

Abstract
Arrestins are a small family of proteins with four isoforms in humans. Remarkably, two arrestins regulate signaling from >800 G protein-coupled receptors (GPCRs) or non-receptor activators by simultaneously binding an activator and one out of hundreds of other signaling proteins. When arrestins are bound to GPCRs or other activators, the affinity for these signaling partners changes. Thus, it is proposed that an activator alters arrestin’s ability to transduce a signal. The comparison of all available arrestin structures identifies several common conformational rearrangements associated with activation. In particular, it identifies elements that are directly involved in binding to GPCRs or other activators, elements that likely engage distinct downstream effectors, and elements that likely link the activator-binding sites with the effector-binding sites.
Keywords: arrestin, structure, activation, GPCR, cell signaling
Arrestin signaling overview
Arrestins (see Glossary) are ~ 45 kDa proteins that control information flow in G protein-coupled receptor (GPCR) signaling. Vertebrates have only four arrestin isoforms [1]. Arrestin-1 and -4 are expressed in photoreceptor cells and termed visual arrestins, whereas arrestin-2 and -3 (a.k.a. β-arrestin1 and β-arrestin2, respectively) are expressed in virtually every cell. The visual arrestins are dedicated interaction partners of rhodopsin and cone opsins, while the non-visual arrestins can interact with >800 GPCRs, other cell surface receptors, and non- receptor partners that include clathrin, clathrin adaptor AP2 and various signaling proteins [2–4]. Of these, GPCRs are the major activators of arrestin. In addition to GPCRs, a naturally abundant small molecule, inositol hexakisphosphate (IP6), has recently been shown to activate arrestin-3. IP6 likely facilitates receptor-independent JNK3 activation in cells [5]. Following activation, non-visual arrestins directly bind to non-receptor partners, including effectors. For example, mitogen-activated protein kinases (MAPKs) and their upstream activators, which regulate critical cellular functions, including proliferation, differentiation, and apoptotic death, are among the better characterized arrestin-regulated signaling proteins [2, 3].
The ability to transmit a signal between a receptor and a non-receptor partner requires that non-visual arrestins have at least three types of elements: (1) receptor-binding sites that sense and select for distinct functional states of the receptor; (2) effector-binding sites that become structurally available when arrestin is associated with a receptor; and (3) allosteric connections that allow communication between the receptor- and effector-binding sites. The prediction that arrestin activation is accompanied by a global conformational change was first made in 1989, long before any structural information became available, based on the high Arrhenius activation energy of forming a high-affinity rhodopsin-arrestin complex [6]. In light of recent advances in the structural characterization of basal and active states of arrestin, here we focus on arrestin activation and signaling from a structural perspective.
Arrestin sensors and activation
Both the activation and phosphorylation of the GPCRs enhance arrestin binding. This was first discovered in the visual system, where simultaneous light-activation and phosphorylation of rhodopsin yields the binding of arrestin-1 that is >20-fold greater than the binding to light-activated unphosphorylated or inactive phosphorylated rhodopsin [7]. These data led to the hypothesis that arrestin has two distinct “sensors”. A phosphate sensor recognizes that the receptor is phosphorylated, while an activation sensor recognizes that the receptor is in activated state. These two sensors are proposed to bind the receptor in two steps to promote a high-affinity receptor-arrestin complex. The original model suggested that either of these sensors can be engaged first, followed by the engagement of the other in the second step [7]. An alternative model suggests that the first phase involves the binding of arrestin phosphate sensor to the phosphorylated receptor elements, while the second phase is the interaction of the arrestin activation sensor with the active receptor [8]. However, this latter model does not explain higher binding of wild type arrestins to active and unphosphorylated GPCRs than to inactive and unphosphorylated GPCRs [7, 9, 10]. Regardless of the order of binding by these sensors, the phosphate and activation sensors are likely common contact sites of the two non-visual arrestins and hundreds of different GPCRs, as well as non-receptor activators. For example, elimination of phosphate-binding residues in non-visual arrestins significantly reduces their interactions with β2-adrenergic and neuropeptide Y receptors [11, 12], whereas mutations in the activation sensor reduce arrestin-2 binding to M2 muscarinic and D2 dopamine receptors [5]. Additional contact sites that vary depending on unique properties of individual receptors are likely also involved in the interaction [13, 14].
Basal conformation of arrestins: ready to act
Crystal structures of all four vertebrate subtypes in the basal state revealed that arrestins are two-domain molecules with a long axis of ~75Å, and a short axis of ~35Å (Figure 1) [15–20]. There are few interactions between the two domains and disruption of these interactions yields arrestins that are more easily activated. One of the interactions stabilizing the relative orientation of the two domains is an unusual “polar core”, which includes five charged side chains that are buried and almost entirely solvent excluded (Figure 1) [16]. Disrupting the polar core by reversing the charges of the participating residues creates arrestin mutants that are more easily activated (sometimes called “pre-activated” arrestins) [21, 22]. These mutants demonstrate enhanced binding to the non-preferred forms of cognate receptors, active unphosphorylated and inactive phosphorylated [7, 23]. The other inter-domain interaction in basal arrestin is termed the “three-element interaction” (Figure 1) [16], which involves two elements of the N-domain (β-strand I and the α-helix) and a short region of the C-terminus (termed C-tail) of arrestin. Disruption of this interaction also “pre-activates” arrestins, as evidenced by the natural splice variant that lacks the C-tail and therefore the three-element interaction [24], and arrestin mutants where key residues that anchor C-tail to the N-domain are altered [9, 10, 23]. Collectively these data led to a model of arrestin activation by the change in conformation involving the movement of the domains relative to each other [25].
Figure 1. The crystal structure of arrestin-2 in the basal state.
