2003;138:757–66. doi: 10.1038/sj.bjp.0705109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Leung T, Chen XQ, Manser E, Lim L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol Cell Biol. 1996;16:5313–27. doi: 10.1128/mcb.16.10.5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Hiroki J, Shimokawa H, Higashi M, Morikawa K, Kandabashi T, Kawamura N, et al. Inflammatory stimuli upregulate Rho-kinase in human coronary vascular smooth muscle cells. J Mol Cell Cardiol. 2004;37:537–46. doi: 10.1016/j.yjmcc.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 360.Amano M, Chihara K, Nakamura N, Kaneko T, Matsuura Y, Kaibuchi K. The COOH terminus of Rho-kinase negatively regulates rho-kinase activity. The Journal of biological chemistry. 1999;274:32418–24. doi: 10.1074/jbc.274.45.32418. [DOI] [PubMed] [Google Scholar]
- 361.Chen XQ, Tan I, Ng CH, Hall C, Lim L, Leung T. Characterization of RhoA-binding kinase ROKalpha implication of the pleckstrin homology domain in ROKalpha function using region-specific antibodies. The Journal of biological chemistry. 2002;277:12680–8. doi: 10.1074/jbc.M109839200. [DOI] [PubMed] [Google Scholar]
- 362.Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, et al. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science. 1997;275:1308–11. doi: 10.1126/science.275.5304.1308. [DOI] [PubMed] [Google Scholar]
- 363.Amano M, Fukata Y, Kaibuchi K. Regulation and functions of Rho-associated kinase. Exp Cell Res. 2000;261:44–51. doi: 10.1006/excr.2000.5046. [DOI] [PubMed] [Google Scholar]
- 364.Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N, et al. Rho-associated kinase of chicken gizzard smooth muscle. The Journal of biological chemistry. 1999;274:3744–52. doi: 10.1074/jbc.274.6.3744. [DOI] [PubMed] [Google Scholar]
- 365.Gong MC, Fuglsang A, Alessi D, Kobayashi S, Cohen P, Somlyo AV, et al. Arachidonic acid inhibits myosin light chain phosphatase and sensitizes smooth muscle to calcium. The Journal of biological chemistry. 1992;267:21492–8. [PubMed] [Google Scholar]
- 366.Shirao S, Kashiwagi S, Sato M, Miwa S, Nakao F, Kurokawa T, et al. Sphingosylphosphorylcholine is a novel messenger for Rho-kinase-mediated Ca2+ sensitization in the bovine cerebral artery: unimportant role for protein kinase C. Circulation research. 2002;91:112–9. doi: 10.1161/01.res.0000026057.13161.42. [DOI] [PubMed] [Google Scholar]
- 367.Doran JD, Liu X, Taslimi P, Saadat A, Fox T. New insights into the structure-function relationships of Rho-associated kinase: a thermodynamic and hydrodynamic study of the dimer-to-monomer transition and its kinetic implications. Biochem J. 2004;384:255–62. doi: 10.1042/BJ20040344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Couzens AL, Saridakis V, Scheid MP. The hydrophobic motif of ROCK2 requires association with the N-terminal extension for kinase activity. Biochem J. 2009;419:141–8. doi: 10.1042/BJ20081376. [DOI] [PubMed] [Google Scholar]
- 369.Riento K, Guasch RM, Garg R, Jin B, Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol. 2003;23:4219–29. doi: 10.1128/MCB.23.12.4219-4229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Ward Y, Yap SF, Ravichandran V, Matsumura F, Ito M, Spinelli B, et al. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J Cell Biol. 2002;157:291–302. doi: 10.1083/jcb.200111026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Blumenstein L, Ahmadian MR. Models of the cooperative mechanism for Rho effector recognition: implications for RhoA-mediated effector activation. The Journal of biological chemistry. 2004;279:53419–26. doi: 10.1074/jbc.M409551200. [DOI] [PubMed] [Google Scholar]
- 372.Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) The Journal of biological chemistry. 1996;271:20246–9. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- 373.Goto H, Kosako H, Tanabe K, Yanagida M, Sakurai M, Amano M, et al. Phosphorylation of vimentin by Rho-associated kinase at a unique amino-terminal site that is specifically phosphorylated during cytokinesis. The Journal of biological chemistry. 1998;273:11728–36. doi: 10.1074/jbc.273.19.11728. [DOI] [PubMed] [Google Scholar]
- 374.Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, et al. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol. 1998;140:647–57. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Fukata Y, Oshiro N, Kinoshita N, Kawano Y, Matsuoka Y, Bennett V, et al. Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J Cell Biol. 1999;145:347–61. doi: 10.1083/jcb.145.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–56. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 377.Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005;4:387–98. doi: 10.1038/nrd1719. [DOI] [PubMed] [Google Scholar]
- 378.Nishioka T, Shohag MH, Amano M, Kaibuchi K. Developing novel methods to search for substrates of protein kinases such as Rho-kinase. Biochim Biophys Acta. 2015;1854:1663–6. doi: 10.1016/j.bbapap.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 379.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–8. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 380.Kitazawa T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. The Journal of biological chemistry. 2000;275:9897–900. doi: 10.1074/jbc.275.14.9897. [DOI] [PubMed] [Google Scholar]
- 381.Kaneko T, Amano M, Maeda A, Goto H, Takahashi K, Ito M, et al. Identification of calponin as a novel substrate of Rho-kinase. Biochemical and biophysical research communications. 2000;273:110–6. doi: 10.1006/bbrc.2000.2901. [DOI] [PubMed] [Google Scholar]
- 382.Vahebi S, Kobayashi T, Warren CM, de Tombe PP, Solaro RJ. Functional effects of rho-kinase-dependent phosphorylation of specific sites on cardiac troponin. Circulation research. 2005;96:740–7. doi: 10.1161/01.RES.0000162457.56568.7d. [DOI] [PubMed] [Google Scholar]
- 383.Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, et al. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7:399–404. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- 384.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, et al. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1842–7. doi: 10.1161/01.ATV.0000142813.33538.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Begum N, Sandu OA, Ito M, Lohmann SM, Smolenski A. Active Rho kinase (ROK-alpha) associates with insulin receptor substrate-1 and inhibits insulin signaling in vascular smooth muscle cells. The Journal of biological chemistry. 2002;277:6214–22. doi: 10.1074/jbc.M110508200. [DOI] [PubMed] [Google Scholar]
- 386.Riento K, Totty N, Villalonga P, Garg R, Guasch R, Ridley AJ. RhoE function is regulated by ROCK I-mediated phosphorylation. EMBO J. 2005;24:1170–80. doi: 10.1038/sj.emboj.7600612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Couzens AL, Gill RM, Scheid MP. Characterization of a modified ROCK2 protein that allows use of N6-ATP analogs for the identification of novel substrates. BMC Biotechnol. 2014;14:2. doi: 10.1186/1472-6750-14-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010;67:545–54. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Julian L, Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. 2014;5:e29846. doi: 10.4161/sgtp.29846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Street CA, Bryan BA. Rho kinase proteins--pleiotropic modulators of cell survival and apoptosis. Anticancer Res. 2011;31:3645–57. [PMC free article] [PubMed] [Google Scholar]
