

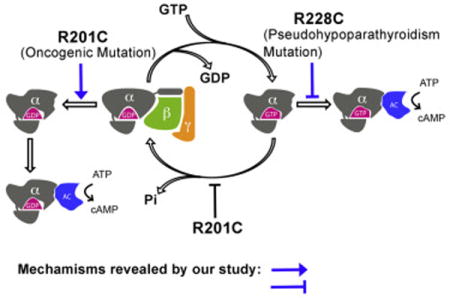
Summary
The single most frequent cancer-causing mutation across all heterotrimeric G proteins is R201C in Gαs. The current model explaining the gain-of-function activity of the R201 mutations is through the loss of GTPase activity and resulting inability to switch off to the GDP state. Here, we find that the R201C mutation can bypass the need for GTP binding by directly activating GDP-bound Gαs through stabilization of an intramolecular hydrogen bond network. Having found that a gain-of-function mutation can convert GDP into an activator, we postulated that a reciprocal mutation might disrupt the normal role of GTP. Indeed, we found R228C, a loss-of-function mutation in Gαs that causes pseudohypoparathyroidism type 1a (PHP-Ia), compromised the adenylyl cyclase-activating activity of Gαs bound to a non-hydrolyzable GTP analog. These findings show that disease-causing mutations in Gαs can subvert the canonical roles of GDP and GTP, providing new insights into the regulation mechanism of G proteins.
Graphical abstract
Frequent pathogenic mutations in G proteins can cause signaling activation by converting GDP into an activator, rather than locking the proteins at a GTP-bound state.
Introduction
GTPase proteins are the transducers of transmembrane receptor cascades serving as timers of signaling through adoption of a transiently active GTP-bound state. Termination of signaling is achieved through intrinsic GTPase activity or heterodimerization with GTPase activating proteins (GAPs) accelerating hydrolysis of GTP to GDP, causing a conformational change producing a GDP-bound species which loses the ability to bind and activate downstream effectors (Gilman, 1995). Inherited and somatic mutations of GTPases are the causal basis of a wide assortment of disease states. The KRAS gene, which encodes the small GTPase K-Ras, is the most frequently activated oncogene in cancer. Mutations at the G12 position of K-Ras lock K-Ras in its GTP-bound active state through disturbing the “arginine finger” which is provided by GAPs thereby disrupting the transition state for GTP hydrolysis (Bourne, 1997; Rodenhuis et al., 1987; Scheffzek, 1997). The most frequently mutated heterotrimeric G protein in cancer is Gαs encoded by GNAS. Gain-of-function mutations in Gαs cause growth hormone (GH) secreting pituitary tumors and other cAMP dependent tumors (Landis et al., 1989; O’Hayre et al., 2013; Vallar et al., 1987). More than half of these mutations in Gαs occur at a single hotspot, R201, which serves as the “arginine finger” in Gαs (O’Hayre et al., 2013). Unlike K-Ras, this “arginine finger” is built into Gαs instead of being provided by GAPs, but in an analogous fashion to Ras mutations, the R201 mutations decrease the GTP hydrolysis rate, thereby maintaining Gαs in a GTP-bound active state (Sprang, 2016).
The analyses of the role of activating mutations in K-Ras and Gαs have presumed the canonical view that GTP is required for the proteins to adopt the active conformation and stimulate downstream effectors, and the GDP-bound state is not of relevance to positive signaling. While this is most certainly appropriate for the small GTPase K-Ras, we wondered if the G protein mutations may influence the GDP state. Since R201 is an intramolecular arginine finger, its presence in the nucleotide pocket in the GDP-bound state could afford a layer of control over the GDP-bound conformation that may be disrupted by the oncogenic R201C hotspot mutation. Indeed, through structural and functional analysis of the R201C Gαs gain-of-function mutation we uncovered the unprecedented ability of the protein to activate its downstream effector adenylyl cyclase while binding to GDP even in the presence of Gβγ subunits. We ascribe this behavior to the involvement of R201 in maintaining GDP-bound Gαs in an off state through destabilization of an intramolecular hydrogen bond network (H-bond network).
Having found that a gain-of-function mutation can convert GDP into an activator, we postulated that a loss-of-function mutation might disrupt the normal role of GTP. Loss-of-function mutations in the H-bond network of Gαs which cause pseudohypoparathyroidism (PHP-Ia) have been ascribed to defects in GTP binding as well as hyper GTPase activity. As our analysis of R201C revealed the paradoxical effect of the H-bond network on the GDP state, we wondered if loss-of-function mutations might destabilize the GTP state. Indeed, we identified R228C Gαs which has WT like ability to bind and hydrolyze GTP yet is compromised in its ability to stimulate adenylyl cyclase when a non-hydrolyzable GTP analog is bound. These studies reveal a new molecular mechanism for the diverse disease-causing mutations in Gαs and uncover the importance of a H-bond network in G protein activation and inactivation.
Results
The GDP dissociation rate of the R201C mutant of Gαs is slower than its GTP hydrolysis rate
The R201C mutation was reported to disrupt the GTPase activity of Gαs (Landis et al., 1989). We confirmed this using a single turnover GTP hydrolysis assay. Wild type (WT) human Gαs and the R201C mutant were overexpressed in E. coli and purified to homogeneity (Figure S1A), and their GTPase activities were measured (Figures 1A and 1B). WT Gαs exhibited an intrinsic GTP hydrolysis rate (kcat) of 1.183 ± 0.074 min−1 at 0 °C. The hydrolysis of GTP by the R201C mutant was too slow to be measured at 0 °C, so instead, it was measured at 20 °C (0.020 ± 0.003 min−1). The R201C mutation does not completely disrupt the GTPase activity of Gαs. In line with this, we found that both WT and R201C Gαs purified from E. coli were in a GDP-bound state (Figure S1B).
Figure 1. Characterization of the R201C mutant of Gαs.
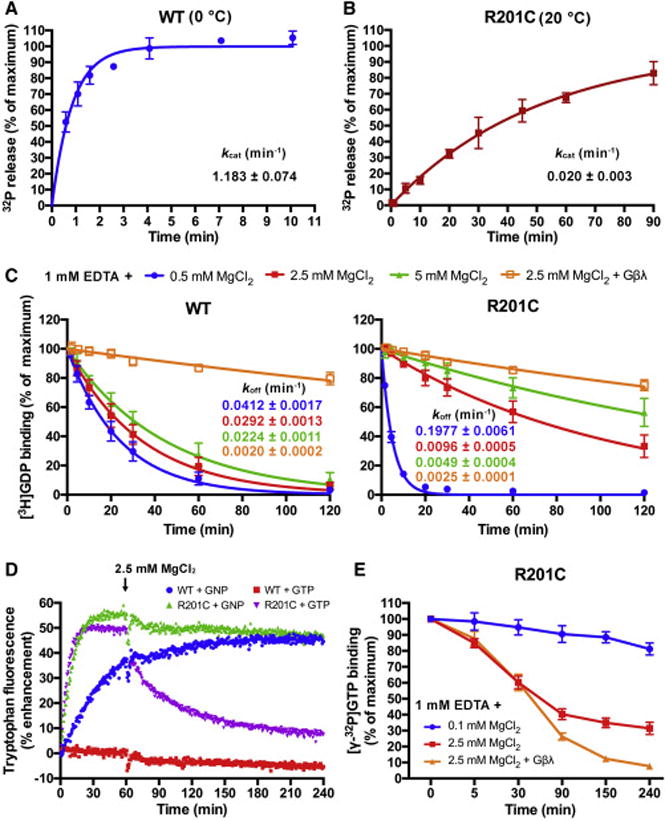
(A and B) The single turnover GTP hydrolysis rates (kcat) of wild type (WT) Gαs and the R201C mutant. Purified Gαs in a Mg2+-free or low Mg2+ buffer was first incubated with [γ-32P]GTP at the indicated temperature, then high concentration of Mg2+ and GTP were added to initiate the hydrolysis. 32PO43− release was quantified using liquid scintillation counting. The data represents the mean ± SD of four (WT) or three (R201C) independent measurements.
(C) Influence of MgCl2 and Gβγ subunits on the rates of GDP dissociation (koff) from WT Gαs and the R201C mutant. Gαs preloaded with [3H]GDP was assayed in a buffer containing 1 mM EDTA, 0.5 mM GDP and the indicated concentration of MgCl2 with or without 1 μM Gβ/γ(C68S). The data represents the mean ± SD of three independent measurements.
(D) The changes in the intrinsic tryptophan fluorescence of WT Gαs and the R201C mutant caused by binding of GNP, or binding and hydrolysis of GTP. 5 μM GDP-bound Gαs was mixed with 0.5 mM GNP or GTP in a buffer containing 1 mM EDTA and 0.1 mM MgCl2 to initiate the nucleotide exchange; after 1 hour, MgCl2 was added to a final concentration of 2.5 mM to decrease the GDP dissociation rates.
(E) Evaluation of the GTP occupancy of the R201C mutant in the presence of excess GTP. The R201C mutant was incubated with 20 nM [γ-32P]GTP and 400 μM GTP in a low Mg2+ buffer until the binding of [γ-32P]GTP to the R201C mutant reached a maximum, and the concentration of bound [γ-32P]GTP was measured and defined as the zero time point. Then MgCl2 or MgCl2 together with Gβ/γ(C68S) was added immediately, and the concentration of bound [γ-32P]GTP was measured at various time points. The data represents the mean ± SD of three independent measurements.
See also Figure S1.
GDP dissociation is the rate-limiting step in the process of GDP-GTP exchange (Gilman, 1987). We evaluated the GDP dissociation rates (koff) of WT Gαs and the R201C mutant in buffers containing different concentrations of MgCl2 and 1 mM EDTA. The concentrations of free Mg2+ were calculated using a method described previously (Higashijima et al., 1987c) (also see methods). Mg2+ was reported to increase the GTP binding affinity of Gα proteins, but had less effect on GDP binding (Higashijima et al., 1987c); in agreement with this, we found that the koff of WT Gαs was only slightly decreased by increasing the free Mg2+ concentration from 1 μM (1 mM EDTA + 0.5 mM MgCl2) to 3.6 mM (1 mM EDTA + 5 mM MgCl2) (Figure 1C). In contrast, the koff of the R201C mutant exhibited a quite different dependence on the Mg2+ concentration: it was nearly 5 times that of WT Gαs at a low Mg2+ concentration (1 mM EDTA + 0.5 mM MgCl2), but was decreased to about 1/3 of that of WT Gαs when the free Mg2+ concentration increased to 1.2 mM (1 mM EDTA + 2.5 mM MgCl2), and further decreased at a free Mg2+ concentration of 3.6 mM (1 mM EDTA + 5 mM MgCl2) (Figure 1C). In the cytoplasm of mammalian cells, the free Mg2+ concentration has been estimated to be 0.5-1 mM, with an additional 4-5 mM Mg2+ being in complex with phosphonucleotides and phosphometabolites, which represents a large Mg2+ pool (Romani, 2011). As a result, under physiological conditions, the R201C mutation is anticipated to significantly decrease the koff of Gαs. The presence of Gβγ subunits reduced the koff of both WT Gαs and the R201C mutant (Figure 1C). The R201C mutant exhibits a GDP-GTPγS exchange rate (kapp) slower than that of WT Gαs in the presence of 2.5 mM MgCl2 and 1 mM EDTA (Figure S1C), consistent with the koff values.