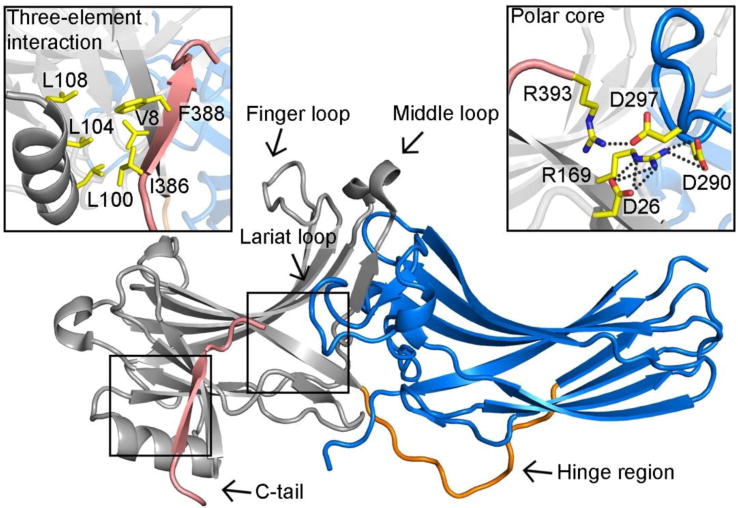
Arrestin-2 (PDB ID: 1G4M) contains two domains, the N- (colored grey) and C-domain (colored blue), which are linked by a hinge region (colored orange). The C-tail (colored pink) of arrestin-2 is attached to the N-domain. One inter-domain contact is the hydrophobic interaction involving β-strand I and the α-helix of the N-domain and the C-tail. It is termed “three-element interaction” and is shown in detail in the upper left box. Another major interaction that holds arrestin-2 in the basal state is the “polar core”, which consists of five charged residues from both domains and the C-tail. It is shown in detail in the upper right box. All participating residues are shown as yellow stick models.
Active arrestins and cellular signaling
Identifying the conformational changes associated with arrestin activation posed a major challenge to the field. The biologically relevant activated state of arrestin is usually stabilized by an interaction with a membrane-spanning cognate GPCR, and methods to determine the structures of complexes between a membrane protein and a binding partner are still not well developed. As a result, a number of different stabilization methods were used to capture arrestin in its active conformation. These included the use of a pre-activated form of arrestin-1 fused to phosphorylated and active rhodopsin [13, 14], a splice variant of arrestin-1 that is more easily activated [24], a receptor phosphopeptide and a stabilizing nanobody for arrestin-2 [26], and a small molecule IP6 that activates arrestin-3 [5], but not highly homologous arrestin-2 [27]. The comparison of all active arrestin structures indicates that common rearrangements are associated with activation.
The largest activation-associated conformational change is a ~20° rotation between the N- and C-domains (Figure 2A) [5, 13, 14, 24, 26]. The domain rotation is accompanied by significant rearrangements of the inter-domain interface, including the disruption of both the polar core and the three-element interaction. Notably, in the basal state of arrestin, the C-tail participates in both the polar core and three-element interaction, and appears to stabilize the orientation of the two domains (Figure 1). The structures of active arrestins confirmed that another hallmark of activation is the release of the C-tail from the N-domain, which destabilizes basal inter-domain interactions.
Figure 2. The global conformational change of arrestin upon activation.
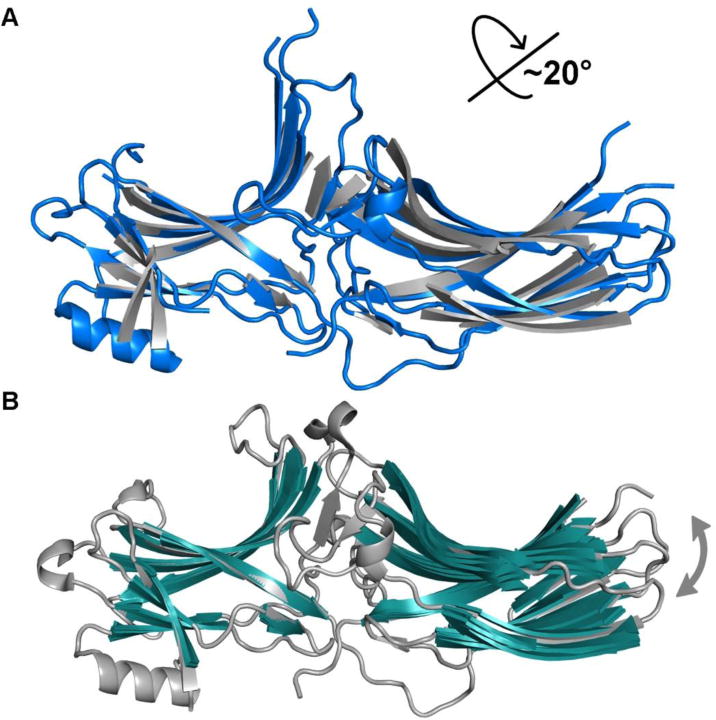
A. Superposition of the N-domain of active (colored blue, PDB entry 4JQI) and basal (colored grey, PDB entries 1G4M) arrestin-2 highlights the 20° inter-domain rotation. B. Superposition of the N-domain of basal arrestin-2 variants (colored grey and green, PDB entries 1G4M, 1G4R, 1ZSH, 1JSY, 2WTR, 3GD1, 3GC3) reveals a narrower range of rotational movements between domains in the various basal states.
Receptor-binding interface
The phosphate sensor
The phosphate sensor of arrestin directly interacts with phosphates either from GPCRs [14, 26] or non-receptor activators, such as IP6 [5]. The lysines and arginines on the N-domain of arrestins directly bind phosphates. This is supported by mutagenesis [28–32] and the structures of arrestin-1 in complex with phosphorylated active rhodopsin [14], arrestin-2 with V2 vasopressin receptor-derived phosphopeptide [26] and arrestin-3 with IP6 [5] (Figure 3A-C). All three structures revealed that highly conserved lysines and arginines form salt bridges with phosphates either from the receptor C-terminus or from IP6 (Figure 3A-C). These charge-charge interactions anchor part of the receptor C-terminus to arrestin, which likely enhances receptor binding [14]. Indeed, high-affinity binding of arrestin-1 requires a minimum of three phosphates per rhodopsin in vivo [33] and in vitro [34].
Figure 3. The phosphate-binding residues of arrestins.