- 391.Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, et al. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol. 1999;147:1023–38. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Amano T, Tanabe K, Eto T, Narumiya S, Mizuno K. LIM-kinase 2 induces formation of stress fibres, focal adhesions and membrane blebs, dependent on its activation by Rho-associated kinase-catalysed phosphorylation at threonine-505. Biochem J. 2001;354:149–59. doi: 10.1042/0264-6021:3540149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Yoneda A, Multhaupt HA, Couchman JR. The Rho kinases I and II regulate different aspects of myosin II activity. J Cell Biol. 2005;170:443–53. doi: 10.1083/jcb.200412043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Wang Y, Zheng XR, Riddick N, Bryden M, Baur W, Zhang X, et al. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circulation research. 2009;104:531–40. doi: 10.1161/CIRCRESAHA.108.188524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Shimizu Y, Thumkeo D, Keel J, Ishizaki T, Oshima H, Oshima M, et al. ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. J Cell Biol. 2005;168:941–53. doi: 10.1083/jcb.200411179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Thumkeo D, Keel J, Ishizaki T, Hirose M, Nonomura K, Oshima H, et al. Targeted disruption of the mouse rho-associated kinase 2 gene results in intrauterine growth retardation and fetal death. Mol Cell Biol. 2003;23:5043–55. doi: 10.1128/MCB.23.14.5043-5055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–4. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 398.Chrissobolis S, Sobey CG. Evidence that Rho-kinase activity contributes to cerebral vascular tone in vivo and is enhanced during chronic hypertension: comparison with protein kinase C. Circulation research. 2001;88:774–9. doi: 10.1161/hh0801.090441. [DOI] [PubMed] [Google Scholar]
- 399.Jin L, Ying Z, Hilgers RH, Yin J, Zhao X, Imig JD, et al. Increased RhoA/Rho-kinase signaling mediates spontaneous tone in aorta from angiotensin II-induced hypertensive rats. The Journal of pharmacology and experimental therapeutics. 2006;318:288–95. doi: 10.1124/jpet.105.100735. [DOI] [PubMed] [Google Scholar]
- 400.Ito K, Hirooka Y, Kishi T, Kimura Y, Kaibuchi K, Shimokawa H, et al. Rho/Rho-kinase pathway in the brainstem contributes to hypertension caused by chronic nitric oxide synthase inhibition. Hypertension. 2004;43:156–62. doi: 10.1161/01.HYP.0000114602.82140.a4. [DOI] [PubMed] [Google Scholar]
- 401.Moriki N, Ito M, Seko T, Kureishi Y, Okamoto R, Nakakuki T, et al. RhoA activation in vascular smooth muscle cells from stroke-prone spontaneously hypertensive rats. Hypertension research: official journal of the Japanese Society of Hypertension. 2004;27:263–70. doi: 10.1291/hypres.27.263. [DOI] [PubMed] [Google Scholar]
- 402.Shin HK, Salomone S, Potts EM, Lee SW, Millican E, Noma K, et al. Rho-kinase inhibition acutely augments blood flow in focal cerebral ischemia via endothelial mechanisms. J Cereb Blood Flow Metab. 2007;27:998–1009. doi: 10.1038/sj.jcbfm.9600406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Mita S, Kobayashi N, Yoshida K, Nakano S, Matsuoka H. Cardioprotective mechanisms of Rho-kinase inhibition associated with eNOS and oxidative stress-LOX-1 pathway in Dahl salt-sensitive hypertensive rats. Journal of hypertension. 2005;23:87–96. doi: 10.1097/00004872-200501000-00017. [DOI] [PubMed] [Google Scholar]
- 404.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Budzyn K, Paull M, Marley PD, Sobey CG. Segmental differences in the roles of rho-kinase and protein kinase C in mediating vasoconstriction. The Journal of pharmacology and experimental therapeutics. 2006;317:791–6. doi: 10.1124/jpet.105.100040. [DOI] [PubMed] [Google Scholar]
- 406.Noma K, Goto C, Nishioka K, Jitsuiki D, Umemura T, Ueda K, et al. Roles of rho-associated kinase and oxidative stress in the pathogenesis of aortic stiffness. Journal of the American College of Cardiology. 2007;49:698–705. doi: 10.1016/j.jacc.2006.06.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Nohria A, Grunert ME, Rikitake Y, Noma K, Prsic A, Ganz P, et al. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circulation research. 2006;99:1426–32. doi: 10.1161/01.RES.0000251668.39526.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Kishi T, Hirooka Y, Masumoto A, Ito K, Kimura Y, Inokuchi K, et al. Rho-kinase inhibitor improves increased vascular resistance and impaired vasodilation of the forearm in patients with heart failure. Circulation. 2005;111:2741–7. doi: 10.1161/CIRCULATIONAHA.104.510248. [DOI] [PubMed] [Google Scholar]
- 409.Kandabashi T, Shimokawa H, Miyata K, Kunihiro I, Eto Y, Morishige K, et al. Evidence for protein kinase C-mediated activation of Rho-kinase in a porcine model of coronary artery spasm. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:2209–14. doi: 10.1161/01.ATV.0000104010.87348.26. [DOI] [PubMed] [Google Scholar]
- 410.Jacobs M, Hayakawa K, Swenson L, Bellon S, Fleming M, Taslimi P, et al. The structure of dimeric ROCK I reveals the mechanism for ligand selectivity. The Journal of biological chemistry. 2006;281:260–8. doi: 10.1074/jbc.M508847200. [DOI] [PubMed] [Google Scholar]
- 411.Tsai CC, Liu HF, Hsu KC, Yang JM, Chen C, Liu KK, et al. 7-Chloro-6-piperidin-1-yl-quinoline-5,8-dione (PT-262), a novel ROCK inhibitor blocks cytoskeleton function and cell migration. Biochemical pharmacology. 2011;81:856–65. doi: 10.1016/j.bcp.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 412.Kale VP, Hengst JA, Desai DH, Dick TE, Choe KN, Colledge AL, et al. A novel selective multikinase inhibitor of ROCK and MRCK effectively blocks cancer cell migration and invasion. Cancer Lett. 2014;354:299–310. doi: 10.1016/j.canlet.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Yamamoto K, Maruyama K, Himori N, Omodaka K, Yokoyama Y, Shiga Y, et al. The novel Rho kinase (ROCK) inhibitor K-115: a new candidate drug for neuroprotective treatment in glaucoma. Invest Ophthalmol Vis Sci. 2014;55:7126–36. doi: 10.1167/iovs.13-13842. [DOI] [PubMed] [Google Scholar]
- 414.Xin YL, Yu JZ, Yang XW, Liu CY, Li YH, Feng L, et al. FSD-C10: A more promising novel ROCK inhibitor than Fasudil for treatment of CNS autoimmunity. Biosci Rep. 2015:35. doi: 10.1042/BSR20150032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Chen J, Yin W, Tu Y, Wang S, Yang X, Chen Q, et al. L-F001, a novel multifunctional ROCK inhibitor, suppresses neuroinflammation in vitro and in vivo: Involvement of NF-kappaB inhibition and Nrf2 pathway activation. European journal of pharmacology. 2017;806:1–9. doi: 10.1016/j.ejphar.2017.03.025. [DOI] [PubMed] [Google Scholar]
- 416.Crews JK, Murphy JG, Khalil RA. Gender differences in Ca(2+) entry mechanisms of vasoconstriction in Wistar-Kyoto and spontaneously hypertensive rats. Hypertension. 1999;34:931–6. doi: 10.1161/01.hyp.34.4.931. [DOI] [PubMed] [Google Scholar]
- 417.Murphy JG, Herrington JN, Granger JP, Khalil RA. Enhanced [Ca2+]i in renal arterial smooth muscle cells of pregnant rats with reduced uterine perfusion pressure. American journal of physiology Heart and circulatory physiology. 2003;284:H393–403. doi: 10.1152/ajpheart.00247.2002. [DOI] [PubMed] [Google Scholar]
