

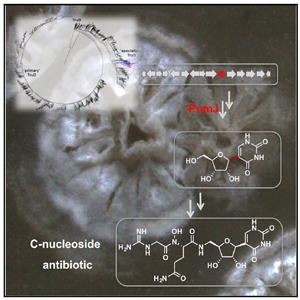
SUMMARY
Pseudouridimycin (PUM) is a selective nucleosideanalog inhibitor of bacterial RNA polymerase with activity against Gram-positive and Gram-negative bacteria. PUM, produced by Streptomyces sp. ID38640, consists of a formamidinylated, N-hydroxylated Gly- Gln dipeptide conjugated to 5′-aminopseudouridine. We report the characterization of the PUM gene cluster. Bioinformatic analysis and mutational knockouts of pum genes with analysis of accumulated intermediates, define the PUM biosynthetic pathway. The work provides the first biosynthetic pathway of a C-nucleoside antibiotic and reveals three unexpected features: production of free pseudouridine by the dedicated pseudouridine synthase, PumJ; nucleoside activation by specialized oxidoreductases and aminotransferases; and peptide-bond formation by amide ligases. A central role in the PUM biosynthetic pathway is played by the PumJ, which represents a divergent branch within the TruD family of pseudouridine synthases. PumJ-like sequences are associated with diverse gene clusters likely to govern the biosynthesis of different classes of C-nucleoside antibiotics.
Graphical abstract
INTRODUCTION
The rise and spread of bacterial pathogens resistant to almost all antibiotics currently in clinical use creates an urgent need for discovery of new antibacterials. Unfortunately, recent decades have witnessed a decline in the number of antibiotics introduced into clinical practice, which is exacerbated by the fact that most newly introduced antibiotics are variants of previous antibiotics, rather than novel classes with novel mechanisms (Monciardini et al., 2014; Butler et al., 2017).
Bacterial RNA polymerase (RNAP), the enzyme responsible for bacterial DNA-dependent RNA synthesis, is the target of two classes of antibacterial drugs in current clinical use: the rifamycins and the lipiarmycins (Srivastava et al., 2011; Ma et al., 2016).
We recently reported the identification and characterization of pseudouridimycin (PUM), a novel antibiotic that inhibits bacterial RNAP through a different binding site and different mechanism than rifamycins and lipiarmycins, that has no cross-resistance with rifamycins and lipiarmycins, and that has a spontaneous resistance rate less than one-tenth that of rifamycins and lipiarmycins (Maffioli et al., 2017). PUM exhibits antibacterial activity against drug-sensitive, drug-resistant, and multi-drug-resistant bacterial pathogens in culture and can clear streptococcal infections in mice. PUM is a nucleoside-analog inhibitor that functions by mimicking UTP. It binds to the RNAP active-center NTP addition site and competes with UTP for occupancy of the NTP addition site, making Watson-Crick H-bonded contacts with an adenine base in DNA template and H-bonded interactions with the nascent RNA product and RNAP. Because most RNAP residues that interact with PUM are essential residues that cannot be substituted without loss of RNAP activity, the PUM resistance spectrum is small and the PUM resistance rate is low.
PUM is produced by Streptomyces sp. and consists of a formamidinylated, N-hydroxylated Gly-Gln dipeptide conjugated to 5′-aminopseudouridine (Figure 1). PUM is part of the broader group of “peptidyl nucleoside” antibiotics (Niu and Tan, 2015; Winn et al., 2010) that interfere with different biological processes, including bacterial peptidoglycan biosynthesis (e.g., pacidamycin and other inhibitors of the bacterial translocase MraY), bacterial teichoic-acid biosynthesis (e.g., tunicamycin), bacterial and fungal protein synthesis (e.g., puromycin), and fungal chitin synthesis (e.g., nikkomycin). PUM is a “C-nucleoside” antibiotic, meaning that the nucleoside base is connected to the nucleoside sugar through a carbon atom of the nucleoside base. In contrast, most previously known nucleoside antibiotics are “N-nucleosides,” with a nitrogen atom of the nucleoside base connecting to the sugar. Indeed, just a few C-nucleosides have been described, including formycin and malayamycin, possessing anti-HIV-1 and antifungal activity, respectively (Li et al., 2008; De Clercq, 2016).
Figure 1. The pum Biosynthetic Cluster and the Proposed Biosynthetic Pathway for Pseudouridimycin.
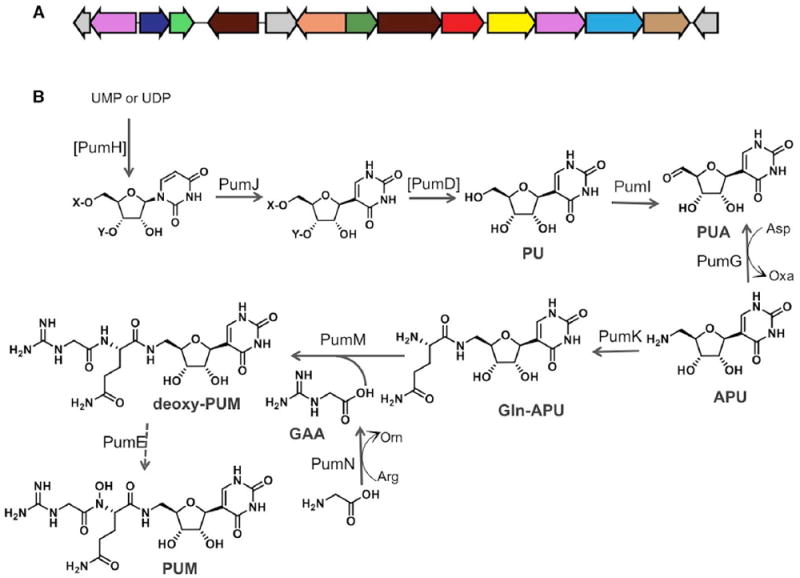
(A) Organization of the pum biosynthetic cluster.
(B) Proposed biosynthetic pathway for pseudouridimycin. Enzymes reported within brackets have not been experimentally determined. X and Y denote possible phosphate substituents on the nucleoside. Abbreviations of pathway intermediates: PU, pseudouridine; PUA, 5′-oxo-PU; APU, 5′-aminopseudouridine; Gln- APU, glutamine-APU; GAA, guanidinoacetic acid; PUM, pseudouridimycin; UMP and UDP (uridine-based nucleotide).
The enzymatic mechanisms for C-nucleoside biosynthesis have thus far remained elusive. Here, we have identified, sequenced, and characterized the PUM biosynthetic locus. The results define the actual pathway of PUM biosynthesis, demonstrate the production of PUM by precursor feeding, and set the stage for engineered overproduction of PUM and PUM analogs. Biosynthesis of PUM depends on the pseudouridine synthase PumJ. Phylogenomic analysis of gene clusters encoding PumJ-like pseudouridine synthases show that C-nucleoside biosynthetic pathways are diverse and taxonomically widespread.
RESULTS
Identification of the PUM Biosynthetic Gene Cluster
In order to identify the PUM gene cluster, we have generated a draft genome of Streptomyces sp. ID38640. The antiSMASH platform identified 26 gene clusters, one of which attracted our attention because of the presence of a pseudouridine synthase and a glycine amidinotransferase, possible candidates for generating the C-nucleoside and the guanidinoacetic acid (GAA) moieties of PUM, respectively (Figure 1A and Table 1). The pum cluster is likely to encompass a genomic segment of about 20 kbp containing 15 CDSs (Coding DNA Sequences) associated with PUM biosynthesis, export, and regulation, and designated pumA-pumO. Bioinformatic analysis enabled assigning a putative function to most gene products, as summarized in Table 1 and discussed below.
Table 1.