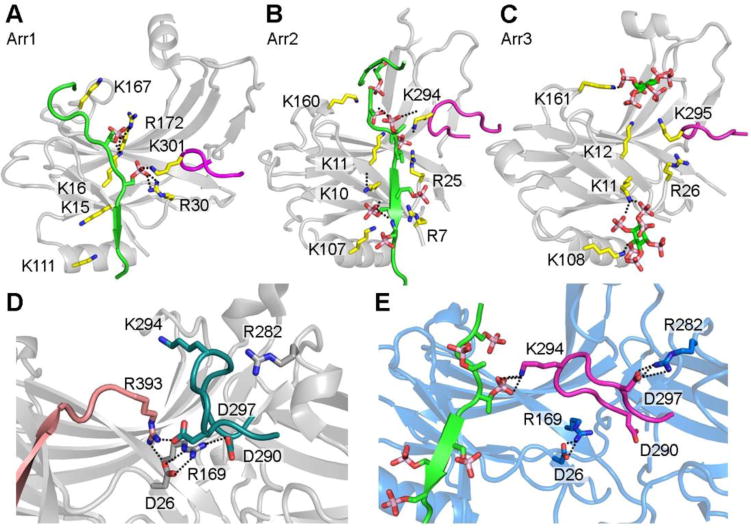
A. Phosphorylated receptor C-terminus binding sites within the arrestin-1 N-domain (PDB entry 5W0P). B. Phospho-peptide binding sites within the arrestin-2 N- domain (PDB entry 4JQI). C. IP6 binding sites within the arrestin-3 N-domain (PDB entry 5TV1). D. Conformation of the lariat loop in basal arrestin-2 (PDB entry 3P2D) with an intact polar core. The lariat loop contributes two charged residues to the polar core (D290 and D297) and also contains one phosphate-binding residue (K294). E. Conformation of the lariat loop in phospho-peptide activated arrestin-2 (PDB entry 4JQI) with the polar core disrupted. The lysine 294 binds to one phosphate from the receptor C-terminus phospho-peptide, which leads to the conformational change in lariat loop and the disruption of the polar core. The salt bridge is shown by dotted line.
Interestingly, although the potential phosphorylated sites of different GPCRs vary, many GPCRs have the common phosphorylation motif Px(x)PxxP, where P is a phosphorylated Ser or Thr residue, and x can be any residue [14]. This creates a spatial pattern of negative charges that interact with the positively charged residues of arrestin. Because the phosphate sensor directly responds to the phosphates regardless of the sequence context, this explains how the two non- visual arrestins bind hundreds of GPCRs with little sequence conservation in the phosphorylated elements. Obviously, this “code” idea has certain limitations: β2-adrenergic receptors were used to discover and characterize both non-visual arrestins [35, 36] but appear to have only a partial phosphorylation code [14]. There is growing evidence that different phosphorylation patterns on a few GPCRs lead to distinct functional outcomes, as posited by the “barcode” hypothesis [37, 38]. One possibility is that different phosphorylation patterns of GPCRs lead to distinct active conformations of arrestins, which in turn favor certain downstream signaling pathways. Indeed, NMR spectroscopy using an F2Y unnatural amino acid incorporated at various sites revealed that receptor-derived phosphopeptides with different phosphorylation patterns caused distinct conformational changes in arrestin [39].
But how does this promote arrestin activation? Comparison of the structures of basal [16- 20, 27] and activated [5, 13, 14, 24, 26] arrestins suggests how triggering the phosphate sensor shifts the equilibrium of arrestin to favor the active conformation. The phosphate-bearing GPCRs (or IP6) bind to arrestin in the same location as the C-tail, so that activators directly compete with the C-tail for the N-domain. This physically disrupts the three-element interaction that stabilized the basal conformation (Figure 1). Moreover, one of the phosphate-binding elements, a conserved lysine (Lys 301 in arrestin-1, Lys 294 in arrestin-2, Lys 295 in arrestin-3), is in a region termed the lariat loop (Figure 3A-C) [5, 14, 26]. The lariat loop contains two aspartic acid residues of the polar core that stabilize the basal conformation (Figure 1, Figure 3D). The binding of the lysine side chain to phosphate displaces the lariat loop, disrupts the polar core, and contributes to arrestin activation (Figure 3D, E). It should be noted that a previous model of activation, in which the polar core was proposed to be directly disrupted by the receptor-attached phosphates, was developed before the structures of active arrestins became available and was mainly based on mutagenesis [25]. A common feature of the previous [25] and current structure- based model of arrestin activation (Figure 3) is that the polar core must be disrupted by the direct action of activator-attached phosphates.
The activation sensor
The activation sensor of arrestins was proposed to differentiate between active and inactive GPCRs [7], although recent data show that it also responds to non-receptor activators, such as IP6 [5]. Nevertheless, the activation sensor can best be conceptualized in the context of GPCR-dependent activation. One common feature of active GPCRs is the emergence of a binding pocket on the intracellular side through the outward shift of TM5 and TM6 [40, 41]. This pocket apparently accommodates the C-terminal helix of the G protein α-subunit and probably the N-terminal helix of GRKs [42–45]. In arrestin, a region termed the finger loop is unstructured in the basal conformation, but upon activation, this element forms an α-helix, and directly inserts into this pocket characteristic for the active GPCR (Figure 4A, B), as revealed by the structures of active rhodopsin in complex with either a peptide derived from the arrestin-1 finger loop [44] or full-length arrestin-1 [13, 14]. This suggests that finger loop is a key part of the activation sensor. In the α-helix the two conserved hydrophobic residues are aligned, which facilitates their direct interaction with the hydrophobic partners in the active receptor core (Figure 4B) [13, 14]. It also places the adjacent residues on the arrestin-1 finger loop in the position favorable to form hydrogen bonds with two highly conserved motifs in rhodopsin α- helix 8 and the intracellular side of TM3, as revealed by the structure of rhodopsin in complex with arrestin-1 finger loop peptide (Figure 4A) [44]. Furthermore, it is likely that the α-helix of the finger loop places three charged residues in positions favorable to form salt bridges with rhodopsin active core [13]. Thus, the ability of the finger loop to form an α-helix is anticipated to be important for receptor association. Indeed, disruption of the α-helix by introducing a proline in the middle dramatically decreases arrestin-3 binding to the muscarinic M2 and dopamine D2 receptors [5]. The helical conformation of the finger loop also appears to assist in arrestin activation by the non-receptor partner IP6 [5]. In this case, the α-helical conformation of the finger loop is stabilized by interactions with the α-helices of other protomers in a trimer, with trimerization stabilizing the active state of arrestin (Figure 4C).
Figure 4. The activation sensor of arrestin.