- 418.Matsui T, Takuwa Y, Johshita H, Yamashita K, Asano T. Possible role of protein kinase C-dependent smooth muscle contraction in the pathogenesis of chronic cerebral vasospasm. J Cereb Blood Flow Metab. 1991;11:143–9. doi: 10.1038/jcbfm.1991.17. [DOI] [PubMed] [Google Scholar]
- 419.Ito A, Shimokawa H, Nakaike R, Fukai T, Sakata M, Takayanagi T, et al. Role of protein kinase C-mediated pathway in the pathogenesis of coronary artery spasm in a swine model. Circulation. 1994;90:2425–31. doi: 10.1161/01.cir.90.5.2425. [DOI] [PubMed] [Google Scholar]
- 420.Ding Q, Chai H, Mahmood N, Tsao J, Mochly-Rosen D, Zhou W. Matrix metalloproteinases modulated by protein kinase Cepsilon mediate resistin-induced migration of human coronary artery smooth muscle cells. Journal of vascular surgery. 2011;53:1044–51. doi: 10.1016/j.jvs.2010.10.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Gilio K, Harper MT, Cosemans JM, Konopatskaya O, Munnix IC, Prinzen L, et al. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. The Journal of biological chemistry. 2010;285:23410–9. doi: 10.1074/jbc.M110.136176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Pula G, Schuh K, Nakayama K, Nakayama KI, Walter U, Poole AW. PKCdelta regulates collagen-induced platelet aggregation through inhibition of VASP-mediated filopodia formation. Blood. 2006;108:4035–44. doi: 10.1182/blood-2006-05-023739. [DOI] [PubMed] [Google Scholar]
- 423.Abdala-Valencia H, Cook-Mills JM. VCAM-1 signals activate endothelial cell protein kinase Calpha via oxidation. Journal of immunology. 2006;177:6379–87. doi: 10.4049/jimmunol.177.9.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Kouroedov A, Eto M, Joch H, Volpe M, Luscher TF, Cosentino F. Selective inhibition of protein kinase Cbeta2 prevents acute effects of high glucose on vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 2004;110:91–6. doi: 10.1161/01.CIR.0000133384.38551.A8. [DOI] [PubMed] [Google Scholar]
- 425.Javaid K, Rahman A, Anwar KN, Frey RS, Minshall RD, Malik AB. Tumor necrosis factor-alpha induces early-onset endothelial adhesivity by protein kinase Czeta-dependent activation of intercellular adhesion molecule-1. Circulation research. 2003;92:1089–97. doi: 10.1161/01.RES.0000072971.88704.CB. [DOI] [PubMed] [Google Scholar]
- 426.Okazaki J, Mawatari K, Liu B, Kent KC. The effect of protein kinase C and its alpha subtype on human vascular smooth muscle cell proliferation, migration and fibronectin production. Surgery. 2000;128:192–7. doi: 10.1067/msy.2000.108062. [DOI] [PubMed] [Google Scholar]
- 427.Campbell M, Trimble ER. Modification of PI3K- and MAPK-dependent chemotaxis in aortic vascular smooth muscle cells by protein kinase CbetaII. Circulation research. 2005;96:197–206. doi: 10.1161/01.RES.0000152966.88353.9d. [DOI] [PubMed] [Google Scholar]
- 428.Liu B, Ryer EJ, Kundi R, Kamiya K, Itoh H, Faries PL, et al. Protein kinase C-delta regulates migration and proliferation of vascular smooth muscle cells through the extracellular signal-regulated kinase 1/2. Journal of vascular surgery. 2007;45:160–8. doi: 10.1016/j.jvs.2006.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Kim J, Min G, Bae YS, Min DS. Phospholipase D is involved in oxidative stress-induced migration of vascular smooth muscle cells via tyrosine phosphorylation and protein kinase C. Exp Mol Med. 2004;36:103–9. doi: 10.1038/emm.2004.15. [DOI] [PubMed] [Google Scholar]
- 430.Thomas AC, Newby AC. Effect of matrix metalloproteinase-9 knockout on vein graft remodelling in mice. Journal of vascular research. 2010;47:299–308. doi: 10.1159/000265564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Rodriguez-Pla A, Bosch-Gil JA, Rossello-Urgell J, Huguet-Redecilla P, Stone JH, Vilardell-Tarres M. Metalloproteinase-2 and -9 in giant cell arteritis: involvement in vascular remodeling. Circulation. 2005;112:264–9. doi: 10.1161/CIRCULATIONAHA.104.520114. [DOI] [PubMed] [Google Scholar]
- 432.Ling S, Little PJ, Williams MR, Dai A, Hashimura K, Liu JP, et al. High glucose abolishes the antiproliferative effect of 17beta-estradiol in human vascular smooth muscle cells. American journal of physiology Endocrinology and metabolism. 2002;282:E746–51. doi: 10.1152/ajpendo.00111.2001. [DOI] [PubMed] [Google Scholar]
- 433.Deuse T, Koyanagi T, Erben RG, Hua X, Velden J, Ikeno F, et al. Sustained inhibition of epsilon protein kinase C inhibits vascular restenosis after balloon injury and stenting. Circulation. 2010;122:S170–8. doi: 10.1161/CIRCULATIONAHA.109.927640. [DOI] [PubMed] [Google Scholar]
- 434.Zhou H, Wang Y, Zhou Q, Wu B, Wang A, Jiang W, et al. Down-Regulation of Protein Kinase C-epsilon by Prolonged Incubation with PMA Inhibits the Proliferation of Vascular Smooth Muscle Cells. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2016;40:379–90. doi: 10.1159/000452553. [DOI] [PubMed] [Google Scholar]
- 435.Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, et al. Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. 2003;108:2304–7. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- 436.Churchill EN, Mochly-Rosen D. The roles of PKCdelta and epsilon isoenzymes in the regulation of myocardial ischaemia/reperfusion injury. Biochem Soc Trans. 2007;35:1040–2. doi: 10.1042/BST0351040. [DOI] [PubMed] [Google Scholar]
- 437.Budas GR, Churchill EN, Mochly-Rosen D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol Res. 2007;55:523–36. doi: 10.1016/j.phrs.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 438.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–5. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Tanaka M, Terry RD, Mokhtari GK, Inagaki K, Koyanagi T, Kofidis T, et al. Suppression of graft coronary artery disease by a brief treatment with a selective epsilonPKC activator and a deltaPKC inhibitor in murine cardiac allografts. Circulation. 2004;110:II194–9. doi: 10.1161/01.CIR.0000138389.22905.62. [DOI] [PubMed] [Google Scholar]
- 440.Young LH, Balin BJ, Weis MT. Go 6983: a fast acting protein kinase C inhibitor that attenuates myocardial ischemia/reperfusion injury. Cardiovasc Drug Rev. 2005;23:255–72. doi: 10.1111/j.1527-3466.2005.tb00170.x. [DOI] [PubMed] [Google Scholar]
- 441.Chou WH, Choi DS, Zhang H, Mu D, McMahon T, Kharazia VN, et al. Neutrophil protein kinase Cdelta as a mediator of stroke-reperfusion injury. The Journal of clinical investigation. 2004;114:49–56. doi: 10.1172/JCI21655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Bright R, Steinberg GK, Mochly-Rosen D. DeltaPKC mediates microcerebrovascular dysfunction in acute ischemia and in chronic hypertensive stress in vivo. Brain Res. 2007;1144:146–55. doi: 10.1016/j.brainres.2007.01.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Koponen S, Kurkinen K, Akerman KE, Mochly-Rosen D, Chan PH, Koistinaho J. Prevention of NMDA-induced death of cortical neurons by inhibition of protein kinase Czeta. J Neurochem. 2003;86:442–50. doi: 10.1046/j.1471-4159.2003.01846.x. [DOI] [PubMed] [Google Scholar]