CDSs from the pum Cluster of Streptomyces sp. ID38640 and Established/Proposed Roles in the PUM Pathway
| CDS | Size (aa) | Protein Family | Homologa [Pathway, Strain, Accession No.] | Identity (%)b | Rolec |
|---|---|---|---|---|---|
| pumA | 134 | alpha/beta hydrolase | [unknown, Streptomyces sp. BoleA5, WP_018563811.1] | 59 | unknown |
| pumB | 396 | MFS (Major Facilitator Superfamily) | [unknown, Streptomyces sp. NRRL S-1813, SOD85851] | 90 | export |
| pumC | 252 | DeoR transcriptional regulator | [unknown, Streptomyces sp. NRRL S-1813, WP_030985690.1] | 95 | regulator |
| pumD | 200 | HAD family hydrolase | [unknown, Streptomyces sp. NRRL S-1813, WP_030985692.1] | 91 | phosphatase |
| pumE | 418 | flavin-dependent oxidoreductase | CetI [Cetoniacytone A, Actinomyces sp. Lu 9419, ACH85569.1]; ORF36 [Micromonospora carbonacea var. africana, ACF94630.1] | 71; 36 | deoxy-PUM N-hydroxylation |
| pumF | 269 | hypothetical protein | Mur33 [muraymycin, Streptomyces sp. NRRL 30471, ADZ45345.1] | 42 | unknown |
| pumG | 440 | PLP-dependent aspartate aminotransferase | CetH [cetoniacytone A, Actinomyces sp. Lu 9419, ACH85568.1]; Mur20 [muraymycin, Streptomyces sp. NRRL 30471, ADZ45332.1] | 63; 59 | PUA aminotransferase |
| pumH | 233 | adenylate kinase | PolQ2 [polyoxin, Streptomyces cacaoi, ABX24486.1] | 42 | phosphorylation of a uridine-based substrate |
| pumI | 535 | glucose-methanol-choline-oxidoreductase | CetG [cetoniacytone A, Actinomyces sp. Lu 9419, ACH85567.1] | 62 | PU 5′ oxidase |
| pumJ | 342 | tRNA pseudouridine synthase, TruD | TruD-like protein [cetoniacytone A, Actinomyces sp. Lu 9419, ACH85566.1] | 59 | pseudouridine synthase |
| pumK | 402 | ATP-grasp domain protein | NikS-like protein [cetoniacytone A, Actinomyces sp. Lu 9419, ACH85565.1]; NikS, [nikkomycin, Streptomyces tendae, CAC11141.1] | 55; 39 | Gln-APU carboxylate-amine ligase |
| pumL | 423 | MFS (Major Facilitator Superfamily) | NocH-like_protein [cetoniacytone A, Actinomyces sp. Lu 941, ACH85564.1] | 65 | export |
| pumM | 477 | D-alanine-D-alanine ligase | [unknown, Kitasatospora aureofaciens, KOG75670] | 78 | GAA and Gln-APU amide ligase |
| pumN | 380 | amidinotransferase | GdnJ [guadinomine, Streptomyces sp. K01-0509, AFU82619.1] | 78 | GAA formation |
| pumO | 210 | hypothetical | [unknown, Streptomyces griseoflavus, KOG58705.1] | 78 | unknown |
Sequences with the highest BLAST score from the MIBiG database of validated biosynthetic gene clusters (Medema et al., 2015) or from the nonredundant GenBank database (no protein name; in italics), excluding sequences from Streptomyces rimosus subsp. rimosus ATCC 10970. Occasionally, an additional sequence is reported.
% identity of the best matching sequence(s).
Established roles from experimental data (in bold type) or predicted from sequence homology. PU, pseudouridine; APU, aminopseudouridine; PUA, pseudouridine aldehyde; Gln-APU, Glutamine-APU; GAA, guanidinoacetic acid.
We developed a method for the genetic manipulation of Streptomyces sp. ID38640 based on intergeneric conjugation from Escherichia coli followed by gene replacement using in vitro mutated pum genes carried by an unstable pWHM3-based replicon (see STAR Methods; Figure S1). The genotypes of the resulting mutants were confirmed by PCR. Following growth and metabolite extraction, the PUM-related compounds produced by the different mutants were analyzed by liquid chromatography-mass spectrometry (LC-MS), and their identity was confirmed by comparison with chemically synthesized or commercially available compounds. Through this methodology, we knocked out seven pum genes and analyzed the corresponding mutants.
Formation of Pseudouridine by the Dedicated Pseudouridine Synthase PumJ
Pseudouridine (PU) is the most abundant modification present in tRNAs and is introduced by a post-transcriptional isomerization carried by four distinct families of PU synthases, designated as TruA to TruD (Hamma and Ferré -D’Amaré, 2006; Li et al., 2016). Encoded by the pum cluster is PumJ, a protein showing 28% amino acid identity to Escherichia coli TruD. Knocking out pumJ in Streptomyces sp. ID38640 abolished PUM production, with accumulation of only a compound with retention time (1.2 min) and m/z value (344 [M + 2TFA-H]-) corresponding to GAA (Figure 2; see also below). Adding 0.2 or 0.5 mg/mL PU to the pumJ mutant fully rescued PUM production, with the accumulation of two PUM-related compounds with the same retention times and m/z values as synthetic 5′aminopseudouridine (APU; 1.2 min and 244 [M + H]+) and Gln-APU (1.7 min and 372 [M + H]+), in addition to residual PU (Figure 2). These results indicate that PumJ is involved in the biosynthesis of PUM and that PU is efficiently taken up by the strain and converted into the pathway intermediates APU and Gln-APU, as well as into the final product. Together with the bioinformatic predictions, these results indicate that PumJ is involved in the N- to C-isomerization of a nucleoside-like intermediate in the PUM pathway and that free PU is a pathway intermediate.
Figure 2. Metabolite Analysis of pumJ.
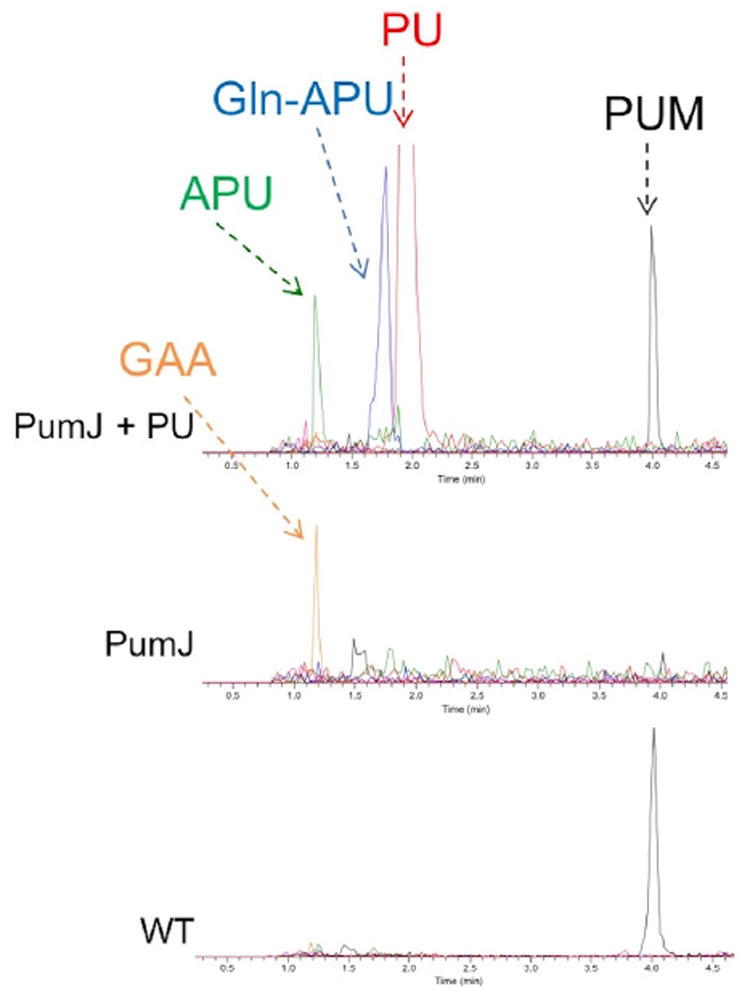
Analyses performed on cultures of the parental strain (wild-type [WT]) and of the pumJ mutant without (PumJ) and with (PumJ + PU) supplementation with 0.5 mg/mL PU. The analyses show extracted ion chromatograms of pseudouridimycin (PUM, m/z 487 [M + H]+, black line), pseudouridine (PU, m/z 245, [M + H]+, red line), aminopseudouridine (APU, m/z 244 [M + H]+, green line), Gln-APU (m/z 372 [M + H]+, blue line) and guanidinoacetic acid (GAA, m/z 344 [M + 2TFA-H]−, orange line).
In a parallel experiment, addition of equivalent concentrations of uridine to the ΔpumJ culture did not lead to the accumulation of any PUM-related metabolite (data not shown), suggesting that the enzymes downstream of PumJ in the pathway can effectively discriminate between C- and N-based nucleosides. It should also be noted that the pumJ mutant does actually produce a small amount (less than 5% of the wild-type level) of a compound with the same retention time, UV spectrum, and m/z signal as PUM (Figure 2). Most likely, this compound is identical to PUM and probably results from the small intracellular amounts of PU present in tRNA, rRNA, and/or small nuclear RNA and released as free nucleoside by RNA turnover.
Formation of Aminopseudouridine by PumI and PumG
We predicted that the conversion of PU into its 5′amino derivative APU requires the concerted action of an oxidoreductase and an aminotransferase. PumI resembles FAD-dependent glucose-methanol-choline oxidoreductases, with 62% identity to CetG (Table 1), an enzyme of unassigned role from the cetoniacytone cluster (Wu et al., 2009). PumI is likely to be involved in PU dehydrogenation to yield the corresponding pseudouridine aldehyde (PUA). Knockout of pumI abolished PUM production, with no significant accumulation of any PU-related intermediates (Figure 3). Considering that GAA accumulates in most pum mutants, we infer that the lack of PU-related intermediates in the pumI mutant is likely due to polar effects of the introduced mutations on the expression of downstream pum genes.
Figure 3. Metabolite Analysis of pumM, pumK, pumG, and pumI Mutants.