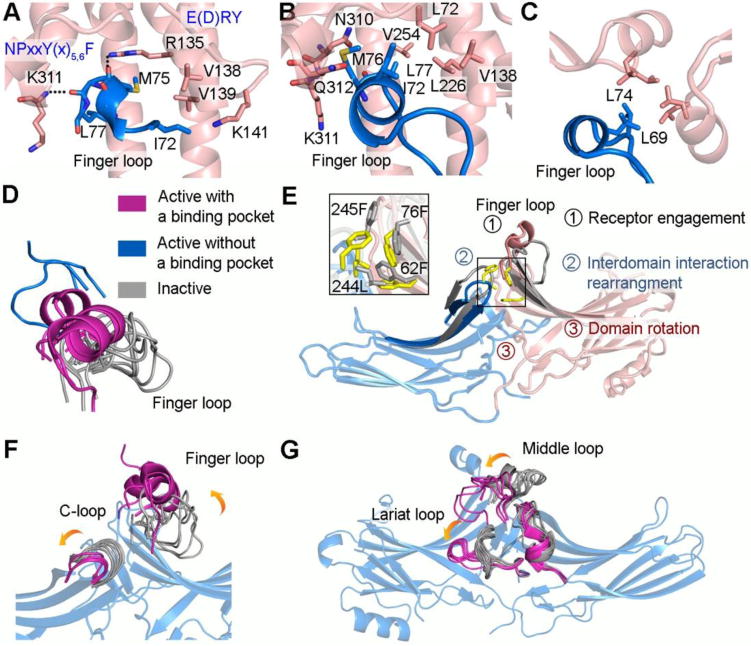
A. The arrestin-1 finger loop peptide (colored blue) directly inserts into rhodopsin active core (colored pink) (PDB entry 4PXF). The center of the finger loop forms a short α-helix. The finger loop binds the receptor via both hydrogen bonds and hydrophobic interactions. The residues in rhodopsin that form hydrogen bonds with arrestin-1 finger loop are from two very conserved motifs, NPxxY(x)5,6F and E(D)RY, respectively. B. The center of the finger loop (colored blue) forms an α-helix and binds the receptor through hydrophobic interactions (colored pink) in the rhodopsin-arrestin complex structure (PDB entry 5W0P). It was also proposed that the finger loop interacts with receptor via salt bridges based on Tango assay. C. A similar hydrophobic contact is observed in the arrestin-3 trimer (PDB entry 5TV1). D. Superposition of β-strands V and VI connected by the finger loop from available arrestin structures. Basal states (colored grey, PDB entries 2WTR, 4ZRG, 3UGU, 3P2D, 3UGX, 1G4R, 3GC3, 3GD1, 1JSY, 1G4M); active with a hydrophobic core (colored magenta, PDB entries 5TV1, 5W0P, 4ZWJ); active without a hydrophobic core (colored blue, PDB entries 4J2Q, 4JQI). E. Superposition of arrestin-3 in the basal (colored grey, PDB entry 3P2D) and active state (colored blue and pink, PDB entry 5TV1) highlights rearrangement of the hydrophobic interactions (colored yellow and grey), which is shown in detail in rectangular box. The proposed model includes three steps: the active receptor core directly engages the finger loop (step 1), leads to the rearrangement of the hydrophobic interactions between two domains (step 2), which allows the domains to rotate (step 3). F-G. The Superposition of β-strands from N- or C- domain of active (colored magenta, PDB entries 4J2Q, 4JQI, 4ZWJ, 5TV1, 5W0P) with basal (colored grey, PDB entries 1G4M, 1G4R, 1JSY, 3P2D, 3GD1, 3GC1, 1ZSH, 1CF1, 3GUX, 3UGU, 4ZRG, 2WTR) arrestins highlights the conformational changes of loops upon activation. The β-strands from arrestin-3 in the active state (PDB entry 5TV1) are shown while in the other structures they are omitted for clarity.
Interestingly, G proteins and G protein-coupled receptor kinases (GRKs), two other major effectors that directly bind active GPCRs, also contain sequence motifs that insert into the active receptor core [46–48]. In G proteins, structures of β2AR-Gs-protein complex [43], A2AR- mini-Gs complex [49], CTR-Gs complex [50] and GLP-1R-Gs complex [51] identified that the C- terminus of the α-subunit forms an α-helix upon interaction with this binding pocket in the active receptor. In GRKs, their N terminus was proposed to form an α-helix and promote receptor binding and kinase activity [45, 52, 53]. The finding that these binding partners all use the same pocket on active GPCR provides the mechanistic basis for the direct competition of arrestins with G proteins for the activated receptor observed in biochemical studies [54, 55], as well as for the reported competition of arrestin-2 with GRK2 [56].
As two non-visual arrestins bind diverse receptors, one critical feature of the activation sensor is the broad receptor specificity. Or to put it another way, regardless of the identity of the receptor, it must be able to engage the arrestin finger loop. We propose that the characteristics of this pocket (hydrophobic on one side, positively charged on the other) allow for relatively promiscuous interactions that are required for the observed binding of arrestins to various GPCRs (Figure 4A, B). Structural work suggests that the arrestin finger loop is on a flexible tether, which may be necessary for binding (Figure 4D). Indeed, the finger loop assumes a variety of conformations in different crystal structures [57]; in some structures the electron density of the finger loop is uninterpretable, which is commonly associated with a dynamic region [24, 58–60] (Figure 4D). Moreover, when arrestin is bound to an activator, the finger loop adopts different angles relative to the rest of the arrestin molecule (Figure 4D) [5, 13, 14, 57]. This flexibility likely allows the finger loop to adapt to the specific binding pocket of each of the >800 active GPCRs. Consistent with this idea, the reduction of flexibility of the finger loop by a proline at the base significantly decreases binding to all receptors [5].
There is currently little experimental evidence indicating how triggering this activation sensor and engaging the finger loop leads to arrestin activation. One model was proposed that binding to the phosphorylated receptor C-terminus releases the finger loop from basal state and allows it to engage the active receptor core [44, 57]. Here we propose a different model explaining how the engagement of the finger loop leads to arrestin activation independently of phosphate binding (Figure 4E). The finger loop is located between β-strands V and VI in the N- domain (Figure 1, Figure 4E, F). These β-strands directly contact the C-domain via hydrophobic residues. Receptor engaging the finger loop induces the conformational changes in it and shifts these two β-strands. This likely results in a “sliding” motion of the two domains due to the rearrangement of the hydrophobic network between them (Figure 4E, F). This motion might facilitate the disruption of inter-domain contacts including the polar core and the three-element interaction. Indeed, the NMR study revealed that the non-phosphorylated active rhodopsin triggers the conformational changes in both regions of arrestin-1 [61]. The angle and extent of finger loop interaction with the receptor could affect the magnitude of the rotation between the two domains of arrestin, so that engagement by different receptors can translate into distinct active states of arrestin.
