

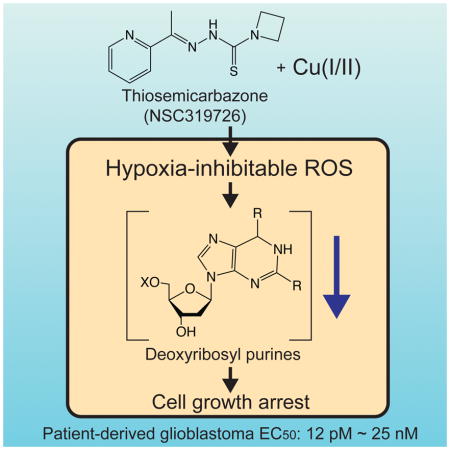
Summary
Transition metals are essential, but deregulation of their metabolism causes toxicity. Here, we report that the compound NSC319726 binds copper to induce oxidative stress and arrest glioblastoma-patient-derived cells at picomolar concentrations. Pharmacogenomic analysis suggested that NSC319726 and 65 other structural analogs exhibit lethality through metal binding. Although NSC319726 has been reported to function as a zinc ionophore, we report here that this compound binds to copper to arrest cell growth. We generated and validated pharmacogenomic predictions: copper toxicity was substantially inhibited by hypoxia, through a HIF-1α–dependent pathway; copper-bound NSC319726 induced the generation of reactive oxygen species and depletion of deoxyribosyl purines, resulting in cell cycle arrest. These results suggest that metal-induced DNA damage may be a consequence of exposure to some xenobiotics, therapeutic agents, as well as other causes of copper dysregulation, and reveal a potent mechanism for targeting glioblastomas.
eTOC Blurb
Shimada et al. report that the compound NSC319726 arrests glioblastoma-patient-derived cells at picomolar concentrations. The compound binds to copper, generates ROS using ambient oxygen, and depletes nucleotide pools. This represents a new strategy for potently blocking the growth of glioblastoma.
Introduction
Transition metals play essential and diverse roles in cell biology (Dudev and Lim, 2008; Waldron and Robinson, 2009; Williams, 2014). Redox-inactive metals such as sodium and potassium signal by changing their distributions quickly, while redox-active metals such as iron embedded within protein active sites act as cofactors to aid in catalytic functions (Chang, 2015). Despite their critical role, excess abundance of transition metals can be toxic to cells. As a result, cellular abundance of transition metals is tightly regulated. Copper is the third most abundant transition metal in biological systems, after iron and zinc. Copper is a redox-active metal like iron, and this property is exploited by many biological processes, such as cytochrome c oxidase, superoxide dismutase, and tyrosinase (Malmström and Leckner, 1998; Solomon et al., 1992). Copper also controls lipolysis in adipocytes by regulating the activity of cAMP-degrading enzyme phosphodiesterase PDE3B (Krishnamoorthy et al., 2016). Also, deregulation of copper metabolism can cause pathologies. For example, genetic diseases such as Wilson’s disease or treatment with some xenobiotic compounds leads to copper accumulation, which causes toxicity, in some cases by triggering oxidative stress (Gaetke and Chow, 2003; Uriu-Adams and Keen, 2005).
In this study, we discovered that a thiosemicarbazone NSC319726 induces copper-dependent cell cycle arrest at picomolar concentrations in glioblastoma (GBM) cells. Using pharmacogenomic and molecular and cell biology approaches, we found that this compound functions in GBM cells as a copper ionophore. Copper bound to NSC319726 causes the generation of reactive oxygen species (ROS), and induces G1 cell cycle arrest.
Results
NSC319726 is a potent growth inhibitor of cancer cells
GBM is one of the most common and lethal cancers (Dunn et al., 2012). To identify mechanisms for treating this intractable cancer, we performed small molecule screening in 13 primary tumor-derived models from GBM patients using stem cell culture conditions in a high-throughput assay format (Quartararo et al., 2015). We discovered that NSC319726 is an extremely potent growth inhibitor, with EC50 ranging from 12 pM to 25 nM across these models (Table 1). NSC319726 induces a cytostatic effect not only in GBM primary-tumor-derived samples, but also in HT-1080 fibrosarcoma cells, albeit at lower potency, indicating a potentially broad spectrum of effectiveness. Since HT-1080 cells grow rapidly and are more amenable to diverse assay formats compared to GBM lines, we initially characterized the mechanism of action of NSC319726 in this cell line. We synthesized the compound, which showed equivalent potency to commercially available batches, confirming the identity of the compound (Fig. S1A–C).
Table 1.
Sensitivity to NSC319726 and TP53 mutation status of patient-derived GBM cells
| GBM line | EC50 (nM) | TP53 mutation |
|---|---|---|
| HF3037 | 0.012 | WT |
| HF2998 | 0.012 | WT |
| HF2885 | 0.014 | WT |
| HF2906 | 0.040 | R175H |
| HF3026 | 0.042 | WT |
| HF2381 | 0.046 | WT |
| HF2876 | 0.095 | WT |
| HF3013 | 0.130 | V272M |
| HF2476 | 0.197 | WT |
| HF2303 | 0.959 | G245S |
| HF3177 | 1.747 | M133T |
| HF2790 | 6.791 | C242F |
| HF2941 | 24.960 | WT |
NSC319726 was originally discovered as a selective lethal compound for cancer cell lines carrying allele specific TP53 mutation, R175H (Yu et al., 2014; Lin et al., 2015). The gain-of-function R175H mutation is one of the most frequent mutations observed in TP53 gene, contributing to various properties of transformed cancers, such as invasion, proliferation, drug resistance (Soussi and Wiman, 2007). p53R175H protein has decreased binding affinity to Zn(II), which is required for DNA-binding, resulting in a change in protein conformation (Xu et al., 2011). NSC319726 was reported to function as zinc-metallochaperone (and thereby named ZMC1), increasing the intracellular Zn(II) pool to correct the p53R175H protein conformation to that of wild-type p53. It was found that cancers carrying p53R175H rely on the “acquired functions” of the mutant protein for its survival, thus NSC319726 kills these addicted cells. Since the binding affinity of p53R175H to Zn(II) is diminished, the mutant protein lacks Zn(II), which is required for DNA-binding. NSC319726 was shown to facilitate the binding of zinc to p53R175H by acting as a zinc ionophore, which enables the mutant protein to recover its wild-type conformation (Yu et al., 2012, 2014).
NSC319726 acts as zinc-metallochaperone in cancers carrying p53R175H. Knockdown of TP53 in these cells block the compound’s efficacy dramatically, strongly indicating the role of correctly folded p53 involved in the mechanism of NSC319726 in these cells. However, other cancer cells carrying wild-type or different mutations of p53 exhibit sensitivity to NSC319726, and the compound’s mechanism of action against these cells is not clear. Therefore, we sought to define the mechanism of action of this compound in these contexts.
First, we explored the hypothesis that NSC319726 might cause zinc accumulation or interfere with p53 activity in GBM and HT-1080 cells. While Zn(II) supplementation slightly increased the compound’s efficacy in these contexts, treatment with a specific Zn(II) chelator, N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine (TPEN) (Verhaegh et al., 1998), did not substantially change its potency or efficacy in HT-1080 cells or GBM models, suggesting that the effect of NSC319726 is largely independent of binding to Zn(II) in these cell models, while Zn(II) supplementation slightly potentiated the effect of the compound (Fig. 1A). Moreover, we did not observe a connection between TP53 mutation status and the sensitivity to NSC319726 across the 13 GBM models (Table 1). Although the latter analysis was performed with too few cells to conclusively find associations between genotype and phenotype, we were motivated to seek additional mechanisms underlying the cytostatic effect of the compound in these cell types.
Figure 1. NSC319726 is a potent growth inhibitor of cancer cells.

(A) The effect of zinc supplementation and chelation on sensitivity to NSC319726. (B) Structures of NSC319726 and similar previously reported nonapoptotic cell-death inducers. (C) The effect of 100 μM Co(II) on NSC319726 toxicity. Error bars in (A) and (C) indicate SEM of technical triplicates. See also Figure S1a-d.
Examination of the structure of NSC319726 revealed that it is similar to three cytotoxic compounds that induce a non-apoptotic form of cell death (Shimada et al., 2016a) (Fig. 1B). These compounds were discovered in a screen specifically for lethal molecules that induce regulated, non-apoptotic cell death (Shimada et al., 2016a). Their lethality in cancer cell lines was inhibited by binding to Co(II), although their mechanism of action was not fully elucidated. Due to the structural similarity, we suspected that NSC319726 might share a similar mechanism of action. Intriguingly, NSC319726’s cytostatic effect was strongly inhibited by supplementation with 100 μM Co(II), which is similar to the effect observed with these three reported compounds (Fig. 1C). Co(II) is an essential transition metal, widely known for its function in a cofactor vitamin B12. However, as holo-B12 supplementation did not suppress NSC319726’s lethality, depletion of Co(II) from B12 is likely not the cause of NSC319726’s cytotoxic effect (Fig. S1D).
Three distinct compound scaffolds share similar lethal mechanisms
While NSC319726 exhibited picomolar potency in some GBM patient-derived models, the difference in potency between the 13 cell lines was >2,000 fold. We previously studied a publicly available large pharmacogenomic dataset to identify biomarkers predicting drug sensitivity, and reported that lethal compounds whose potency vary across different cells were informative for study of molecular mechanisms (Shimada et al., 2016b).