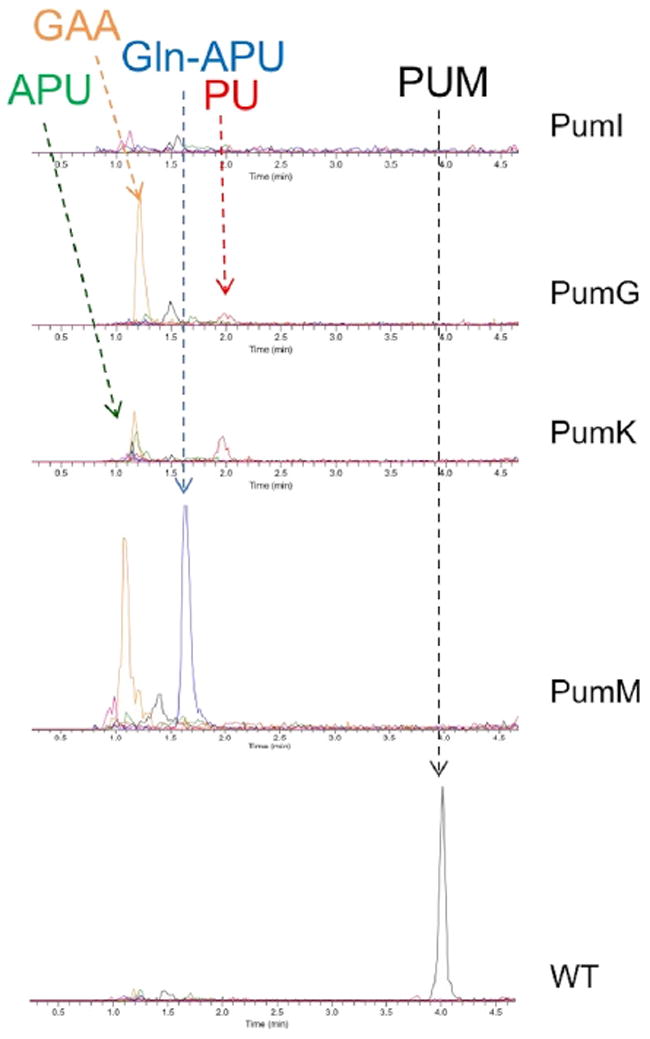
Analyses performed on cultures of the parental strain (wild-type [WT]) and of the pumM, pumK, pumG, and pumI mutants. The analyses show extracted ion chromatograms of pseudouridimycin (PUM, m/z 487 [M + H]+, black line), pseudouridine (PU, m/z 245, [M + H]+, red line), aminopseudouridine (APU, m/z 244 [M + H]+, green line), Gln-APU (m/z 372 [M + H]+, blue line), and guanidinoacetic acid (GAA, m/z 344 [M + 2TFA-H]−, orange line).
PumG is highly related to pyridoxal phosphate-dependent aspartate aminotransferases that act on basic amino acids and their derivatives. Related proteins (Table 1) are CetH, encoded by the cetoniacytone cluster, and Mur20, involved in the biosynthesis of the uridine-based nucleoside antibiotic muraymycin (Cheng et al., 2011). LC-MS analysis of the ΔpumG mutant showed complete loss of PUM and accumulation of a small amount of a compound with retention time (2.0 min) and m/z value (245 [M + H]+) identical to those of PU, as well as a significant amount of GAA (Figure 3).
Addition of 0.4 mg/mL PU to cultures of the ΔpumI strain did not rescue PUM production (data not shown), indicating that PumI acts downstream of PU in the biosynthetic pathway. However, PUA and APU were also unable to rescue PUM production in the pumI mutant. The data presented below are consistent with the possibility that PUA and APU are not taken up by the cells. Additional experiments will be necessary to confirm that no uptake of these precursors occurred.
Formation of the Peptide Bonds by the Carboxylate-Amine Ligases PumK and PumM
PUM contains two amide bonds, one linking the Gln moiety to APU and the other joining the Gln amino group to a modified glycine. Contrary to expectations derived from other peptidyl nucleoside antibiotic pathways (Trautman and Crawford, 2017), the pum cluster does not encode non-ribosomal peptide synthetases (NRPS). Instead, it encodes PumK and PumM, proteins that show similarity to ATP-dependent carboxylate-amine ligases containing an ATP-grasp domain. These enzymes activate carboxylate groups as acyl phosphates prior to amide bond formation (Li et al., 2004; Wu et al., 2009). PumK shows 39% identity to NikS, involved in the formation of the related peptidyl nucleoside antibiotic nikkomycin in Streptomyces tendae, as well as 55% identity to the uncharacterized NikS-like protein from the cetoniacytone pathway (Table 1). The N-terminal domain of PumM is similar to D-Ala-D-Ala ligases and related ATP-grasp enzymes (Table 1). These proteins also catalyze the ATP-assisted ligation of a carboxylate-containing molecule to a nucleophile via the formation of an acyl phosphate intermediate (Fawaz et al., 2011).
Deleting pumK yielded a strain incapable of producing PUM but accumulating APU as well as PU (Figure 3). Deleting pumM led to a strain accumulating Gln-APU (Figure 3), demonstrating that PumM extends Gln-APU through the formation of an additional amide bond. Both mutants also accumulated GAA (Figure 3).
It should be noted that addition of Gln-APU to a culture of the pumK mutant did not restore PUM production (data not shown), whereas this intermediate accumulated in the pumM mutant. In a parallel experiment in which Gln-APU was added to wild-type and pumK cultures, we observed time-dependent decrease of Gln-APU in the former but not in the latter culture (Figure S2A). While we cannot rule out polar effects of the introduced mutations on the expression of other pumM (the pumK mutant accumulates GAA, indicating a functional pumN gene; see below), the results from adding PUA, APU, and Gln-APU to mutant cultures are also consistent with the possibility that uptake of these pathway intermediates might involve the presence of dedicated transporters, whose expression is regulated by one or more products of the PUM pathway. Anyway, genetic complementation experiments will be necessary to rule out possible polar effects of these and other pum knockout mutations.
Formamidylation by the Amidinotransferase PumN
pumN encodes a protein with 78% identity to the glycine amidinotransferase from the biosynthetic pathway of guadinomine, a type III secretion inhibitor synthesized through a polyketide synthase assembly line that utilizes GAA as the starter unit (Holmes et al., 2012). Knockout of pumN abolished PUM production with the accumulation of APU and Gln-APU (Figure 4). Based on these results, PumN is likely to generate GAA through the transfer of the amidino group from arginine to the acceptor glycine. GAA would then be the cosubstrate of PumM. Consistently, supplementing the medium with 0.5 mg/mL GAA enabled the pumN mutant to convert most of the accumulated Gln-APU into PUM (Figure 4). Thus, the accumulation of GAA in most pum knockout mutants and the ability of exogenously added GAA to rescue production in the pumN mutant establish that GAA is a free intermediate in the PUM pathway.
Figure 4. Metabolite Analysis of pumE and pumN Mutants.

Analyses performed on cultures of the parental strain (wild-type [WT]), of the pumE mutant and of the pumN mutant without (PumN) and with (PumN + GAA) supplementation with 0.5 mg/mL guanidinoacetic acid. The analyses show extracted ion chromatograms of pseudouridimycin (PUM, m/z 487 [M + H]+, black line), pseudouridine (PU, m/z 245, [M + H]+, red line), amino-pseudouridine (APU, m/z 244 [M + H]+, green line), Gln-APU (m/z 372 [M + H]+, blue line), guanidinoacetic acid (GAA, m/z 344 [M + 2TFA-H]−, orange line), and deoxy-PUM (m/z 471 [M + H]+, pink line).
See also Figures S2 and S3.
Careful inspection of Figure 4 indicated that the GAA-supplemented pumN mutant actually produced more PUM than the wild-type, suggesting that GAA might be limiting PUM production. Triplicate experiments of GAA-supplemented cultures of the wild-type and the pumN mutant resulted in equivalent titers (152 ± 4 and 168 ± 8 mg/L PUM, respectively), which represent 1.5- and 1.7-fold, respectively, the level produced by unsupplemented wild-type cultures. Further experiments indicated that best production is obtained supplementing the medium with 0.5 mg/mL GAA, while 0.1 mg/mL GAA had minimal impact on PUM levels (data not shown). The above results confirmed that the GAA is limiting PUM production in the wild-type strain. Similar results were observed with Streptomyces sp. ID38673, another independently isolated PUM producer (Maffioli et al., 2017). Also in this case, addition of GAA to the wild-type culture resulted in PUM titers 80% higher than the unsupplemented controls (Figure S2B).
We then investigated whether substrates different from GAA could be incorporated into Gln-APU by the pumN mutant. However, addition of 3-guanidinopropanoic acid, 4-guanidinobutanoic acid, or creatine did not lead to the formation of any PUM-related metabolite, with the supplemented mutant accumulating APU and Gln-APU in equivalent amounts to the unsupplemented cultures (Figure S3). These results indicate that either GAA analogs are inefficiently taken up by the cells or, more likely, that PumM has narrow substrate specificity.