Other arrestin-GPCR interaction sites and interplay between the two sensors
Additional sites, mainly the loops on the concave side of arrestin, also contribute to receptor binding, as suggested by the rhodopsin-arrestin-1 complex structure [13, 14]. Interestingly, the comparison of active and basal arrestin structures reveals that these flexible loops display large shifts, some of which are uniform in all subtypes (Figure 4 F, G). The middle loop (termed 139-loop in arrestin-1) shifts towards the N-domain (Figure 4G) [5, 13, 14, 26, 62]. The lariat loop, which supplies two negative charges to the polar core in the basal state, turns almost 90° counter-clockwise and moves towards the N-domain, thereby destabilizing the polar core (Figure 4G). The C-loop twists away from the interface between the N- and C-domains, which in turn adjusts the positions of the two β-strands that it connects (Figure 4F). Some of these contacts require the active conformations of both arrestin and receptor. Importantly, the conformational change in the middle and C-loop creates a cleft to accommodate the intercellular loop 2 of rhodopsin [13]. These rearrangements support the role of the phosphate and activation sensor in stabilization of the active arrestin-GPCR complex. However, it is important to note that structures of active arrestins show the end result, but do not reveal the sequence of events that lead to it. Moreover, a recent study suggests that the edge of arrestin C-domain engages the lipid membrane and facilitates receptor binding [63].
The engagement of effectors
Following activator binding, non-visual arrestins change their ability to engage signaling effectors. But how do the conformational changes elicited by the activators affect the binding of numerous non-receptor partners? Controlling the accessibility of effector binding sites is a common way for a signaling protein to switch a specific pathway on and off. Considering that most effectors show enhanced binding to arrestin in an activated state [3], one possibility is that the effectors bind across the two domains of arrestin, with domain rotation aligning different parts of the arrestin molecule to create (or destroy) the effector-binding site. Different rotation angles could create a range of unique effector-binding interfaces (Figure 5A). Consistent with this model, there is growing evidence that arrestins engage effectors through multiple contact points that span both domains [64–66]. Unfortunately, these considerations have to remain largely theoretical, as the binding sites of very few effectors have been identified (discussed in [67]).
Figure 5. Switch regions of arrestin-3.
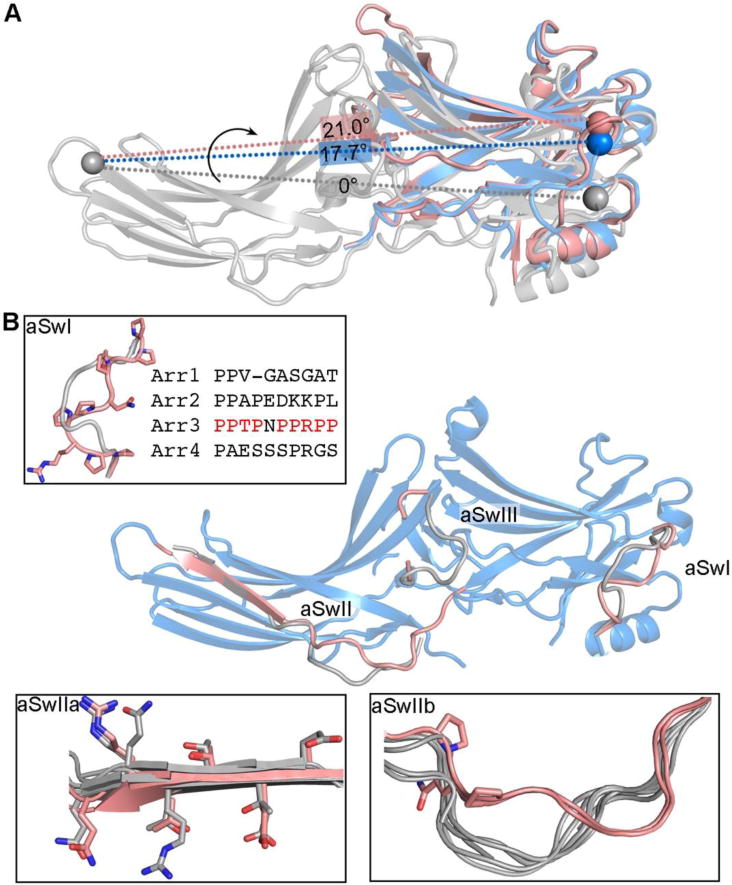
A. The inter-domain twist realigns effector-binding sites that span two domains. The spheres mark equivalent residues. The alignment of two potential effector bindings sites (spheres) is different in basal arrestin-3 (colored grey, PDB entry 3P2D), active arrestin-3 (colored blue, PDB entry 3P2D) and active arrestin-2 (colored pink, PDB entry 4JQI) due to different rotational angle between two domains. For an effector, which engages multiple binding sites spanning both domains, the change in the alignment of these binding sites can lead to the change of its affinity for arrestin. B. Conformations of the arrestin-3 switch regions in the active state (colored pink, PDB entry 5TV1) differs from the one in the basal state (colored grey, PDB entry 3P2D). The switch regions are enlarged in the insets.
In addition to the domain rotation, regions that are predicted to engage the effectors also undergo conformational changes upon arrestin activation [5, 13, 14, 24, 26]. These structural elements may act as molecular switches that bind effectors and initiate downstream signaling [5], and are termed arrestin switch regions (aSw) based upon their proposed functional similarity to nucleotide-dependent switch regions in GTP and ATP regulated signaling proteins (Figure 5B).
Arrestin switch region I (aSwI) is unique to arrestin-3 (residues 89-97). It is a nine- residue segment with seven prolines containing two PPXP motifs, which are consensus recognition sites for SH3 domains (Figure 5B) [18, 68]. In IP6-activated arrestin-3, the backbone of aSwI shifted significantly from its location in the basal state, with a maximal displacement of 5.8 Å [5, 69]. There was also an unusual pair of tandem cis bonds (Pro94-Pro95 and Pro95- Arg96) in the best-fitting model [5].
Switch region II (aSwII) is found in all four arrestin isoforms and contains two parts, the hinge region (aSwIIa) that connects the N- and C- domains and the entire β-strand XI (aSwIIb) that extends from the hinge region (Figure 5B). The aSwIIa also contains an additional poly-proline motif that might serve as recognition site for SH3 domains [27]. ASwIIa shows a nearly identical conformation in all available active structures [5, 13, 14, 24, 26], while its conformation in basal arrestins varies [15–20]. A notable feature of aSwIIb is a register-shifted β-strand XI that correlates with activation (Figure 5B) [5, 26]. This β-strand may have some inherent flexibility as this register-shift is also observed in several structures of arrestin-2 determined without an activator [17, 18, 27, 60]. Biochemical studies support a role for this register shift associated with arrestin-3 activation in effector binding. When a disulfide bond was introduced to trap β-strand XI in the registered-shifted position found in the activated state, purified arrestin-3 showed enhanced binding to the downstream effector JNK3 [5].