Based on the measured rate constants for the individual steps in the GTPase cycle it is possible to calculate the fraction of R201C Gαs bound to each nucleotide. In the presence of excess GTP, when the cycle of GTP binding, hydrolysis, and GDP release reaches equilibrium, the fraction of Gα proteins occupied by GTP is less than koff/(koff + kcat). In the presence of 2.5 mM MgCl2 and 1 mM EDTA, less than 32% of R201C is calculated to be in the GTP state without stimulation by guanine nucleotide exchange factors (GEFs), and Gβγ subunits can further lower the ratio to ~ 11%.
To experimentally measure the differences between WT Gαs and the R201C mutant in terms of both the GDP-GTP exchange and GTP hydrolysis steps we turned to intrinsic tryptophan fluorescence that has been used to monitor nucleotide exchange of G proteins, such as Gαo (Higashijima et al., 1987b) and Gαt (Phillips and Cerione, 1988). During replacement of GDP by GTP, three regions of G proteins, named switch I, II and III, undergo significant conformational changes (Lambright et al., 1994), resulting in an increase in the intrinsic tryptophan fluorescence; thus the change in tryptophan fluorescence can be used to quantify the ratio of Gαs that associates with GTP. GDP-bound WT Gαs and the R201C mutant were incubated with excess GTP or its non-hydrolysable analog GNP (Guanosine 5′-[β,γ-imido]triphosphate) (500 μM) in a buffer containing 0.1 μM free Mg2+ (1 mM EDTA + 0.1 mM MgCl2) to facilitate nucleotide exchange; after 1 hour, the concentration of MgCl2 was increased to 2.5 mM (about 1 mM free Mg2+ in the buffer, which is close to the concentration of cytoplasmic free Mg2+).
In the presence of GNP, the tryptophan fluorescence of WT Gαs increased nearly 40% but had not reached its maximum following a 1-hour incubation; the fluorescence of the R201C mutant increased about 55% to reach its maximum after 30 minutes, much faster than that of WT Gαs, indicating a faster rate of GNP binding (Figure 1D). The changes of the fluorescence before additional MgCl2 was added were consistent with the GDP dissociation data at a low Mg2+ concentration (0.5 mM MgCl2 + 1 mM EDTA) (Figure 1C).
In the presence of GTP, the fluorescence of WT Gαs did not increase but slightly decreased over time, which can be explained by the fast kcat and slow koff of WT Gαs; the slight decrease of tryptophan fluorescence is probably due to fluorescence quenching. Before MgCl2 concentration was increased, the fluorescence of the R201C mutant in the presence of GTP increased similarly to that in the presence of GNP, consistent with the fast koff and relatively slow kcat of the R201C mutant at a low Mg2+ concentration; but after the MgCl2 concentration was increased to 2.5 mM, the R201C fluorescence significantly decreased over time, and was close to the fluorescence of WT Gαs after 4 hours (Figure 1D), since the koff of the R201C mutant under this condition is slower than the kcat. This data supports our calculation that the R201C mutant is not locked in a GTP-bound state even in the presence of excess GTP, despite its significant loss of GTPase activity.
To validate the nucleotide state predicted based on tryptophan fluorescence we turned to a [γ-32P]GTP binding assay (Figure 1E). The R201C mutant was pre-incubated in a low Mg2+ buffer (1 mM EDTA + 0.1 mM MgCl2) with 400 μM GTP that is close to the physiological concentration of GTP (Traut, 1994); 20 nM [γ-32P]GTP was added as an internal standard. After the binding of [γ-32P]GTP to the R201C mutant reached a maximum, the concentration of free Mg2+ was increased to about 1.1 mM (1 mM EDTA + 2.5 mM MgCl2) and the changes of bound [γ-32P]GTP with time were measured. The bound [γ-32P]GTP decreased to about 30% of the maximum after 4 hours, which can be explained by the faster GTP hydrolysis than GDP dissociation. When Gβγ subunits were added together with MgCl2 (Gβγ:Gαs = 1.5:1, molar ratio), the bound [γ-32P]GTP further decreased to below 10% of the maximum after 4 hours, which supports the finding that Gβγ subunits decrease the rate of GDP dissociation (Figure 1C). In contrast, when the free Mg2+ concentration was kept at 0.1 μM (1 mM EDTA + 0.1 mM MgCl2), the bound [γ-32P]GTP only slowly decreased to about 80% of the maximum, which may be due to the instability of the R201C mutant in the low Mg2+ buffer.
These in vitro assays demonstrate that the R201C mutant is not locked in the GTP state, instead, without GEF stimulation it would be mainly in the GDP state in cells considering that the presence of Gβγ subunits and millimolar Mg2+ dramatically decrease the rate of GDP dissociation. This conclusion is supported by a previously published cellular study, in which the authors showed an increase of the adenylyl cyclase-activating activity of the R201C mutant when β-adrenergic receptor (β-AR) was stimulated by isoproterenol (Landis et al., 1989). If R201C was in a persistent GTP-bound state, such a β-AR agonist would not be able to stimulate R201C Gαs signaling.
Crystal structure of GDP-bound Gαs(R201C/C237S)
The GDP dissociation assay indicates that the R201C mutation not only decreases the GTP hydrolysis rate of Gαs, but may also changes the protein conformation to affect the Mg2+ and nucleotide binding properties. We attempted to solve the crystal structure of the R201C mutant in a GDP-bound state. Failure to obtain suitably diffracting crystals led us to consider modifications to the protein to aid crystallization. We performed a screen for oxidizable cysteine residues, since free cysteines on the protein surface often complicate crystallization and found that mutating C237 to serine enabled crystallization of the R201C mutant. Gαs(C237S) and Gαs(R201C/C237S) behaved the same as WT Gαs and the R201C mutant, respectively, in the GDP dissociation assay (Figures S2A and S2B), GTPγS binding assay (Figures S1C and S2C) and tryptophan fluorescence assay (Figures 1D and S2D).
The structure of Gαs(R201C/C237S) was determined by molecular replacement and refined to 1.7 Å (Table S1). The overall structure is shown in Figure 2A. Three switch regions, and the two mutant residues C201 and S237 are highlighted. In the nucleotide binding pocket, a Mg2+ ion coordinates with the β phosphate of GDP, the side chain of S54, and four water molecules; one of the four water molecules interacts with D223 at the N terminus of switch II through two hydrogen bonds (Figures 2B and S3A).
Figure 2. Crystal structure of GDP-bound Gαs(R201C/C237S).
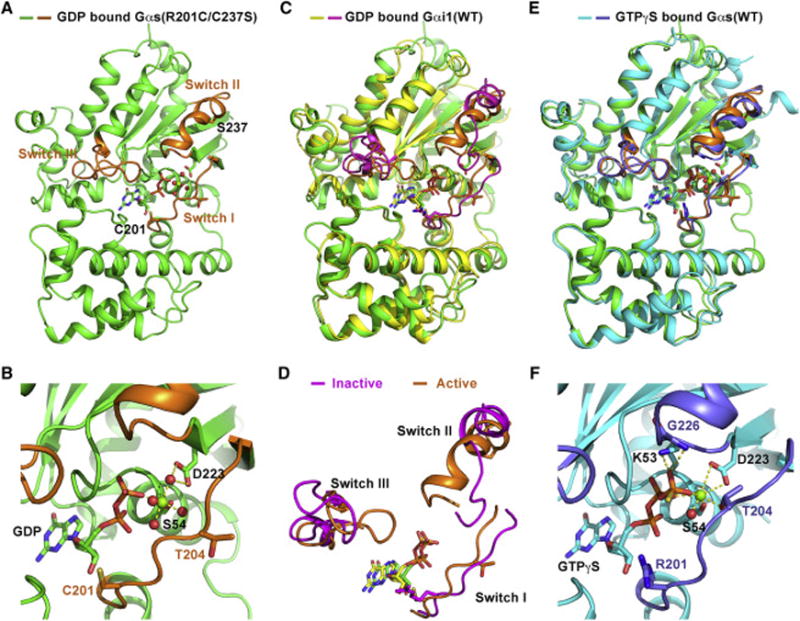
(A) The overall structure of GDP-bound Gαs(R201C/C237S). The switch I, II and III regions are colored orange. GDP and the side chains of residues C201 and S237 are showed as sticks.
(B) Structural details of nucleotide binding pocket in our structure. Water molecules and Mg2+ are shown as red and green spheres, respectively. Hydrogen bonds are represented by yellow dash lines.
(C) Alignment of our structure with the structure of GDP-bound Gαi1(WT) (colored yellow) in the crystal structure of Gαi1/Gβ1/γ2 heterotrimer (PDB code 1GP2). The switch regions of GDP-bound Gαi1(WT) are colored light magenta.
(D) The conformational differences between the switch regions in our structure (active) and that in the structure of GDP-bound Gαi1(WT) (inactive).
(E) Alignment of our structure with the crystal structure of GTPγS-bound Gαs(WT) (colored cyan, PDB code 1AZT). The switch regions of GTPγS-bound Gαs(WT) are colored slate.
(F) The local structure of the nucleotide binding pocket of GTPγS-bound Gαs(WT). All structural figures were made using PyMOL.
See also Figures S2, S3, and Table S1.
We attempted to overlay our structure with the crystal structure of a G protein in the GDP-bound state. No structure of GDP-bound Gαs has been reported, so instead, we used the structure of GDP-bound wild type Gαi1 in the crystal structure of Gαi1/Gβ1/γ2 heterotrimer (PDB code: 1GP2) (Wall et al., 1995), in which switch II and III of Gαi1 are stabilized by Gβ1/γ2 in a fully inactive conformation. Gαi1 shares a sequence identity of 41% with Gαs used in our study. The conformation of the switch regions in our GDP-bound Gαs(R201C/C237S) structure is quite different from the inactive conformation (Figure 2C). Specifically, in our structure the N terminus of switch II is well folded as an α-helix, and closely interacts with switch III; but in the inactive conformation represented by the GDP-bound Gαi1(WT) structure, the N terminus of switch II is unstructured, and is far from switch III (Figure 2D). There are two structures of GDP-bound WT Gαi1 monomer have also been reported, one is in a Mg2+-free state (Mixon et al., 1995), and the other is in a Mg2+-bound state (Coleman and Sprang, 1998). We didn’t choose them as representative of the inactive structure of Gαi1 because switch II and III in both the two structures are disordered and invisible; in addition, without the inhibition by Gβγ subunits, GDP-bound Gαs has considerable activity to activate adenylyl cyclase (Sunahara et al., 1997a), indicating that GDP-bound Gα proteins alone are not in a fully inactive state.
We next aligned our structure with a structure of GTPγS-bound Gαs (PDB code: 1AZT), which was the first crystal structure of Gαs solved, representing the active conformation of Gαs (Sunahara et al., 1997b). Surprisingly, the conformation of the switch regions in our structure is very similar to that in the structure of GTPγS-bound Gαs (Figure 2E). The local conformation of the nucleotide binding pocket in our structure is also nearly the same as that in the GTPγS-bound structure (Figure 2F), suggesting that our GDP-bound structure of Gαs(R201C/C237S) is in an active conformation.
The above analysis suggests that despite being bound to GDP, Gαs(R201C/C237S) is in an active conformation. This finding may provide an explanation for the GDP dissociation rates of the R201C mutant at different Mg2+ concentrations. In the presence of millimolar free Mg2+, GDP-bound Gαs(R201C) prefers to adopt an active-like conformation, in which GDP is not easily released, so its GDP dissociation rate is much slower than that of WT Gαs (Figure 1C); Mg2+ stabilizes the active-like conformation through forming water-mediated hydrogen bonds with D223, and through coordinating with GDP to inhibit GDP release (Figure 2B). Once the free Mg2+ concentration is lowered to micromolar range, GDP-bound Gαs(R201C) can no longer maintain an active-like conformation. Based on the crystal structure of GDP-bound WT Gαi1 (PDB code: 1GP2), in the inactive conformation, the side chains of R201 and E50 form two hydrogen bonds to block GDP dissociation (Figure 4C); the R201C mutation facilitates GDP dissociation through disrupting the two hydrogen bonds at low Mg2+ concentrations.