The NCI-60 dataset contains basal gene expression profiles of 60 cell lines, as well as drug sensitivity for 6,529 compounds (Shoemaker, 2006). Of these, we focused on 2,565 cell-line-selective compounds, clustered them into 18 groups based on the similarity of the drug sensitivity across 60 cell lines, and found that each cluster represents distinct mechanisms (e.g., DNA targeting agents, tyrosine kinase inhibitors, and ferroptosis) (Shimada et al., 2016b). NSC319726 was included in the 2,565 cell line-selective compounds tested in the NCI-60 dataset, and these analyses revealed that NSC319726 clustered together with 82 other compounds (Fig. 2A). While these 83 compounds were clustered based on the similarity of the sensitivity profile across cell lines, the compounds in this cluster also share substantial structural similarities and are characterized by four core scaffolds (Fig. 2B). The first two classes contain a semicarbazone and a heteroatom (either nitrogen or sulfur), suggesting that these scaffolds bind to metal ions. Notably, NSC319726 and three structural analogs are also nitro- or thio-semicarbazones (Fig. 2B). As for the latter two classes, 8-hydroxyquinoline compounds are known to form metal complexes (Prachayasittikul et al., 2013), while phenothiazines are not reported to bind to metals. Whether these compounds are capable of binding to metals or not, these 83 compounds were suspected to induce similar lethality compared to the other 2,565 cell-line-selective lethal compounds, based on their growth inhibition (GI50) profiles. Among these, thio-/nitro-semicarbazones were more potent than 8-hydroxyquinolines or phenothiazines, and notably, NSC319726 was one of the most potent compounds (Fig. 2C).
Figure 2. NSC319726, 8-hydroxyquinoline, and phenothiazine share similar mechanisms of action against cancer cells.

(A) Left: relative GI50 profiles across the NCI-60 panel against 18 compound clusters representing distinct mechanisms of action of 2,565 cell-line-selective lethal compounds. Right: relative GI50 profiles of 83 compounds in the cluster represented by NSC319726. (B) Four core scaffolds represented in the same cluster as NSC319726. (C) Median sensitivity of the 83 compounds across the NCI-60 panel. See also Figure S2 for the 83 compounds’ structures.
NSC319726 induces copper-dependent toxicity
The NCI-60 data analysis suggested that semicarbazones, including NSC319726, bind to metal ions to induce lethality in cancer cells. We sought to identify the metal species responsible for this effect. Thiosemicarbazones have been long studied as a potential anticancer agent, and their binding to transition metal ions, particularly iron, zinc, and copper, is suggested to be the key for their lethal effect (Crim and Petering, 1967; Sartorelli et al., 1977). To find out if this is the case, we assessed if any specific metal-binding is important for semicarbazones. A cell-permeable metal-binding compound acting as an ionophore decreases the concentration gradient across the plasma membrane. It may deplete or overload the metal, depending on the ratio between its intracellular and extracellular concentrations. In either scenario, supplementation of the corresponding metal ion should alter the effect of NSC319726. We co-treated HT-1080 cells with NSC319726 and various metal ions in a concentration series, and found that only Cu(II) dramatically enhanced the growth inhibitory effect of the compound. This effect of Cu(II) was diminished by the addition of Co(II) (Fig. 3A,B). While Fe(II) and Fe(III) supplementation also induced growth inhibition that was inhibited by Co(II) co-treatment (Fig. 3A), and the iron-specific chelator deferoxamine suppressed NSC319726’s effect (Fig. S1E), the effect of iron supplementation was independent of NSC319726. Moreover, iron supplementation was previously found to be protective against NSC319726 in TOV112D cells, similar to Co(II) supplementation in HT-1080 cells (Yu et al., 2012). Due to this idiosyncratic effect of iron in different cells and the compound-independent nature of Co(II) inhibition of iron toxicity in HT-1080 cells, we concluded that iron toxicity was not the key mechanism of NSC319726 lethality in GBM and HT-1080 cells.
Figure 3. NSC319726 induces copper toxicity.

(A) The effect of various metal supplementation on the sensitivity to NSC319726 was seen in HT-1080 cells. (B) The effect of Cu(II) and/or Co(II) supplementation on HT-1080 cells’ sensitivity to NSC319726. Color code is the same as (A). In (A) and (B), testing metals were treated at 2-fold dilution series with 100 μM as highest concentration while Co(II) and NSC319726 were co-supplemented at fixed concentrations, 100 μM and 31 nM, respectively. (C) The effect of Cu(II) modulation on the sensitivity to NSC319726, 7Br8HQ, and MB. (D) Intracellular metal quantification upon NSC319726/7Br8HQ/MB treatment. (E) Intracellular metal quantification upon NSC319726 and/or Co(II) supplementation. (F) Time-course measurement of copper and zinc for 0, 3, 9, 24 h. Values in (D)-(F) were normalized to inorganic phosphate quantity of the same samples. Error bars in (B) and (C) are technical triplicates, (D)-(F) are SEM of biological triplicates. See also Figure S1e.
To ensure that NSC319726’s interaction with copper was relevant to its lethality, we treated cells with a dilution series of NSC319726 in media supplemented with either Cu(II), in the absence or presence of the Cu(II)–specific chelator, triethylenetetramine (TETA) (Fig. 3C) (Lu, 2010). Consistent with the hypothesis that NSC319726 binds copper to induce its growth inhibitory effect, the copper chelator reversed the synergy between Cu(II) and NSC319726. Two other compounds were tested in similar conditions: 7-bromo-8-hydroxyquinoline (7Br8HQ) and methylene blue (MB), representing the 8-hydroxyquinoline and phenothiazine scaffolds, respectively. 7Br8HQ was potentiated by Cu(II) and unchanged by TETA; MB was unchanged by either (Fig. 3C).
To evaluate the relationship between these three compounds and the cellular accumulation of metals, we directly measured the abundance of iron, copper, and zinc in HT-1080 cells using inductively-coupled plasma mass spectrometry (ICP-MS). Consistent with previous and earlier findings, Cu(II) and Zn(II) were significantly increased upon treatment with either NSC319726 or 7Br8HQ, whereas treatment with MB had no effect. Moreover, NSC319726 specifically increased cellular copper abundance, while 7Br8HQ increased the level of all three metal ions. Taken together, these findings indicate that NSC319276 can function as a specific copper and zinc ionophore, 7Br8HQ is a non-specific transition metal ionophore, and MB is not a metal chelator (Fig. 3D). The increase in copper and zinc abundance was suppressed by the addition of Co(II), suggesting that NSC319726 binds to Co(II) when the latter is present in excess (Fig. 3E). Copper levels significantly increased as quickly as 3 h after NSC319726 treatment, and steadily increased up to 24 h post treatment (Fig. 3F).
The metal quantification and cell viability assays after perturbation of metal content suggest a couple of important features: NSC319726 specifically binds to both copper and zinc, of which copper is specifically responsible for NSC319726’s cytotoxicity; 7Br8HQ binds to several transition metals; the effect of MB is independent of copper. Second, NSC319726 inhibited cell growth and killed cells completely when Cu(II) was further added to the cell culture while 7Br8HQ induced complete lethality without copper supplementation. This discrepancy is possibly due to the difference in metal binding specificity of the two molecules. Metals that 7Br8HQ binds to but NSC319726 does not, such as iron, may induce complete lethality achieved by 7Br8HQ. Thus, 7Br8HQ seems to induce two mechanisms of lethality: a copper-dependent mechanism that is comparable to NSC319726’s toxicity and a copper-independent mechanism that is orthogonal to NSC319726’s toxicity.
Transcriptome analysis uncovered gene signatures of sensitivity and resistance to NSC319726
In a prior NCI-60 analysis, we found that drug sensitivity profiles are indicative of mechanisms of action and that the transcriptome can be used to successfully discover biomarkers of sensitivity (Shimada et al., 2016b). Using the same approach, we looked for pathways (entries in Gene Ontology, KEGG, Reactome, BioCarta, and ‘hallmarks’ in MSigDB (Subramanian et al., 2005)) that serve as biomarkers for the sensitivity of cells to compounds in the cluster containing NSC319726 (Fig. S2A). We discovered pathways that are significantly upregulated in cell lines sensitive or resistant to this cluster (high significance, corresponding to the x-axis of Fig. S2B), which are also uniquely associated with this cluster among the 18 clusters shown in Fig. 2A (high specificity, corresponding to y-axis of Fig. S2B).
We then measured gene expression changes after 24 h of 10 μM NSC319726 treatment in the most sensitive (HF3037) and resistant (HF2941) GBM patient-derived cell models. We asked if any of the pathways with high significance and high specificity in the NCI-60 dataset were up- or down-regulated in a meaningful way. We assumed that if genes in a pathway overexpressed in NSC319726-resistant vs sensitive cells in the NCI-60 panel were also upregulated in GBM cells upon NSC319726 treatment, the pathway is likely to contribute to cellular resistance to NSC319726. We found two pathways satisfying these criteria, namely ‘TNFα signaling via NF-κB’ and ‘Hypoxia’ (Fig. 4A, Fig. S2C,D). These two pathways were also more upregulated in resistant GBM cells (HF2941) than sensitive ones (HF3037) in the basal state in both the NCI-60 and the GBM cell panels, but they were also even more expressed upon NSC319726 treatment in the two GBM cells; thus, we suspected that these pathways were functionally relevant to the mechanism that cells use to acquire resistance to NSC319726. We then evaluated how hypoxia might contribute to resistance further.
