

Abstract
Lipid-derived electrophiles (LDEs) are reactive metabolites, which can covalently modify proteins and DNA, and regulate diverse cellular processes. 2-trans-hexadecenal (2-HD) is a byproduct of sphingolipid metabolism, involved in cytoskeletal reorganization, DNA damage and apoptosis. In addition, loss of ALDH3A2, an enzyme removing 2-HD in cells, is responsible for the Sjörgen-Larsson Syndrome (SJS), suggesting that accumulation of 2-HD could lead to pathogenesis. However, the targets and the precise mechanisms of 2-HD are not well characterized. Herein, we report alkyne-2-HD derivative as a bioorthogonal probe to explore the functions of 2-HD. We identified more than 500 potential cellular targets. Among them, the pro-apoptotic protein Bax can be covalently modified by 2-HD directly at the conserved Cys62 residue. Our work provided new chemical tools to explore the cellular functions of LDEs, and revealed new mechanistic insights of the deregulation of lipid metabolism in diseases.
Graphical Abstract
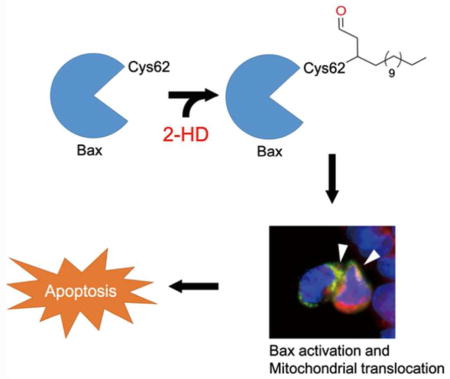
Lipid-derived electrophiles (LDEs), such as 4-hydroxy-2-nonenal (HNE), 15-deoxy-Δ12, 14-prostaglandin J2 (15d-PGJ2), and 2-trans-hexadecenal (2-HD), are generated in cells through lipid metabolic pathways or peroxidation of polyunsaturated lipids.1, 2 They harbor chemically reactive moieties, such Michael acceptors, and react with nucleophiles in cells, including cysteine residues, resulting in covalent modifications of the target proteins. Such covalent and irreversible modifications can alter the functions of protein, and regulate many cellular processes.1, 3 Therefore, misregulation of the metabolic pathways and accumulations of LDEs are often involved in diseases, such as inflammation, genotoxicity, and tissue degeneration.4 Previous work has shown that HNE, a byproduct of lipid oxidation and oxidative stress, can covalently modify many proteins in cells, including key kinases, and is a critical regulator of cellular redox-responsive pathways. 1, 3
2-Trans-hexadecenal (2-HD) is a metabolite in sphingolipid metabolism pathway, where sphingosine-1-phosphate (S1P) is degraded by S1P lyase to generate phosphoethanolamine and 2-HD. 2-HD can be further oxidized to palmitic acid by fatty aldehyde dehydrogenases, such as ALDH3A2,5 which serves as an important scavenging pathway and recycles sphingolipid to generate palmitate, the key intermediate of lipid biosynthesis. It has been noted that 2-HD could induce cytoskeletal reorganization and mitochondria apoptosis.6, 7 Importantly, loss-of-function mutations or deletion of ALDH3A2 in human resulted in an autosomal inherited disease, Sjögren-Larsson Syndrome (SJS), characterized by dry, scaly skin (ichthyosis), muscle spasm and neurological disorders.5, 8 In addition, 2-HD is responsible for ceramide-induced apoptosis in cells by activating the pro-apoptotic protein Bax.6 However, the detailed mechanisms and whether 2-HD exhibits its activities by covalently modifying target proteins are not known.
Previously, chemoproteomic approaches, such as using LDEs to compete with a cysteine-modifying activity-based protein profiling (ABPP) probe, have been reported.1 Such approaches successfully identified cellular targets of HNE and 15d-PGJ. However, it was unsuccessful for 2-HD. The discrepancy could be resulted from the weaker reactivity of 2-HD, which might be insufficient to compete with the highly reactive ABPP probes. In addition, the detoxification enzymes in cells, such as ALDH3A2, which remove 2-HD efficiently, might limit the efficiency of probe competition. Clearly, alternative approaches have to be developed to study the mechanisms of 2-HD in physiological and pathological conditions.