Figure 4. An intramolecular hydrogen bond network between the P-loop, switch III and switch II helps stabilize the active conformation of Gαs.
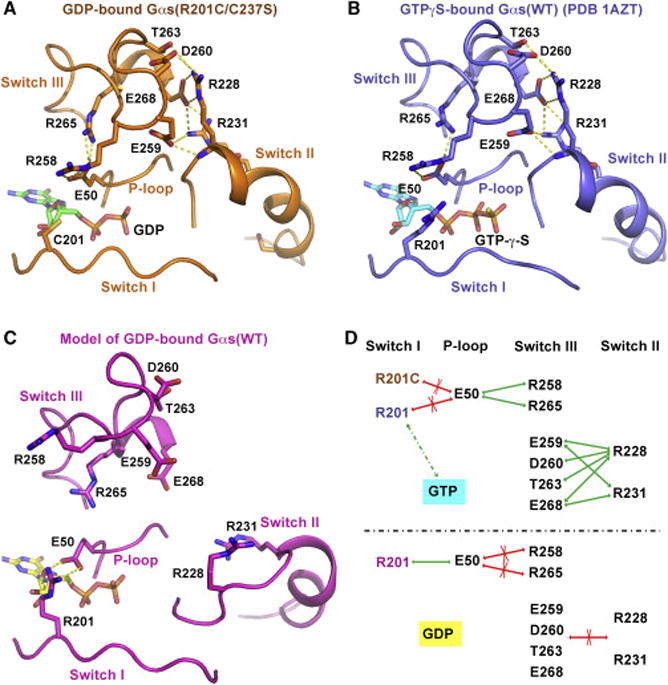
(A and B) In the crystal structure of GDP-bound Gαs(R201C/C237S) (A) and that of GTPγS-bound Gαs(WT) (B), E50 in the P-loop interacts with R258 and R265 in switch III through hydrogen bonding, while E259, D260, T263 and E268 in switch III accept hydrogen bonds from R228 and R231 in switch II. The hydrogen bonds are showed as yellow dash lines.
(C) A model of GDP-bound Gαs(WT) in an inactive conformation based on the crystal structure of GDP-bound Gαi1(WT) (PDB code: 1GP2) was built by SWISS-MODEL (Biasini et al., 2014). In this model, R201 in switch I donates two hydrogen bonds to E50 in the P-loop, blocking the interaction between E50 and R258, R265.
(D) A schematic diagram of the H-bond network. The green arrows represent the hydrogen bonds; the red arrows with a cross indicate the interaction is impaired; the green dash line represents the dynamic interaction between R201 and GTP.
See also Figures S3 and S4.
It should be noted that the active conformation of the GDP-bound Gαs(R201C) is not a perfect mimic of the canonical active state, as it lacks several stabilizing interactions observed in GNP-bound Gαs. In the latter, Mg2+ interacts with D223 directly, and forms a hydrogen bond with the side chain of T204 in switch I; the γ-phosphate accepts hydrogen bonds from the main chain amide of G226 and the side chain of K53 to further stabilize the active conformation (Figure 2F).
GDP-bound Gαs(R201C) effectively binds to and activates adenylyl cyclase
We next determined whether the GDP-bound Gαs(R201C) observed crystallographically can activate its downstream effector, adenylyl cyclase. The switch II region of Gαs is responsible for binding and activation of adenylyl cyclase. This same switch region is also involved in binding to Gβγ subunits. In its inactive conformation, switch II prefers to bind to Gβγ subunits, while in its active conformation, it prefers adenylyl cyclase. The ability of Gαs to bind adenylyl cyclase is strongly influenced by the presence of Gβγ subunits, so we included Gβγ subunits in our analysis. Five isoforms of Gβ and 12 isoforms of Gγ in mammalian cells have been identified (Khan et al., 2013); among them, the Gβ1/Gγ2 complex was reported to be one of the combinations that interact with Gαs (Rasmussen et al., 2011). Prenylation of Gγ at residue C68 is responsible for attaching the Gβγ complex to cell membranes (Muntz et al., 1992). The mutation C68S creates a soluble form of the Gβ1/Gγ2 complex. Nine isoforms of mammalian membrane-bound adenylyl cyclase have been identified; each isoform consists of two transmembrane domains and a cytoplasmic domain (Hanoune and Defer, 2001). The cytoplasmic domain, which can be further divided into C1 and C2 domains, is the catalytic domain that can be activated by Gαs. The full length as well as the cytoplasmic domain are difficult to overexpress in E. coli, but the C1 domain of adenylyl cyclase V (VC1) and C2 domain of adenylyl cyclase II (IIC2) can be overexpressed in E. coli. VC1 and IIC2 form a complex in the presence of Forskolin (FSK), and this complex can be activated by Gαs (Sunahara et al., 1997a). We expressed and purified the recombinant human VC1 and IIC2, as well as the Gβ1/Gγ2(C68S) complex, to reconstitute Gαs activity assays.
We used gel filtration to evaluate the ability of Gαs to bind to adenylyl cyclase (VC1/IIC2) in the presence of Gβ1/Gγ2(C68S) (Figure 3A). VC1/IIC2 and Gβ1/Gγ2(C68S) were injected separately as controls (Figure 3A, upper panel). After incubation with both VC1/IIC2 and Gβ1/Gγ2(C68S), WT Gαs in the GDP-bound state selectively bound Gβ1/Gγ2(C68S) to form a ternary complex, while in the GNP-bound state, it mainly formed a complex with VC1/IIC2 (Figure 3A, middle panel). In contrast, in the GDP-bound state, though most Gαs(R201C) associated with Gβ1/Gγ2(C68S), a small fraction formed a complex with VC1/IIC2; in the GNP-bound state, Gαs(R201C) exclusively bound to VC1/IIC2 (Figure 3A, bottom panel). The results of WT Gαs are consistent with the current view that Gα in GDP-bound state forms a ternary complex with Gβγ subunits, while in GTP-bound state, Gα binds to its effectors (Sprang, 2016). But the results of Gαs(R201C) indicate that the R201C mutation enables GDP-bound Gαs to bind to adenylyl cyclase even in the presence of Gβ1/Gγ2(C68S), though the binding is weaker than that between GNP-bound Gαs and adenylyl cyclase.
Figure 3. Comparison of the activities of WT Gαs and the R201C mutant to bind to and activate adenylyl cyclase.
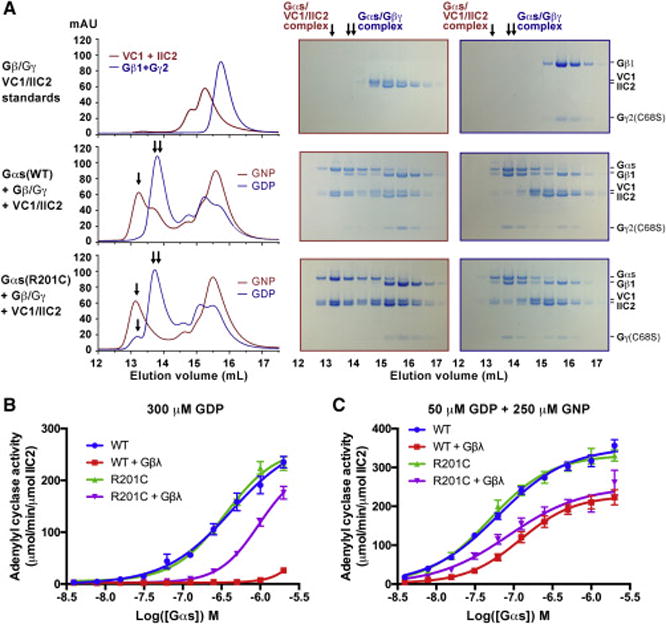
(A) GDP-bound Gαs(R201C) can partly bind to adenylyl cyclase even in the presence of Gβ1/γ2(C68S). WT Gαs or the R201C mutant was first incubated with GDP or GNP, and then incubated with Gβ1/γ2(C68S), adenylyl cyclase (consisting of VC1 and IIC2) and Forskolin (FSK). The mixture was then separated by gel filtration and analyzed by SDS-PAGE. The PAGE gels surrounded by red outlines and that surrounded by blue outlines correspond to the gel filtration chromatograms with red and blue colors, respectively. The single arrow (↓) and double arrow (↓↓) indicate the peak position of the Gαs/VC1/IIC2 complex and that of the Gαs/Gβ1/γ2(C68S) complex, respectively.
(B and C) Activation of adenylyl cyclase by Gαs in the absence or presence of Gβ1/γ2(C68S) complex. GDP-bound Gαs was first incubated with GDP (B) or a mixture of GDP and GNP (C), and then mixed with Gβ1/γ2(C68S) complex (or buffer), adenylyl cyclase (VC1/IIC2), FSK. After adding ATP, the reaction was carried out at 30 °C for 10 minutes. Production of cAMP was evaluated by the LANCE Ultra cAMP kit. The data represents the mean ± SE of six independent measurements.
Next, we evaluated the ability of Gαs to activate adenylyl cyclase. Production of cAMP catalyzed by adenylyl cyclase was measured by a time-resolved fluorescence resonance energy transfer (TR-FRET) assay. In the absence of Gβγ subunits, WT Gαs and the R201C mutant in the GDP-bound state showed similar activity (Figure 3B); but in the presence of Gβγ subunits, WT Gαs was significantly inhibited, while the R201C mutant was only modestly inhibited (Figure 3B). After incubation with GNP, both WT Gαs and the R201C mutant showed higher activities, and the activities also decreased in the presence of Gβγ subunits (Figure 3C), but again, the R201C mutant showed a higher activity. These results demonstrate a strong inhibitory effect of Gβγ subunits on the adenylyl cyclase-activating activity of GDP-bound Gαs, and prove that GDP-bound Gαs(R201C) can partly bind to and activate adenylyl cyclase even in the presence of Gβγ subunits.
An intramolecular hydrogen bond network stabilizes the active conformation of Gαs
How does the R201C mutation result in activation of GDP-bound Gαs even in the presence of Gβγ subunits? An NMR study of Gαi1 indicates that GDP-bound Gα subunits dynamically exist in two conformational states: a ground state that is preferred by G protein-coupled receptors (GPCRs), and an excited state that is similar to the GTP-bound state (Goricanec et al., 2016). The R201C mutation may shift the equilibrium towards the excited state. After analyzing the structures of GDP-bound Gαs(R201C/C237S) and GTPγS-bound WT Gαs (PDB code: 1AZT), we identified an intramolecular hydrogen bond network (H-bond network) between the P-loop, switch III and switch II in both structures. Residue E50 in the P-loop accepts hydrogen bonds from residues R258 and R265 in switch III. Residues E259, D260, T263 and E268 in switch III form hydrogen bonds with residues R228 and R231 in switch II (Figures 4A, 4B, S3B and S3C). We also built a model of the inactive state of Gαs based on the structure of GDP-bound Gαi1 (PDB code: 1GP2), using SWISS-MODEL (Biasini et al., 2014). In this model, residue R201 in switch I donates two hydrogen bonds to E50 in the P-loop; switch III and switch II are far from each other (Figure 4C).