N-Hydroxylation by the Oxidoreductase PumE
PumE is 36% identical to ORF36 from the everninomicin cluster of Micromonospora carbonacea var. africana, which catalyzes amino sugar oxidation to give the corresponding hydroxylamine (Hu et al., 2008), and 71% identical to the uncharacterized CetI protein from the cetoniacytone pathway (Table 1). Like ORF36, PumE belongs to the broad family of flavin-dependent oxidoreductases (Table 1). LC-MS analysis of a ΔpumE mutant culture showed loss of the characteristic PUM peak and production of a metabolite with m/z 471 [M + H]+, thus 16 amu less than PUM and with the same retention time as synthetic deoxy- PUM (Figure 4). Amide nitrogens are less reactive than amine nitrogens, and amine hydroxylation is common in natural products (Huijbers et al., 2014). We are not aware of amide hydroxylation in the biosynthesis of microbial products. Nevertheless, we were unable to identify specific accumulation in the pumM and pumN mutants of a compound with an m/z value (388 [M + H]+), consistent with that of N-hydroxy-Gln-APU. It should be noted that the pumE mutant accumulates N-deoxy-PUM to a lesser extent than Gln-APU. Formally, we cannot rule out the possibility that N-hydroxy-Gln-APU is actually formed, but it is too instable to be detected under our conditions and that N-deoxyPUM is actually a shunt metabolite formed by GAA condensation to the less favorable but more abundant substrate Gln-APU in the pumE mutant. However, the simplest interpretation of our data is that PumE performs an amide bond hydroxylation in N-deoxyPUM.
The PUM Biosynthetic Pathway
Overall, the compounds accumulated by the pum mutants, the results from feeding experiments and bioinformatics predictions provide sufficient evidence to formulate a likely biosynthetic pathway for PUM. The exact mechanism of PU formation is currently unknown, but some candidate genes were identified in the cluster. PumH resembles adenylate kinases (Table 1) and shows 42% identity to PolQ2 from the polyoxin gene cluster, whose role has not, however, been established yet (Chen et al., 2009). PumD belongs to the HAD-like hydrolase family that includes proteins with phosphatase activity (Kuznetsova et al., 2006). Thus, it is tempting to speculate that formation of PU involves phosphorylation of a uridine-based substrate catalyzed by PumH, followed by the PumJ-catalyzed N- to C-isomerization and finally phosphate release by PumD. According to this hypothesis, exogenously added PU should be able to rescue PUM production in a pumD mutant.
Accordingly, the PUM biosynthetic pathway is likely to proceed as reported in Figure 1B. A uridine-based nucleotide (probably UMP or UDP) is phosphorylated by the kinase PumH (possibly at the 30 position, to mimic a tRNA-embedded moiety), isomerized to the corresponding C-nucleotide by the PU synthase PumJ, and dephosphorylated by the hydrolase PumD to yield free PU. The dehydrogenase PumI then converts PU to its 5′-dehydro derivative PUA and the aminotransferase PumG yields APU, possibly using aspartate as the amino donor. Then, the amide ligase PumK condenses glutamine with APU to yield Gln-APU, while the amidinotransferase PumN produces GAA from arginine and glycine. GAA is then condensed to Gln- APU by PumM to generate N-deoxy-PUM, which is N-hydroxylated by PumE to yield the final product (Figure 1B). Alternatively, as explained above, Gln-APU is N-hydroxylated by PumE to yield N-OH-Gln-APU, which is the substrate of PumM. Accordingly, nine genes in the pum cluster have a possible role in the PUM pathway.
PUM Export, Self-Resistance, and Regulation
Additional genes from the pum cluster are (Table 1): pumA, pumF, and pumO, which encode proteins of unknown function; pumB and pumL, specifying transporters of the major facilitator superfamily; and pumC, coding for a DeoR-like transcriptional regulator. Upstream of pumA is a gene coding for an acetyltransferase, and further downstream of pumO are genes coding for a protein of unknown function and for a glycine N-methyltransferase. None of these three genes is likely to be involved in PUM biosynthesis, export, or regulation, delimiting the borders of the pum cluster as shown in Figure 1A. PumF is encoded by other pum-like clusters (see below) and a PumF-like sequence is also present in the muraymycin gene cluster (Cheng et al., 2011; Table 1), suggesting that this protein may also play a role in PUM biosynthesis. The pum cluster does not encode any protein with an obvious role in PUM resistance. The Streptomyces sp. 38640 genome contains a single set of genes encoding the subunits of RNAP (M. Sosio et al., unpublished data). None of the substitutions conferring PUM resistance in either E. coli or Streptococcus pneumoniae (Maffioli et al., 2017), is present in the β and β′ subunits of RNAP (data not shown), suggesting that the enzyme in the producing strain is actually sensitive to PUM. Thus, resistance might simply depend on efficient export of the molecule outside of the cell. However, knockouts of pumB and pumL will be necessary to establish their possible involvement in export and/or resistance.
Interplay between PUM Biosynthesis and Other Secondary Metabolite Pathways
Close inspection of the LC-MS profile of both Streptomyces sp. ID38640 and ID38673 revealed the consistent appearance of metabolites unrelated to PUM eluting at 7.1 and 7.64 min and showing major m/z signals of 601 [M + H]+ and 841 [M + H]+, respectively (Figure S4). The fragmentation profiles, the UV spectra, and the difference in m/z values among the various congeners of the m/z 601 species were superimposable to those reported for desferrioxamine, a siderophore produced by several Streptomyces spp. (Barona-Gómez et al., 2006). Similarly, the m/z 841 species were identified as lydicamycin, a polyketide containing tetramic acid and amidinopyrrolidine moieties (Furumai et al., 2002). Consistently, the Streptomyces sp. ID38640 genome harbors two biosynthetic gene clusters (BGCs) that are closely related to the experimentally determined desferrioxamine (Barona-Gómez et al., 2004) or hypothesized lydicamycin (Komaki et al., 2015) gene clusters (Figures S5A and S5B).
When analyzing the LC-MS profiles of the pum mutants, we noticed that pumN produced increased levels of lydicamycin compared with the wild-type strain, and that addition of GAA to the wild-type or to the pumN mutant had no effect on the lydicamycin levels (Figure S5C). Lydicamycin biosynthesis is predicted to involve a type I polyketide synthase that utilizes an arginine- derived metabolite as starter unit (Komaki et al., 2015). We also observed that the pumE mutant produced increased levels of desferrioxiamine (Figure S5D). The latter consists of alternating diamine and dicarboxylic acid building blocks linked by amide bonds and its formation requires FAD-dependent N-hydroxylation. Thus, the PUM pathway might drain amino acids (arginine) and FAD or some other cofactors from the lydicamycin and desferrioxamine pathways, respectively, although we cannot exclude that regulatory circuits are at work.
Occurrence of PumJ-Related Sequences in Biosynthetic Gene Clusters
The proposed biosynthetic pathway points to PU as a key, early intermediate in PUM biosynthesis. Consequently, a central role in the pathway is played by PumJ. As mentioned, PumJ is only 28% identical to E. coli TruD, which carries out C-isomerization of uracil-13 in tRNAs (Ericsson et al., 2004). In contrast to TruD, which acts on an RNA molecule, PumJ most likely acts on a suitably modified nucleotide. The difference in substrate (large, folded polynucleotide versus a mononucleotide) and cellular function (nucleic acid metabolism versus secondary metabolism) is consistent with the observed sequence divergence of PumJ from TruD. We then wondered whether other sequences could be identified in microbial genomes with higher similarity to PumJ than to TruD, and whether these PumJ-related sequences would be associated with likely BGCs.
We constructed a phylogenetic tree of ~7,000 TruD homologs found across public bacterial genomes, rooted with a set of TruB enzymes, a related protein family (Hamma and Ferré-D’Amaré, 2006), is shown in Figure 5. PumJ clades with other sequences in a separate branch that is clearly divergent from the experimentally established TruD from E. coli and its close relatives (primary TruD in Figure 5). Next, we analyzed the CDSs flanking the pumJ homologs by extracting the corresponding genomic regions (30 kbp left and right) and performing a MultiGeneBlast search with the pum cluster as query. This led to the selection of 38 interesting BGCs encoding PumJ homologs, whose borders were manually curated. This analysis identified three main, broad BGC families containing PumJ homologs.
Figure 5. Phylogenomic Analysis of PumJ Sequences.
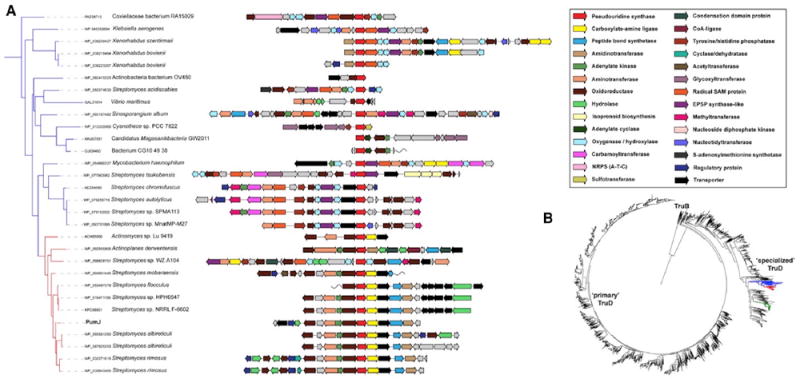
(A) Enlargement of the phylogenetic tree (panel B) with PumJ-related sequences from families 1 and 2 and the corresponding gene clusters, each centered on the PumJ homolog. The predicted enzymatic functions are color-coded as indicated in the side panel. The pum cluster is indicated as “PumJ.” Clusters from families 1 and 2 are shown in greater detail as Figures S6 and S7. Selected BGCs from family 3 are reported in Figure 6.