- 444.Bright R, Sun GH, Yenari MA, Steinberg GK, Mochly-Rosen D. epsilonPKC confers acute tolerance to cerebral ischemic reperfusion injury. Neurosci Lett. 2008;441:120–4. doi: 10.1016/j.neulet.2008.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Posti JP, Salo P, Ruohonen S, Valve L, Muszkat M, Sofowora GG, et al. A polymorphism in the protein kinase C gene PRKCB is associated with alpha2-adrenoceptor-mediated vasoconstriction. Pharmacogenet Genomics. 2013;23:127–34. doi: 10.1097/FPC.0b013e32835d247f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Khalil RA, Lajoie C, Morgan KG. In situ determination of [Ca2+]i threshold for translocation of the alpha-protein kinase C isoform. The American journal of physiology. 1994;266:C1544–51. doi: 10.1152/ajpcell.1994.266.6.C1544. [DOI] [PubMed] [Google Scholar]
- 447.Liou YM, Morgan KG. Redistribution of protein kinase C isoforms in association with vascular hypertrophy of rat aorta. The American journal of physiology. 1994;267:C980–9. doi: 10.1152/ajpcell.1994.267.4.C980. [DOI] [PubMed] [Google Scholar]
- 448.Campen MJ, Shimoda LA, O’Donnell CP. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. J Appl Physiol (1985) 2005;99:2028–35. doi: 10.1152/japplphysiol.00411.2005. [DOI] [PubMed] [Google Scholar]
- 449.Snow JB, Gonzalez Bosc LV, Kanagy NL, Walker BR, Resta TC. Role for PKCbeta in enhanced endothelin-1-induced pulmonary vasoconstrictor reactivity following intermittent hypoxia. American journal of physiology Lung cellular and molecular physiology. 2011;301:L745–54. doi: 10.1152/ajplung.00020.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Snow JB, Kanagy NL, Walker BR, Resta TC. Rat strain differences in pulmonary artery smooth muscle Ca(2+) entry following chronic hypoxia. Microcirculation. 2009;16:603–14. doi: 10.1080/10739680903114268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Allahdadi KJ, Duling LC, Walker BR, Kanagy NL. Eucapnic intermittent hypoxia augments endothelin-1 vasoconstriction in rats: role of PKCdelta. American journal of physiology Heart and circulatory physiology. 2008;294:H920–7. doi: 10.1152/ajpheart.01264.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Snow JB, Kitzis V, Norton CE, Torres SN, Johnson KD, Kanagy NL, et al. Differential effects of chronic hypoxia and intermittent hypocapnic and eucapnic hypoxia on pulmonary vasoreactivity. J Appl Physiol (1985) 2008;104:110–8. doi: 10.1152/japplphysiol.00698.2005. [DOI] [PubMed] [Google Scholar]
- 453.Zhu S, White RE, Barman SA. Role of phosphodiesterases in modulation of BKCa channels in hypertensive pulmonary arterial smooth muscle. Ther Adv Respir Dis. 2008;2:119–27. doi: 10.1177/1753465808091327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Dakshinamurti S, Mellow L, Stephens NL. Regulation of pulmonary arterial myosin phosphatase activity in neonatal circulatory transition and in hypoxic pulmonary hypertension: a role for CPI-17. Pediatr Pulmonol. 2005;40:398–407. doi: 10.1002/ppul.20290. [DOI] [PubMed] [Google Scholar]
- 455.Littler CM, Morris KG, Jr, Fagan KA, McMurtry IF, Messing RO, Dempsey EC. Protein kinase C-epsilon-null mice have decreased hypoxic pulmonary vasoconstriction. American journal of physiology Heart and circulatory physiology. 2003;284:H1321–31. doi: 10.1152/ajpheart.00795.2002. [DOI] [PubMed] [Google Scholar]
- 456.Littler CM, Wehling CA, Wick MJ, Fagan KA, Cool CD, Messing RO, et al. Divergent contractile and structural responses of the murine PKC-epsilon null pulmonary circulation to chronic hypoxia. American journal of physiology Lung cellular and molecular physiology. 2005;289:L1083–93. doi: 10.1152/ajplung.00472.2004. [DOI] [PubMed] [Google Scholar]
- 457.Chang K, Xiao D, Huang X, Longo LD, Zhang L. Chronic hypoxia increases pressure-dependent myogenic tone of the uterine artery in pregnant sheep: role of ERK/PKC pathway. American journal of physiology Heart and circulatory physiology. 2009;296:H1840–9. doi: 10.1152/ajpheart.00090.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Xiao D, Zhu R, Zhang L. Gestational hypoxia up-regulates protein kinase C and inhibits calcium-activated potassium channels in ovine uterine arteries. Int J Med Sci. 2014;11:886–92. doi: 10.7150/ijms.9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. The Journal of clinical investigation. 1999;103:945–52. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–66. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 461.Meier M, King GL. Protein kinase C activation and its pharmacological inhibition in vascular disease. Vasc Med. 2000;5:173–85. doi: 10.1177/1358836X0000500307. [DOI] [PubMed] [Google Scholar]
- 462.Thallas-Bonke V, Thorpe SR, Coughlan MT, Fukami K, Yap FY, Sourris KC, et al. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes. 2008;57:460–9. doi: 10.2337/db07-1119. [DOI] [PubMed] [Google Scholar]
- 463.Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. The New England journal of medicine. 1994;331:1480–7. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 464.Huang Q, Yuan Y. Interaction of PKC and NOS in signal transduction of microvascular hyperpermeability. The American journal of physiology. 1997;273:H2442–51. doi: 10.1152/ajpheart.1997.273.5.H2442. [DOI] [PubMed] [Google Scholar]
- 465.Williams B, Gallacher B, Patel H, Orme C. Glucose-induced protein kinase C activation regulates vascular permeability factor mRNA expression and peptide production by human vascular smooth muscle cells in vitro. Diabetes. 1997;46:1497–503. doi: 10.2337/diab.46.9.1497. [DOI] [PubMed] [Google Scholar]
- 466.Pomero F, Allione A, Beltramo E, Buttiglieri S, D’Alu F, Ponte E, et al. Effects of protein kinase C inhibition and activation on proliferation and apoptosis of bovine retinal pericytes. Diabetologia. 2003;46:416–9. doi: 10.1007/s00125-003-1044-5. [DOI] [PubMed] [Google Scholar]
- 467.Lizotte F, Pare M, Denhez B, Leitges M, Guay A, Geraldes P. PKCdelta impaired vessel formation and angiogenic factor expression in diabetic ischemic limbs. Diabetes. 2013;62:2948–57. doi: 10.2337/db12-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A. 1992;89:11059–63. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Yokota T, Ma RC, Park JY, Isshiki K, Sotiropoulos KB, Rauniyar RK, et al. Role of protein kinase C on the expression of platelet-derived growth factor and endothelin-1 in the retina of diabetic rats and cultured retinal capillary pericytes. Diabetes. 2003;52:838–45. doi: 10.2337/diabetes.52.3.838. [DOI] [PubMed] [Google Scholar]
- 470.Jakus V, Rietbrock N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiol Res. 2004;53:131–42. [PubMed] [Google Scholar]
- 471.Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal. 1998;10:529–42. doi: 10.1016/s0898-6568(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 472.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–45. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 473.Gao L, Mann GE. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovascular research. 2009;82:9–20. doi: 10.1093/cvr/cvp031. [DOI] [PubMed] [Google Scholar]