Clickable analogues of 2-HD as chemical probes for target identification
Toward this end, we have synthesized the “clickable” terminal alkyne-containing analogues of the saturated aldehyde and 2-HD (probe 1 and 2), which serve as a non-reactive control and an electrophile, respectively (Figure 1A and 1B). Initial labeling experiments were carried out with probe 1 and 2 in HEK293A cells to identify the endogenous cellular targets by chemoproteomic methods. We filtered mass spectrometry results with duplicates, and with >3-fold enrichment of identified total spectra with probe 2 labeling versus control probe 1. The results have shown that probe 2 could label more than 500 endogenous proteins compared to saturated aldehyde probe 1 (Table 1 and Supplementary Table S1). Among them, we have identified several targets, including the apoptosis regulator Bax protein (more than 12 unique peptides out of total 20 peptides identified). Other protein targets include mTOR, CNOT1, CTNND1, BASP1, and ATAD3A etc. Our results suggest that 2-HD could potentially modify many cellular proteins, and the functions of these modifications might be relevant to 2-HD’s physiological and pathological functions. We focused our further studies on Bax, as 2-HD has been shown to induce apoptosis, although the detailed mechanisms remain elusive.
Figure 1. Clickable analogues of 2-trans-hexadecenal as a chemical probe.

(A) Structures of the clickable terminal alkyne-containing analogues of the saturated aldehyde and 2-trans-hexadecenal (2-HD) (probe 1 and 2). (B) Scheme of profiling 2-HD target in cells. (C) Labeling of HA-Bax with Probe 2. Probe 2 could label HA-Bax dose-dependently (D) Time-dependent labeling of HA-Bax in cell lysate with probe 2 (25 μM). (E) Probe 2 (10 μM) could label recombinant His-Bax (50 ng) in vitro. 2-HD displaces probe 2 in dose dependent manner in labeling of Bax (See methods for detailed procedures).
Table 1.
Selected potential protein targets labeled by probe 2 in cell lysates. The number of total matched peptide spectra was listed for each protein
| Protein | Exp-1 | Exp-2 | ||
|---|---|---|---|---|
|
| ||||
| Control | Probe 2 | Control | Probe 2 | |
| Brain acid soluble protein 1 | 10 | 33 | 1 | 12 |
| ATP-binding cassette sub-family B member 7 | 12 | 31 | 5 | 12 |
| Enhancer of mRNA-decapping protein 4 | 10 | 31 | 5 | 16 |
| CCR4-NOT transcription complex subunit 1 | 9 | 30 | 7 | 22 |
| Serine/threonine-protein kinase mTOR | 0 | 27 | 7 | 23 |
| Sister chromatid cohesion protein PDS5 homolog A | 7 | 23 | 3 | 16 |
| Apoptosis regulator BAX | 5 | 20 | 5 | 12 |
| ATPase family AAA domain-containing protein 3A | 7 | 20 | 1 | 8 |
| Isocitrate dehydrogenase [NAD] subunit beta, mitochondrial | 5 | 19 | 0 | 15 |
| Catenin delta-1 | 0 | 18 | 0 | 13 |
| Large proline-rich protein BAG6 | 0 | 17 | 0 | 27 |
| Dolichol-phosphate mannosyltransferase subunit 1 | 0 | 16 | 2 | 10 |
| Exocyst complex component 4 | 0 | 16 | 1 | 10 |
| Peptidyl-prolyl cis-trans isomerase D | 0 | 16 | 0 | 16 |
| Probable ATP-dependent RNA helicase DDX20 | 0 | 16 | 1 | 11 |
| AH receptor-interacting protein | 0 | 15 | 0 | 11 |
2-HD covalently modifies Bax
Bax is a Bcl-2 family pro-apoptotic protein primarily found in cytosol, and translocate to mitochondrial outer membrane (MOM) upon activation.9 Bax can be activated by many upstream apoptosis inducing agents, such as oxidative stress 10, ceramides 11 and small molecules etc.12 Activated Bax forms oligomeric pores in MOM, leading to the release of cytochrome c, and the activation of downstream apoptotic pathway. The C terminus of helix α9 anchors Bax in the membrane9 or folds into in a hydrophobic groove on the surface of cytosolic Bax.13 Recent structural studies suggested that the oligomerization of the Bax is triggered when BH3 domain of one monomer is engaged with the canonical binding groove of another activated Bax. However, Bax activation with lipids and small molecules might operate through different mechanisms, as they require cysteine residues but not BH3 domains.12
To validate our findings that 2-HD could covalently modify Bax, we transfected HA-tagged Bax into HCT116 Bax/Bak double knockout (DKO) cells, and carried out probe labeling experiments. We observed that probe 2 could covalently label Bax, while the control probe 1 did not (Figure 1C). We further tested the dose-dependency and time-dependency of the modification, and have observed that covalent modification could be achieved with 10 μM of 2 (Figure 1C), and within 1h (Figure 1D). Taken together, our results have confirmed that 2-HD could covalently modify Bax protein.