Based on the analysis, we developed a model to explain the effect of the R201C mutation on the functional state of Gαs (Figure 4D). In the GDP-bound state, ammonium η1 and η2 of R201 tend to make an end-on salt-bridge with E50 to preclude the interactions between E50 and R258, R265 (Figure 4D, bottom). The R201C mutation results in the loss of ammonium η1 and η2 of R201 thus freeing E50 to interact with R258 and R265, bringing switch III close to switch II to facilitate their interactions (Figure 4D, top). This H-bond network stabilizes Gαs in an active-like conformation, decreasing its affinity for Gβγ subunits to facilitate its interaction with adenylyl cyclase. GTP binding may attract R201 to disrupt its interaction with E50, playing a similar role as the R201C mutation (Figure 4D, top). Most residues in this network are highly conserved across the α-subunits of human heterotrimeric G proteins (Figure S4) (Flock et al., 2015).
An arginine mimic lacking N(ε) at position 201 corrects GDP-misactivation of Gαs(R201C)
The above model indicates the importance of the interactions between E50 and ammonium η1 and η2 of R201 in maintaining GDP-bound Gαs in an inactive state. To test this model, we sought to replace the side chain of R201C with a close mimic of native arginine which contains N(η1) and N(η2) but lacking N(ε) which is not predicted to be involved in the H-bond network. The cysteine at position 201 offers an opportunity for site specific alkylation to introduce such an arginine analog. Upon reaction with cysteine residues, acrylamidine creates an arginine mimetic with an amidine functionality lacking N(ε) (Figure 5A) (Le et al., 2013). We first tested the reactivity of acrylamidine towards WT Gαs. In the construct of WT Gαs we used in this study, there are 7 native cysteine residues, but the liquid chromatography and mass spectrometry (LC/MS) analysis indicated that only one of them could be modified by acrylamidine, and when C237 was mutated to serine (C237S), no adduct peak could be detected (Figure 5B). This suggests that C237 is the only cysteine residue that can be modified by acrylamidine in WT Gαs. Next, we tested the ability of acrylamidine to modify Gαs(R201C), and two adduct peaks were detected: one was Gαs(R201C) modified by one molecule of acrylamidine, while the other was modified by two molecules of acrylamidine, indicating that C201 was also modified in addition to C237. In the R201C/C237S mutant, C201 is the only residue that can be modified by acrylamidine as only one adduct peak was detected (Figure 5B).
Figure 5. Modification of C201 by acrylamidine corrected the adenylyl cyclase-activating activity and restored the single turnover GTPase activity of the R201C mutant.
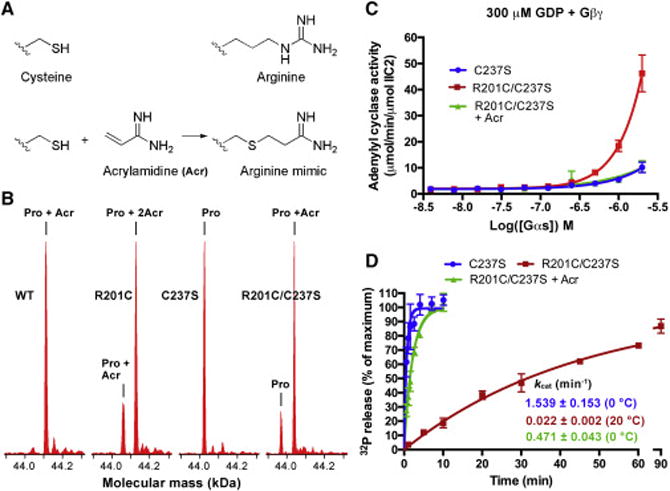
(A) The structures of the side chain of cysteine and arginine, and the reaction between acrylamidine (Acr) and the side chain of cysteine.
(B) Analysis of the modification of Gαs using liquid chromatography and mass spectrometry (LC/MS). Before analysis, 6 μM Gαs was incubated with 500 μM acrylamidine at room temperature for 2 hours. According to the molecular mass spectra, WT Gαs was completely modified by one molecule of Acr, and mutating C237 to serine blocked this modification; introduction of the R201C mutation enabled Gαs to be modified by one additional Acr.
(C) Modification of C201 by acrylamidine decreased the adenylyl cyclase-activating activity of the R201C/C237S mutant. The adenylyl cyclase activity was measured in the presence of GDP and Gβ1/γ2(C68S). The data represents the mean ± SE of three independent measurements.
(D) Modification of C201 by acrylamidine increased the single turnover GTPase activity of the R201C/C237S mutant to a level close to that of the C237S mutant. The data represents the mean ± SD of at least three independent measurements.
We evaluated the effect of modifying C201 with acrylamidine on the adenylyl cyclase-activating activity of GDP-bound Gαs(R201C/C237S). In the presence of GDP and Gβ1/Gγ2(C68S), the unmodified Gαs(R201C/C237S) showed significantly higher activity than Gαs(C237S) (Figure 5C), consistent with our finding that Gαs(R201C) has a higher activity than WT Gαs (Figure 3B). After Gαs(R201C/C237S) was modified by acrylamidine (free acrylamidine was removed by gel filtration), its ability to activate adenylyl cyclase was lowered to the same level as that of Gαs(C237S) (Figure 5C). We conclude that site specific modification of C201 with acrylamidine can effectively restore the canonical role of GDP to Gαs disrupted by the R201C mutation, supporting the role of N(η1) and N(η2) in restraining the GDP-bound form in a state that does not activate adenylyl cyclase in the presence of Gβγ subunits. We also demonstrated that this modification can partly restore the GTPase activity of Gαs(R201C/C237S). The kcat of Gαs(C237S) is 1.539 ± 0.153 min−1 (measured at 0 °C), slightly higher than that of WT Gαs (Figure 5D). The unmodified R201C/C237S mutant showed a slow kcat (0.022 ± 0.002 min−1) even at 20 °C, similar to that of Gαs(R201C). Following acrylamidine modification of the R201C/C237S mutant its kcat increased to 0.471 ± 0.043 min−1 (measured at 0 °C), about 30% of that of Gαs(C237S).
Mutations of the H-bond network destabilize the active state
Loss-of-function mutations in the H-bond network of Gαs have been identified in PHP-Ia and characterized by loss of the ability to stimulate Gαs (Lemos and Thakker, 2015). Two biochemical mechanisms have been characterized to explain their inactivating effects. (1) Mutations such as R231H (Iiri et al., 1997) and R265H (Leyme et al., 2014), impair Gαs activation through destabilization of GTP binding. (2) A second class of mutations, represented by R258A, turn off GTP-bound Gαs very quickly by increasing the intrinsic GTP hydrolysis rate of Gαs (Warner and Weinstein, 1999; Warner et al., 1998).
The finding that the gain-of-function mutation R201C stabilizes the H-bond network to activate GDP-bound Gαs lead us to investigate whether a third potential mechanism for the loss-of-function mutations could be due to destabilization of the active conformation in GTP-bound Gαs causing a decrease in effector stimulation. To test this mechanism, we generated three Gαs mutants: R265H, R258A, and R228C, the first two which have been characterized previously while R228C is a novel mutation recently identified in PHP-Ia (Tam et al., 2014). Residue R228 mediates the switch III-switch II interactions together with residue R231 (Figure 4).
We expressed and purified these mutants, and first tested their properties with respect to mechanisms (1) and (2). The R228C, R258A and R265H mutants all exhibited increased steady-state GTP hydrolysis rates, which were 1.5-, 4-, and 7.5-fold that of WT Gαs, respectively (Figure 6A). Since the GTP hydrolysis cycle consists of a slower nucleotide exchange step (replacement of GDP by GTP) (Brandt and Ross, 1985; 1986; Higashijima et al., 1987a) followed by a GTP hydrolysis step we hypothesized that mutations R228C, R258A and R265H accelerate nucleotide exchange. WT Gαs and the mutants in a GDP-bound state were incubated with [35S]GTPγS and excess GTPγS to measure GTPγS binding. All three Gαs mutants increased the apparent rate of GTPγS binding to Gαs (kapp) (Figures 6B and 6F). Among them, R265H showed the fastest binding rate, followed by R258A and R228C, consistent with their steady state GTP hydrolysis rates. Rates of GTP binding to G proteins are thought to be limited by GDP dissociation rates (Gilman, 1987). We confirmed that mutations R228C and R258A facilitate GDP dissociation (Figure S5). The rate of GDP dissociation from the R265H mutant was too fast to be quantified using the [3H]GDP assay.
Figure 6. Mutations of key residues in the H-bond network facilitated nucleotide exchange while decreased the adenylyl cyclase-activating activity of Gαs.
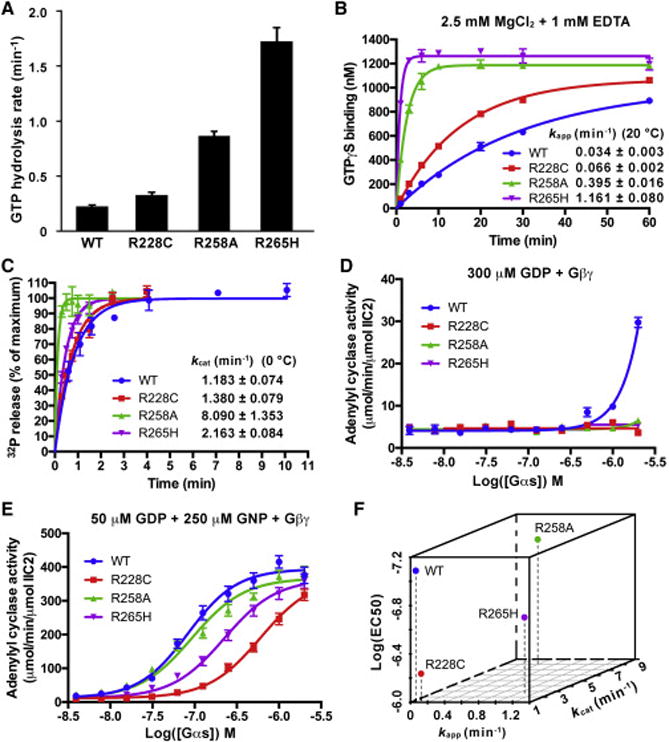
(A) The steady state GTP hydrolysis rates of WT Gαs and the R228C, R258A and R265H mutants were determined at 37 °C. The data represents the mean ± SD of three independent measurements.
(B) The rates of GTPγS binding to WT Gαs and the R228C, R258A and R265H mutants were determined by mixing GDP-bound Gαs with a mixture of [35S]GTPγS and GTPγS in a buffer containing 2.5 mM MgCl2 and 1 mM EDTA. The data represents the mean ± SD of two independent measurements.
(C) The single turnover GTPase activities of WT Gαs and the R228C, R258A and R265H mutants were determined at 0 °C. The data represents the mean ± SD of four (WT) or three (R228C, R258A and R265H) independent measurements. The data of WT Gαs used here was also used in Figure 1A.
(D and E) Activation of adenylyl cyclase by Gαs in the presence of Gβ1/γ2(C68S). GDP-bound Gαs was first incubated with GDP (D) or a mixture of GDP and GNP (E), and then mixed with Gβ1/γ2(C68S), adenylyl cyclase (VC1/IIC2) and FSK. The reaction was initiated by adding ATP. The data represents the mean ± SE of three (D) or nine (E) independent measurements.