Figure 4. HIF-1α-dependent response to hypoxia induces resistance to NSC319726.

(A) Summary of the pathway enrichment analysis. (B) Effect of hypoxia on NSC319726-induced lethality. Error bars are SEM from sigmoidal curve fitting of a representative experiment. (C) Effect of siHIF-1α on HIF-1α protein level (left) and cellular sensitivity to NSC319726. (D) Chemical induction of HIF-1α and cellular sensitivity to NSC319726. (E) Schematic relationship between NSC319726 and hypoxia. Error bars in (C) and (D) indicate SEM of technical triplicates. See also Figures S3–4.
Hypoxia suppresses NSC319726 through a HIF-1α dependent pathway
We first tested whether hypoxia made cells resistant to NSC319726 in four GBM models. Cells grown in hypoxia (1.5% O2) were more resistant to NSC319726 than cells grown in normoxia (ambient, 21% O2) by 100-fold in all four cell models tested (Fig. 4B, Fig. S3A). As the transcriptional response to hypoxia is regulated in part by hypoxia-inducible factor 1α (HIF-1α), and copper can stabilize HIF-1α by inhibiting its prolyl hydroxylation and subsequent degradation (Martin et al., 2005), we suspected that HIF-1α stabilization might provide a cellular defense mechanism to NSC319726. A potent inhibitor of the NSC319726 lethality, Co(II), also stabilizes HIF-1α by inhibiting the interaction between HIF-1α and von Hippel-Lindau, an E3 ligase ubiquitylating and promoting HIF-1α degradation in response to oxygen, which may also contribute to the resistance (Yuan et al., 2003). We confirmed that HIF-1α protein was significantly stabilized upon both NSC319726 and Co(II) treatment, supporting the hypothesis that HIF-1α may mediate cellular responses to NSC319726 (Fig. S3B).
To explore more directly the role of HIF-1α in response to NSC319726, HIF-1α expression was inhibited by siRNA knockdown in HT-1080 cells. Inhibition of HIF-1α expression restored sensitivity to NSC319726 under hypoxic conditions, explaining the resistance previously observed (Fig. 4C). However, HIF-1α mediated resistance was achieved only in hypoxic conditions because HIF-1α knockdown marginally sensitizes cells to NSC319726 in normoxic conditions, and treatment of dimethyloxalylglycine (DMOG), a compound that stabilizes HIF-1α in normoxic conditions, did not alter NSC319726 sensitivity under normoxia (Fig. 4D, Fig. S3B). These results indicate that the extent of the stimuli that NSC319726 triggers leading to cytostasis vary between hypoxia and normoxia; although HIF-1α tries to protect cells from NSC319726, the lethal stimuli under normoxia are greater than what stabilized HIF-1α can handle (Fig. 4E).
NSC319726 induces reactive oxygen species leading to cell cycle arrest
To further explore what causes cytostasis upstream of HIF-1α upon NSC319726 treatment, we re-analyzed gene expression changes in two GBM cell models treated with 10 μM NSC319726. By comparing their expression profiles with those from 1,698 experiments from chemical and genetic perturbations collected in the Molecular Signatures Database (MSigDB), we found that NSC319726 treatment of HF3037 and HF2941 GBM cell models induced gene expression profiles similar to T24 bladder cancer cells treated with hypericin-mediated photodynamic therapy (Fig. S4) (Buytaert et al., 2007). Photodynamic therapy is chemotherapy consisting of the administration of photosensitizing chemicals such as hypericin. The photosensitizing compounds, preferentially taken up by cancers, absorb near infrared light and transfer its energy to oxygen molecules, generating singlet oxygen that kills cells (Karioti and Bilia, 2010).
Based on the similar transcriptome profiles, we hypothesized that NSC319726 generates ROS, comparable to photodynamic therapy. Semicarbazones were previously reported to bind to and activate copper, facilitating redox cycling between Cu(I) and Cu(II), leading to more efficient ROS generation (Jansson et al., 2010); we suspected that NSC319726-bound copper was more susceptible to redox changes. Notably, NSC319726-bound copper propagated ROS from hydrogen peroxide in a cell free solution more efficiently than the free unbound form (Fig. 5A, Fig. S5A), supporting this hypothesis.
Figure 5. NSC319726 induces reactive oxygen species generation.
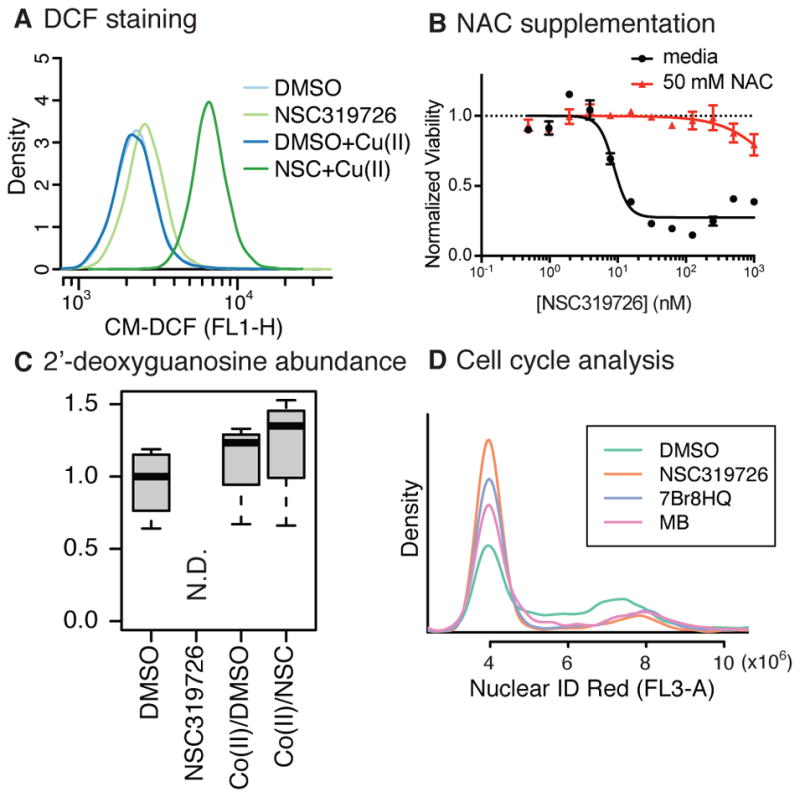
(A) ROS measurement using CM-H2DCFDA upon NSC319726 and/or Cu(II) treatment. (B) The effect of N-acetylcysteine (NAC) supplementation on the lethality of NSC319726. (C) Abundance of 2′-deoxyguanosine upon NSC319726 and/or Co(II) supplementation. (D) Cell cycle analysis using Nuclear ID Red staining upon treatment of NSC319726/7Br8HQ/MB. Error bars in (B) indicate SEM of technical triplicates. See also Figures S5–8.
To determine which ROS are involved in this cytotoxicity, cells were treated with both NSC319726 and one of several antioxidants quenching different ROS; we found that N-acetylcysteine rescued cells at 50 mM. While N-acetylcysteine is not a very specific scavenger, other antioxidants (100 μM trolox for lipophilic ROS, and 100 μM 4-hydroxy-TEMPO for superoxide) showed little effect on NSC319726 lethality, indicating that lipophilic hydroxyl radicals and superoxide were likely not involved (Fig. S5B,C). Moreover, a pan-caspase inhibitor zVAD or a necroptosis inhibitor Necrostatin-1 did not suppress the cytotoxic effect of NSC319726, indicating that known regulated cell death phenotypes, such as apoptosis or necroptosis, were not involved (Fig. S5D). Intriguingly, other scaffolds, 7Br8HQ and MB, also induce ROS generation detectable by CM-H2DCFDA, and suppressed by 50 mM N-acetylcysteine (Fig. 5B, Fig. S5C–E). These shared features suggest the three compounds evoke ROS-induced cytostasis through a similar mechanism, and explain why these molecules co-segregated in the clustering analysis.
To further understand the implication of NSC319726-mediated ROS production on cellular metabolism, and to glean insight into how these changes could result in cell cycle arrest, we performed untargeted metabolomic profiling of cells treated with DMSO, NSC319726, DMSO/Co(II), and NSC319726/Co(II) for six hours. Across these four conditions, only NSC319726 treatment induced lethality. Among the 505 metabolites identified and quantified, we looked for metabolites whose abundance was changed significantly over the other conditions, and the analysis revealed that purine deoxyribonucleosides/deoxyribonucleotides, namely, 2′-deoxyadenosine and its phosphate conjugates, 2′-deoxyguanosine, and 2′-deoxyinosine were completely depleted upon NSC319726 treatment (Fig. 5C, Fig. S6) while pyrimidine deoxyribonucleosides and purine ribonucleosides/ribonucleotides were unaffected. There are two possibilities explaining this observation: first, purine bases were selectively oxidized by NSC319726-copper complex, consistent with prior works showing 8-oxodeoxyguanosine formation upon NSC319726 treatment (Angelé-Martínez et al., 2014; Yu et al., 2014). However, this cannot explain why deoxyribosyl-purines were only depleted while ribosyl-purines were intact. The second possibility is that ribonucleotide reductase (RNR) activity is inhibited by NSC319726. RNR is an enzyme catalyzing the formation of deoxyribonucleotides from ribonucleotides. Triapine, a thiosemicarbazone, was reported to inhibit RNR activity (Finch et al., 2000). Whatever the mechanisms of deoxyribosyl-purine depletion may be, we found G1 cell cycle arrest in HT-1080 cells 24 h after NSC319726 treatment. As expected from NCI-60 analysis, 7Br8HQ and MB treatments also induced G1-arrest in HT-1080 cells after 24 h (Fig. 5D), possibly induced by the same ROS as generated by NSC319726 treatment.