(B) A phylogenetic tree of ~7,000 TruD sequences. The tree was rooted with a set of TruB enzymes. The branches representing families 1, 2, and 3 are shown in red, blue, and green, respectively.
See also Figures 6, S6, and S7.
The first family, found mostly in Streptomyces spp. (Figure 5A, indicated in red in the phylogenetic tree of Figure 5B; see also Figure S6), contains a number of near-identical copies of the pum cluster (e.g., the one from Streptomyces rimosus NRRL WC-3930), some of which have gene cassettes adjacent to the pum-like segment that encode redox enzymes that may or may not be involved in further modifying a possible PUM-like compound. The most divergent variant within this family is the BGC from Actinoplanes derwentensis, which encodes standalone NRPS-like domain proteins (condensation, adenylation, and thiolation) in place of the peptide bond synthetases found in the pum cluster, as well as two hydrolases of the same class as PumD surrounded by core pum genes and not at the edge of the cluster. One interesting note is the BGC from Actinomyces sp. Lu 9419: it contains three CDSs (named CetI, CetG, and CetH) that are closely related to PumE, PumI, and PumG, respectively, and the PumJ, PumK, and PumL homologs adjacent to them (Table 1), which have probably been incorrectly implicated in cetoniacytone A biosynthesis (Wu et al., 2009). Most likely, these genes are part of a pum-like cluster that looks incomplete. One possible explanation is that the cet and pum BGCs were inserted into the same genomic island, and that the pum cluster eventually degenerated in Actinomyces sp. Lu 9419.
The second family (Figure 5A, indicated in blue in the phylogenetic tree of Figure 5B; see also Figure S7) is more distantly related to pum and has a broad taxonomic distribution. It is found in the actinobacteria Streptomyces, Sinosporangium, and Mycobacterium, as well as in Xenorhabdus and members of the Coxiellaceae (both γ-Proteobacteria). Most of these BGC share a core set of enzymes in addition to PumJ: an EPSP-synthaselike enzyme (putatively related to UDP-N-acetylglucosamine 1-carboxyvinyltransferase), a radical SAM enzyme, a tyrosine phosphatase, an adenylate kinase (except for the Streptomyces tsukubensis cluster), and oxygenases. Several of them also have homologs of the PumK carboxylate-amine ligase, the PumM peptide bond synthetase and the PumG aminotransferase. On top of this shared core, they exhibit some interesting variations: e.g., some encode one or more carbamoyltransferases and/or a nucleotidyltransferase; the BGC from Streptomyces tsukubensis encodes a glycosyltransferase and has an isoprenoid biosynthesis- related cassette right next to it (which may or may not be part of the cluster); and the cluster from the Coxiellaceae bacterium RA15029 encodes an NRPS-like protein (with domain architecture A-T-C).
The third family (Figure 6, indicated with the green branch in the TruD phylogeny in Figure 5B) is found mainly in myxobacteria and represents the most divergent family compared with the PUM biosynthetic gene cluster. Virtually all of its members encode homologs of the sarcosine oxidase alpha and beta subunits, as well as adenylate cyclase homologs, which suggest a possible cyclic structure of the nucleosides. This is corroborated by the presence of transcriptional regulators with cyclic nucleotide-binding GAF domains in most clusters, which indicate a possible regulatory feedback loop. Besides these gene products, the clusters encode a variety of enzymes, including oxidoreductases, acyltransferases, glycosyltransferases, and methyltransferases. Notably, in the genome of Myxococcus xanthus, the putative cyclic nucleoside biosynthetic gene cluster is located directly adjacent to the myxovirescin (also known as TA antibiotic) biosynthetic gene cluster. It is tempting to speculate that its product acts as a signal to regulate myxovirescin biosynthesis. In fact, the predicted cyclic nucleotide-binding regulator mentioned above has been termed TaR2 in patent literature, and has been tentatively implicated in the regulation of myxovirescin (Paitan et al., 1999). However, the evidence for this remains unclear. Interestingly, expression of TaR2 has been shown to be upregulated 6-fold in yellow, high-swarming variants of Myxococcus xanthus (Furusawa et al., 2011).
Figure 6. Organization of Family 3 BGCs.

Selected BGCs from Figure 5 are reported. The predicted functions of the CDSs are color-coded as in the side panel.
In summary, PumJ-related sequences are associated with likely BGCs found in a broad variety of bacterial taxa. On the basis of cluster architecture, none of the BGCs from Figure 5 is likely to be associated with the C-nucleoside antibiotic formycin, which contains a modified purine. Compounds related to malayamycin, which contains a cyclized and an N-carbamylated APU moiety with no peptide bonds, might be synthesized by BGC clusters from Streptomyces tsukubensis, Streptomyces chromofuscus, Streptomyces autolyticus, and Streptomyces sp. SPMA113, which encode PumJ-, PumI-, and PumG-related sequences, as well as a carbamoyltransferase, but not amide-forming enzymes (Figure S7). Overall, the results from Figure 5 suggest that additional compounds within the family of C-nucleoside metabolites await discovery.
DISCUSSION
In this work, we have identified and characterized the cluster responsible for PUM production in Streptomyces sp. ID38640, and established important paradigms for the biosynthesis of C-nucleoside antibiotics. Several biosynthetic pathways for peptidyl nucleoside antibiotics have been characterized (Chen et al., 2016; Niu and Tan, 2015; Walsh and Zhang, 2011), including those for the formation of the cytosine derivative blasticidin S, the adenine derivative puromycin (protein synthesis inhibitors), and the uridine-based peptides nikkomycin and polyoxin (inhibitors of fungal chitin synthesis), and pacidamycin and mureidomycins (inhibitors of the translocase MraY). These pathways have been shown to start with a nucleotide, which can be modified to its 5′-amino derivative before formation of carboxamido linkage to the peptide moiety (Walsh and Zhang, 2011). To our knowledge, the work reported here represents the first experimental characterization of a pathway for a C-nucleoside antibiotic. While this work was in progress, Palmu et al. (2017) reported the characterization of the gene cluster involved in the biosynthesis of showdomycin, which consists of maleimide C-linked to ribose. Its biosynthesis has been proposed to start with glutamine and to involve a C-specific sugar transferase (Palmu et al., 2017). The nucleobase of showdomycin is unrelated to nucleic acids and so this molecule represents a special case of C-nucleosides. Indeed, the showdomycin gene cluster bears no resemblance to the pum gene cluster. Just a few C-nucleosides have been described, including formycin and malayamycin, possessing anti-HIV-1 and antifungal activity, respectively (Li et al., 2008; De Clercq, 2016).
One remarkable feature of the PUM pathway is the use of an enzyme resembling pseudouridine synthases. PumJ is distantly related to the TruD family of pseudouridine synthases, with only 28% sequence identity to E. coli TruD. This is not surprising, since pseudouridine synthases involved in tRNA modification act in a sequence-specific manner and must thus recognize the target U for isomerization in the context of a large macromolecule. In contrast, PumJ is likely to recognize a suitably modified nucleotide. Given the central role of U-based nucleotides as precursors for many essential cellular pathways, it is likely that PumJ has evolved to recognize a suitably modified UDP-related metabolite for N- to C-isomerization. Further work will be necessary to establish the actual substrate of PumJ.
PumJ homologs have been detected in several BGCs from Actinobacteria and Proteobacteria. While most clusters from Figures 5 and S6 are likely to specify the synthesis of PUM or a highly related metabolite, those from Figures S7 and 6 are substantially divergent and may specify the biosynthesis of other C-nucleoside antibiotics. Given the large number of pathways affected by nucleoside antibiotics, the products of BGCs containing PumJ-like sequences might act on targets other than RNAP. Actinobacterial genomes do not appear to encode bona fide TruD sequences, except those highly related to PumJ and associated with likely BGCs (http://pfam.xfam.org/family/trud#tabview=tab7). The genome of the PUM producer itself encodes one TruA and one TruB homolog, but no sequence related to bona fide TruD (M. Sosio, unpublished data). Thus, it is tempting to speculate that the TruD family of pseudouridine synthases has been recruited for secondary metabolism in actinomycetes and possibly in other bacteria.
We found that PUM production in the pumJ knockout mutant can be rescued by feeding PU, but addition of uridine does not lead to any PUM-related metabolite, suggesting that enzymes downstream of PU effectively discriminate between C- and N-based nucleosides. Such discrimination is likely to involve PumI and/or PumG, and possibly the amide ligase PumK. Interestingly, some nucleoside antibiotics (e.g., pacidamycin) contain a 5′-aminouridine moiety, whose formation requires PacI, a FAD-dependent oxidoreductase that generates the 5′-aldehyde (Rackham et al., 2010; Zhang et al., 2010). However, PacI acts on phosphorylated substrates and shows no relatedness to PumI, suggesting a different evolution for C- and N- nucleoside dehydrogenating enzymes, possibly associated with substrate discrimination.