(F) Visualization of the effects of mutations R228C, R258A and R265H on the apparent GTPγS binding rate (kapp), intrinsic GTPase activity (kcat) and the adenylyl cyclase-activating activity (EC50) of Gαs.
See also Figures S4, S5.
We next tested whether GTPase activity is elevated in the mutants (mechanism (2)) (Figures 6C and 6F). The GTP hydrolysis rate (kcat) of R258A is 8.090 ± 1.353 min−1, consistent with the reported data (Warner and Weinstein, 1999). The R265H mutant also showed a higher GTPase activity, with its kcat twice that of WT Gαs. The R228C mutant showed a similar GTPase activity to that of the WT. These results indicate that mechanism (2) is involved in the inactivating effects of the R258A and R265H mutations, but cannot explain how R228C decreases Gαs activity.
As a result of the ability of R228C Gαs to bind and hydrolyze GTP at close to wild-type efficiency, we asked if destabilization of the active state might be responsible for its loss-of-function behavior. We compared the adenylyl cyclase-activating activities of these mutants with that of WT Gαs in the presence of Gβ1/Gγ2(C68S). When 300 μM GDP was present, the activities of all three mutants were undetectable using the TR-FRET assay even at a concentration of 2 μM, while WT Gαs showed much higher activity at this concentration (Figure 6D). When GNP was added, the activities of these mutants were increased significantly, but were still lower than that of WT Gαs (Figures 6E and 6F). Among these, R228C severely decreased the activity of Gαs, providing a new mechanism to explain the loss-of-function behavior in which the stimulation of effector is diminished despite its retention of near WT levels of GTP binding and GTPase activity.
Discussion
GPCRs and G proteins comprise the largest family of signal transducing proteins in the human genome. G proteins are the targets of somatic and inherited mutations as well as cell penetrating toxins. The most frequent cancer-causing mutation among all heterotrimeric G proteins (and GPCRs) is position R201 in Gαs, leading to its constitutive activation, driving cAMP pathways (O’Hayre et al., 2013). This same arginine residue is ADP-ribosylated by the cholera toxin causing constitutive activation, elevation of cAMP levels, and activation of the cystic fibrosis transmembrane conductance regulator (CFTR) (De Haan and Hirst, 2004). The guanidine moiety of R201 is critical for GTP hydrolysis and its disruption by mutation (R201C/H) or ADP-ribosylation leads to a decrease in GTPase hydrolysis rate (Landis et al., 1989).
Mutations in oncogenes are often not confined to a single dominant hot-spot (e.g. RAS mutations at G12, Q61 both disrupt GTPase activity and are found frequently). Mutations which hyper-activate biochemical functions such as kinase activities in oncogenes such BRAF in contrast are often dominated by a single hotspot (V600E) due to the difficulty of enhancing in comparison to compromising catalytic activity. Our biochemical and structural data reveal that the R201C Gαs mutant not only exhibits decreased GTPase activity, but also is capable of activating adenylyl cyclase when bound to GDP even in the presence of Gβγ subunits. This new activation mechanism may explain the highly focused Gαs mutations in cancer. Although not a subject of our studies, several studies of the activation of R201 by ADP-ribosylation, suggest that mechanisms in addition to the loss of GTPase activity may also be involved. In particular, ADP-ribosylation of R201 was shown to disrupt Gαs binding to Gβγ (Kahn and Gilman, 1984). Such a result cannot be explained by the GTPase-inhibiting effect of ADP-ribosylation, but can be explained by our data.
One potential confounding aspect of the R201C mutant is the presence of two effects (1) a decrease in GTPase rate leading to increased stability of the activator-GTP, and (2) the ability to be active in the GDP state. To support the notion that pathophysiology can be driven by a non-canonical nucleotide state we would need to identify a disease-causing mutation in which the only effect is due to a non-canonical nucleotide effect. Therefore, we pursued the study of the mutations found in PHP-1a patients. The hypomorphic mutations of Gαs which cause PHP-1a have been identified at multiple residues with no dominant hot-spots, consistent with the notion that many different residues can be targeted to disrupt adenylyl cyclase stimulation by Gαs. Biochemical studies including those described here identified PHP-1a-causing Gαs mutations with increased GTPase activity which explain their hypomorphic activity. We did not find support (although we did not search exhaustively) for one mechanism proposed in the literature, that of an inability to bind GTP (Leyme et al., 2014). One of the hypomorphic mutations identified recently in patients, and biochemically analyzed here, R228C (Tam et al., 2014), could bind and hydrolyze GTP with nearly the same biochemical constants of WT Gαs, prompting our search for a new mechanism explaining the loss of G protein function. Based on our finding that the R201C mutation could subvert the normal inability of GDP to activate adenylyl cyclase, we asked if R228C was compromised in its ability to activate adenylyl cyclase when bound to GTP analogs. Indeed, even with GNP bound to R228C, the protein showed significantly reduced ability to stimulate adenylyl cyclase, again demonstrating the ability of a disease mutation to alter the normal nucleotide control of a G protein, in this case with GTP. That disease-causing mutations in Gαs can subvert the roles of both GDP and GTP provides new insights into the plasticity of these central switches in pathophysiology.
Our finding that the R201C mutant is not in a persistent GTP state sheds light on the future development of inhibitors of this oncogenic mutant. The best characterized small molecule inhibitor of heterotrimeric G proteins is the natural product YM-254890 which binds to GDP-bound Gαq to inhibit Gαq activation (Nishimura et al., 2010). Our results suggest that YM-254890 analogs which bind to Gαs rather than Gαq would be capable of treating patients with R201C mutation. Prior to our work, such a therapeutic strategy would not seem viable since the prior literature suggests only the GTP state predominates. Similar logic has been applied to find small molecule inhibitors of K-Ras (G12C) which bind to the GDP state of the oncogenic mutant in cells (Ostrem et al., 2013), when prior work had suggested oncogenic K-Ras mutants are uniformly in a persistent GTP state. These two examples of gain-of-function mutations in GTPases, suggest that the widespread view that such proteins are “locked” in the GTP state, which is widely appreciated to be recalcitrant to high affinity small molecule binding, is not correct, providing an opportunity for drug discovery against the GDP state.
STAR★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Kevan Shokat (Kevan.Shokat@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
WT Gαs, all the mutants of Gαs, the C1 domain (residues 442-658, VC1) of human ADCY5 (adenylyl cyclase V) and the C2 domain (residues 871-1082, IIC2) of human ADCY2 (adenylyl cyclase II) were overexpressed in Escherichia coli BL21(DE3) cultured in Terrific Broth (TB) Medium. Human GNB1 (Gβ1) and GNG2 (Gγ2) were co-expressed in Sf9 insect cells cultured in Sf-900™ III SFM medium at 28 °C.
METHOD DETAILS
Protein expression and purification
The gene of residues 7-380 of the short isoform of human Gαs (GNAS, accession number in PubMed: NP_536351) with a stop codon at its end was cloned into the NdeI/XhoI site of a modified pET15b vector, in which a Drice cleavage site (AspGluValAsp↓Ala) was inserted between the thrombin cleavage site and the NdeI site. The plasmid was transformed into Escherichia coli BL21(DE3). The transformed cells were grown in TB medium supplemented with 50 μg/mL carbenicillin at 37 °C until OD600 reached 0.5, and then cooled to 22 °C followed by addition of 40 μM β-D-thiogalactopyranoside (IPTG). After overnight incubation, the cells were harvested by centrifugation, resuspended in lysis buffer (150 mM NaCl, 25 mM Tris 8.0, 1 mM MgCl2), and then lysed by a microfluidizer. The cell lysate was centrifuged at 14000 g for 1 hour at 4 °C. The supernatant was incubated with TALON Resin (Clontech Laboratories, Inc.) at 4 °C for 1 hour, then the resin was washed by 500 mM NaCl, 25 mM Tris 8.0, 1 mM MgCl2 and 5 mM imidazole 8.0. Gαs was eluted by 25 mM Tris 8.0, 1 mM MgCl2, 250 mM imidazole 8.0, 10% glycerol and 0.1 mM GDP. After adding 5 mM Dithiothreitol (DTT), the eluate was incubated with Drice at 4 °C overnight to remove the hexahistidine tag and loaded into a Source-15Q column (GE Healthcare). Gαs was eluted by a linear gradient from 100% buffer A (25 mM Tris 8.0, 1 mM MgCl2) to 40% Buffer B (25 mM Tris 8.0, 1 M NaCl, 1 mM MgCl2). The peak fractions were pooled, supplemented with 5 mM DTT and 0.1 mM GDP, and then concentrated and purified by gel filtration (Superdex 200 increase, 10/30, GE Healthcare) with buffer C (150 mM NaCl, 20 mM HEPES 8.0, 5 mM MgCl2 and 1 mM EDTA-Na 8.0). The peak fractions were pooled and concentrated for biochemical assay. All mutants of Gαs were expressed and purified with the same protocol. The C2 domain of human ADCY2 (residues 871-1082, IIC2) was also expressed and purified with the same protocol, except that no GDP was added during purification.
Residues D628 and S645 in the C1 domain (residues 443-659) of mouse ADCY5 (adenylyl cyclase V) were mutated to glutamic acid and arginine, respectively, resulting a sequence that is the same as the C1 domain of human ADCY5 (residues 442-658). The gene of this sequence was cloned into the NdeI/XhoI site of a pET29b vector. The transformed Escherichia coli BL21(DE3) cells were cultured in TB medium supplemented with 50 μg/mL kanamycin at 37 °C until OD600 reached 0.5, and then cooled to 22 °C followed by addition of 40 μM IPTG. After incubation at 22 °C for 4-5 hours, the cells were harvested, lysed. After centrifugation, the supernatant was purified by TALON Resin with the same protocol described above. The eluate was mixed with 5 mM DTT and further purified by gel filtration (Superdex 200 increase, 10/30, GE Healthcare) with buffer C (150 mM NaCl, 20 mM HEPES 8.0, 5 mM MgCl2 and 1 mM EDTA-Na 8.0).
Human Gβ1 with a hexahistidine tag at its N terminus and human Gγ2(C68S) were cloned into pFastBac™ Dual expression vector (Invitrogen). The plasmid was transformed into DH10Bac competent cells to generate bacmid DNA, which was then used to generate baculoviruses in Sf9 insect cells (Invitrogen). Sf9 cells grown in Sf-900™ III SFM medium with a density of 1.8×106 cells/mL was infected by the baculoviruses. Forty-eight hours later, the cells were harvested by centrifugation, and resuspended in lysis buffer (150 mM NaCl, 25 mM Tris 8.0, 1 mM MgCl2) supplemented with cOmplete™ Protease Inhibitor Cocktail (Sigma-aldrich). The cells were disrupted by a microfluidizer. After centrifugation, the supernatant was purified by TALON Resin and gel filtration (Superdex 200 increase, 10/30, GE Healthcare) with the same buffers used for Gαs purification.
HPLC analysis
Wide type Gαs or the R201C mutant purified by gel filtration was concentrated to 11.5 mg/mL (about 0.25 mM). EDTA-Na 8.0 was added to a final concentration of 10 mM. The protein was then denatured by heating at 98 °C for 5 minutes. The bound nucleotides were released from the proteins and then analyzed by HPLC using a method described previously (Hertz et al., 2013). After centrifuging at 20,000 g for 10 minutes at room temperature, 20 μL of the supernatant was injected into a C-18 column (Agilent ZORBAX 300SB-C18) in a HPLC system (Waters) and eluted by a linear gradient from 100% buffer A (5 mM tetrabutylammonium bromide, 25 mM KH2PO4 pH 6.5, 5% acetonitrile) to 65% buffer B (5 mM tetrabutylammonium bromide, 25 mM KH2PO4 pH 6.5, 60% acetonitrile) over 40 minutes using a flow rate of 1 mL/min. As standards, GMP, GDP and GTP in the gel filtration buffer were also treated with EDTA and heat before injection.