Next, we carried out the labeling experiments with recombinant Bax protein to confirm that 2-HD could directly modify Bax protein in vitro. The streptavidin blot showed covalent modification of recombinant Bax (Figure 1E). No modification was observed with either DMSO or control probe 1. Furthermore, we performed competition experiments using 2-HD at various doses. We observed dose-dependent competition of probe 2 by 2-HD in modifying Bax (Figure 1E and Supplementary Figure S1A). To confirm that the cysteine residues are modified by probe 2, we pre-treated cell lysates or Bax protein with N-ethylmaleimide or iodoacetamide to cap the cysteine thiols. Labeling of Bax by Probe 2 was completely abolished with NEM and iodoacetamide (Supplementary Figure S1B).
Next, we tested weather 2-HD-induced apoptosis is Bax dependent. We treated the HCT116 wild type (WT) and Bax/Bak DKO cells with 25 or 50μM of probe 2. HCT116 WT cells were more sensitive to probe 2, compared to DKO cells, whereas probe 1 has no effect on either of them, confirming that probe 2 induces Bax-dependent cell death (Figure 2A and 2B). Probe 2 has similar activities as 2-HD in both labeling and cell death assays (Supplementary Figure S2A), suggesting that it is indeed a suitable probe to study 2-HD functions. Its been reported previously that some LDEs, such as 4-HNE, can induce cell death by oxidative stress. 14 We measured reactive oxygen species (ROS) levels by treating HCT116 WT and DKO cells with probe 2 and 4-HNE alkyne as a positive control. We observed increased ROS levels with probe 2 in a Bax-dependent manner, which could be resulted from Bax activation and apoptosis related ROS elevation (Figure 2C).15 However, 4-HNE-induced ROS and cell death are Bax-independent (Figure 2C). Although 4-HNE could non-specifically label Bax at high concentrations (100 μM) (Supplementary Figure S2B and S2C), such modification does not lead to Bax-dependent apoptosis. In addition, Bax was not identified as a target of 4-HNE in previous studies.1, 16 Therfore, the effect on Bax-dependent cell death is a unique activity of 2-HD, but not other LDEs.
Figure 2. 2-HD covalently modifies the conserved Cys62 of Bax and induces Bax-dependent cell death.

(A) Probe 2 induces Bax-dependent cell death in HCT116 wild type cells, compared to HCT116 Bax/Bak DKO cells. (B) Quantification of cell death effects induced by Probe 2. Data are represented as mean +/− SD, n=3, **, P<0.01, ***, P<0.001. (C) Probe 2 induces Bax-dependent increase of ROS, while 4-HNE induce Bax-independent ROS. Data are represented as mean +/− SD, n=3, **, P<0.01. (D) Cys62 is the major site of 2-HD modification on Bax. Probe 2 partially labels Bax C62S and does not label C62/126S (2CS) mutant, while labeling wild type and C126S mutant efficiently. (E) Detection of the probe-labeled peptide containing Cys62 by mass spectrometry. Observed the m/z of 1054.635, which matches the probe-modified Cys62 in peptide KLSECLK.
2-HD covalently modifies the conserved Cys62 of Bax
Bax protein has two conserved cysteine residues (Cys62 and Cys126), which could be the sites of modification. We mutated these cysteine residues to serine, singly or in combination (C62S, C126S, or C62/126S (2CS)), and tested whether probe 2 can covalently label the wild type (WT) or the mutant Bax. Interestingly, probe 2 could label WT and C126S mutant, but labeling was partially reduced with C62S, and almost abolished with 2CS mutants (Figure 2D). We also noticed that at higher concentration of probe 2 (50 μM), C62S mutant can be labeled, but not the 2CS mutant (Supplementary Figure S2D). Similar results can be obtained when using anti-Bax antibody (Supplementary Figure S2E). Our results indicated that Cys62 might be the primary site of 2-HD modification, and at higher doses of 2-HD, Cys126 could also be modified. Previous structural studies of the Bax suggest that Cys62 is located in α2 helix of pro-apoptotic BH3 domain, which is required for Bax conformational change.9 Therefore, it is likely that covalent modification of Cys62 is responsible for 2-HD-mediated Bax activation. Consistently, it has been reported that Cys62 is required in small molecules induced apoptosis.10 To further confirm that Cys62 is indeed the site of covalent modification, we have analyzed Bax protein after treatment with probe 2 with mass spectrometry. We have observed the m/z of 1054.635, which matches the probe-modified Cys62 in peptide KLSECLK (Figure 2E). Such modified peptide was not observed with controls. We did not observe other probe-modified peptides in this study, confirming that Cys62 could be the major site of covalent modification.