The ability of rescuing blocked mutants by feeding pathway intermediates provides an opportunity for generating PUM analogs by mutasynthesis and precursor feeding. In addition, the supply of the inexpensive chemical GAA and, possibly, the block of competing pathways can offer opportunities for increasing PUM production.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
“Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Margherita Sosio (msosio@naicons.com)”
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial Strains and Growth Conditions
Escherichia coli strains XL1Blue and NEB 5-alpha were used as general cloning hosts and ET12567/pUB307 as the donor strain in conjugations. The E. coli strains were cultured in LB medium at 37°C, with the appropriate selection (50 μg/mL ampicillin, 50 μg/mL apramycin, 12.5 μg/mL tetracycline, 25 μg/mL kanamycin, or 34 μg/mL chloramphenicol). Spores of Streptomyces sp. ID38640 were collected from cultures grown on MS medium (20 g/L mannitol, 20 g/L soy flour, 20 g/L agar), resuspended at 109 spores/mL in 0.9% NaCl and stored at -80 °C. Spores were used for mating experiments.
Streptomyces sp. ID38640 has been deposited in the Deutsche Sammlung von Mikroorganismen und Zellkulturen patent depository collection with accession number DSMZ: DSM-26212. Streptomyces sp. ID38640, Streptomyces sp. ID38673 and the mutant strains were cultured as described (Maffioli et al., 2017).
METHOD DETAILS
DNA Isolation and Manipulations
Genetic procedures such as plasmid DNA isolation (EuroGold Plasmid Miniprep KIT - EuroClone), restriction endonuclease digestion (EcoRI-HF, BamHI-HF, XbaI, BglII – NewEngland Biolabs Inc), alkaline phosphatase treatment (NewEngland Biolabs Inc), DNA ligations (Quick Ligation Kit – NewEngland Biolabs Inc), blund-ends DNA generation [DNA Polymerase I, Large (Klenow) Fragment] and genomic DNA extraction (GenElute Bacterial Genomic DNA kit, Sigma-Aldrich) were performed according to the manufacturers’ protocols. DNA fragments were excised from agarose gels and residual agarose was removed with the GenElute Gel Extraction Kit, Sigma-Aldrich. PCR was carried out using DreamTaq Green PCR Master Mix (ThermoScientific) and PCRBIO HiFi Polymerase (PCRBiosystems) according to the manufacturers’ procedures.
Construction of Knockout Mutants
The general procedure for mutant construction is illustrated in Figure S1. It made use of pWHM3-oriT, a derivative of pWHM3 (Vara et al., 1989) kindly provided by Prof. Gilles van Wezel (Leiden University, The Netherlands). In the vector, the XbaI site was removed after restriction digestion, filling in of 5’ overhangs with DNA Polymerase I, Large (Klenow) Fragment, followed by blunt ends ligation. For disruption of a target pum gene, two fragments (A and B) of ~1.5-kbp each were amplified from genomic DNA using primers containing EcoRI and XbaI (fragment A) and XbaI and BamHI (fragment B) tails. After restriction endonuclease digestion, the fragments were cloned into the EcoRI-BamHI site of the vector. In the resulting plasmid, the XbaI fragment containing the apramycin resistance gene from pSET152 (Bierman et al., 1992) was inserted at the XbaI site within the PCR-amplified pum segment (Figure S1) to generate the knockout plasmid, which was introduced into E. coli ET12567/pUB307. The resulting strain was used as donor for conjugation with Streptomyces sp. ID38640 spores following minor modifications of described procedures (Tocchetti et al., 2013). In each mating experiment, the spore suspension was heat-shocked at 50 °C for 10 min and mixed in a 5:1 donor to recipient ratio. Then, 100 μL of the conjugation mixture were spread on MS plates supplemented with 10 mM MgCl2. The plates were incubated 16-20 hr at 30 °C and then each plate was overlaid with 3 mL LB medium containing 0.3% agar, 10 μg/mL nalidixic acid and 50 μg/mL apramycin. After about 10 days, the ex-conjugants were transferred to freshMS plates containing 50 μg/mL apramycin (Ap) and 50 μg/mL thiostrepton (Thio). After PCR confirmation of the presence of the knockout plasmid, two independent colonies were grown for 3 days in AF containing 50 μg/mL Ap and 50 μg/mL Thio, followed by three rounds of sub-culturing (5% inoculum) for 3 days in AF medium with Ap only. Then, the culture was appropriately diluted and plated on Ap-containing MS medium. Individual colonies were tested for Thio sensitivity. From each conjugation experiment, several ApR and ThioS colonies were selected and their genotype was established by PCR. Table S1 lists, for each pum gene, the primers used for constructing the knockout plasmids and for PCR analysis of the ex-conjugants.
Metabolite Analysis
Streptomyces sp. ID38640 and the mutant strains were cultured as described (Maffioli et al., 2017). Mycelium from BTT medium was inoculated in 50 mL Erlenmeyer flask containing 15 mL of seed medium (20 g/L dextrose monohydrate, 2 g/L yeast extract, 8 g/L soybean meal, 1 g/L NaCl, and 4 g/L CaCO3, pH 7.3), and incubated 72 hr at 28°C. Then, a 5-mL culture was transferred into 3x 100 mL of production medium (10 g/L dextrose monohydrate, 24 g/L maize dextrin, 8 g/L soy peptone, 5 g/L yeast extract, and 1 g/L NaCl, pH 7.2) in 500-mL buffled flasks. Where required, guanidineacetic acid, 3-guanidinopropanoic, 4-guanidinobutanoic acid, creatine, pseuduridine or uridine (Sigma-Aldrich) were added to production medium from filter-sterilized 10 mg/mL stock solutions. For metabolite analysis, 0.5 mL of the culture was centrifuged at 3000 rpm for 10 min and the supernatant was filtered through a 0.2-μM filter membrane (EuroClone) before LC-MS analysis.
HPLC analyses were performed using an YL Instrument 9300 liquid chromatographer (Young Lin Instrument Co., Anyang-si, Korea) equipped with a Waters Symmetry Shield –RP18, 5 μm, 4.6 × 250 mm column (Waters, Milford, Massachusetts, USA). Elution was performed at 1 mL/min and 40°C with a linear gradient from 0 to 12.5% phase B in 25 min. Phases A and B were 2mM heptafluorobutiryc acid in water and acetonitrile, respectively. UV detection was at 262 nm and a standard PUM solution was used for quantification.
LC-MS analyses were performed with on a Dionex UltiMate 3000 coupled with an LCQ Fleet (Thermo scientific) mass spectrometer equipped with an electrospray interface (ESI) and a tridimensional ion trap. The column was an Atlantis T3 C18 5 μm × 4.6 mm × 50 mm maintained at 40 °C at a flow rate of 0.8 mL/min. Phases A and B were 0.05% trifluoroacetic acid in water and acetonitrile, respectively. The gradient started at 0% organic phase B up to 25% in 4 minutes, followed by a 2 min-wash at 90% and a 3 min-reequilibration at0%phase B. UV-VIS signals (190-600 nm) were acquired using the diode array detector. The m/z range was 120-1500 and the ESI conditions were as follows: spray voltage of 3500 V, capillary temperature of 275 °C, sheath gas flow rate at 35 units and auxiliary gas flow rate at 15 units.
Synthesis of Pathway Intermediates
APU was prepared from commercially available β-D-pseudouridine as described (Maffioli et al., 2017). Gln-APU was prepared essentially as described (Maffioli et al., 2017), by condensing protected APU with Fmoc-protected Gln. The identity of intermediates and final product was established by LC-MS analysis: FmocGln-acetonide-APU, 6.05 min and m/z 634 [M+H]+; Gln-acetonide-APU, 4.33 min and m/z 412 [M+H]+; and Gln-APU, 1.75 min and m/z 372 [M+H]+. PUA was prepared by oxidizing acetonide-protected PU (40 mg, 0.14 mmol), prepared as described (Maffioli et al., 2017), with 2-iodoxybenzoic acid (0.21 mmol) in acetonitrile (7 mL) at 80 °C. After 2 hr, conversion was complete (monitoring by LC-MS: acetonide-protected PU, 6.05 min and m/z 285 [M+H]+; and acetonide-protected PUA, 5.57 min and m/z 283 [M+H]+). The suspension was filtered and the solvent removed under reduced pressure. The crude mixture was suspended in water (5 mL), centrifuged (16000 rpm for 2 minutes) and the pellet discarded. Trifluoroacetic acid (100 μL) was added and the solution stirred at room temperature for 1 hr until complete acetonide deprotection (PUA, at 1.32 min and m/z 243 [M+H]+). The pH was adjusted to 6 with 1M NaOH (1.5 mL) and the solvent removed under reduced pressure. The crude material was used as such for feeding experiments.
Bioinformatic Analyses
A draft genome sequence of Streptomyces sp. ID38640 was generated through Illumina technology by CeBiTec (Universität Bielefeld, Germany). Identification of biosynthetic gene clusters was performed using the antiSMASH v3.0.1 tool using default settings (Weber et al., 2015). BLAST analysis of individual CDSs was performed against the MIBiG database of known gene clusters (Medema et al., 2015) and against the Protein Databank. For identification of gene clusters containing PumJ homologs, we performed first a Blastp search of PumJ against NCBI nr, with settings of 1000 target sequences, then extracted all 1000+ genomic regions of nonredundant hits (30 kb left and right of the pumJ homolog), and performed a MultiGeneBlast search on all these genomic regions with the pum cluster as query. Next, we selected 38 homologous and interesting gene clusters from these hits, manually curated all their gene cluster borders and analyzed them further in BiG-SCAPE (https://git.wageningenur.nl/medema-group/BiG-SCAPE, Navarro-Muñoz et al., unpublished data).