Calculation of the free Mg2+ concentration
The concentrations of free Mg2+ were calculated following a method described previously (Higashijima et al., 1987c). In our in vitro assays, the total Mg2+ concentration is presumed to be the sum of the concentrations of free Mg2+, Mg2+ in complex with EDTA, GDP, GTP (or GNP, GTPγS), and Mg2+ bound to Gαs. Because the concentration of Gαs used in each assay was much lower than the total Mg2+ concentration, the contribution of Gαs to the free Mg2+ concentration was neglected in the calculation. The following equation was used to calculate the free Mg2+ concentration:
in which fi = [Mg2+]free/(Kdi + [Mg2+]free), and Kdi is the dissociation constant of the corresponding binding reaction. For the binding between EDTA and Mg2+, the Kd is 1 μM at pH 7.6 (Higashijima et al., 1987c), and we adopted this number in our calculation. The Kd value for GDP-Mg2+ binding or for GTP-Mg2+ binding has not been reported, so instead, we used the Kd values for ADP-Mg2+ binding (670 ± 50 μM) and for ATP-Mg2+ binding (35 ± 3 μM) reported previously (Gout et al., 2014) as the approximate Kd for GDP-Mg2+ binding and for GTP-Mg2+ binding, respectively. We also presumed that the Kd for GNP-Mg2+ binding and for GTPγS-Mg2+ binding are the same as that for ATP-Mg2+ binding.
Steady state GTPase assay
WT Gαs and its mutants purified by gel filtration were concentrated to 8.5 mg/mL and then adjusted to 3 μM (WT) or 0.6 μM (R228C, R258A, R265H) in 20 mM HEPES 7.5, 150 mM NaCl, 1 mM EDTA-Na 8.0. The proteins were 1:1 (v:v) diluted with the reaction buffer (20 mM HEPES 7.5, 150 mM NaCl, 20 mM MgCl2 and 1 mM GTP), and incubated at 37 °C. After 20, 30, 48, 70 or 100 minutes, 100 μL of the sample was removed to measure the inorganic phosphate (Pi) concentration by PiColorLock™ Phosphate Detection kit (Innova Biosciences). A standard curve was made using the 0.1 mM Pi stock in the kit.
Tryptophan fluorescence
Gαs at a concentration of 8.5 mg/mL in 20 mM HEPES 8.0, 150 mM NaCl were adjusted to 10 μM in the dilution buffer (20 mM HEPES 7.5, 150 mM NaCl, 1 mM EDTA-Na 8.0, 2 mM DTT and 50 uM GDP), transferred to a costar 96-well black microplate with a volume of 50 μL/well. After adding 50 μL of GTP or GNP stock (1 mM GTP or GNP in the dilution buffer plus 0.2 mM MgCl2) to each well, the plate was immediately read by a SpectraMax M5 plate reader (Molecular Devices) at room temperature using an excitation wavelength of 290 nm and an emission wavelength of 340 nm.
Gel filtration
GDP-bound WT Gαs or the R201C mutant (30 μM) was incubated with 1 mM GDP, 45 μM VC1, 60 μM IIC2, 39 μM Gβ1/γ2(C68S) complex, 100 μM Forskolin (FSK), 2 mM DTT on ice for 2 hours. Then 200 μL of the mixture was separated by gel filtration (Superdex 200 increase, 10/30, GE Healthcare) with buffer D (150 mM NaCl, 20 mM HEPES 8.0, 5 mM MgCl2, 1 mM EDTA-Na 8.0, 2 mM DTT and 25 μM FSK). The fractions were analyzed by SDS-PAGE and stained by Coomassie blue.
GNP-bound Gαs was incubated with the same components except that GDP was replaced by GNP. To generate GNP-bound Gαs(WT) or Gαs(R201C), the protein purified by Source-15Q column was concentrated to about 12 mg/mL and then diluted to about 1 mg/mL with a buffer containing 20 mM HEPES 8.0, 150 mM NaCl, 1 mM EDTA, 5 mM DTT and 0.5 mM GNP. After incubation at room temperature for 1 hour, 20 mM MgCl2 was added to stabilize the protein. The GNP-bound protein was purified by gel filtration (Superdex 200 increase, 10/30, GE Healthcare) with buffer C (150 mM NaCl, 20 mM HEPES 8.0, 5 mM MgCl2 and 1 mM EDTA-Na 8.0).
Adenylyl cyclase activity assay
WT Gαs and the mutants at a concentration of 8.5 mg/mL (about 190 μM) in 20 mM HEPES 8.0, 150 mM NaCl, 5 mM MgCl2, 1 mM EDTA-Na 8.0 were diluted to a series concentrations (8 μM, 4 μM, 2 μM, 1 μM, 0.5 μM, 0.25 μM, 125 nM, 62.5 nM, 31 nM, 15.6 nM) in buffer E (1× PBS 7.4, 0.1% BSA, 1 mM EDTA-Na 8.0, 2 mM DTT) plus 0.2 mM GDP. 5 μL of each sample was then mixed with 5 μL of GDP stock (buffer E plus 1 mM GDP) or GNP stock (buffer E plus 1 mM GNP) in a non-skirted 96-well PCR plate (USA Scientific). After incubation at room temperature for 1.5 hour to allow nucleotide exchange, 2 μL of MgCl2 stock (20 mM MgCl2, 1× PBS 7.4, 0.1% BSA) was added, followed by addition of 4 μL of AC stock (10 μM VC1, 5 nM IIC2, 150 μM FSK, 1× PBS 7.4, 0.1% BSA) or AC/Gβγ stock (AC stock plus 20 μM Gβ1/γ2(C68S)). After incubation at room temperature for 1 hour, the samples were placed on ice for 5 minutes. cAMP production was initiated by addition of 4 μL of ATP stock (1 mM ATP, 1× PBS 7.4, 0.1% BSA). The reaction was carried out at 30 °C for 10 minutes in a PCR machine, and stopped by heating at 95 °C for 3 minutes.
The cAMP concentrations were measured by the LANCE Ultra cAMP kit (PerkinElmer). A cAMP standard curve was generated in the same plate using the 50 μM cAMP standard in the kit. Before the measurement, the samples were diluted by stimulation buffer (1× PBS 7.4, 0.1% BSA) to 1/60, 1/120, 1/240 or 1/480 to make sure the cAMP concentrations were in the dynamic range of the cAMP standard curve. 20 μL of each diluted sample was mixed with 10 μL of 4X Eu-cAMP tracer and 10 μL of 4X ULight-anti-cAMP in a white, opaque Optiplate-384 microplate, incubated for 1 hour at room temperature, and the time-resolved fluorescence resonance energy transfer (TR-FRET) signals were read on a Spark 20M plate reader.
The cAMP standard curve was fitted by the software GraphPad Prism using the following equation in which “Y” is the TR-FRET signal and “X” is the log of cAMP standard concentration (M):
After obtained the values of the four parameters “Bottom”, “Top”, “LogIC50” and “HillSlope”, we used this equation to convert the TR-FRET signals of the samples into cAMP production values. The cAMP production curves were fitted by the following equation to calculated LogEC50:
in which “Y” is the cAMP production value, “X” is the log of Gαs concentration (M).
GDP dissociation assay
Gαs at a concentration of 8.5 mg/mL (about 190 μM) in 20 mM HEPES 8.0, 150 mM NaCl, 5 mM MgCl2, 1 mM EDTA-Na 8.0 was diluted to 400 nM in an EDTA buffer (20 mM HEPES 7.5, 150 mM NaCl, 1 mM EDTA-Na 8.0, 2 mM DTT). [3H]GDP (1 mCi/mL, 24 μM) was added to a final concentration of 1.2 μM. After incubation at 20 °C for 3 hours, the same volume of assay buffer (1 mM, 5 mM or 10 mM MgCl2 in the EDTA buffer plus 1 mM GDP, or 5 mM MgCl2 and 2 μM Gβ/γ(C68S) in EDTA buffer plus 1 mM GDP) was added to initiate [3H]GDP dissociation. At various points, 20 μL of the sample was removed and mixed with 380 μL of ice-cold wash buffer (20 mM HEPES 7.5, 150 mM NaCl, 20 mM MgCl2). The mixture was immediately filtered through a mixed cellulose membrane (25 mm, 0.22 μm) held by a microanalysis filter holder (EMD Millipore). The membrane was washed by ice-cold wash buffer (500 μL × 3), put in a 6-mL plastic vial and air-dried (room temperature 1.5 h). 5 mL of CytoScint™-ES Liquid Scintillation Cocktail (MP Biomedicals) was added to each vial. After incubation overnight at room temperature, the vial was used for liquid scintillation counting with a LS 6500 Multi-Purpose Scintillation Counter (Beckman Coulter).
The GDP dissociation curves were fitted by the software GraphPad Prism using the following equation to calculate the dissociation rates (k):
in which “Y” is the radioactivity (Counts per minute) of the sample at time “X” (minutes), and Y0 is the calculated radioactivity of the sample at the time point 0.
GTPγS binding assay
Gαs at a concentration of 8.5 mg/mL (about 190 μM) in 20 mM HEPES 8.0, 150 mM NaCl, 5 mM MgCl2, 1 mM EDTA-Na 8.0 was diluted to 10 μM with dilution buffer (20 mM HEPES 7.5, 150 mM NaCl, 2.5 mM MgCl2, 1 mM EDTA-Na 8.0, 2 mM DTT). GTPγS binding was initiated by diluting the sample to 2 μM with the reaction buffer (50 nM [35S]GTPγS and 100 μM GTPγS in dilution buffer) at room temperature. At various time points, 10 μL of the sample was removed and mixed with 390 μL of ice-cold wash buffer (20 mM HEPES 7.5, 150 mM NaCl, 20 mM MgCl2). The mixture was filtered through a mixed cellulose membrane (25 mm, 0.22 μm). The membrane was washed by ice-cold wash buffer (500 μL × 3), put in a 6-mL plastic vial and air-dried (room temperature 1.5 h). 5 mL of CytoScint™-ES Liquid Scintillation Cocktail (MP Biomedicals) was added to each vial. After incubation overnight at room temperature, the vial was used for liquid scintillation counting with a LS 6500 Multi-Purpose Scintillation Counter (Beckman Coulter).
A standard curve was generated using [35S]GTPγS. The radioactive activity (Counts per minute) of the samples were converted to the GTPγS concentration. The GTPγS binding curves were fitted by the software GraphPad Prism using the following equation to calculate the apparent GTPγS binding rates (kapp):
in which “Y” is the concentration of GTPγS that bound to Gαs at time “X” (minutes).