ASwIII is an extension of the lariat loop and likely becomes more dynamic upon activation in both arrestin-2 and -3, as suggested by a loss of interpretable electron density in the active structures (Figure 5B) [5, 26]. The lariat loop contributes two charged residues to the polar core and also contains one phosphate-binding residue (Figure 3A-C). The activation of arrestin triggers the conformational changes in the lariat loop (Figure 4G), which likely directly increases its dynamics, thus turning on specific signaling.
Finally, the C-tail, which is invariably released upon receptor binding [61, 70–72], should be considered aSwIV. It contains the binding sites for clathrin [73] and clathrin adaptor AP2 [74], two key players in the receptor endocytosis. C-tail release makes clathrin- and AP2-binding sites accessible, thus promoting receptor internalization.
The connection between activator- and effector-binding sites
The key to arrestin function as a signal transducer is that the activator (GPCR or IP6) binds on one side of arrestin, and elicits conformational rearrangements on the other side that enhance effector binding. This requires allosteric connections between the activator and the effector binding sites. Although underlying mechanism is not clear, there are several connections between the activator-binding surface and the switch regions that can be traced structurally (Figure 6). The binding of phosphate to the α-helix I likely affect the adjacent aSwI (Figure 6A). Engaging the phosphate-binding lysine in the lariat loop is likely involved in boosting the local dynamic of the aSwIII, which extends from lariat loop (Figure 6B). Since aSwIIa is the hinge region connecting the N- and C- domain, the rotation between these two domains likely directly results in the conformational change in aSwIIa and possibly adjacent aSwIIb. The molecular basis and physiological implications of these connections need to be tested experimentally.
Figure 6. The connection between activator- and effector-binding sites.
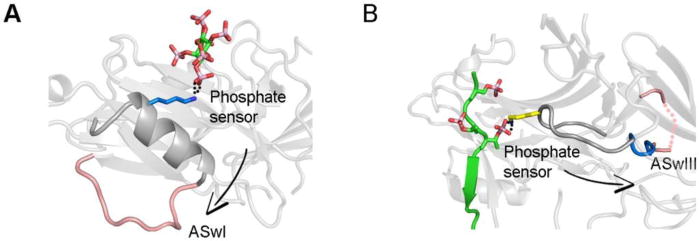
A. Engagement of the α-helix 1 by the phosphate likely affects the conformation of aSwI (PDB entry 5TV1). B. Engagement of the lysine in the lariat loop likely enhances the dynamics of aSwIII (PDB entry 4JQI).
Dynamic nature of arrestins
One common functional feature of most signaling pathways, including arrestin-dependent, is that there is basal signaling even in the absence of an activator. The mechanism of basal signaling is also suggested by recent structural studies. Like most signaling proteins, arrestins are dynamic, likely changing conformation even in the absence of activating stimuli. Moreover, it is clear that the concept of “active arrestin” is misleading because arrestins can adopt multiple active conformations [75]. This can be visualized by superpositioning all available crystal structures of arrestin-2 while aligning one of the two domains (Figure 2B). This comparison reveals that even in basal arrestin, a range of inter-domain angles can be achieved, i.e. even in the basal state the orientation of the two domains is not fixed by the inter-domain interactions. Importantly, arrestin perhaps has an even greater number of conformational options after activation as the change in inter-domain angles can be as big as 21°, as evidenced by a similar superposition of arrestins activated by distinct stimuli, e.g. receptor phosphopeptide [26] or small molecule activator IP6 (Figure 5A) [5]. This conformational flexibility of active arrestins is reflected in recent NMR [61] and EPR [62] studies, which reveal dynamics. For example, numerous NMR peaks of 13C-methyl-arrestin-1 disappeared upon binding to the phosphorylated active rhodopsin, which is consistent with a conformational exchange in active arrestin-1 [61]. Distance measurements within doubly spin-labeled arrestin-1 showed relatively broad distributions both in free and rhodopsin-bound state [62], suggesting high conformational flexibility. The same phenomenon was observed in non-visual arrestins [72]. The flexibility likely allows arrestin to sample multiple conformations suitable for distinct downstream effectors [75] and/or helps a specific receptor in a particular active state to stabilize arrestin in a defined conformation that selects for a particular downstream effector.
Concluding remarks
The comparison of recent crystal structures of active arrestin-1, -2, and -3 with basal conformations of these proteins revealed the molecular mechanisms of arrestin activation. Most importantly, a large domain rotation of the active arrestin changes the alignment of effector- binding sites. Within these sites, elements termed arrestin switch regions additionally change conformation upon arrestin activation, suggesting a multi-layered mechanism of regulating the interaction between arrestin and effectors. The analysis of these structures also reveals potential allosteric connections from the activator-binding surface to the effector-binding regions. Notably, a single GPCR activated by different agonists or bearing different phosphorylation patterns can lead to distinct functional outcomes. One of the challenges in the field is identifying how activation of arrestin promotes signaling toward a preferred effector.
Trends box.
Free arrestins have very similar conformations in crystal structures, with the relative orientation of the two domains held by several intra-molecular interactions. These interactions are destabilized by activators: active phosphorylated GPCRs and inositol-6-phosphate (IP6), an inositol metabolite abundant in the cytoplasm. The latest high-resolution structure of the non- visual arrestin-3 with IP6 suggest molecular mechanisms of arrestin activation that induce characteristic rearrangements in the arrestin molecule.
Arrestin-1, -2, and -3 activated by different means demonstrate a global conformational rearrangemnet. In particular, several elements of activated arrestins dramatically change their conformation from basal to active, which is very similar in all arrestins subtypes. These elements (that we propose to call “arrestin switches”, by analogy with G protein switch regions) are likely docking sites for the signaling and trafficking proteins that preferentially bind active arrestins.
As arrestins bind numerous protein partners, narrowing the search of their docking sites to arrestin switch regions will greatly facilitate the identification of the elements and individual residues specific for each interaction.
Identification of the binding sites of non-receptor signaling proteins paves the way to the design of signaling-biased arrestins, where particular capabilities are specifically disabled or enhanced, which would be novel potent tools for targeted manipulation of cell signaling for research and therapy.
Outstanding questions box.
Do all arrestins in complex with different GPCRs assume similar conformations?
How is the functional outcome of arrestin binding to differentially phosphorylated active GPCRs determined by the arrestin conformation and/or its pose on the receptor?
Where the binding sites of numerous non-receptor partners are localized on arrestins?
Are there other than IP6 non-receptor activators of arrestins?