NSC319726 does not exhibit neurotoxicity in differentiated SH-SY5Y cells
One major concern toward clinical application for GBM treatment is neurotoxicity. Thus, we utilized SH-SY5Y human neuroblastoma cell model to assess neurotoxicity of NSC319726 (Fig. S7). Undifferentiated SH-SY5Y cells were more sensitive to all four lethal compounds with distinct mechanisms compared to the differentiated cells. Among them, a non-specific kinase inhibitor staurosporine was only 4-fold selective between undifferentiated and differentiated cells, but DNA damaging agents, i.e., NSC319726 and type-II topoisomerase inhibitor doxorubicin, were much more selective and differentiated cells did not show substantial toxicity up to the highest tested concentrations. Selective lethality in GBM and neuroblastoma over differentiated and non-growing neurons is a desired characteristic of NSC319726 for an anti-GBM agent.
Discussion
Although thiosemicarbazones have been long recognized as metal binding molecules and potential anticancer agents, their mechanism of action is complicated because abundant transition metals in cells, i.e., iron, zinc, and copper, seem all relevant in their mechanisms. However, the fact that various thiosemicarbazones showed similar drug sensitivity profiles in NCI-60 cell line panel indicates that their mechanism of action is similar overall. In this study, we focused on characterizing the mechanism of action of one of the most potent, NSC319726. NSC319726 functions as a copper ionophore that generates ROS to deplete deoxyribosyl-purines and induce growth inhibition in multiple cancer cell models (Fig. 6).
Figure 6. Model of NSC319726–induced toxicity.
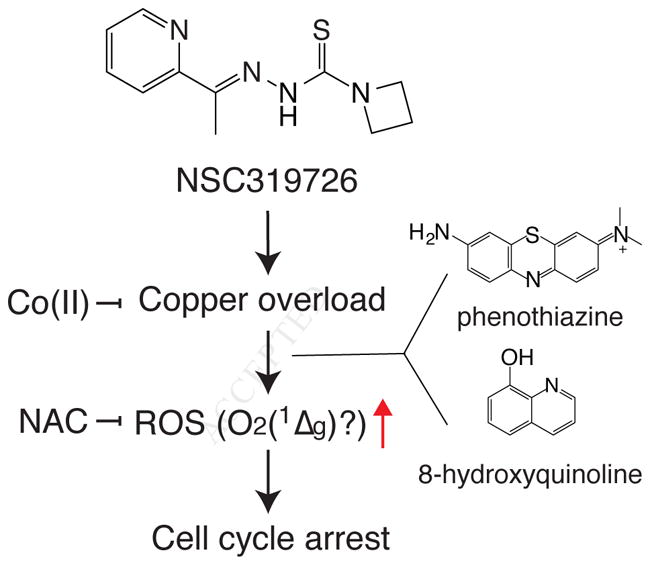
Current model of NSC319726’s mechanism of action. NSC319726 binds to copper to induce ROS, possibly singlet oxygen, and arrests cell cycle. While two other scaffolds, 8-hydroxyquinoline and phenothiazine, do not generate ROS through copper binding, they also generate NAC inhibitable ROS to induce cell cycle arrest.
To identify molecular pathways involved in NSC319726’s lethality, we adopted a pharmacogenomic approach. While large pharmacogenomic efforts such as the NCI-60 project are often considered noisy and inconsistent, we previously showed that proper filtering of the data provides useful signals (Shimada et al., 2016b). In this study, we illustrated how such data can characterize the mechanism of action of a lethal compound. Combining the NCI-60 data with RNA-seq experiments to detect gene expression changes upon NSC319726 treatment, we identified biomarkers to sensitivity, which are upregulated. This approach can serve to identify gene sets that are not only biomarkers, but functionally relevant to mechanisms of action. Our discovery of HIF-1α making cell resistant to thiosemicarbazone-induced cytotoxicity has not been reported before, however, structurally related bis(thiosemicarbazone) complexed with copper(II) was reported to be accumulated in hypoxic regions in vivo and thus is exploited as a hypoxia imaging agent (Price et al., 2011). Notably, its hypoxia-selective biodistribution is highly correlated with the Cu(II/I) redox potential (Dearling et al., 2002). Susceptibility to switch between Cu(I) and Cu(II) is also the key for toxicity of thiosemicarbazone-copper complex, suggesting that these two phenomena are related. Since a potential anticancer activity of bis(thiosemicarbazone) has been also suggested (Palanimuthu et al., 2013), hypoxic cells should be tolerant to both scaffolds assuming that toxic mechanism of thiosemicarbazone and bis(thiosemicarbazone) is similar. On the other hand, normoxic cells are more susceptible to thiosemicarbazone-induced cytotoxicity, while they may excrete bis(thiosemicarbazone) from cells more efficiently and achieve hypoxia-selective accumulation of the compound.
To fully connect how the NSC319726-copper complex induces cytostatic effect, the identification of the precise ROS species generated upon NSC319726-copper treatment targeting purine deoxyribonucleosides is required. We also want to note that although NSC319726-bound Cu(II) is susceptible to propagate free radicals from hydrogen peroxide more efficiently than unbound Cu(II) (Fig. S5A), hydroxyl radicals may not be the same ROS generated in vivo. In regard to the exact ROS, it is tempting to propose that singlet oxygen is generated in response to any of three compounds (NSC319726, 7Br8HQ, and MB). However, unlike photodynamic therapy, the source of energy to generate singlet oxygen for this mechanism is unknown. There has been evidence reported that small-molecule-bound copper behaves differently to target DNA, possibly through generation of singlet oxygen (Devasagayam et al., 1991; Frelon et al., 2003; Jose et al., 2011; Li and Trush, 1993; Macomber et al., 2007). We also have evidence supporting the hypothesis that NSC319726-copper complex induces singlet oxygen: (i) transcriptome analysis against the Molecular Signature Database revealed that the genes up- and down-regulated upon NSC319726 treatment were most significantly correlated with that in hypericin-mediated photodynamic therapy that induces single oxygen (Fig. 4C), (ii) among multiple ROS scavengers, specific antioxidants targeting hydroxyl radicals, lipid peroxides, and superoxide were not effective, but only N-acetylcysteine suppressed NSC319726 cytotoxicity, but only at higher concentrations, (iii) lethal stimuli generated upon NSC319726 treatment are oxygen pressure dependent, suggesting that oxygen molecules may directly contribute to ROS production, and (iv) 2′-deoxyguanosine is a known target of singlet oxygen (Jose et al., 2011; Li and Trush, 1993; Ravanat and Cadet, 1995). While these multiple lines of evidence point toward singlet oxygen, in the absence of a plausible mechanism for the generation of singlet oxygen, this remains speculative.
However and whatever ROS may be generated upon NSC319726 treatment, its picomolar potency to sensitive GBM cells is a valuable property. Towards clinical application, it may be noteworthy that NSC319726’s effectiveness may depend on the sex of the patients. Although our analysis based on 13 patient-derived-GBM models was not powered for this analysis (p-value of 0.073 using Wilcoxon’s rank sum test), NSC319726 was more effective against GBM cells from female patients than GBM cells from male patients (Experimental Model and Subject Details describes the genders of the patients). While there are still certain obstacles to translating this finding into a therapeutic agent, e.g., it was unclear whether NSC319726 penetrates the blood-brain barrier, and what the oxygen tension is in various GBM tissues, a sophisticated drug delivery system to selectively administer drugs to GBM cells along with oxygen might overcome such issues. Irrespective, these findings suggest that copper dysregulation can induce oxidative stress and DNA damage, leading to cell cycle arrest. This may have applications in a variety of pathological and therapeutic contexts.
Significance
Transition metals are essential for many cellular functions; deregulation of their metabolism causes toxicity that may be exploited for therapeutic gain. In this study, we report that the compound NSC319726 binds copper to induce oxidative stress and arrest glioblastoma-patient-derived cells at picomolar to nanomolar concentrations. This effect was discovered through screening compounds in 13 patient-derived glioblastoma cell models. While NSC319726 has been reported to function as a zinc ionophore at micromolar concentrations in some cancer cell lines, we found that it was not responsible for NSC319726’s cytotoxicity to GBM cells. Therefore, we decided to adopt a data-driven pharmacogenomic approach to identify likely mechanism of action of NSC319726; through analysis of drug sensitivity data and basal transcriptome of the NCI-60 human cancer cell line panel, we hypothesized that NSC319726 and 65 other structural analogs exhibit lethality through metal binding, consistent with their chemical structures and reported activities. Supplementing multiple metal ions, we finally found that NSC319726 binds copper to arrest cell growth in GBM cells. Furthermore, pharmacogenomic analysis of both the NCI-60 data and transcriptomic changes of GBM cells upon NSC319726 treatment suggested that the copper toxicity induced by NSC319726 was substantially inhibited by hypoxia, through a HIF-1α–dependent pathway; we confirmed this hypoxia-dependent lethality in glioblastoma patient samples using a hypoxia chamber. Treatment with copper-bound NSC319726 resulted in the generation of reactive oxygen species and a depletion of deoxyribosyl-purines, including 2′-deoxyguanosine, resulting in G1 cell cycle arrest. These data suggest that DNA damage is the primary consequence of copper-mediated oxidative stress, subsequently inducing cell cycle arrest. Thus, copper dysregulation and metal-induced DNA damage may be a consequence of exposure to some therapeutic agents or xenobiotics. Moreover, this study highlights the power of a data-driven pharmacogenomic approach to characterize mechanism of action of small molecules.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information about reagents and resources used in this study, please contact Brent Stockwell (bstockwell@columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and tissue culture
Collection of 13 patient-derived GBM samples were executed by Henry Ford Hospital as approved by the Institutional Review Board. The use of patient-derived GBM cell culture was executed as approved by the Institutional Review Boards of Columbia University and Henry Ford Hospital. Of 13 cells, 7 are from female patients (HF3037, HF2998, HF2885, HF2906, HF3026, HF2476, HF2790) and 6 from male (HF2381, HF2876, HF3013, HF2303, HF3177, HF2941).