- 474.Straub SV, Girouard H, Doetsch PE, Hannah RM, Wilkerson MK, Nelson MT. Regulation of intracerebral arteriolar tone by K(v) channels: effects of glucose and PKC. American journal of physiology Cell physiology. 2009;297:C788–96. doi: 10.1152/ajpcell.00148.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Rainbow RD, Hardy ME, Standen NB, Davies NW. Glucose reduces endothelin inhibition of voltage-gated potassium channels in rat arterial smooth muscle cells. The Journal of physiology. 2006;575:833–44. doi: 10.1113/jphysiol.2006.114009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Liu Y, Terata K, Rusch NJ, Gutterman DD. High glucose impairs voltage-gated K(+) channel current in rat small coronary arteries. Circulation research. 2001;89:146–52. doi: 10.1161/hh1401.093294. [DOI] [PubMed] [Google Scholar]
- 477.Hien TT, Turczynska KM, Dahan D, Ekman M, Grossi M, Sjogren J, et al. Elevated Glucose Levels Promote Contractile and Cytoskeletal Gene Expression in Vascular Smooth Muscle via Rho/Protein Kinase C and Actin Polymerization. The Journal of biological chemistry. 2016;291:3552–68. doi: 10.1074/jbc.M115.654384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Nakano S, Uchida K, Kigoshi T, Azukizawa S, Iwasaki R, Kaneko M, et al. Circadian rhythm of blood pressure in normotensive NIDDM subjects. Its relationship to microvascular complications. Diabetes Care. 1991;14:707–11. doi: 10.2337/diacare.14.8.707. [DOI] [PubMed] [Google Scholar]
- 479.Palmas W, Pickering T, Teresi J, Schwartz JE, Eguchi K, Field L, et al. Nocturnal blood pressure elevation predicts progression of albuminuria in elderly people with type 2 diabetes. J Clin Hypertens (Greenwich) 2008;10:12–20. doi: 10.1111/j.1524-6175.2007.07170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Bonaa KH, Bjerve KS, Straume B, Gram IT, Thelle D. Effect of eicosapentaenoic and docosahexaenoic acids on blood pressure in hypertension. A population-based intervention trial from the Tromso study. The New England journal of medicine. 1990;322:795–801. doi: 10.1056/NEJM199003223221202. [DOI] [PubMed] [Google Scholar]
- 481.Chin JP, Gust AP, Nestel PJ, Dart AM. Marine oils dose-dependently inhibit vasoconstriction of forearm resistance vessels in humans. Hypertension. 1993;21:22–8. doi: 10.1161/01.hyp.21.1.22. [DOI] [PubMed] [Google Scholar]
- 482.Hui R, Robillard M, Falardeau P. Inhibition of vasopressin-induced formation of diradylglycerols in vascular smooth muscle cells by incorporation of eicosapentaenoic acid in membrane phospholipids. Journal of hypertension. 1992;10:1145–53. doi: 10.1097/00004872-199210000-00006. [DOI] [PubMed] [Google Scholar]
- 483.Haller H, Baur E, Quass P, Behrend M, Lindschau C, Distler A, et al. High glucose concentrations and protein kinase C isoforms in vascular smooth muscle cells. Kidney international. 1995;47:1057–67. doi: 10.1038/ki.1995.152. [DOI] [PubMed] [Google Scholar]
- 484.Igarashi M, Wakasaki H, Takahara N, Ishii H, Jiang ZY, Yamauchi T, et al. Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways. The Journal of clinical investigation. 1999;103:185–95. doi: 10.1172/JCI3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, Way KJ, et al. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes. 2006;55:691–8. doi: 10.2337/diabetes.55.03.06.db05-0771. [DOI] [PubMed] [Google Scholar]
- 486.Sampson SR, Cooper DR. Specific protein kinase C isoforms as transducers and modulators of insulin signaling. Mol Genet Metab. 2006;89:32–47. doi: 10.1016/j.ymgme.2006.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Bakker W, Sipkema P, Stehouwer CD, Serne EH, Smulders YM, van Hinsbergh VW, et al. Protein kinase C theta activation induces insulin-mediated constriction of muscle resistance arteries. Diabetes. 2008;57:706–13. doi: 10.2337/db07-0792. [DOI] [PubMed] [Google Scholar]
- 488.Amadio M, Bucolo C, Leggio GM, Drago F, Govoni S, Pascale A. The PKCbeta/HuR/VEGF pathway in diabetic retinopathy. Biochemical pharmacology. 2010;80:1230–7. doi: 10.1016/j.bcp.2010.06.033. [DOI] [PubMed] [Google Scholar]
- 489.Gogula SV, Divakar C, Satyanarayana C, Kumar YP, Lavanaya VS. Computational investigation of pkcbeta inhibitors for the treatment of diabetic retinopathy. Bioinformation. 2013;9:1040–3. doi: 10.6026/97320630091040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Lu T, Chai Q, Yu L, d’Uscio LV, Katusic ZS, He T, et al. Reactive oxygen species signaling facilitates FOXO-3a/FBXO-dependent vascular BK channel beta1 subunit degradation in diabetic mice. Diabetes. 2012;61:1860–8. doi: 10.2337/db11-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Ritter O, Jelenik T, Roden M. Lipid-mediated muscle insulin resistance: different fat, different pathways? J Mol Med (Berl) 2015;93:831–43. doi: 10.1007/s00109-015-1310-2. [DOI] [PubMed] [Google Scholar]
- 492.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. The Journal of biological chemistry. 2002;277:50230–6. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 493.Bezy O, Tran TT, Pihlajamaki J, Suzuki R, Emanuelli B, Winnay J, et al. PKCdelta regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. The Journal of clinical investigation. 2011;121:2504–17. doi: 10.1172/JCI46045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, et al. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. 2009;15:1298–306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Klymenko K, Novokhatska T, Kizub I, Parshikov A, Dosenko V, Soloviev A. PKC-delta isozyme gene silencing restores vascular function in diabetic rat. J Basic Clin Physiol Pharmacol. 2014:1–9. doi: 10.1515/jbcpp-2013-0147. [DOI] [PubMed] [Google Scholar]
- 496.Ishida K, Matsumoto T, Taguchi K, Kamata K, Kobayashi T. Protein kinase C delta contributes to increase in EP3 agonist-induced contraction in mesenteric arteries from type 2 diabetic Goto-Kakizaki rats. Pflugers Archiv: European journal of physiology. 2012;463:593–602. doi: 10.1007/s00424-012-1088-9. [DOI] [PubMed] [Google Scholar]
- 497.Sobhia ME, Grewal BK, Ml SP, Patel J, Kaur A, Haokip T, et al. Protein kinase C inhibitors: a patent review (2008 – 2009) Expert Opin Ther Pat. 2013;23:1297–315. doi: 10.1517/13543776.2013.805205. [DOI] [PubMed] [Google Scholar]
- 498.Mehta NN, Sheetz M, Price K, Comiskey L, Amrutia S, Iqbal N, et al. Selective PKC beta inhibition with ruboxistaurin and endothelial function in type-2 diabetes mellitus. Cardiovasc Drugs Ther. 2009;23:17–24. doi: 10.1007/s10557-008-6144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Aiello LP, Vignati L, Sheetz MJ, Zhi X, Girach A, Davis MD, et al. Oral protein kinase c beta inhibition using ruboxistaurin: efficacy, safety, and causes of vision loss among 813 patients (1,392 eyes) with diabetic retinopathy in the Protein Kinase C beta Inhibitor-Diabetic Retinopathy Study and the Protein Kinase C beta Inhibitor-Diabetic Retinopathy Study 2. Retina. 2011;31:2084–94. doi: 10.1097/IAE.0b013e3182111669. [DOI] [PubMed] [Google Scholar]
- 500.Batchuluun B, Inoguchi T, Sonoda N, Sasaki S, Inoue T, Fujimura Y, et al. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis. 2013;232:156–64. doi: 10.1016/j.atherosclerosis.2013.10.025. [DOI] [PubMed] [Google Scholar]