Does arrestin binding to receptors other than GPCRs induce similar conformational rearrangements?
Acknowledgments
This work was supported in part by NIH grants to VVG (GM077561, GM109955 (these two R01 grants were replaced by R35 GM122491), EY011500, DA043680) and TMI (GM095633, GM120569, DA043680).
Glossary
- Arrestins
44-48 kDa proteins that specifically bind active phosphorylated GPCRs. Vertebrates have four subtypes: arrestin-1 and -4 are specialized visual, predominantly expressed in photoreceptor cells in the retina, whereas arrestin-2 and -3 are non-visual, expressed in virtually every cell. These two arrestins bind hundreds of different GPCRs and dozens of non- receptor protein partners, mediating numerous signaling pathways.
- Arrestin switch regions (ASw)
elements of arrestin that significantly change conformation upon activation. Possible docking sites of arrestin binding partners that preferentially bind active or inactive arrestins.
- GPCRs
G protein-coupled receptors. Mammals have from ~800 (humans and other primates, bats) to ~3,400 (elephants) different subtypes, including ~400 non-odorant GPCRs in every mammalian species. Common core of all GPCRs consists of seven trans-membrane helices (TM) (hence the synonym of GPCRs, seven trans-membrane domain receptors, or 7TMRs), whereas extracellular N-terminus and loops, as well as intracellular C-terminus and loops widely vary in size. Three classes of proteins preferentially bind active GPCRs: heterotrimeric G proteins, which were the first discovered signal transducers, GPCR kinases (GRKs), and arrestins.
- GRKs
GPCR kinases that preferentially phosphorylate active GPCRs. This reason of this specificity is believed to be that GRKs are activated by physical binding to active GPCRs.
- Polar core
an arrangement of five interacting charged side chains (two Arg and three Asp) between the two domains in all arestins serving as the key component of the phosphate sensor. Receptor-attached phosphates destabilize the polar core, which is one of the signals for arrestins to transition into high-affinity receptor-binding conformation (which is believed to be an active state of arrestins).
- Three-element interaction
the interaction of the β-strand I, α-helix I in the N-domain and β- strand XX of the C-terminus of all arrestins, mediated by large hydrophobic side chains. Holds arrestins in their basal conformation. Activators (active phosphorylated GPCRs and IP6) disrupt the three-element interaction and induce the displacement of the C-terminus, which is one of the hallmarks of arrestin activation.
- Finger loop
the loop between β-strands V and VI on the receptor-binding side of arrestins. This loop directly engages GPCRs via the cavity between their trans-membrane helices that opens upon receptor activation, likely serving as a critical part of the activation sensor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Here we use the systematic names of arrestins, where the number after the dash indicates the order of cloning: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin).
References
- 1.Indrischek H, et al. Uncovering missing pieces: duplication and deletion history of arrestins in deuterostomes. BMC Evol Biol. 2017;17:163. doi: 10.1186/s12862-017-1001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharm Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peterson YK, Luttrell LM. The Diverse Roles of Arrestin Scaffolds in G Protein- Coupled Receptor Signaling. Pharmacol Rev. 2017;69:256–297. doi: 10.1124/pr.116.013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao K, et al. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Q, et al. Structural basis of arrestin-3 activation and signaling. Nat Commun. 2017;8:1427. doi: 10.1038/s41467-017-01218-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schleicher A, et al. Kinetics, binding constant, and activation energy of the 48-kDa protein-rhodopsin complex by extra-metarhodopsin II. Biochemistry. 1989;28:1770–1775. doi: 10.1021/bi00430a052. [DOI] [PubMed] [Google Scholar]
- 7.Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin: Sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 8.Shukla AK, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kovoor A, et al. Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J Biol Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- 10.Celver J, et al. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 11.Gimenez LE, et al. Role of receptor-attached phosphates in binding of visual and non- visual arrestins to G protein-coupled receptors. J Biol Chem. 2012;287:9028–9040. doi: 10.1074/jbc.M111.311803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gimenez LE, et al. Mutations in arrestin-3 differentially affect binding to neuropeptide Y receptor subtypes. Cell Signal. 2014;26:1523–1531. doi: 10.1016/j.cellsig.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang Y, et al. Crystal structure of rhodopsin bound to arrestin determined by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou XE, et al. Structural Identification of Phosphorylation Codes for Arrestin Recruitment by G protein-Coupled Receptors. Cell. 2017;170:457–469. doi: 10.1016/j.cell.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Granzin J, et al. X-ray crystal structure of arrestin from bovine rod outer segments. Nature. 1998;391:918–921. doi: 10.1038/36147. [DOI] [PubMed] [Google Scholar]
- 16.Hirsch JA, et al. The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 17.Han M, et al. Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane Translocation. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 18.Milano SK, et al. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry. 2002;41:3321–3328. doi: 10.1021/bi015905j. [DOI] [PubMed] [Google Scholar]
- 19.Sutton RB, et al. Crystal structure of cone arrestin at 2.3A: evolution of receptor specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 20.Zhan X, et al. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J Mol Biol. 2011;406:467–478. doi: 10.1016/j.jmb.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurevich VV, Benovic JL. Visual arrestin binding to rhodopsin: diverse functional roles of positively charged residues within the phosphorylation-recignition region of arrestin. J Biol Chem. 1995;270:6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- 22.Gurevich VV, Benovic JL. Mechanism of phosphorylation-recognition by visual arrestin and the transition of arrestin into a high affinity binding state. Mol Pharmacol. 1997;51:161–169. doi: 10.1124/mol.51.1.161. [DOI] [PubMed] [Google Scholar]
- 23.Gurevich VV. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- 24.Kim YJ, et al. Crystal structure of pre-activated arrestin p44. Nature. 2013;497:142–146. doi: 10.1038/nature12133. [DOI] [PubMed] [Google Scholar]
- 25.Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. Trends Pharmacol Sci. 2004;25:59–112. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Shukla AK, et al. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milano SK, et al. Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding. J Biol Chem. 2006;281:9812–9823. doi: 10.1074/jbc.M512703200. [DOI] [PubMed] [Google Scholar]
- 28.Gurevich VV, Benovic JL. Visual arrestin binding to rhodopsin: diverse functional roles of positively charged residues within the phosphorylation-recignition region of arrestin. J Biol Chem. 1995;270:6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- 29.Hanson SM, Gurevich VV. The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem. 2006;281:3458–3462. doi: 10.1074/jbc.M512148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vishnivetskiy SA, et al. An additional phosphate-binding element in arrestin molecule: implications for the mechanism of arrestin activation. J Biol Chem. 2000;275:41049–41057. doi: 10.1074/jbc.M007159200. [DOI] [PubMed] [Google Scholar]