Single turnover GTPase assay
Gαs (WT, C237S, R201C/C237S-acrylamidine, R228C, R258A, R265H) at a concentration of 8.5 mg/mL (about 190 μM) in 20 mM HEPES 8.0, 150 mM NaCl was diluted to 1 μM in EDTA buffer (20 mM HEPES 7.5, 150 mM NaCl, 5 mM EDTA-Na 8.0, 2 mM DTT). After incubation with 10 nM [γ-32P]GTP (6476 Ci/mmol) on ice for 1.5 hour, 4 μL of the sample was removed and mixed with 580 μL of ice-cold 5% activated charcoal in 20 mM H3PO4, pH 2.4 (time point 0), while 29 μL of the sample was mixed with 116 μL of reaction buffer (20 mM HEPES 7.5, 150 mM NaCl, 20 mM MgCl2, 0.25 mM GTP) on ice. At various time points, 20 μL of the sample was removed and mixed with 580 μL of ice-cold 5% (wt/vol) activated charcoal in 20 mM H3PO4, pH 2.4 to stop the reaction. The activated charcoal slurry was then centrifuged for 5 min at 20,000 × g (4 °C), and 500 μL of the supernatant was carefully removed and mixed with 4.5 mL of CytoScint™-ES Liquid Scintillation Cocktail (MP Biomedicals) in a 6-mL vial. The vial was used for liquid scintillation counting with a LS 6500 Multi-Purpose Scintillation Counter (Beckman Coulter). For each sample, the background [32P]Pi detected at time point 0 was subtracted from the data.
To measure the single turnover GTP hydrolysis rates of the R201C and R201C/C237S mutants, the proteins were diluted to 1 μM in a low Mg2+ buffer (20 mM HEPES 7.5, 150 mM NaCl, 1 mM EDTA-Na 8.0, 0.1 mM MgCl2, 2 mM DTT) and incubated with 10 nM [γ-32P]GTP (6476 Ci/mmol) at room temperature for 30 minutes before reaction buffer was added. The release of 32PO43− was measured at 20 °C.
The data was fitted by the software GraphPad Prism using the following equation to calculate the single turnover GTP hydrolysis rates (kcat):
in which “Y” is the radioactivity of the [32P]Pi released from the protein at time “X” (minutes).
[γ-32P]GTP binding assay
The R201C mutant of Gαs at a concentration of 8.5 mg/mL (about 190 uM) in 20 mM HEPES 8.0, 150 mM NaCl was diluted to 10 μM in dilution buffer (20 mM HEPES 7.5, 150 mM NaCl, 0.1 mM MgCl2, 1 mM EDTA-Na 8.0, 2 mM DTT). GTP binding was initiated by mixing 1 volume of the protein with 4 volumes of the reaction buffer (dilution buffer + 25 nM [γ-32P]GTP + 500 μM GTP) at room temperature for 1 hour. 10 μL of the sample (1.238 Ci/) was removed to measure the concentration of bound [γ-32P]GTP. This concentration was defined as the concentration at the zero time point. Then the sample was mixed with 1/10 volume of dilution buffer or MgCl2 buffer (dilution buffer + 26.4 mM MgCl2) or MgCl2/Gβγ (dilution buffer + 26.4 mM MgCl2 + 30 μM Gβγ). After that, at each time point (5 min, 0.5 hour, 1.5 hours, 2.5 hours and 4 hours), 11 μL of the sample was removed and added into 390 μL of ice-cold wash buffer (20 mM HEPES 7.5, 150 mM NaCl, 20 mM MgCl2). The mixture was filtered through mixed cellulose membranes (25 mm, 0.22 μm), washed by ice-cold wash buffer (500 μL × 3), and air-dried (room temperature 1.5 hours). The membrane was put in a 6-mL plastic vial in which 5 mL of CytoScint™-ES Liquid Scintillation Cocktail (MP Biomedicals) was added. After incubation overnight at room temperature, the vial was used for liquid scintillation counting with a LS 6500 Multi-Purpose Scintillation Counter (Beckman Coulter). A standard curve was generated using [γ-32P]GTP.
Synthesis of acrylamidine
Acrylamidine was synthesized following the procedure in published literatures (Le et al., 2013; Zuev and Sheridan, 2004). A mixture of 760 mg of NH4Cl in 10 mL anhydrous toluene was stirred at 0 °C under argon atmosphere. Then 6.6 mL of trimethylaluminum solution in toluene (2.0 M) was added carefully. The reaction was stirred at room temperature (about 20 °C) for 2 hours. After addition of 0.6 mL of acrylonitrile, the reaction was warmed to 80 °C and stirred for 18 hours. The reaction was then transferred to a slurry of 5 g of silica gel in 15 mL of CHCl3, stirred on ice for 1 hour, and filtered and washed with methanol. The filtrate was evaporated to give the product as a white solid (509 mg, 80%). Our NMR data matched the reported data (Zuev and Sheridan, 2004). 1H NMR (400 MHz, DMSO-d6): δ 9.22 (broad s, 1.5 H), 8.87 (broad s, 1.5 H), 6.57 (d, 1 H, J=17.6 Hz), 6.35 (dd, 1 H, J=17.6, 11.1 Hz), 6.12 (d, 1H, 11.1 Hz).
Crystallization
Gαs(R201C/C237S) that was purified by gel filtration was concentrated to 10 mg/mL and mixed with 1 mM GDP. For crystallization, 0.1 μL of the protein was mixed with 0.1 μL of the well buffer containing 0.1 M Tris 8.5, 25% PEG8000, 10 mM TCEP hydrochloride. Crystals were grown at 20 °C in a 96-well plate using the hanging-drop vapour-diffusion method, transferred to a cryoprotectant solution (the well buffer plus 25% glycerol), and flash-frozen in liquid nitrogen.
Data collection and structure determination
The data set was collected at the Advanced Light Source beamline 8.2.2 with X-ray at a wavelength of 0.999907 Å. Then the data set was indexed and integrated using iMosflm (Battye et al., 2011), scaled with Scala (Evans, 2006) and solved by molecular replacement using Phaser (McCoy et al., 2007) in CCP4 software suite (Winn et al., 2011). The crystal structure of GTPγS-bound Gαs (PDB code: 1AZT) was used as the initial model. The structure was manually refined with Coot (Emsley et al., 2010) and PHENIX (Adams et al., 2010). Data collection and refinement statistics can be found in Table S1 (related to Figure 2). In the Ramachandran plot of the final structure, 97.64% and 2.06% of the residues are in the favored regions and allowed regions, respectively, while one residue, V65 in a loop, is calculated as an outlier.
QUANTIFICATION AND STATISTICAL ANALYSIS
The curves in Figures 1A, 1B, 1C, 3B, 3C, 5C, 5D, 6B, 6C, 6D, 6E, S1C, S2A, S2B, S2C and S5 were fitted by GraphPad Prism. The data in Figure 6A was analyzed by Excel. All the details can be found in the figure legends of these figures and in the METHOD DETAILS. The data collection and refinement statistics of the crystal structure of the GDP-bound Gαs(R201C/C237S) can be found in Table S1 (related to Figure 2).
DATA AND SOFTWARE AVAILABILITY
Data Resources
The crystal structure of GDP-bound Gαs(R201C/C237S) has been deposited in the PDB under ID code 6AU6.
Supplementary Material
Figure S1: Nucleotide occupancy and GTPγS binding rates of WT Gαs and the R201C mutant. Related to Figure 1.
(A) Purified, recombinant wild type (WT) Gαs and the R201C mutant were examined by SDS– PAGE and visualized by Coomassie blue staining.
(B) HPLC analysis of the nucleotide occupancy of Gαs. WT Gαs or the R201C mutant purified from E. coli were denatured to release the bound nucleotide. The supernatant was analyzed by HPLC. The curves showed here represent the UV absorbance at 254 nm.
(C) The rates of GTPγS binding to WT Gαs and the R201C mutants were determined by mixing GDP-bound Gαs with a mixture of [35S]GTPγS and GTPγS in a buffer containing 2.5 mM MgCl2 and 1 mM EDTA. The data represents the mean ± SD of three independent measurements.
Figure S2: The mutant Gαs(R201C/C237S) exhibited similar behavior to the R201C mutant. Related to Figure 2.
(A and B) Influence of MgCl2 concentration on the rates of GDP dissociation from the C237S and the R201C/C237S mutants. Gαs preloaded with [3H]GDP was assayed in a buffer containing 1 mM EDTA, 0.5 mM GDP and 0.5 mM or 5 mM MgCl2. The data represents the mean ± SD of three independent measurements.
(C) Rates of GTPγS binding to the C237S and the R201C/C237S mutants in the presence of 2.5 mM MgCl2 and 1 mM EDTA at room temperature. The data represents the mean ± SD of three independent measurements.
(D) The changes in the intrinsic tryptophan fluorescence of the C237S and the R201C/C237S mutants in response to GNP binding or GTP binding and hydrolysis. 5 μM GDP-bound Gαs was mixed with 0.5 mM GNP or GTP in a buffer containing 1 mM EDTA and 0.1 mM MgCl2 to initiate the nucleotide exchange; after 1 hour, MgCl2 was added to a final concentration of 2.5 mM to decrease the GDP dissociation rates.
Figure S3: GDP and water molecules in the nucleotide-binding pocket and key residues in the hydrogen bond network of Gαs are well defined in the 1.7 Å structure of Gαs(R201C/C237S). Related to Figures 2 and 4.
(A) GDP and the sidechains of C201, T204 and D223 are showed as sticks. The Mg2+ and the water molecules coordinated with the Mg2+ are showed as green and red spheres, respectively.
(B) The sidechains of E50 in the P-loop and R258 and R265 in switch III are showed as sticks.
(C) The sidechains of E259, D260, T263 and E268 in switch III and R228 and R231 in switch II are showed as sticks. The 2mFo-DFc electron density map of the structure is contoured at 2.0 σ and colored blue.
Figure S4: Key residues in the intramolecular H-bond network of Gαs are highly conserved in other 15 human Gα proteins. Related to Figures 4 and 6.
The protein sequence of human Gαs (GNAS2) was aligned with that of other 15 human Gα proteins. The key residues in the intramolecular hydrogen bond network are showed here. Residues that are not conserved in other Gα proteins are colored yellow.
Figure S5: Effects of the R228C and R258A mutations on the rate of GDP dissociation from Gαs. Related to Figure 6.
Gαs preloaded with [3H]GDP was assayed in a buffer containing 1 mM EDTA, 2.5 mM MgCl2 and 0.5 mM GDP. The data represents the mean ± SD of three (WT) or two (R228C, R258A) independent measurements. The data of WT Gαs used here was also used in Figure 1C.
Highlights.