Briefly, GBM cells were defrosted and grown in T-25, T-75, or T-175 tissue-culture-treated flasks as non-adhesive neurospheres in a serum-free, DMEM/F12 base media (Invitrogen #11330-057) supplemented with N-2 (Invitrogen #17502048), Bovine Serum Albumin (Sigma-Aldrich #A4919), and recombinant human EGF and FGF (PeproTech #AF-100-15 and # 100-18B, respectively). GBMs that failed to grow in suspension as neurospheres were supplemented with laminin (Sigma-Aldrich #L2020), added directly to the flask, at a concentration of 1 μL/cm2. Cells were cultured at 37°C, 100% humidity, and 5% CO2. Neurospheres were dissociated via Dulbecco’s PBS and mechanical pipetting, while Accutase was used to detach GBMs grown on laminin. Upon dissociation, cells were counted using a Beckman Coulter Vi-Cell Cell Viability Analyzer and re-plated in the afore-described conditions.
HT-1080 cells were acquired from ATCC. Cells were submitted for authentication to ATCC (sample #STRA3235) and verified as an exact match for ATCC CCL-121 (HT-1080) on March 13th 2017. The cells grew at 37°C under 100% humidity, 5% CO2. For maintenance, HT-1080 cells were cultured in 1% non-essential amino acid and 10% fetal bovine serum containing Dulbecco modified Eagle’s MEM (Gibco, #11995). One million cells were seeded per 50 mL in a T-175 flask, and cells were trypsinized and reseeded at 1:10 dilution to a new flask every two days.
SH-SY5Y cell line was obtained from ATCC and passaged a few times before use. For maintenance, cells were cultured in DMEM/F12 (ThermoFisher 10565042) supplemented with GlutaMAX, 10% heat-inactivated FBS, and Penicillin Streptomycin (Pen Strep, ThermoFisher 15070063). When reaching 90% confluency, cells were trypsinized and replated at 1:3 dilution. For differentiation, cells were plated at 40% confluency with maintenance media for 12 h, replaced with D1 media (DMEM/F12 + GlutaMAX + 5% FBS + 10 μM retinoic acid + Pen Strep) for 5 days (fresh media change every 2 days), followed by D2 media (DMEM/F12 + GlutaMAX + 50μg/ml BDNF + Pen Strep) for 3 days before drug treatment. Retinoic acid and BDNF were purchased from Sigma-Aldrich (B3795-10UG and R2625-100mg, respectively).
METHOD DETAILS
Synthesis of NSC319726
Methyl hydrazinecarbodithioate (1)
To a cooled solution of potassium hydroxide (6.60 g, 0.1 mol, 1.0 eq) in water (8 mL) and isopropanol (6.7 mL) was added 80% hydrazine hydrate (6.26 g, 6.06 mL, 0.1 mol, 1.0 eq). Ice-cooled carbon disulfide (7.61 g, 6.04 mL, 0.1 mol, 1.0 eq) was added dropwise to the stirred solution over 90 min, while keeping the temperature of the reaction mixture below 10°C. The bright yellow mixture was stirred for an additional 1 h, after which ice-cooled methyl iodide (14.2 g, 6.23 mL, 0.1 mol, 1.0 eq) was added dropwise over a 2 h period and the mixture was allowed to stir overnight while slowly warming to room temperature. The white precipitate was collected by filtration and washed with ice-cold water. The crude product was recrystallized from dichloromethane to give methyl hydrazinecarbodithioate (1): 2.8 g, 23% yield. The spectroscopic data matched those reported in the literature (Klayman et al., 1979a).
Methyl 2-(1-(pyridin-2-yl)ethylidene)hydrazine-1-carbodithioate (2)
Methyl hydrazinecarbodithioate (1, 2.8 g, 22.95 mmol) was dissolved in isopropanol (7 mL) and 2-acetylpyridine was added (2.79 g, 2.59 mL, 23.07 mmol, 1.005 eq). The reaction mixture turned yellow followed by precipitation of a yellow solid. The reaction mixture was stirred for an additional 2 h and cooled overnight. The precipitate was collected by filtration, washed with isopropanol and air-dried to yield methyl 2-(1-(pyridin-2-yl)ethylidene)hydrazine-1-carbodithioate (2): 4.72 g, 91% yield as a 63:37 mixture of E/Z-isomers. The spectroscopic data matched those reported in the literature (Klayman et al., 1979a).
(E)-N′-(1-(pyridin-2-yl)ethylidene)azetidine-1-carbothiohydrazide (NSC319726) (Klayman et al., 1979b)
Methyl 2-(1-(pyridin-2-yl)ethylidene)hydrazine-1-carbodithioate (2, 2.25 g, 10 mmol, 1.0 eq) was suspended in methanol (25 mL) and azetidine (0.57 g, 674 μL, 10 mmol, 1.0 eq) was added and the mixture was stirred at 50°C for 6 h. The solid dissolved and after 20 min a white solid precipitated. After cooling to room temperature the solid was collected by filtration to give the product as a fine white powder, NSC319726: 1.81 g, 77% yield.
1H NMR (400 MHz, DMSO-d6): δ 10.19 (s, 1H), 8.59 (dt, J = 4.9, 1.2 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.82 (td, J = 7.7, 1.8 Hz, 1H), 7.38 (ddd, J = 7.4, 4.8, 1.2 Hz, 1H), 4.58 (bs, 2H), 4.13 (bs, 2H), 2.35 (s, 3H), 2.25 (p, J = 7.8 Hz, 2H) ppm.
13C NMR (100 MHz, DMSO-d6): δ 175.8, 155.0, 148.6, 148.5, 136.7, 123.8, 119.8, 56.6, 53.1, 16.0, 11.6 ppm.
HRMS (ESI+, m/z): calcd for C11H15N4S [M+H]+: 235.1017, found: 235.1020.
Cell viability assay
When cellular viability assays were performed in 384-well plates (Corning #3712), we often treated cells with two components. A compound tested in a 2-fold dilution series (denoted as L as they are often lethal compounds), and a modulator tested at a single concentration (denoted as M here). M may interfere with cellular signaling responding to the stimuli L and consequently it may affect the phenotype. M was already treated to the cellular suspension before seeding cells (e.g., when M is a compound, it was added to the cell suspension just before seeding the cells to plates; when siRNAs, cells were first transfected as described in the following section, and in two days transfected cells expressing the siRNAs were reseeded in 384 well plates). 1,000 (HT-1080) or 2,000 (GBMs) cells treated with M were seeded per 36 μl in each well of 384 well plates (Corning, #3712). Compounds examined in assays were first dissolved in appropriate solutions (dimethylsulfoxide, ethanol, or culture media, depending on the compounds’ solubility), and then diluted to make 10x concentrations of the tested concentrations in the culture media. 4 μl of the 10x solutions were added to each well, which results in treating cells with 1x concentration of the compounds. After cells were incubated with the compounds for 48 h, 10 μl per well of 50% Presto Blue (Thermo Fisher Scientific) in culture media was added to each plate, the plates were further incubated for eight hours, and the fluorescence (ex/em: 530/590) was measured using a Victor plate reader (Perkin Elmer). The raw fluorescence acquired from the plate reader was normalized using the following equation: VL|M = (IM,L − I0)/(IM − I0), where IM,L, IM, and I0 correspond to fluorescence intensity from wells containing cells treated with M and L, wells containing cells treated with M only, and wells containing media, M, and L (but no cells), respectively. The resulting value (VL|M) is the normalized viability. Note that VL|M = 0 and 1 indicate that L completely kills the cells or that L does not induce any growth inhibitory effect, respectively. When only a lethal compound was treated to cells in dilution series, we still use this equation but in this case M is the vehicle control. The assays were done in biological triplicates; a representative single experiment was chosen to show dose-concentration curves for viability. In a plot, points are the mean of normalized viability tested in technical triplicates.
Sequencing of TP53 gene of 13 GBM primary cells
TP53 primers were designed using the Consensus CDS (CCDS) available on the NCBI website (CCDS11118.1), with forward primer TGAAGCTCCCAGAATGCCAG and reverse primer TCTCGGAACATCTCGAAGCG. The total sequence covered 846 bp, including both primers. The forward primer extended from nucleotide 183 to 202, with the reverse primer extending from nucleotide 1009 to 1028.