QUANTIFICATION AND STATISTICAL ANALYSIS
See individual sections above for details on the statistics used for analysis.
DATA AND SOFTWARE AVAILABILITY
The software used in this study are listed in the Key Resources Table.
The sequence of the pum cluster has been deposited in the NCBI under the accession number GenBank MG266907. The sequences of the lydicamycin and desferroxamine gene clusters have been deposited in the NCBI under the accession number GenBank MG459168 and MG459167, respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli NEB-alpha | BioLabs | Cat# C2987H |
| Escherichia coli XL1Blue | Agilent/Stratagene | Cat# 200229 |
| Escherichia coli ET12567/pUB307 | Tocchetti et al., 2013 | N/A |
| Streptomyces sp. ID38640 | Maffioli et al., 2017 | DSMZ: DSM 26212 |
| Streptomyces sp. ID38673 | Maffioli et al., 2017 | N/A |
| Streptomyces ΔpumE | this paper | N/A |
| Streptomyces ΔpumG | this paper | N/A |
| Streptomyces ΔpumI | this paper | N/A |
| Streptomyces ΔpumJ | this paper | N/A |
| Streptomyces ΔpumK | this paper | N/A |
| Streptomyces ΔpumM | this paper | N/A |
| Streptomyces ΔpumN | this paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ampicillin | Fluka | Cat# 10044 |
| Apramycin sulfate | SIGMA | Cat# A2024-5G |
| chloramphenicol | SIGMA | Cat# C0378 |
| Creatine | SIGMA | Cat# 1150320 |
| Kanamycin sulfate | Fluka | Cat# 60615 |
| guanidine acetic acid | SIGMA | Cat# G11608 |
| Nalidixic acid | SIGMA | Cat# 1451000 |
| β-D-pseudouridine | Berry&Associates | Cat# PYA11080 |
| tetracycline | Fluka | Cat# 87128 |
| thiostrepton | SIGMA | Cat# T8902 |
| 3-guanidinopropanoic | SIGMA | Cat# G6868 |
| 4-guanidinobutanoic acid | SIGMA | Cat# G6503 |
| Uridine | SIGMA | Cat# U3750 |
| AminopseudoUridine | Maffioli et al., 2017 | N/A |
| Gln-APU | Maffioli et al., 2017 | N/A |
| PUA | this paper | N/A |
| Trifluoroacetic acid | SIGMA | Cat# T62200 |
| heptafluorobutiryc acid | SIGMA | Cat# 52411 |
| Acetonitrile HPLC grade | SIGMA | Cat# 39998 |
| Critical Commercial Assays | ||
| GenElute Bacterial Genomic DNA kit | SIGMA | Cat# NA2110 |
| GenElute Gel Extraction Kit | SIGMA | Cat# NA1111 |
| EuroGold Plasmid Miniprep KIT | EuroClone | Cat# EMR500200 |
| Quick Ligation Kit | NewEngland Biolabs Inc | Cat# M22005 |
| pEASY-T1 Cloning Kit | Transgen | Cat# CT101 |
| DreamTaq Green PCR Master Mix | ThermoScientific | Cat# K1082 |
| PCRBIO HiFi Polymerase | PCRBiosystems | Cat# PB 10.41-02 |
| Deposited Data | ||
| pum gene cluster | this paper | https://www.ncbi.nlm.nih.gov/nuccore/MG266907 |
| lydicamycin gene cluster | this paper | https://www.ncbi.nlm.nih.gov/nuccore/MG459168 |
| Desferroxamine gene cluster | this paper | https://www.ncbi.nlm.nih.gov/nuccore/MG459167 |
| Recombinant DNA | ||
| pWHM3-oriT | G. van Wezel, unpublished | N/A |
| pWHM3-oriT ΔXbaI | This paper | N/A |
| pSET152 | Bierman et al., 1992 | N/A |
| Software and Algorithms | ||
| antiSMASH v3.0.1 | Weber et al., 2015 | https://antismash.secondarymetabolites.org/ |
| MIBiG database | Medema et al., 2015 | https://mibig.secondarymetabolites.org/ |
| BiG-SCAPE | https://git.wageningenur.nl/medema-group/BiG-SCAPE, | |
| Other | ||
| YL 9300 HPLC | Young Lin | N/A |
| Symmetry Shield RP18, 5 μm, 4.6 × 250 mm column | Waters | Cat# WAT200670 |
| HPLC UltiMate 3000 | Dionex | N/A |
| LCQ Fleet mass spectrometer | Thermo scientific | N/A |
| Atlantis T3 C18 5 μm × 4.6 mm × 50 mm column | Waters | Cat# 186003744 |
SIGNIFICANCE.
We characterized the gene cluster responsible for the biosynthesis of pseudouridimycin, a nucleoside-analog inhibitor of bacterial RNA polymerase. This work represents the first report on the biosynthesis of a C-nucleoside antibiotic and provides important paradigms: the use of the specialized pseudouridine synthase most likely acting on a monomeric substrate; free pseudouridine as a pathway intermediate; oxidoreductase and aminotransferase acting on a C-nucleoside and unrelated to those acting on N-nucleosides; and peptide-bond formation through use of amide ligases instead of non-ribosomal peptide synthetases. In addition, we show that the pseudouridine synthase PumJ is a suitable probe to search genomes for biosynthetic gene clusters likely to specify the formation of C-nucleosides unrelated to pseudouridimycin.
In Brief.
Sosio et al. describe the biosynthetic pathway for the C-nucleoside antibiotic pseudouridimycin. Biosynthesis proceeds through formation of pseudouridine by the pseudouridine synthase PumJ, with specialized oxidoreductase, aminotransferase, and amide ligases leading to the final compound. Microbial genomes harbor diverse gene clusters encoding PumJ-related sequences.
Highlights.
Pseudouridimycin as a C-nucleoside antibiotic inhibiting bacterial RNA polymerase
First biosynthetic pathway for a C-nucleoside antibiotic elucidated
Free pseudouridine as a pathway intermediate
Dedicated pseudouridine synthase, oxidoreductases, aminotransferases, amide ligases
Acknowledgments
This work was supported by NIH grants GM041376 and AI104660 to R.H.E. and partially supported from MIUR Regione Lombardia grant 30190679 to NAICONS. We thank Gilles van Wezel for providing plasmid pWHM3-oriT and helpful advice on gene replacement and Arianna Tocchetti for valuable advice.
Footnotes
AUTHORS CONTRIBUTIONS
M. Sosio and S.D. designed work, analyzed data, and wrote the paper; E.G. and S.P. generated mutants and analyzed data; M.I. identified metabolites and analyzed data; M. Sosio and M.H.M performed bioinformatics analyses; A.B., M. Simone, and S.I.M. provided synthetic standards and analyzed data; R.H.E. provided financial support and analyzed data.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and one table and can be found with this article online at https://doi.org/10.1016/j.chembiol.2018.02.008.
DECLARATION OF INTERESTS
M. Sosio, M.I., M. Simone, S.I.M., and S.D. are employees and/or shareholders of NAICONS, which owns an intellectual property for pseudouridimycin.