- 501.Huang H, Lee DH, Zabolotny JM, Kim YB. Metabolic actions of Rho-kinase in periphery and brain. Trends Endocrinol Metab. 2013;24:506–14. doi: 10.1016/j.tem.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Knipe RS, Tager AM, Liao JK. The Rho kinases: critical mediators of multiple profibrotic processes and rational targets for new therapies for pulmonary fibrosis. Pharmacological reviews. 2015;67:103–17. doi: 10.1124/pr.114.009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Sawada N, Liao JK. Rho/Rho-associated coiled-coil forming kinase pathway as therapeutic targets for statins in atherosclerosis. Antioxid Redox Signal. 2014;20:1251–67. doi: 10.1089/ars.2013.5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, et al. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circulation research. 2003;92:411–8. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- 505.Mukai Y, Shimokawa H, Matoba T, Kandabashi T, Satoh S, Hiroki J, et al. Involvement of Rho-kinase in hypertensive vascular disease: a novel therapeutic target in hypertension. FASEB J. 2001;15:1062–4. doi: 10.1096/fj.00-0735fje. [DOI] [PubMed] [Google Scholar]
- 506.Kataoka C, Egashira K, Inoue S, Takemoto M, Ni W, Koyanagi M, et al. Important role of Rho-kinase in the pathogenesis of cardiovascular inflammation and remodeling induced by long-term blockade of nitric oxide synthesis in rats. Hypertension. 2002;39:245–50. doi: 10.1161/hy0202.103271. [DOI] [PubMed] [Google Scholar]
- 507.Higashi M, Shimokawa H, Hattori T, Hiroki J, Mukai Y, Morikawa K, et al. Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: effect on endothelial NAD(P)H oxidase system. Circulation research. 2003;93:767–75. doi: 10.1161/01.RES.0000096650.91688.28. [DOI] [PubMed] [Google Scholar]
- 508.Funakoshi Y, Ichiki T, Shimokawa H, Egashira K, Takeda K, Kaibuchi K, et al. Rho-kinase mediates angiotensin II-induced monocyte chemoattractant protein-1 expression in rat vascular smooth muscle cells. Hypertension. 2001;38:100–4. doi: 10.1161/01.hyp.38.1.100. [DOI] [PubMed] [Google Scholar]
- 509.Takeda K, Ichiki T, Tokunou T, Iino N, Fujii S, Kitabatake A, et al. Critical role of Rho-kinase and MEK/ERK pathways for angiotensin II-induced plasminogen activator inhibitor type-1 gene expression. Arteriosclerosis, thrombosis, and vascular biology. 2001;21:868–73. doi: 10.1161/01.atv.21.5.868. [DOI] [PubMed] [Google Scholar]
- 510.Hishikawa K, Nakaki T, Marumo T, Hayashi M, Suzuki H, Kato R, et al. Pressure promotes DNA synthesis in rat cultured vascular smooth muscle cells. The Journal of clinical investigation. 1994;93:1975–80. doi: 10.1172/JCI117189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Numaguchi K, Eguchi S, Yamakawa T, Motley ED, Inagami T. Mechanotransduction of rat aortic vascular smooth muscle cells requires RhoA and intact actin filaments. Circulation research. 1999;85:5–11. doi: 10.1161/01.res.85.1.5. [DOI] [PubMed] [Google Scholar]
- 512.Zeidan A, Nordstrom I, Albinsson S, Malmqvist U, Sward K, Hellstrand P. Stretch-induced contractile differentiation of vascular smooth muscle: sensitivity to actin polymerization inhibitors. American journal of physiology Cell physiology. 2003;284:C1387–96. doi: 10.1152/ajpcell.00508.2002. [DOI] [PubMed] [Google Scholar]
- 513.Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004;68:75–103. doi: 10.1016/j.mvr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 514.Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- 515.Guilluy C, Sauzeau V, Rolli-Derkinderen M, Guerin P, Sagan C, Pacaud P, et al. Inhibition of RhoA/Rho kinase pathway is involved in the beneficial effect of sildenafil on pulmonary hypertension. British journal of pharmacology. 2005;146:1010–8. doi: 10.1038/sj.bjp.0706408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, et al. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med. 2005;171:494–9. doi: 10.1164/rccm.200405-637OC. [DOI] [PubMed] [Google Scholar]
- 517.Jernigan NL, Walker BR, Resta TC. Chronic hypoxia augments protein kinase G-mediated Ca2+ desensitization in pulmonary vascular smooth muscle through inhibition of RhoA/Rho kinase signaling. American journal of physiology Lung cellular and molecular physiology. 2004;287:L1220–9. doi: 10.1152/ajplung.00196.2004. [DOI] [PubMed] [Google Scholar]
- 518.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, et al. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. American journal of physiology Lung cellular and molecular physiology. 2004;287:L656–64. doi: 10.1152/ajplung.00090.2003. [DOI] [PubMed] [Google Scholar]
- 519.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, et al. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. American journal of physiology Lung cellular and molecular physiology. 2004;287:L665–72. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 520.Abe K, Shimokawa H, Morikawa K, Uwatoku T, Oi K, Matsumoto Y, et al. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circulation research. 2004;94:385–93. doi: 10.1161/01.RES.0000111804.34509.94. [DOI] [PubMed] [Google Scholar]
- 521.Morgado M, Cairrao E, Santos-Silva AJ, Verde I. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell Mol Life Sci. 2012;69:247–66. doi: 10.1007/s00018-011-0815-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Fan HC, Fernandez-Hernando C, Lai JH. Protein kinase C isoforms in atherosclerosis: pro- or anti-inflammatory? Biochemical pharmacology. 2014;88:139–49. doi: 10.1016/j.bcp.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 523.Huang W, Bansode RR, Bal NC, Mehta M, Mehta KD. Protein kinase Cbeta deficiency attenuates obesity syndrome of ob/ob mice by promoting white adipose tissue remodeling. J Lipid Res. 2012;53:368–78. doi: 10.1194/jlr.M019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Harja E, Chang JS, Lu Y, Leitges M, Zou YS, Schmidt AM, et al. Mice deficient in PKCbeta and apolipoprotein E display decreased atherosclerosis. FASEB J. 2009;23:1081–91. doi: 10.1096/fj.08-120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Huang C, Chang JS, Xu Y, Li Q, Zou YS, Yan SF. Reduction of PKCbetaII activity in smooth muscle cells attenuates acute arterial injury. Atherosclerosis. 2010;212:123–30. doi: 10.1016/j.atherosclerosis.2010.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Monti M, Donnini S, Giachetti A, Mochly-Rosen D, Ziche M. deltaPKC inhibition or varepsilonPKC activation repairs endothelial vascular dysfunction by regulating eNOS post-translational modification. J Mol Cell Cardiol. 2010;48:746–56. doi: 10.1016/j.yjmcc.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Jialal I, Machha A, Devaraj S. Small interfering-RNA to protein kinase C-delta reduces the proinflammatory effects of human C-reactive protein in biobreeding diabetic rats. Horm Metab Res. 2013;45:326–8. doi: 10.1055/s-0032-1327643. [DOI] [PubMed] [Google Scholar]
- 528.Wetsel WC, Khan WA, Merchenthaler I, Rivera H, Halpern AE, Phung HM, et al. Tissue and cellular distribution of the extended family of protein kinase C isoenzymes. J Cell Biol. 1992;117:121–33. doi: 10.1083/jcb.117.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Haller H, Quass P, Lindschau C, Luft FC, Distler A. Platelet-derived growth factor and angiotensin II induce different spatial distribution of protein kinase C-alpha and -beta in vascular smooth muscle cells. Hypertension. 1994;23:848–52. doi: 10.1161/01.hyp.23.6.848. [DOI] [PubMed] [Google Scholar]
- 530.Ohanian V, Ohanian J, Shaw L, Scarth S, Parker PJ, Heagerty AM. Identification of protein kinase C isoforms in rat mesenteric small arteries and their possible role in agonist-induced contraction. Circulation research. 1996;78:806–12. doi: 10.1161/01.res.78.5.806. [DOI] [PubMed] [Google Scholar]