Upon neurosphere dissociation or laminin detachment, RNA was collected from untreated GBM cells with a Qiagen QIAshredder (#79656) and RNeasy Mini Kit (#74106) as per the protocol provided with the kit, analyzed for quality via Nanodrop Lite spectrophotometer, and cDNA was generated from each sample through PCR reaction with the aforementioned TP53 primers.
cDNA samples were sent to GENEWIZ for sequencing, and GBM TP53 sequences were returned aligned with the actual, expected TP53 sequence. Upon visual inspection, mismatched pairs of nucleotides were identified, highlighted, and matched to the translated amino acid they code for.
Screening for lethal compounds in 13 GBM primary cells
Lethal compounds were tested within GBM patient models as per the high-throughput screening protocol established in Quartararo et al. (Quartararo et al., 2015). In brief, immediately upon detachment of adhesive cells or dissociation of neurospheres, GBMs were seeded in 384-well, white, opaque, tissue-culture-treated plates (PerkinElmer, #6007688) at a density of 1,000 cells per well, with 26 μL of growth media in each well. 24 h later, inhibitors were added in a 17-point, two-fold dilution series. The final concentration range was 0.8 nM to 50 μM. 72 h post inhibitor addition, a one-to-one mixture of DMEM/F12 and CellTiter-Glo (CTG) was added, and luminescence was quantified with a PerkinElmer Victor X5 plate reader. A Biomek FX liquid handling system was used to seed cells, add inhibitors, and add the DMEM/F12:CTG luminescence mix, with cell-seeded plates maintained in a 37°C,100% humidity, 5% CO2 environment during incubation and post inhibitor addition.
Percent viability was calculated using growth media only wells as 0% viable, and 0.5% DMSO treated cells as 100% viable. GraphPad Prism 6.0e was used to calculate the EC50 values for each compound with four-parameter Hill equation, or “log(inhibitor) vs. response- Variable slope (four parameters)”.
Screening for a responsible metal in NSC319726-induced cell death
1,000 HT-1080 cells/32 μL per well were seeded in a 384-well plate. The cells were grown overnight under 5% CO2 at 37°C. On the next day, 4μL/well of 10x metal chloride and 4 μL/well 310 nM NSC319726 with or without 1 mM Co(II) were concomitantly added. When the solutions were added, cells were treated with 31 nM NSC319726, with or without 100 μM Co(II) and with 2-fold 12-point dilution series of metal chloride with the highest concentration of 100μM. The cells were incubated for 48 h followed by addition of Presto Blue, incubation for 5 h and subsequent fluorescence measurement. Note that metal salts were tested in a dilution series and NSC319726 was tested at a fixed concentration. Therefore, as described in ‘Cell Viability Assay’ section above, the normalized viability of one corresponds to the viability of cells treated with DMSO, NSC319726, DMSO/Co(II), or NSC319726/Co(II), equivalent to the condition of interest, but only without metals.
Inductively coupled plasma-mass spectrometry (ICP-MS) for metal quantification
Cells were plated in 6-well plates and cultured for one day. The vehicle or compounds were added to the cells the following day and incubated for specified times. NSC319726, 8HQ and MB were added for 24 h. Cobalt chloride was added either alone or in combination with NSC319726 for 24 h. The plates were then washed 2 times with PBS containing 1 mM EDTA and 2 times with PBS alone. After the addition of 215 μL concentrated nitric acid (BDH Aristar Ultra), the plates were sealed with parafilm and incubated overnight. The samples (150 μL) were then diluted in 2 mL 2% nitric acid (ultrapure nitric acid diluted in MilliQ water) and analyzed on a Thermo Fisher iCAP Qc ICP mass spectrometer in Kinetic Energy Discrimination (KED) mode against a calibration curve of known copper, zinc, iron, cobalt and phosphorus concentrations, with gallium (20 μg/L, Inorganic Ventures) as an internal standard. Each experiment was carried out thrice and each condition was repeated in at least triplicate.
RNA-seq experiment
Sample collection. Poly-A pull-down was performed to enrich mRNAs from total RNA samples (200 ng - 1 μg per sample, RIN>8 required) and mRNAs were proceeded to library preparation by using Illumina TruSeq RNA prep kit. Libraries were then sequenced using Illumina HiSeq2000 at Columbia Genome Center. Samples were multiplexed in each lane, which yields targeted number of single-end/paired-end 100bp reads for each sample, as a fraction of 180 million reads for the whole lane. The data analysis of the acquired sequencing was performed as described later in Quantification and Statistical Analysis.
HIF-1α knockdown
Gene knockdown was performed using siRNA reverse transfection. Non-targeting control or HIF-1α-targeting siRNAs (GE Dharmacon) were dissolved in RNase-free water to make 10 μM stocks. 6 μL of siRNA solution and 15 μL of Lipofectamine RNAiMAX (Thermo Fisher Scientific) were added to 500 μL OptiMEM media (Thermo Fisher) in each eppendorf tube, and placed on a well in 6-well plate. Three wells per each siRNA were prepared. After the plates were incubated for 15 minutes at 37°C, 120,000 HT-1080 cells per 1.5 mL in each well (i.e., 80,000 cells per mL) were seeded. Cells were incubated to allow knockdown proceeded for 48 h. Cells were trypsinized and reseeded for further western blotting and viability assays.
Hypoxia induction
In order to perform assays at 1.5% O2, a H35 HypOxystation (HypOxygen) was used. Upon seeding cells, plates designated for hypoxia testing were manually transferred into the station, and inhibitors were added 24 h later by hand with the plates still inside the Hypoxia chamber. For cell viability assay, 48 h (HT-1080) or 72 h (GBM) post inhibitor addition, plates were removed and DMEM/F12:CTG was added as described above.
Western blotting
300,000 HT-1080 cells were seeded per well in a 6-well plate. The cells were treated with 100 μM Co(II) chloride, 100 μM DMOG, or media. The plates were grown under either hypoxia (1.5% O2) or normoxia (21% O2) and under 5% CO2 at 37°C for 24 h. As soon as the plates were taken out of the incubator or hypoxia chamber, they were harvested at normoxia and ambient temperature. Procedure for cell lysis, SDS–PAGE, and western blotting, was described previously (Dixon et al., 2012). Antibodies used were: anti-human α-tubulin (Santa Cruz Biotechnology, sc-32293, 1:10,000 dilution), and anti-human HIF-1α (Abcam, ab51608).
ROS detection in cell culture
HT-1080 cells were trypsinized and diluted to a final density of 100,000 per mL. The cells were then incubated with 1 μM of CM-H2DCFDA for 30 min, spun down at 660 x g for 3 mins, and the supernatant was replaced with fresh media containing lethal compounds and/or Cu(II). Then, the cells were incubated for 30 mins, spun down, and the supernatant was replaced with fresh media. Fluorescence of samples was measured at the FL1-H channel using the Accuri C6 flow cytometer (BD Bioscences). Flow cytometry data were analyzed and visualized using flowCore package (Hahne et al., 2009) and R.
ROS detection in cell free system
Phosphate Buffered Saline (PBS), L-Cysteine (Sigma-Aldrich, #C7352), and H2O2 (Sigma-Aldrich, #516813, 50 wt. % in H2O) were used to generate a master mix (MM) at a pH of 7.4. A clear 96 well plate (Sigma-Aldrich, #CLS3596) had the following conditions added per row, with a technical triplicate of wells for each condition: PBS only, H2DCFDA only, MM only, MM with CuCl2, MM with NSC319726, and MM with both CuCl2 and NSC319726. The final volume per well was 200 μL, with the following concentrations of components per relevant well: H2DCFDA at 5 μM, Cysteine at 100 μM, H2O2 at 100 μM, NSC319276 at 10 μM, and CuCl2 at 10 μM. The plate was incubated at 37°C for minutes and read using a Victor X5 plate reader (ex/em: 485/535). The experiment was done in biological duplicates. Data was analyzed using Microsoft Excel.
Cell cycle assay
300,000 HT-1080 cells per well were plated in 6-well plates and treated with 1 μM NSC319726, 50 μM 7Br8HQ, or 50 μM MB for 12 h. The cells were trypsinized and spun down at 700 rcf. The pellets were triturated and fixed with ice-cold 70% ethanol for 30 mins. The cells were then spun down at 1,500 x g. After a PBS wash, the cells were incubated in 500 μL FxCycle PI/RNase staining solution at room temperature for 30 mins while protected from light. Cells were immediately analyzed on the Accuri C6 flow cytometer (BD Biosciences), and the fluorescence intensity on the FL2-A or FL3-A channel was monitored. Flow cytometry data were analyzed and visualized using flowCore package and R.
Metabolomic profiling
HT-1080 cells were seeded into 10 million cells per T-175 flask in four flasks. Immediately, two flasks were treated with 0.1% DMSO and two with 5 μM NSC319726 and incubated for six hours. The cells were trypsinized and collected into 15 mL conical tubes. 500 μL from each tube was saved for cell counting using ViCell (Beckman Coulter), and the rest of the tubes were spun down at 1,500 rpm for 5 mins. Pellets were washed gently with 5mL PBS. 500 μL PBS was added to the pellets, triturated, and transferred to cryovials. The tubes were spun down at 1,500 rpm for 5mins, and the supernatant was removed. The pellet was flash frozen in a dry ice-isopropanol bath and stored at −80°C. Sample collection was performed in biological quadruplicates. After the last sample collection, the samples were shipped in dry ice to Metabolon for further sample preparation. At Metabolon, 505 metabolites from the samples were identified and quantified using LC/MS and GC/MS as described previously (Skouta et al., 2014).