Loss of Cys62 modification or 2CS mutation compromises Bax activation
To further test whether loss of Cys62 modification or 2CS mutation might compromise 2-HDinduced Bax activation, we transfected HCT116 Bax/Bak DKO cells with wild type (WT) HA-Bax, C62S, C126S or 2CS mutant, and then treated the cells with probe 2 for 16 h. Confocal microscopy showed translocation of WT HA-Bax and HA-Bax C126S into mitochondria, whereas HA-Bax (C62S) or HA-Bax (2CS) mutant had no significant change (Figure 3A and 3B). We measured the activated Bax using anti-Bax 6A7 antibody, which recognizes Bax in an activated conformation. Consistently, Bax activation is only observed in cells transfected with WT Bax and C126S mutant, but not with the C62S or 2CS mutant (Figure 3C and 3D). Control probe 1 has no effect even at high concentration (50 μM) (Supplementary Figure S3A). Taken together, our results suggested that probe 2 could induce Bax activation through covalent modification of Cys62. Furthermore, we tested the effect of probe 2 in cell death with HCT116 Bax/Bak DKO cells transfected with Bax WT, C62S, C126S or the 2CS mutant. DKO cells transfected with Bax 2CS mutant remain resistant to probe 2 induced cell death, while cells expressing Bax WT or C126S are sensitive to probe 2. Interestingly, we have observed that DKO cells expressing Bax (C62S) mutant are resistant to lower dose of probe 2 (25 μM), while high dose of probe 2 (50 μM) can still induce significant cell death (Figure 3E and 3F). These observation is consistent with our labeling experiments at higher dose of probe 2 could label Bax C62S mutant. Therefore, low dose of 2-HD could induce cell death by primarily labeling Cys62, and at higher doses, both Cys62 and Cys126 could be modified, leading to conformational change and activation of Bax. To evaluate the effects of endogenous 2-HD, we tested effect of ceramides (C2 and C6) in Bax/Bak DKO cells transfected with HA-Bax WT and mutants. It has been previously reported that ceramides act as a precursor for 2-HD biosynthesis in cells.5, 6 We noticed that Bax-WT and C126S transfected cells were sensitive to C2 and C6 ceramides, whereas C62S and 2CS mutants rescued the effect (Supplementary Figure S3B and S3C). The effect was much clearer with increased concentration of ceramides. Our results suggest that exogenous and endogenous 2-HD could induce cell death through Bax modification.
Figure 3. Covalent modification of Cys62 is critical for 2-HD-induced Bax activation, translocation and cell death.

(A) A confocal microscopy analysis of Bax translocation to mitochondria. Mitochondria and Nuclei were fluorescently stained with MitoTracker and DAPI. Scale bar, 10 μm. (B) Quantitative analysis of the images. Data are represented as mean +/− SD, n=3, **, P<0.01. (C) Detection of the activated Bax (anti-Bax 6A7) by western blot. (D) Quantification of the activated Bax by densitometry. Data are represented as mean +/− SD, n=3, **, P<0.01. (E) Colony formation assay on Bax WT or mutant transfected HCT116 Bax/Bak DKO cells. (F) The colonies were quantified by densitometric reading. Data are represented as mean +/− SD, n=3, **, P<0.01, ***, P<0.001.
In Sjögren-Larsson Syndrome (SJS), loss-of-function mutations of ALDH3A2 might lead to the accumulation of 2-HD, and therefore, enhanced apoptosis. To test this hypothesis, we used CHO-K1A cells, which were selected to have ALDH3A2 deficiency, and have been used as a SJS model system.17 Indeed, we found that the ALDH3A2-defecient CHO-K1A cells are more sensitive to 2-HD or probe 2-induced apoptosis, compared to wild type CHO cells (Supplementary Figure S3D). Under basal culture conditions, we also observed that CHOK1A cells have lower cell viability compared to WT CHOK1 cells (Supplementary Figure S3E), consistent with the notion that accumulation of 2-HD might affect cell viability. In addition, probe 2 induces more efficient PARP-1 cleavage in CHO-K1A, compared to control cells (Supplementary Figure S3F). Taken together, our results suggest that reactive 2-HD induces more profound cell death in cells lacking the ALDH3A2. Therefore, accumulation of 2-HD in SJS patient, might enhance aberrant cell death, and contribute partly to SJS pathogenesis (Supplementary Figure S4).