The oncogenic Gαs mutation R201C allows GDP-bound Gαs to activate adenylyl cyclase
GDP-bound Gαs(R201C/C237S) adopts an active state in its crystal structure
The R201C mutation activates Gαs through stabilizing an intramolecular H-bond network
Loss-of-function mutations R228C and R265H destabilize the GTP active state of Gαs
Acknowledgments
We would like to thank the staff at A.L.S. beamline 8.2.2. We also would like to thank Drs. Henry Bourne, Davide Ruggero, and Aashish Manglik for helpful comments, and Xi Liu for assistance in protein expression. This work was supported by the Howard Hughes Medical Institute and R01CA190408 to K.M.S. Q.H. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-[2229-15]).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
K.M.S. conceived the project. Q.H. and K.M.S designed the experiments. Q.H. performed the experiments. K.M.S and Q.H. analyzed the data and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne HR. G proteins. The arginine finger strikes again. Nature. 1997;389:673–674. doi: 10.1038/39470. [DOI] [PubMed] [Google Scholar]
- Brandt DR, Ross EM. GTPase activity of the stimulatory GTP-binding regulatory protein of adenylate cyclase, Gs. Accumulation and turnover of enzyme-nucleotide intermediates. J Biol Chem. 1985;260:266–272. [PubMed] [Google Scholar]
- Brandt DR, Ross EM. Catecholamine-stimulated GTPase cycle. Multiple sites of regulation by beta-adrenergic receptor and Mg2+ studied in reconstituted receptor-Gs vesicles. J Biol Chem. 1986;261:1656–1664. [PubMed] [Google Scholar]
- Coleman DE, Sprang SR. Crystal structures of the G protein Gi alpha 1 complexed with GDP and Mg2+: a crystallographic titration experiment. Biochemistry. 1998;37:14376–14385. doi: 10.1021/bi9810306. [DOI] [PubMed] [Google Scholar]
- De Haan L, Hirst TR. Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms (Review) Mol Membr Biol. 2004;21:77–92. doi: 10.1080/09687680410001663267. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr. D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Gilman AG. G Proteins and Regulation of Adenylate Cyclase (Nobel Lecture) Angewandte Chemie International Edition in English. 1995;34:1406–1419. [Google Scholar]
- Goricanec D, Stehle R, Egloff P, Grigoriu S, Plückthun A, Wagner G, Hagn F. Conformational dynamics of a G-protein α subunit is tightly regulated by nucleotide binding. Proc Natl Acad Sci USA. 2016;113:E3629–E3638. doi: 10.1073/pnas.1604125113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout E, Rébeillé F, Douce R, Bligny R. Interplay of Mg2+, ADP, and ATP in the cytosol and mitochondria: unravelling the role of Mg2+ in cell respiration. Proc Natl Acad Sci USA. 2014;111:E4560–E4567. doi: 10.1073/pnas.1406251111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, Shokat KM. A Neo-Substrate that Amplifies Catalytic Activity of Parkinson’s-Disease-Related Kinase PINK1. Cell. 2013;154:737–747. doi: 10.1016/j.cell.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashijima T, Ferguson KM, Smigel MD, Gilman AG. The effect of GTP and Mg2+ on the GTPase activity and the fluorescent properties of Go. J Biol Chem. 1987a;262:757–761. [PubMed] [Google Scholar]
- Higashijima T, Ferguson KM, Sternweis PC, Ross EM, Smigel MD, Gilman AG. The effect of activating ligands on the intrinsic fluorescence of guanine nucleotide-binding regulatory proteins. J Biol Chem. 1987b;262:752–756. [PubMed] [Google Scholar]
- Higashijima T, Ferguson KM, Sternweis PC, Smigel MD, Gilman AG. Effects of Mg2+ and the beta gamma-subunit complex on the interactions of guanine nucleotides with G proteins. J Biol Chem. 1987c;262:762–766. [PubMed] [Google Scholar]
- Iiri T, Farfel Z, Bourne HR. Conditional activation defect of a human Gsalpha mutant. Proceedings of the National Academy of Sciences. 1997;94:5656–5661. doi: 10.1073/pnas.94.11.5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn RA, Gilman AG. ADP-ribosylation of Gs promotes the dissociation of its alpha and beta subunits. J Biol Chem. 1984;259:6235–6240. [PubMed] [Google Scholar]
- Khan SM, Sleno R, Gora S, Zylbergold P, Laverdure JP, Labbe JC, Miller GJ, Hebert TE. The Expanding Roles of G Subunits in G Protein-Coupled Receptor Signaling and Drug Action. Pharmacological Reviews. 2013;65:545–577. doi: 10.1124/pr.111.005603. [DOI] [PubMed] [Google Scholar]
- Lambright DG, Noel JP, Hamm HE, Sigler PB. Structural determinants for activation of the alpha-subunit of a heterotrimeric G protein. Nature. 1994;369:621–628. doi: 10.1038/369621a0. [DOI] [PubMed] [Google Scholar]
- Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340:692–696. doi: 10.1038/340692a0. [DOI] [PubMed] [Google Scholar]
- Le DD, Cortesi AT, Myers SA, Burlingame AL, Fujimori DG. Site-specific and regiospecific installation of methylarginine analogues into recombinant histones and insights into effector protein binding. J Am Chem Soc. 2013;135:2879–2882. doi: 10.1021/ja3108214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos MC, Thakker RV. GNAS mutations in Pseudohypoparathyroidism type 1a and related disorders. Hum Mutat. 2015;36:11–19. doi: 10.1002/humu.22696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyme A, Marivin A, Casler J, Nguyen LT, Garcia-Marcos M. Different biochemical properties explain why two equivalent Gα subunit mutants cause unrelated diseases. J Biol Chem. 2014;289:21818–21827. doi: 10.1074/jbc.M114.549790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, Sprang SR. Tertiary and quaternary structural changes in Gi alpha 1 induced by GTP hydrolysis. Science. 1995;270:954–960. doi: 10.1126/science.270.5238.954. [DOI] [PubMed] [Google Scholar]
- Muntz KH, Sternweis PC, Gilman AG, Mumby SM. Influence of gamma subunit prenylation on association of guanine nucleotide-binding regulatory proteins with membranes. Molecular Biology of the Cell. 1992;3:49–61. doi: 10.1091/mbc.3.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura A, Kitano K, Takasaki J, Taniguchi M, Mizuno N, Tago K, Hakoshima T, Itoh H. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc Natl Acad Sci USA. 2010;107:13666–13671. doi: 10.1073/pnas.1003553107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hayre M, Vázquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, Gutkind JS. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer. 2013;13:412–424. doi: 10.1038/nrc3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips WJ, Cerione RA. The intrinsic fluorescence of the alpha subunit of transducin. Measurement of receptor-dependent guanine nucleotide exchange. J Biol Chem. 1988;263:15498–15505. [PubMed] [Google Scholar]
- Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the β2 adrenergic receptor– Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenhuis S, van de Wetering ML, Mooi WJ, Evers SG, van Zandwijk N, Bos JL. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N Engl J Med. 1987;317:929–935. doi: 10.1056/NEJM198710083171504. [DOI] [PubMed] [Google Scholar]
- Romani AMP. Cellular magnesium homeostasis. Arch Biochem Biophys. 2011;512:1–23. doi: 10.1016/j.abb.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffzek K. The Ras-RasGAP Complex: Structural Basis for GTPase Activation and Its Loss in Oncogenic Ras Mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- Sprang SR. Invited review: Activation of G proteins by GTP and the mechanism of Gα-catalyzed GTP hydrolysis. Biopolymers. 2016;105:449–462. doi: 10.1002/bip.22836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunahara RK, Dessauer CW, Whisnant RE, Kleuss C, Gilman AG. Interaction of Gsa with the Cytosolic Domains of Mammalian Adenylyl Cyclase. J Biol Chem. 1997a;272:22265–22271. doi: 10.1074/jbc.272.35.22265. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Tesmer JJ, Gilman AG, Sprang SR. Crystal structure of the adenylyl cyclase activator Gsalpha. Science. 1997b;278:1943–1947. doi: 10.1126/science.278.5345.1943. [DOI] [PubMed] [Google Scholar]
- Tam VH, Chen SP, Mak CM, Fung LM, Lee CY, Chan AY. A novel mutation in pseudohypoparathyroidism type 1a in a Chinese woman and her son with hypocalcaemia. Hong Kong Medical Journal. 2014;20:258–260. doi: 10.12809/hkmj134025. [DOI] [PubMed] [Google Scholar]
- Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- Vallar L, Spada A, Giannattasio G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature. 1987;330:566–568. doi: 10.1038/330566a0. [DOI] [PubMed] [Google Scholar]
- Wall MA, Coleman DE, Lee E, Iñiguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR. The structure of the G protein heterotrimer Giα1β1γ2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- Warner DR, Weinstein LS. A mutation in the heterotrimeric stimulatory guanine nucleotide binding protein alpha-subunit with impaired receptor-mediated activation because of elevated GTPase activity. Proceedings of the National Academy of Sciences. 1999;96:4268–4272. doi: 10.1073/pnas.96.8.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner DR, Weng G, Yu S, Matalon R, Weinstein LS. A novel mutation in the switch 3 region of Gsalpha in a patient with Albright hereditary osteodystrophy impairs GDP binding and receptor activation. J Biol Chem. 1998;273:23976–23983. doi: 10.1074/jbc.273.37.23976. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuev PS, Sheridan RS. Singlet vinylcarbenes: spectroscopy and photochemistry. J Am Chem Soc. 2004;126:12220–12221. doi: 10.1021/ja046309q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Nucleotide occupancy and GTPγS binding rates of WT Gαs and the R201C mutant. Related to Figure 1.
(A) Purified, recombinant wild type (WT) Gαs and the R201C mutant were examined by SDS– PAGE and visualized by Coomassie blue staining.
(B) HPLC analysis of the nucleotide occupancy of Gαs. WT Gαs or the R201C mutant purified from E. coli were denatured to release the bound nucleotide. The supernatant was analyzed by HPLC. The curves showed here represent the UV absorbance at 254 nm.
(C) The rates of GTPγS binding to WT Gαs and the R201C mutants were determined by mixing GDP-bound Gαs with a mixture of [35S]GTPγS and GTPγS in a buffer containing 2.5 mM MgCl2 and 1 mM EDTA. The data represents the mean ± SD of three independent measurements.
Figure S2: The mutant Gαs(R201C/C237S) exhibited similar behavior to the R201C mutant. Related to Figure 2.
(A and B) Influence of MgCl2 concentration on the rates of GDP dissociation from the C237S and the R201C/C237S mutants. Gαs preloaded with [3H]GDP was assayed in a buffer containing 1 mM EDTA, 0.5 mM GDP and 0.5 mM or 5 mM MgCl2. The data represents the mean ± SD of three independent measurements.
(C) Rates of GTPγS binding to the C237S and the R201C/C237S mutants in the presence of 2.5 mM MgCl2 and 1 mM EDTA at room temperature. The data represents the mean ± SD of three independent measurements.
(D) The changes in the intrinsic tryptophan fluorescence of the C237S and the R201C/C237S mutants in response to GNP binding or GTP binding and hydrolysis. 5 μM GDP-bound Gαs was mixed with 0.5 mM GNP or GTP in a buffer containing 1 mM EDTA and 0.1 mM MgCl2 to initiate the nucleotide exchange; after 1 hour, MgCl2 was added to a final concentration of 2.5 mM to decrease the GDP dissociation rates.
Figure S3: GDP and water molecules in the nucleotide-binding pocket and key residues in the hydrogen bond network of Gαs are well defined in the 1.7 Å structure of Gαs(R201C/C237S). Related to Figures 2 and 4.
(A) GDP and the sidechains of C201, T204 and D223 are showed as sticks. The Mg2+ and the water molecules coordinated with the Mg2+ are showed as green and red spheres, respectively.
(B) The sidechains of E50 in the P-loop and R258 and R265 in switch III are showed as sticks.
(C) The sidechains of E259, D260, T263 and E268 in switch III and R228 and R231 in switch II are showed as sticks. The 2mFo-DFc electron density map of the structure is contoured at 2.0 σ and colored blue.
Figure S4: Key residues in the intramolecular H-bond network of Gαs are highly conserved in other 15 human Gα proteins. Related to Figures 4 and 6.
The protein sequence of human Gαs (GNAS2) was aligned with that of other 15 human Gα proteins. The key residues in the intramolecular hydrogen bond network are showed here. Residues that are not conserved in other Gα proteins are colored yellow.
Figure S5: Effects of the R228C and R258A mutations on the rate of GDP dissociation from Gαs. Related to Figure 6.
Gαs preloaded with [3H]GDP was assayed in a buffer containing 1 mM EDTA, 2.5 mM MgCl2 and 0.5 mM GDP. The data represents the mean ± SD of three (WT) or two (R228C, R258A) independent measurements. The data of WT Gαs used here was also used in Figure 1C.