References
- Barona-Gómez F, Wong U, Giannakopulos AE, Derrick PJ, Challis GL. Identification of a cluster of genes that directs desferrioxamine biosynthesis in Streptomyces coelicolor M145. J Am Chem Soc. 2004;126:16282–16283. doi: 10.1021/ja045774k. [DOI] [PubMed] [Google Scholar]
- Barona-Gómez F, Lautru S, Francou FX, Leblond P, Pernodet JL, Challis GL. Multiple biosynthetic and uptake systems mediate siderophore- dependent iron acquisition in Streptomyces coelicolor A3(2) and Streptomyces ambofaciens ATCC 23877. Microbiology. 2006;152:3355–3366. doi: 10.1099/mic.0.29161-0. [DOI] [PubMed] [Google Scholar]
- Bierman MI, Stein KL, Snyder JV. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene. 1992;116:43–49. doi: 10.1016/0378-1119(92)90627-2. [DOI] [PubMed] [Google Scholar]
- Butler MS, Blaskovich MA, Cooper MA. Antibiotics in the clinical pipeline at the end of 2015. J Antibiot. 2017;70:3–24. doi: 10.1038/ja.2016.72. [DOI] [PubMed] [Google Scholar]
- Chen W, Huang T, He X, Meng Q, You D, Bai L, Li J, Wu M, Li R, Xie Z, et al. Characterization of the polyoxin biosynthetic gene cluster from Streptomyces cacaoi and engineered production of polyoxin H. J Biol Chem. 2009;284:10627–10638. doi: 10.1074/jbc.M807534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Qi J, Wu P, Wan D, Liu J, Feng X, Deng Z. Natural and engineered biosynthesis of nucleoside antibiotics in Actinomycetes. J Ind Microbiol Biotechnol. 2016;43:401–417. doi: 10.1007/s10295-015-1636-3. [DOI] [PubMed] [Google Scholar]
- Cheng L, Chen W, Zhai L, Xu D, Huang T, Lin S, Zhou X, Deng Z. Identification of the gene cluster involved in muraymycin biosynthesis from Streptomyces sp. NRRL 30471. Mol Biosyst. 2011;7:920–927. doi: 10.1039/c0mb00237b. [DOI] [PubMed] [Google Scholar]
- De Clercq E. C-nucleosides to be revisited. J Med Chem. 2016;59:2301–2311. doi: 10.1021/acs.jmedchem.5b01157. [DOI] [PubMed] [Google Scholar]
- Ericsson UB, Andersson ME, Engvall B, Nordlund P, Hallberg BM. Expression, purification, crystallization and preliminary diffraction studies of the tRNA pseudouridine synthase TruD from Escherichia coli. Acta Crystallogr D Biol Crystallogr. 2004;60:775–776. doi: 10.1107/S0907444904003506. [DOI] [PubMed] [Google Scholar]
- Fawaz MV, Topper ME, Firestine SM. The ATP-grasp enzymes. Bioorg Chem. 2011;39:185–191. doi: 10.1016/j.bioorg.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa G, Dziewanowska K, Stone H, Settles M, Hartzell P. Global analysis of phase variation in Myxococcus xanthus. Mol Microbiol. 2011;81:784–804. doi: 10.1111/j.1365-2958.2011.07732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furumai T, Eto K, Sasaki T, Higuchi H, Onaka H, Saito N, Fujita T, Naoki H, Igarashi Y. TPU-0037-A, B, C and D, novel lydicamycin congeners with anti-MRSA activity from Streptomyces platensis TP-A0598. J Antibiot (Tokyo) 2002;55:873–880. doi: 10.7164/antibiotics.55.873. [DOI] [PubMed] [Google Scholar]
- Hamma T, Ferré-D’Amaré AR. Pseudouridine synthases. Chem Biol. 2006;13:1125–1135. doi: 10.1016/j.chembiol.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Holmes TC, May AE, Zaleta-Rivera K, Ruby JG, Skewes-Cox P, Fischbach MA, DeRisi JL, Iwatsuki M, Ōmura S, Khosla C. Molecular insights into the biosynthesis of guadinomine: a type III secretion system inhibitor. J Am Chem Soc. 2012;134:17797–17806. doi: 10.1021/ja308622d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Al-Mestarihi A, Grimes CL, Kahne D, Bachmann BO. A unifying nitrososynthase involved in nitrosugar biosynthesis. J Am Chem Soc. 2008;130:15756–15757. doi: 10.1021/ja8051415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers MM, Montersino S, Westphal AH, Tischler D, van Berkel WJ. Flavin dependent monooxygenases. Arch Biochem Biophys. 2014;544:2–17. doi: 10.1016/j.abb.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Komaki H, Ichikawa N, Hosoyama A, Fujita N, Igarashi Y. Draft genome sequence of marine-derived Streptomyces sp. TP-A0598, a producer of anti-MRSA antibiotic lydicamycins. Stand Genomic Sci. 2015;10:58. doi: 10.1186/s40793-015-0046-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova E, Proudfoot M, Gonzalez CF, Brown G, Omelchenko MV, Borozan I, Carmel L, Wolf YI, Mori H, Savchenko AV, et al. Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J Biol Chem. 2006;281:36149–36161. doi: 10.1074/jbc.M605449200. [DOI] [PubMed] [Google Scholar]
- Li W, Csukai M, Corran A, Crowley P, Solomon PS, Oliver RP. Malayamycin, a new streptomycete antifungal compound, specifically inhibits sporulation of Stagonospora nodorum (Berk) Castell and Germano, the cause of wheat glume blotch disease. Pest Manag Sci. 2008;64:1294–1302. doi: 10.1002/ps.1632. [DOI] [PubMed] [Google Scholar]
- Li X, Ma S, Yi C. Pseudouridine: the fifth RNA nucleotide with renewed interests. Curr Opin Chem Biol. 2016;33:108–116. doi: 10.1016/j.cbpa.2016.06.014. [DOI] [PubMed] [Google Scholar]
- Li Y, Zeng H, Tan H. Cloning, function, and expression of sanS: a gene essential for nikkomycin biosynthesis of Streptomyces ansochromogenes. Curr Microbiol. 2004;49:128–132. doi: 10.1007/s00284-004-4260-4. [DOI] [PubMed] [Google Scholar]
- Ma C, Yang X, Lewis PJ. Bacterial transcription as a target for antibacterial drug development. Microbiol Mol Biol Rev. 2016;80:139–160. doi: 10.1128/MMBR.00055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffioli SI, Zhang Y, Degen D, Carzaniga T, Del Gatto G, Serina S, Monciardini P, Mazzetti C, Guglierame P, Candiani G, et al. Antibacterial nucleoside-analog inhibitor of bacterial RNA polymerase. Cell. 2017;169:1240–1248.e23. doi: 10.1016/j.cell.2017.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Kottmann R, Yilmaz P, Cummings M, Biggins JB, Blin K, de Bruijn I, Chooi YH, Claesen J, Coates RC, et al. Minimum information about a biosynthetic gene cluster. Nat Chem Biol. 2015;11:625–631. doi: 10.1038/nchembio.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monciardini P, Iorio M, Maffioli SI, Sosio M, Donadio S. Discovering new bioactive molecules from microbial sources. Microb Biotechnol. 2014;7:209–220. doi: 10.1111/1751-7915.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu G, Tan H. Nucleoside antibiotics: biosynthesis, regulation, and biotechnology. Trends Microbiol. 2015;23:110–119. doi: 10.1016/j.tim.2014.10.007. [DOI] [PubMed] [Google Scholar]
- Paitan Y, Orr E, Ron EZ, Rosenberg E. A NusG-like transcription anti-terminator is involved in the biosynthesis of the polyketide antibiotic TA of Myxococcus xanthus. FEMS Microbiol Lett. 1999;170:221–227. doi: 10.1111/j.1574-6968.1999.tb13377.x. [DOI] [PubMed] [Google Scholar]
- Palmu K, Rosenqvist P, Thapa K, Ilina Y, Siitonen V, Baral B, Mäkinen J, Belogurov G, Virta P, Niemi J, Metsä-Ketelä M. Discovery of the showdomycin gene cluster from Streptomyces showdoensis ATCC 15227 yields insight into the biosynthetic logic of C-nucleoside antibiotics. ACS Chem Biol. 2017;12:1472–1477. doi: 10.1021/acschembio.7b00078. [DOI] [PubMed] [Google Scholar]
- Rackham EJ, Grüschow S, Ragab AE, Dickens S, Goss RJM. Pacidamycin biosynthesis: identification and heterologous expression of the first uridyl peptide antibiotic gene cluster. ChemBioChem. 2010;11:1700–1709. doi: 10.1002/cbic.201000200. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Talaue M, Liu S, Degen D, Ebright RY, Sineva E, Chakraborty A, Druzhinin SY, Chatterjee S, Mukhopadhyay J, et al. New target for inhibition of bacterial RNA polymerase: ‘switch region’. Curr Opin Microbiol. 2011;14:532–543. doi: 10.1016/j.mib.2011.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocchetti A, Maffioli SI, Iorio M, Alt S, Mazzei E, Brunati C, Sosio M, Donadio S. Capturing linear intermediates and C-terminal variants during maturation of the thiopeptide GE2270. Chem Biol. 2013;20:1067–1077. doi: 10.1016/j.chembiol.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Trautman EP, Crawford JM. A new nucleoside antibiotic chokes bacterial RNA polymerase. Biochemistry. 2017;56:4923–4924. doi: 10.1021/acs.biochem.7b00680. [DOI] [PubMed] [Google Scholar]
- Vara J, Lewandowska-Skarbek M, Wang YG, Donadio S, Hutchinson CR. Cloning of genes governing the deoxysugar portion of the erythromycin biosynthesis pathway in Saccharopolyspora erythraea (Streptomyces erythreus) J Bacteriol. 1989;171:5872–5881. doi: 10.1128/jb.171.11.5872-5881.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CT, Zhang W. Chemical logic and enzymatic machinery for biological assembly of peptidyl nucleoside antibiotics. ACS Chem Biol. 2011;6:1000–1007. doi: 10.1021/cb200284p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M, Goss RJ, Kimura K, Bugg TD. Antimicrobial antibiotics targeting cell wall assembly: recent advances in structure-function studies and nucleoside biosynthesis. Nat Prod Rep. 2010;27:279–304. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]
- Wu X, Flatt PM, Xu H, Mahmud T. Biosynthetic gene cluster of cetoniacytone A, an unusual aminocyclitol from the endosymbiotic bacterium Actinomyces sp. Lu 9419. ChemBioChem. 2009;10:304–314. doi: 10.1002/cbic.200800527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Ostash B, Walsh CT. Identification of the biosynthetic gene cluster for the pacidamycin group of peptidyl nucleoside antibiotics. Proc Natl Acad Sci USA. 2010;107:16828–16833. doi: 10.1073/pnas.1011557107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.