Neurotoxicity assays
For 96-well plate assays, SH-SY5Y cells were plated at 5K/well. For undifferentiated condition, cells were treated with drugs 24 h after seeding, and for differentiated condition, cells were treated 3 days after switching to D2 media. In both conditions, four drugs were treated at 9-point 2-fold dilution series and cells were incubated for 48 h before viability was measured using CellTiter-Glo (Promega, G7572). Images were taken by IncuCyte (EssenBioscience) at 10X after drug treatment for 48 h.
QUANTIFICATION AND STATISTICAL ANALYSIS
Pharmacogenomic analysis of the NCI-60 data
The NCI-60 data analysis was done as described previously (Shimada et al., 2016b). Briefly, 2,565 cell-line-selective lethal compounds were clustered into 18 groups based on growth inhibition (GI50) profiles in the NCI-60 panel using Gaussian finite mixture model and hierarchical clustering. The former was performed using mclust R package (Scrucca et al., 2016). Each cluster represents distinct mechanisms of action. The Spearman correlation between each compound cluster’s GI50 profile and each gene’s expression across the panel was computed. This correlation matrix (18 compound clusters x 11,642 genes) was used for pathway enrichment analysis (see below).
Processing of raw sequences from RNA-seq
Raw sequences from RNA-seq of GBM cells were acquired as described above. RTA (Illumina) was used for base calling and bcl2fastq (version 1.8.4) for converting BCL to fastq format, coupled with adaptor trimming. The reads were mapped to a reference genome (Human: NCBI/build37.2) using Tophat (version 2.0.4) with 4 mismatches and 10 maximum multiple hits (Trapnell et al., 2009). The relative abundance (aka expression level) of genes and splice isoforms were estimated using cufflinks (version 2.0.2) with default settings (Trapnell et al., 2010).
Analysis of RNA-seq data
Normalized data (FPKM values) of the eight samples (HF3037 and HF2941 cells treated with DMSO or NSC319726, each with biological duplicate) were provided by Columbia University Genome Center. A pseudo-count one was added to all the FPKM values, which were then log–transformed, i.e., xg = log10(ng + 1). For each cell line, the expression change of each gene g is computed by averaging the log-expressions between the duplicates and subtract the expressions induced by NSC319726 from the ones induced by DMSO. These expression profiles were then examined for pathway enrichment analysis as described above to identify pathways (i.e., gene sets) that were up- or down-regulated upon NSC319726 treatment.
Pathway enrichment analysis
Pathway enrichment analysis is the core of our transcriptome analysis to understand the mechanism of lethality of NSC319726. From the normalized NCI-60 and GBM data, we looked for pathways overrepresenting positively or negatively correlated genes in the NCI-60 data and up- or down-regulated genes upon NSC319726 treatment in the GBM transcriptome (Fig. 4A, Fig. S2). We utilized the Broad Institute’s Molecular Signature Database (MSigDB) as collection of pathways (Subramanian et al., 2005). In the first analysis (Fig. S2), we limited our analysis to Gene Ontology, Canonical pathways, BIOCARTA, KEGG, REACTOME, and ‘Hallmarks’ in MSigDB. In the second analysis (Fig. S4), we used 3,400 ‘chemical and genetic perturbation’ gene sets in MSigDB that are collections of up- and down-regulated genes from previously published experiments; we looked for experimental conditions that induce similar gene expression changes to our NSC319726 treatment in GBM cells.
To compute the significance of overrepresentation of a pathway, we developed a method using one-sided Fisher’s exact test. First, genes were sorted based on correlation coefficients for the NCI-60 data, or log fold changes for the GBM RNA-seq data (NSC319726 treatment over vehicle treatment). Second, we selected top and bottom n% of sorted genes (n=5,10,15,…, 95), which gives 38 ways of gene selection. Third, we assessed the significance of overrepresentation of a pathway for each of these 38 selections. Finally, p-value that gives the lowest of the 38 selections was set as the significance of the enrichment of this pathway. When the lowest p-value was given by “top” or “bottom” genes, the pathway was considered up- or down-regulated, respectively. The p-values were further converted to signed log p-values, namely log10(p-value) multiplied by 1 or −1 when the pathway was up- or down-regulated, respectively; large positive or negative signed log p-values indicate significant up- or down-regulation.
To further assess the mechanism of action of the compound cluster containing NSC319726 and other thiosemicarbazones (we call it cluster g here), we looked for pathways that are not only significantly but also exclusively associated with the cluster here. To find such exclusive association, we calculated pathways’ “specificity” for the cluster g (Shimada et al., 2016b). Specificity of a pathway in a compound cluster across multiple clusters becomes positive only when the pathway is most significantly associated with the cluster g in the same direction (up- or down-regulated) across the 18 clusters. Specificity (Sp) is mathematically defined as Sp = min(Δslpg)·sign(slpg), where Δslpg is a vector containing difference of signed log p-values (slp) between cluster g and the other 17 clusters, min() returns a minimum value of the vector, and sign() is a sign function.
Predicting functionally relevant pathways
As summarized in Fig. S2A, we suspected that pathways i) enriched in genes positively correlated with drug-sensitivity profiles (i.e., genes more expressed in resistant cells) from NCI-60 analysis and ii) whose transcriptional activity is upregulated upon NSC319726 treatment are more likely to be involved in protecting cells from the compound, while pathways i) enriched in negatively correlated genes as well as ii) whose transcriptional activity is downregulated upon the compound treatment are more likely to sensitize cells to the lethal stimuli. In both NCI60 and GBM transcriptome analyses, we set specificity of ±5 and significance of ±10 as thresholds for calling them specific and significant, respectively (Fig. S2B,C), and we eventually found two pathways (HALLMARK_TNFA_SIGNALING_VIA_NFKB and HALLMARK_HYPOXIA) likely support the cells to survive from the NSC319726’s lethality.
DATA AND SOFTWARE AVAILABILITY
Normalized RNA-seq data of patient-derived glioblastoma cells, normalized metabolomic profiling data of HT-1080 cells, and chemical structures of 83 compounds whose drug sensitivity profiles across NCi-60 cell lines are similar to NSC319726, are available at Mendeley Data (http://dx.doi.org/10.17632/2d8wzkcn3v.1).
Supplementary Material
Scheme 1.

Synthesis of NSC319726
Highlights.
Picomolar NSC319726 arrests growth of glioblastoma-patient-derived cells
NSC319726 binds to copper to induce oxidative stress using ambient oxygen
NSC319726-derived ROS target purine deoxyribonucleosides to arrest cell cycle
Acknowledgments
We thank Anna Kaplan of Columbia University, Tom Mikkelsen of Henry Ford Hospital, and Christopher Chang of University of California, Ray Pagliarini at Novartis Institutes for BioMedical Research, for helpful discussions. This study was supported by funding from the Accelerate Brain Cancer Cure (ABC2) foundation, NIH grant R35CA209896 (to BRS). LK was supported by NIH grant R01GM079465 and Howard Hughes Medical Institutes.
Footnotes
Declaration of interests
The authors declare no conflict of interest.