- 31.Peterhans C, et al. Functional map of arrestin binding to phosphorylated opsin, with and without agonist. Sci Rep. 2016;6:2868. doi: 10.1038/srep28686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostermaier MK, et al. Functional map of arrestin-1 at single amino acid resolution. Proc Natl Acad Sci U S A. 2014;111:1825–1830. doi: 10.1073/pnas.1319402111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mendez A, et al. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. doi: 10.1016/s0896-6273(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 34.Vishnivetskiy SA, et al. Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem. 2007;282:32075–32083. doi: 10.1074/jbc.M706057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lohse MJ, et al. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 36.Attramadal H, et al. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- 37.Nobles KN, et al. Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal. 2011;4:ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tobin AB, et al. Location, location, location…site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci. 2008;29:413–420. doi: 10.1016/j.tips.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang F, et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nat Commun. 2015;6:8202. doi: 10.1038/ncomms9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farrens DL, et al. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 41.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choe HW, et al. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 43.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szczepek M, et al. Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat Commun. 2014;5:4801. doi: 10.1038/ncomms5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Homan KT, Tesmer JJG. Structural insights into G protein-coupled receptor kinase function. Curr Opin Cell Biol. 2014;27:25–31. doi: 10.1016/j.ceb.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rasmussen SG, et al. Crystal structure of the ≈í‚â§2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Komolov KE, et al. Structural and Functional Analysis of a ≈í‚â§2-Adrenergic Receptor Complex with GRK5. Cell. 2017;169:407–421. doi: 10.1016/j.cell.2017.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He Y, et al. Molecular assembly of rhodopsin with G protein-coupled receptor kinases. Cell Res. 2017;27:728–747. doi: 10.1038/cr.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carpenter B, et al. Erratum: Structure of the adenosine A2A receptor bound to an engineered G protein. Nature. 2016;538:542. doi: 10.1038/nature19803. [DOI] [PubMed] [Google Scholar]
- 50.Liang YL, et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature. 2017;546:118–123. doi: 10.1038/nature22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature. 2017;546:248–253. doi: 10.1038/nature22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pao CS, et al. Role of the amino terminus of G protein-coupled receptor kinase 2 in receptor phosphorylation. Biochemistry. 2009;48:7325–7333. doi: 10.1021/bi900408g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang CC, et al. Activation of G protein-coupled receptor kinase 1 involves interactions between its N-terminal region and its kinase domain. Biochemistry. 2011;50:1940–1949. doi: 10.1021/bi101606e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilden U. Duration and amplitude of the light-induced cGMP hydrolysis in vertebrate photoreceptors are regulated by multiple phosphorylation of rhodopsin and by arrestin binding. Biochemistry. 1995;34:1446–1454. doi: 10.1021/bi00004a040. [DOI] [PubMed] [Google Scholar]
- 55.Krupnick JG, et al. Mechanism of quenching of phototransduction: binding competition between arrestin and transducin for phosphorhodopsin. J Biol Chem. 1997;272:18125–18131. doi: 10.1074/jbc.272.29.18125. [DOI] [PubMed] [Google Scholar]
- 56.Pan L, et al. The nature of the arrestin x receptor complex determines the ultimate fate of the internalized receptor. J Biol Chem. 2003;278:11623–11632. doi: 10.1074/jbc.M209532200. [DOI] [PubMed] [Google Scholar]
- 57.Scheerer P, Sommer ME. Structural mechanism of arrestin activation. Curr Opin Struct Biol. 2017;45:160–169. doi: 10.1016/j.sbi.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 58.Granzin J, et al. Crystal structure of p44, a constitutively active splice variant of visual arrestin. Journal of molecular biology. 2012;416:611–618. doi: 10.1016/j.jmb.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 59.Granzin J, et al. Structural evidence for the role of polar core residue Arg175 in arrestin activation. Sci Rep. 2015;5:15808. doi: 10.1038/srep15808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kang DS, et al. Structure of an arrestin2-clathrin complex reveals a novel clathrin binding domain that modulates receptor trafficking. J Biol Chem. 2009;284:29860–29872. doi: 10.1074/jbc.M109.023366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhuang T, et al. Involvement of Distinct Arrestin-1 Elements in Binding to Different Functional Forms of Rhodopsin. Proc Nat Acad Sci USA. 2013;110:942–947. doi: 10.1073/pnas.1215176110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim M, et al. Conformation of receptor-bound visual arrestin. Proc Nat Acad Sci USA. 2012;109:18407–18412. doi: 10.1073/pnas.1216304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lally CC, et al. C-edge loops of arrestin function as a membrane anchor. Nat Commun. 2017;8:14258. doi: 10.1038/ncomms14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahmed MR, et al. Ubiquitin ligase parkin promotes Mdm2-arrestin interaction but inhibits arrestin ubiquitination. Biochemistry. 2011;50:3749–3763. doi: 10.1021/bi200175q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Song X, et al. How does arrestin assemble MAPKs into a signaling complex? J Biol Chem. 2009;284:685–695. doi: 10.1074/jbc.M806124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhan X, et al. Arrestin-3 binds the MAP kinase JNK3α2 via multiple sites on both domains. Cell Signal. 2014;26:766–776. doi: 10.1016/j.cellsig.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gurevich VV, Gurevich EV. Synthetic biology with surgical precision: targeted reengineering of signaling proteins. Cell Signal. 2012;24:1899–1908. doi: 10.1016/j.cellsig.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luttrell LM, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor- Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 69.Zhan X, et al. Arrestin-3 binds the MAP kinase JNK3α2 via multiple sites on both domains. Cell Signal. 2014;26:766–776. doi: 10.1016/j.cellsig.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hanson SM, et al. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci U S A. 2006;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palczewski K, et al. Phosphorylated rhodopsin and heparin induce similar conformational changes in arrestin. J Biol Chem. 1991;266:18649–18654. [PubMed] [Google Scholar]
- 72.Zhuo Y, et al. Identification of receptor binding-induced conformational changes in non-visual arrestins. J Biol Chem. 2014;289:20991–21002. doi: 10.1074/jbc.M114.560680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goodman OB, Jr, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 74.Laporte SA, et al. The 2-adrenergic receptor/arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Nat Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gurevich VV, Gurevich EV. Extensive shape shifting underlies functional versatility of arrestins. Curr Opin Cell Biol. 2014;27:1–9. doi: 10.1016/j.ceb.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]