LDEs at physiological levels might play important roles in feedback regulation of cell signaling pathways. However, misregulation of lipid metabolism and elevated levels of LDEs have been linked to pathological conditions. In addition, LDEs form stable adducts with the proteins, thus irreversibly altering the properties of those proteins. Recently, some of these LDEs, such as 4-HNE1, 16 and bromomethyl ketone18 were extensively profiled to identify their cellular targets using chemoproteomic methods. In this report, we synthesized and characterized an alkyne-containing analogue of reactive lipid electrophile 2-HD. In particular, we found that 2-HD can covalently modify the conserved Cys62 in pro-apoptotic protein Bax. Covalent modification of Cys62 is required for 2-HD-induced Bax activation and apoptosis. Cys62 is located in the α2 helix of BH3 domain, which could be easily accessible by 2-HD and other small molecules. Therefore, covalent modifications of Cys62 might serve as a molecular switch that induces Bax conformational changes and its activation. In addition to Cys62, it has been reported that Cys126 is critical for Bax activation induced by some small molecules, including BAM7 and OICR766A,12 while the Bax C126A mutant was resistant to OICR766A-induced activation, the Bax C62A mutant can be activated. On the other hand, neither Cys62 nor Cys126 is required for Bax activation induced by etoposide or staurosporine.12 Therefore, the covalent modification on Cys62 might be a unique feature of 2-HD, and Bax activation induced by small molecules could have multiple mechanisms. Previously Cys126 is also reported to be palmitoylated.19 Therefore, lipid metabolism could be linked to cellular apoptosis through multiple mechanisms, although the functions of palmitoylation of Cys126 need further investigation.
In addition to Bax, we have identified other unique cellular targets of 2-HD, including mTOR, ABCB7, PPID, which have not been shown to be modified by 4-HNE or palmitoylation chemical reporters.20–22 Our results suggest that 2-HD could affect a wide range of cellular processes. Therefore, deregulation of sphingolipid metabolism and accumulation of 2-HD might be relevant to many pathological conditions. Further mechanistic studies are needed to elucidate the functions of these modifications and whether such modifications are involved in SJS pathogenesis. Taken together, our work has provided new chemical tools to study the functions of LDEs, and new insights into the roles of lipid metabolism in regulating cellular processes and diseases.
METHODS
Details of materials, methods and experimental procedures were described in Supporting Information.
Supplementary Material
Acknowledgments
This work was supported by a Research Scholar Award from the American Cancer Society (124929-RSG-13-291-01-TBE), and grants from the National Institutes of Health (R01 CA181537, R01 DK107651) to X.W, P41 GM104603 to C. L. and R01 GM107675 to R. Z. We thank R. Youle and L. Zhang for the Bax/Bak double knockout cells, and helpful discussion. We thank Taplin Mass Spec Core at Harvard Medical School for proteomic studies.
Footnotes
Author Contributions
X.W. conceived the concepts, designed the experiments and supervised the studies. G.K, J.R designed and performed the experiments with the help of P.C, Y.S, B.C J. N. and M.D. M.Y & C.L helped with Mass spectrometry studies for protein modification with probe. R.Z provided the SJS cell models. G.K, J.R, P.C, Y.S and X.W analyzed the data. G.K, J.R. and X.W wrote the manuscript with input from all co-authors.
Competing Financial Interests
Authors declare no competing financial interests.
This supporting material is available free of charge via the internet at http://pubs.acs.org
The Supporting Information file contains details on methods, compound characterization, and additional results in Supplementary Figure S1–S4, and Supplementary Table S1.