Author Contributions
Conceptualization, K.S. and B.R.S.; Methodology, K.S., E.R., M.E.S., L.K., P.H.B., Y.S., and C.E.Q.; Software, K.S.; Formal Analysis, K.S.; Investigation, K.S., E.R., M.E.S., L.K., P.H.B., Y.S., C.E.Q., and N.C.P.; Resources, A.C.D., P.H.B., and B.R.S.; Data Curation, K.S., E.R., M.E.S., L.K., C.E.Q., and N.C.P.; Writing – Original Draft, K.S. and B.R.S.; Writing – Review & Editing, K.S., M.E.S., L.K., P.H.B., C.E.Q., D.R.C., D.C.L., and B.R.S.; Visualization, K.S.; Supervision, B.R.S.; Project Administration, K.S.; Funding Acquisition, B.R.S.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angelé-Martínez C, Goodman C, Brumaghim J. Metal-mediated DNA damage and cell death: mechanisms, detection methods, and cellular consequences. Metallomics. 2014;6:1358–1381. doi: 10.1039/c4mt00057a. [DOI] [PubMed] [Google Scholar]
- Buytaert E, Matroule JY, Durinck S, Close P, Kocanova S, Vandenheede JR, de Witte PA, Piette J, Agostinis P. Molecular effectors and modulators of hypericin-mediated cell death in bladder cancer cells. Oncogene. 2007;27:1916–1929. doi: 10.1038/sj.onc.1210825. [DOI] [PubMed] [Google Scholar]
- Chang CJ. Searching for harmony in transition-metal signaling. Nat Chem Biol. 2015;11:744–747. doi: 10.1038/nchembio.1913. [DOI] [PubMed] [Google Scholar]
- Crim JA, Petering HG. The Antitumor Activity of Cu(II)KTS, the Copper(II) Chelate of 3-Ethoxy-2-oxobutyraldehyde Bis(thiosemicarbazone) Cancer Res. 1967;27:1278–1285. [PubMed] [Google Scholar]
- Dearling JL, Lewis JS, Mullen GE, Welch MJ, Blower PJ. Copper bis(thiosemicarbazone) complexes as hypoxia imaging agents: structure-activity relationships. JBIC J Biol Inorg Chem. 2002;7:249–259. doi: 10.1007/s007750100291. [DOI] [PubMed] [Google Scholar]
- Devasagayam TPA, Steenken S, Obendorf MSW, Schulz WA, Sies H. Formation of 8-hydroxy(deoxy)guanosine and generation of strand breaks at guanine residues in DNA by singlet oxygen. Biochemistry (Mosc) 1991;30:6283–6289. doi: 10.1021/bi00239a029. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudev T, Lim C. Metal Binding Affinity and Selectivity in Metalloproteins: Insights from Computational Studies. Annu Rev Biophys. 2008;37:97–116. doi: 10.1146/annurev.biophys.37.032807.125811. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26:756–784. doi: 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch RA, Liu MC, Grill SP, Rose WC, Loomis R, Vasquez KM, Cheng YC, Sartorelli AC. Triapine (3-aminopyridine-2-carboxaldehyde- thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem Pharmacol. 2000;59:983–991. doi: 10.1016/s0006-2952(99)00419-0. [DOI] [PubMed] [Google Scholar]
- Frelon S, Douki T, Favier A, Cadet J. Hydroxyl Radical Is Not the Main Reactive Species Involved in the Degradation of DNA Bases by Copper in the Presence of Hydrogen Peroxide. Chem Res Toxicol. 2003;16:191–197. doi: 10.1021/tx025650q. [DOI] [PubMed] [Google Scholar]
- Gaetke LM, Chow CK. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology. 2003;189:147–163. doi: 10.1016/s0300-483x(03)00159-8. [DOI] [PubMed] [Google Scholar]
- Hahne F, LeMeur N, Brinkman RR, Ellis B, Haaland P, Sarkar D, Spidlen J, Strain E, Gentleman R. flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics. 2009;10:106. doi: 10.1186/1471-2105-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson PJ, Sharpe PC, Bernhardt PV, Richardson DR. Novel Thiosemicarbazones of the ApT and DpT Series and Their Copper Complexes: Identification of Pronounced Redox Activity and Characterization of Their Antitumor Activity. J Med Chem. 2010;53:5759–5769. doi: 10.1021/jm100561b. [DOI] [PubMed] [Google Scholar]
- Jose GP, Santra S, Mandal SK, Sengupta TK. Singlet oxygen mediated DNA degradation by copper nanoparticles: potential towards cytotoxic effect on cancer cells. J Nanobiotechnology. 2011;9:9. doi: 10.1186/1477-3155-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karioti A, Bilia AR. Hypericins as Potential Leads for New Therapeutics. Int J Mol Sci. 2010;11:562–594. doi: 10.3390/ijms11020562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klayman DL, Bartosevich JF, Griffin TS, Mason CJ, Scovill JP. 2-Acetylpyridine thiosemicarbazones. 1 A new class of potential antimalarial agents. J Med Chem. 1979a;22:855–862. doi: 10.1021/jm00193a020. [DOI] [PubMed] [Google Scholar]
- Klayman DL, Scovill JP, Bartosevich JF, Mason CJ. 2-Acetylpyridine thiosemicarbazones. 2 N4, N4-Disubstituted derivatives as potential antimalarial agents. J Med Chem. 1979b;22:1367–1373. doi: 10.1021/jm00197a017. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy L, Cotruvo JA, Jr , Chan J, Kaluarachchi H, Muchenditsi A, Pendyala VS, Jia S, Aron AT, Ackerman CM, Wal MNV, et al. Copper regulates cyclic-AMP-dependent lipolysis. Nat Chem Biol. 2016 doi: 10.1038/nchembio.2098. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Trush MA. DNA damage resulting from the oxidation of hydroquinone by copper: role for a Cu(II)/Cu(I) redox cycle and reactive oxygen generation. Carcinogenesis. 1993;14:1303–1311. doi: 10.1093/carcin/14.7.1303. [DOI] [PubMed] [Google Scholar]
- Lin H, Yu X, Eng OS, Buckley B, Kong ANT, Bertino JR, Carpizo DR, Gounder MK. A sensitive liquid chromatography–mass spectrometry bioanalytical assay for a novel anticancer candidate – ZMC1. Biomed Chromatogr. 2015;29:1708–1714. doi: 10.1002/bmc.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J. Triethylenetetramine Pharmacology and Its Clinical Applications. Mol Cancer Ther. 2010;9:2458–2467. doi: 10.1158/1535-7163.MCT-10-0523. [DOI] [PubMed] [Google Scholar]
- Macomber L, Rensing C, Imlay JA. Intracellular Copper Does Not Catalyze the Formation of Oxidative DNA Damage in Escherichia coli. J Bacteriol. 2007;189:1616–1626. doi: 10.1128/JB.01357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmström BG, Leckner J. The chemical biology of copper. Curr Opin Chem Biol. 1998;2:286–292. doi: 10.1016/s1367-5931(98)80071-9. [DOI] [PubMed] [Google Scholar]
- Martin F, Linden T, Katschinski DM, Oehme F, Flamme I, Mukhopadhyay CK, Eckhardt K, Tröger J, Barth S, Camenisch G, et al. Copper-dependent activation of hypoxia-inducible factor (HIF)-1: implications for ceruloplasmin regulation. Blood. 2005;105:4613–4619. doi: 10.1182/blood-2004-10-3980. [DOI] [PubMed] [Google Scholar]
- Palanimuthu D, Shinde SV, Somasundaram K, Samuelson AG. In Vitro and in Vivo Anticancer Activity of Copper Bis(thiosemicarbazone) Complexes. J Med Chem. 2013;56:722–734. doi: 10.1021/jm300938r. [DOI] [PubMed] [Google Scholar]
- Prachayasittikul V, Prachayasittikul S, Ruchirawat S, Prachayasittikul V. 8-Hydroxyquinolines: a review of their metal chelating properties and medicinal applications. Drug Des Devel Ther. 2013;7:1157–1178. doi: 10.2147/DDDT.S49763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price KA, Crouch PJ, Volitakis I, Paterson BM, Lim S, Donnelly PS, White AR. Mechanisms Controlling the Cellular Accumulation of Copper Bis(thiosemicarbazonato) Complexes. Inorg Chem. 2011;50:9594–9605. doi: 10.1021/ic201334q. [DOI] [PubMed] [Google Scholar]
- Quartararo CE, Reznik E, deCarvalho AC, Mikkelsen T, Stockwell BR. High-Throughput Screening of Patient-Derived Cultures Reveals Potential for Precision Medicine in Glioblastoma. ACS Med Chem Lett. 2015;6:948–952. doi: 10.1021/acsmedchemlett.5b00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravanat JL, Cadet J. Reaction of Singlet Oxygen with 2′-Deoxyguanosine and DNA. Isolation and Characterization of the Main Oxidation Products. Chem Res Toxicol. 1995;8:379–388. doi: 10.1021/tx00045a009. [DOI] [PubMed] [Google Scholar]
- Sartorelli AC, Agrawal KC, Tsiftsoglou AS, Colleen Moore E. Characterization of the biochemical mechanism of action of α-(N)-heterocyclic carboxaldehyde thiosemicarbazones. Adv Enzyme Regul. 1977;15:117–139. doi: 10.1016/0065-2571(77)90012-7. [DOI] [PubMed] [Google Scholar]
- Scrucca L, Fop M, Murphy TB, Raftery AE. mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 2016;8:289–317. [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, Stockwell BR. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016a;12:497–503. doi: 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Hayano M, Pagano NC, Stockwell BR. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol. 2016b;23 doi: 10.1016/j.chembiol.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, et al. Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models. J Am Chem Soc. 2014;136:4551–4556. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon EI, Baldwin MJ, Lowery MD. Electronic structures of active sites in copper proteins: contributions to reactivity. Chem Rev. 1992;92:521–542. [Google Scholar]
- Soussi T, Wiman KG. Shaping Genetic Alterations in Human Cancer: The p53 Mutation Paradigm. Cancer Cell. 2007;12:303–312. doi: 10.1016/j.ccr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uriu-Adams JY, Keen CL. Copper, oxidative stress, and human health. Mol Aspects Med. 2005;26:268–298. doi: 10.1016/j.mam.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Verhaegh GW, Parat MO, Richard MJ, Hainaut P. Modulation of p53 protein conformation and DNA-binding activity by intracellular chelation of zinc. Mol Carcinog. 1998;21:205–214. doi: 10.1002/(sici)1098-2744(199803)21:3<205::aid-mc8>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Waldron KJ, Robinson NJ. How do bacterial cells ensure that metalloproteins get the correct metal? Nat Rev Microbiol. 2009;7:25–35. doi: 10.1038/nrmicro2057. [DOI] [PubMed] [Google Scholar]
- Williams RJP. The natural selection of the chemical elements. Cell Mol Life Sci CMLS. 2014;53:816–829. doi: 10.1007/s000180050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011;7:285–295. doi: 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
- Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-Specific p53 Mutant Reactivation. Cancer Cell. 2012;21:614–625. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Blanden AR, Narayanan S, Jayakumar L, Lubin D, Augeri D, Kimball SD, Loh SN, Carpizo DR. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget. 2014;5:8879–8892. doi: 10.18632/oncotarget.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Hilliard G, Ferguson T, Millhorn DE. Cobalt Inhibits the Interaction between Hypoxia-inducible Factor-α and von Hippel-Lindau Protein by Direct Binding to Hypoxia-inducible Factor-α. J Biol Chem. 2003;278:15911–15916. doi: 10.1074/jbc.M300463200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.