- 531.Watanabe M, Hachiya T, Hagiwara M, Hidaka H. Identification of type III protein kinase C in bovine aortic tissue. Arch Biochem Biophys. 1989;273:165–9. doi: 10.1016/0003-9861(89)90175-6. [DOI] [PubMed] [Google Scholar]
- 532.Zhao M, Xia L, Chen GQ. Protein kinase cdelta in apoptosis: a brief overview. Arch Immunol Ther Exp (Warsz) 2012;60:361–72. doi: 10.1007/s00005-012-0188-8. [DOI] [PubMed] [Google Scholar]
- 533.Khalil RA, Lajoie C, Resnick MS, Morgan KG. Ca(2+)-independent isoforms of protein kinase C differentially translocate in smooth muscle. The American journal of physiology. 1992;263:C714–9. doi: 10.1152/ajpcell.1992.263.3.C714. [DOI] [PubMed] [Google Scholar]
- 534.Suzuki T, Elias BC, Seth A, Shen L, Turner JR, Giorgianni F, et al. PKC eta regulates occludin phosphorylation and epithelial tight junction integrity. Proc Natl Acad Sci U S A. 2009;106:61–6. doi: 10.1073/pnas.0802741106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Goodnight JA, Mischak H, Kolch W, Mushinski JF. Immunocytochemical localization of eight protein kinase C isozymes overexpressed in NIH 3T3 fibroblasts. Isoform-specific association with microfilaments, Golgi, endoplasmic reticulum, and nuclear and cell membranes. The Journal of biological chemistry. 1995;270:9991–10001. doi: 10.1074/jbc.270.17.9991. [DOI] [PubMed] [Google Scholar]
- 536.Akimoto K, Mizuno K, Osada S, Hirai S, Tanuma S, Suzuki K, et al. A new member of the third class in the protein kinase C family, PKC lambda, expressed dominantly in an undifferentiated mouse embryonal carcinoma cell line and also in many tissues and cells. The Journal of biological chemistry. 1994;269:12677–83. [PubMed] [Google Scholar]
- 537.Gailly P, Gong MC, Somlyo AV, Somlyo AP. Possible role of atypical protein kinase C activated by arachidonic acid in Ca2+ sensitization of rabbit smooth muscle. The Journal of physiology. 1997;500(Pt 1):95–109. doi: 10.1113/jphysiol.1997.sp022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Hurd PJ, Bannister AJ, Halls K, Dawson MA, Vermeulen M, Olsen JV, et al. Phosphorylation of histone H3 Thr-45 is linked to apoptosis. The Journal of biological chemistry. 2009;284:16575–83. doi: 10.1074/jbc.M109.005421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N, et al. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature. 2010;464:792–6. doi: 10.1038/nature08839. [DOI] [PubMed] [Google Scholar]
- 540.Parker CA, Takahashi K, Tao T, Morgan KG. Agonist-induced redistribution of calponin in contractile vascular smooth muscle cells. The American journal of physiology. 1994;267:C1262–70. doi: 10.1152/ajpcell.1994.267.5.C1262. [DOI] [PubMed] [Google Scholar]
- 541.Howcroft TK, Lindquist RR. The protein kinase C inhibitor 1-(5-isoquinolinesulfonyl)-2-methylpiperazine dihydrochloride (H-7) inhibits PMA-induced promiscuous cytolytic activity but not specific cytolytic activity by a cloned cytolytic T lymphocyte. Biochemical and biophysical research communications. 1991;179:720–5. doi: 10.1016/0006-291x(91)91876-e. [DOI] [PubMed] [Google Scholar]
- 542.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, et al. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. The Journal of biological chemistry. 1993;268:9194–7. [PubMed] [Google Scholar]
- 543.Grandage VL, Everington T, Linch DC, Khwaja A. Go6976 is a potent inhibitor of the JAK 2 and FLT3 tyrosine kinases with significant activity in primary acute myeloid leukaemia cells. Br J Haematol. 2006;135:303–16. doi: 10.1111/j.1365-2141.2006.06291.x. [DOI] [PubMed] [Google Scholar]
- 544.Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- 545.Peterman EE, Taormina P, 2nd, Harvey M, Young LH. Go 6983 exerts cardioprotective effects in myocardial ischemia/reperfusion. Journal of cardiovascular pharmacology. 2004;43:645–56. doi: 10.1097/00005344-200405000-00006. [DOI] [PubMed] [Google Scholar]
- 546.Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN, et al. The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615. HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005;65:7462–9. doi: 10.1158/0008-5472.CAN-05-0071. [DOI] [PubMed] [Google Scholar]
- 547.Rovedo MA, Krett NL, Rosen ST. Inhibition of glycogen synthase kinase-3 increases the cytotoxicity of enzastaurin. J Invest Dermatol. 2011;131:1442–9. doi: 10.1038/jid.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Slosberg ED, Yao Y, Xing F, Ikui A, Jirousek MR, Weinstein IB. The protein kinase C beta-specific inhibitor LY379196 blocks TPA-induced monocytic differentiation of HL60 cells the protein kinase C beta-specific inhibitor LY379196 blocks TPA-induced monocytic differentiation of HL60 cells. Mol Carcinog. 2000;27:166–76. [PubMed] [Google Scholar]
- 549.Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochemical and biophysical research communications. 1986;135:397–402. doi: 10.1016/0006-291x(86)90008-2. [DOI] [PubMed] [Google Scholar]
- 550.Meggio F, Donella Deana A, Ruzzene M, Brunati AM, Cesaro L, Guerra B, et al. Different susceptibility of protein kinases to staurosporine inhibition. Kinetic studies and molecular bases for the resistance of protein kinase CK2. Eur J Biochem. 1995;234:317–22. doi: 10.1111/j.1432-1033.1995.317_c.x. [DOI] [PubMed] [Google Scholar]
- 551.Deng B, Xie S, Wang J, Xia Z, Nie R. Inhibition of protein kinase C beta(2) prevents tumor necrosis factor-alpha-induced apoptosis and oxidative stress in endothelial cells: the role of NADPH oxidase subunits. Journal of vascular research. 2012;49:144–59. doi: 10.1159/000332337. [DOI] [PubMed] [Google Scholar]
- 552.Tamaoki T. Use and specificity of staurosporine, UCN-01, and calphostin C as protein kinase inhibitors. Methods Enzymol. 1991;201:340–7. doi: 10.1016/0076-6879(91)01030-6. [DOI] [PubMed] [Google Scholar]
- 553.Evenou JP, Wagner J, Zenke G, Brinkmann V, Wagner K, Kovarik J, et al. The potent protein kinase C-selective inhibitor AEB071 (sotrastaurin) represents a new class of immunosuppressive agents affecting early T-cell activation. The Journal of pharmacology and experimental therapeutics. 2009;330:792–801. doi: 10.1124/jpet.109.153205. [DOI] [PubMed] [Google Scholar]
- 554.Naylor TL, Tang H, Ratsch BA, Enns A, Loo A, Chen L, et al. Protein kinase C inhibitor sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res. 2011;71:2643–53. doi: 10.1158/0008-5472.CAN-10-2525. [DOI] [PubMed] [Google Scholar]
- 555.Millward MJ, House C, Bowtell D, Webster L, Olver IN, Gore M, et al. The multikinase inhibitor midostaurin (PKC412A) lacks activity in metastatic melanoma: a phase IIA clinical and biologic study. Br J Cancer. 2006;95:829–34. doi: 10.1038/sj.bjc.6603331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Gekeler V, Boer R, Uberall F, Ise W, Schubert C, Utz I, et al. Effects of the selective bisindolylmaleimide protein kinase C inhibitor GF 109203X on P-glycoprotein-mediated multidrug resistance. Br J Cancer. 1996;74:897–905. doi: 10.1038/bjc.1996.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. The Journal of biological chemistry. 1991;266:15771–81. [PubMed] [Google Scholar]