References
- 1.Wang C, Weerapana E, Blewett MM, Cravatt BF. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods. 2014;11:79–85. doi: 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Codreanu SG, Liebler DC. Novel Approaches to identify protein adducts produced by lipid peroxidation. Free Radical Res. 2015;49:881–887. doi: 10.3109/10715762.2015.1019348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative Stress: An Essential Factor in the Pathogenesis of Gastrointestinal Mucosal Diseases. Physiol Rev. 2014;94:329–354. doi: 10.1152/physrev.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakahara K, Ohkuni A, Kitamura T, Abe K, Naganuma T, Ohno Y, Zoeller RA, Kihara A. The Sjogren-Larsson syndrome gene encodes a hexadecenal dehydrogenase of the sphingosine 1-phosphate degradation pathway. Mol Cell. 2012;46:461–471. doi: 10.1016/j.molcel.2012.04.033. [DOI] [PubMed] [Google Scholar]
- 6.Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, Green DR. Sphingolipid Metabolism Cooperates with BAK and BAX to Promote the Mitochondrial Pathway of Apoptosis. Cell. 2012;148:988–1000. doi: 10.1016/j.cell.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar A, Byun HS, Bittman R, Saba JD. The Sphingolipid Degradation Product Trans-2-Hexadecenal Induces Cytoskeletal Reorganization and Apoptosis in a JNK-Dependent Manner. Cell Signal. 2011;23:1144–1152. doi: 10.1016/j.cellsig.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizzo WB. Sjogren-Larsson syndrome: Molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90:1–9. doi: 10.1016/j.ymgme.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westphal D, Dewson G, Czabotar PE, Kluck RM. Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta. 2011;1813:521–531. doi: 10.1016/j.bbamcr.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 10.Nie C, Tian C, Zhao L, Petit PX, Mehrpour M, Chen Q. Cysteine 62 of Bax Is Critical for Its Conformational Activation and Its Proapoptotic Activity in Response to H(2)O(2)-induced Apoptosis. J Biol Chem. 2008;283:15359–15369. doi: 10.1074/jbc.M800847200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mignard V, Lalier L, Paris F, Vallette FM. Bioactive lipids and the control of Bax pro-apoptotic activity. Cell Death Dis. 2014;5:e1266. doi: 10.1038/cddis.2014.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brahmbhatt H, Uehling D, Al-awar R, Leber B, Andrews D. Small molecules reveal an alternative mechanism of Bax activation. Biochem J. 2016;473:1073–1083. doi: 10.1042/BCJ20160118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, Kluck RM, Adams JM, Colman PM. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 14.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog in lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 15.Buccellato LJ, Tso M, Akinci OI, Chandel NS, Budinger GR. Reactive oxygen species are required for hyperoxia-induced Bax activation and cell death in alveolar epithelial cells. J Biol Chem. 2004;279:6753–6760. doi: 10.1074/jbc.M310145200. [DOI] [PubMed] [Google Scholar]
- 16.Vila A, Tallman KA, Jacobs AT, Liebler DC, Porter NA, Marnett LJ. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem Res Toxicol. 2008;21:432–444. doi: 10.1021/tx700347w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.James PF, Zoeller RA. Isolation of animal cell mutants defective in long-chain fatty aldehyde dehydrogenase. Sensitivity to fatty aldehydes and Schiff’s base modification of phospholipids: implications for Sj-ogren-Larsson syndrome. J Biol Chem. 1997;272:23532–23539. doi: 10.1074/jbc.272.38.23532. [DOI] [PubMed] [Google Scholar]
- 18.Abo M, Weerapana E. A Caged Electrophilic Probe for Global Analysis of Cysteine Reactivity in Living Cells. J Am Chem Soc. 2015;137:7087–7090. doi: 10.1021/jacs.5b04350. [DOI] [PubMed] [Google Scholar]
- 19.Frohlich M, Dejanovic B, Kashkar H, Schwarz G, Nussberger S. S-palmitoylation represents a novel mechanism regulating the mitochondrial targeting of BAX and initiation of apoptosis. Cell Death Dis. 2014;5:e1057. doi: 10.1038/cddis.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng B, DeRan M, Li X, Liao X, Fukata M, Wu X. 2-Bromopalmitate Analogues as Activity-Based Probes To Explore Palmitoyl Acyltransferases. J Am Chem Soc. 2013;135:7082–7085. doi: 10.1021/ja311416v. [DOI] [PubMed] [Google Scholar]
- 21.Chen B, Zheng B, DeRan M, Jarugumilli GK, Fu J, Brooks YS, Wu X. ZDHHC7-Mediated S-Palmitoylation of Scribble Regulates Cell Polarity. Nat Chem Biol. 2016;12:686–693. doi: 10.1038/nchembio.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan P, Han X, Zheng B, DeRan M, Yu J, Jarugumilli GK, Deng H, Pan D, Luo X, Wu X. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat Chem Biol. 2016;12:282–289. doi: 10.1038/nchembio.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.