- 558.Wilkinson SE, Parker PJ, Nixon JS. Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem J. 1993;294(Pt 2):335–7. doi: 10.1042/bj2940335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Reynolds NJ, McCombie SW, Shankar BB, Bishop WR, Fisher GJ. SCH 47112, a novel staurosporine derivative, inhibits 12-O-tetradecanoylphorbol-13-acetate-induced inflammation and epidermal hyperplasia in hairless mouse skin. Arch Dermatol Res. 1997;289:540–6. doi: 10.1007/s004030050236. [DOI] [PubMed] [Google Scholar]
- 560.Manetta A, Emma D, Gamboa G, Liao S, Berman M, DiSaia P. Failure to enhance the in vivo killing of human ovarian carcinoma by sequential treatment with dequalinium chloride and tumor necrosis factor. Gynecol Oncol. 1993;50:38–44. doi: 10.1006/gyno.1993.1161. [DOI] [PubMed] [Google Scholar]
- 561.Castle NA, Haylett DG, Morgan JM, Jenkinson DH. Dequalinium: a potent inhibitor of apamin-sensitive K+ channels in hepatocytes and of nicotinic responses in skeletal muscle. European journal of pharmacology. 1993;236:201–7. doi: 10.1016/0014-2999(93)90590-e. [DOI] [PubMed] [Google Scholar]
- 562.Meotti FC, Luiz AP, Pizzolatti MG, Kassuya CA, Calixto JB, Santos AR. Analysis of the antinociceptive effect of the flavonoid myricitrin: evidence for a role of the L-arginine-nitric oxide and protein kinase C pathways. The Journal of pharmacology and experimental therapeutics. 2006;316:789–96. doi: 10.1124/jpet.105.092825. [DOI] [PubMed] [Google Scholar]
- 563.Navarro-Nunez L, Lozano ML, Martinez C, Vicente V, Rivera J. Effect of quercetin on platelet spreading on collagen and fibrinogen and on multiple platelet kinases. Fitoterapia. 2010;81:75–80. doi: 10.1016/j.fitote.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 564.Noh KM, Hwang JY, Shin HC, Koh JY. A novel neuroprotective mechanism of riluzole: direct inhibition of protein kinase C. Neurobiol Dis. 2000;7:375–83. doi: 10.1006/nbdi.2000.0297. [DOI] [PubMed] [Google Scholar]
- 565.Ogiwara T, Negishi T, Chik CL, Ho AK. Differential effects of two protein kinase C inhibitors, calphostin C and Go6976, on pineal cyclic nucleotide accumulation. J Neurochem. 1998;71:1405–12. doi: 10.1046/j.1471-4159.1998.71041405.x. [DOI] [PubMed] [Google Scholar]
- 566.Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, et al. Rottlerin, a novel protein kinase inhibitor. Biochemical and biophysical research communications. 1994;199:93–8. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 567.Kraft AS, Smith JB, Berkow RL. Bryostatin, an activator of the calcium phospholipid-dependent protein kinase, blocks phorbol ester-induced differentiation of human promyelocytic leukemia cells HL-60. Proc Natl Acad Sci U S A. 1986;83:1334–8. doi: 10.1073/pnas.83.5.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Khan WA, Dobrowsky R, el Touny S, Hannun YA. Protein kinase C and platelet inhibition by D-erythro-sphingosine: comparison with N, N-dimethylsphingosine and commercial preparation. Biochemical and biophysical research communications. 1990;172:683–91. doi: 10.1016/0006-291x(90)90728-6. [DOI] [PubMed] [Google Scholar]
- 569.Kim HL, Im DS. N, N-dimethyl-D-erythro-sphingosine increases intracellular Ca2+ concentration via Na+-Ca2+-exchanger in HCT116 human colon cancer cells. Arch Pharm Res. 2008;31:54–9. doi: 10.1007/s12272-008-1120-y. [DOI] [PubMed] [Google Scholar]
- 570.Zarate CA, Jr, Singh JB, Carlson PJ, Quiroz J, Jolkovsky L, Luckenbaugh DA, et al. Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord. 2007;9:561–70. doi: 10.1111/j.1399-5618.2007.00530.x. [DOI] [PubMed] [Google Scholar]
- 571.Osada H, Sonoda T, Tsunoda K, Isono K. A new biological role of sangivamycin; inhibition of protein kinases. J Antibiot (Tokyo) 1989;42:102–6. doi: 10.7164/antibiotics.42.102. [DOI] [PubMed] [Google Scholar]
- 572.Ringvold HC, Khalil RA. Protein Kinase C as Regulator of Vascular Smooth Muscle Function and Potential Target in Vascular Disorders. Adv Pharmacol. 2017;78:203–301. doi: 10.1016/bs.apha.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Tumey LN, Bhagirath N, Brennan A, Brooijmans N, Lee J, Yang X, et al. 5-Vinyl-3-pyridinecarbonitrile inhibitors of PKCtheta: optimization of enzymatic and functional activity. Bioorg Med Chem. 2009;17:7933–48. doi: 10.1016/j.bmc.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 574.Cywin CL, Dahmann G, Prokopowicz AS, 3rd, Young ER, Magolda RL, Cardozo MG, et al. Discovery of potent and selective PKC-theta inhibitors. Bioorg Med Chem Lett. 2007;17:225–30. doi: 10.1016/j.bmcl.2006.09.056. [DOI] [PubMed] [Google Scholar]
- 575.Whitson EL, Bugni TS, Chockalingam PS, Concepcion GP, Feng X, Jin G, et al. Fibrosterol sulfates from the Philippine sponge Lissodendoryx (Acanthodoryx) fibrosa: sterol dimers that inhibit PKCzeta. J Org Chem. 2009;74:5902–8. doi: 10.1021/jo900844r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Lahn M, Sundell K, Moore S. Targeting protein kinase C-alpha (PKC-alpha) in cancer with the phosphorothioate antisense oligonucleotide aprinocarsen. Annals of the New York Academy of Sciences. 2003;1002:263–70. doi: 10.1196/annals.1281.029. [DOI] [PubMed] [Google Scholar]
- 577.Levesque L, Dean NM, Sasmor H, Crooke ST. Antisense oligonucleotides targeting human protein kinase C-alpha inhibit phorbol ester-induced reduction of bradykinin-evoked calcium mobilization in A549 cells. Mol Pharmacol. 1997;51:209–16. doi: 10.1124/mol.51.2.209. [DOI] [PubMed] [Google Scholar]
- 578.Kim J, Thorne SH, Sun L, Huang B, Mochly-Rosen D. Sustained inhibition of PKCalpha reduces intravasation and lung seeding during mammary tumor metastasis in an in vivo mouse model. Oncogene. 2011;30:323–33. doi: 10.1038/onc.2010.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Ferreira JC, Koyanagi T, Palaniyandi SS, Fajardo G, Churchill EN, Budas G, et al. Pharmacological inhibition of betaIIPKC is cardioprotective in late-stage hypertrophy. J Mol Cell Cardiol. 2011;51:980–7. doi: 10.1016/j.yjmcc.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Stebbins EG, Mochly-Rosen D. Binding specificity for RACK1 resides in the V5 region of beta II protein kinase C. The Journal of biological chemistry. 2001;276:29644–50. doi: 10.1074/jbc.M101044200. [DOI] [PubMed] [Google Scholar]
- 581.Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, et al. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci U S A. 2001;98:11114–9. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Gray MO, Karliner JS, Mochly-Rosen D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. The Journal of biological chemistry. 1997;272:30945–51. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- 583.Yonezawa T, Kurata R, Kimura M, Inoko H. PKC delta and epsilon in drug targeting and therapeutics. Recent Pat DNA Gene Seq. 2009;3:96–101. doi: 10.2174/187221509788654205. [DOI] [PubMed] [Google Scholar]
- 584.Braun MU, Mochly-Rosen D. Opposing effects of delta- and zeta-protein kinase C isozymes on cardiac fibroblast proliferation: use of isozyme-selective inhibitors. J Mol Cell Cardiol. 2003;35:895–903. doi: 10.1016/s0022-2828(03)00142-1. [DOI] [PubMed] [Google Scholar]
- 585.Sweitzer SM, Wong SM, Peters MC, Mochly-Rosen D, Yeomans DC, Kendig JJ. Protein kinase C epsilon and gamma: involvement in formalin-induced nociception in neonatal rats. The Journal of pharmacology and experimental therapeutics. 2004;309:616–25. doi: 10.1124/jpet.103.060350. [DOI] [PubMed] [Google Scholar]