

Abstract
The asmarines are a family of cytotoxic natural products whose mechanism of action is unknown. Here we used chemical synthesis to reverse engineer the asmarines and understand the functions of their individual components. We found that the potent asmarine analog “delmarine” arrested the mammalian cell cycle in G1 phase, and that both cell cycle arrest and cytotoxicity was rescued by co-treatment with ferric and ferrous salts. Cellular iron deprivation was clearly indicated by changes in iron-responsive protein markers, and cytotoxicity occurred independent of radical oxygen species (ROS) production. Chemical synthesis allowed for annotation of the distinct structural motifs required for these effects, especially the unusual diazepine, which we found enforced an iron-binding tautomer without distortion of the NCNO dihedral angle out of plane. With this information and a correlation of cytotoxicity with logP, we could replace the diazepine by lipophilic group appendage to N9, which avoided steric clash with the N6-alkyl required to access the aminopyridine. This study transformed the asmarines, scarce marine metabolites, into easily synthesized, modular chemotypes that may complement or succeed iron-selective binders in clinical trials and use.
INTRODUCTION
The resources and pressures present in nature differ from those present in rational drug design.1, 2 As a result, secondary metabolites occupy areas of chemical space distinct from drugs, even when their functions overlap.2 The structural complexity of secondary metabolites, however, often impedes reverse engineering to annotate function for every atom or group.3 Here we assign a mechanism of action to the asmarine metabolite family and dissect individual structural motifs to show why they are necessary and how they can be altered.
The asmarines (e.g. 1a and 1b, Figure 1) are a small family of cytotoxic natural products that were first isolated off the coast of Eritrea in the late 1990’s.4–7 These compounds contain a unique N-hydroxydiazepine motif connected to a clerodane core. It is likely that the asmarines originate biosynthetically from the related agelasine family of natural products (e.g. 2, Figure 1), which in turn arise from adenylation of a clerodane pyrophosphate (3).8, 9 Interestingly, despite high structural similarity between the asmarines and agelasines, these two classes of natural products appear to be unrelated with respect to their cytotoxic mechanisms (vide infra).
Figure 1.
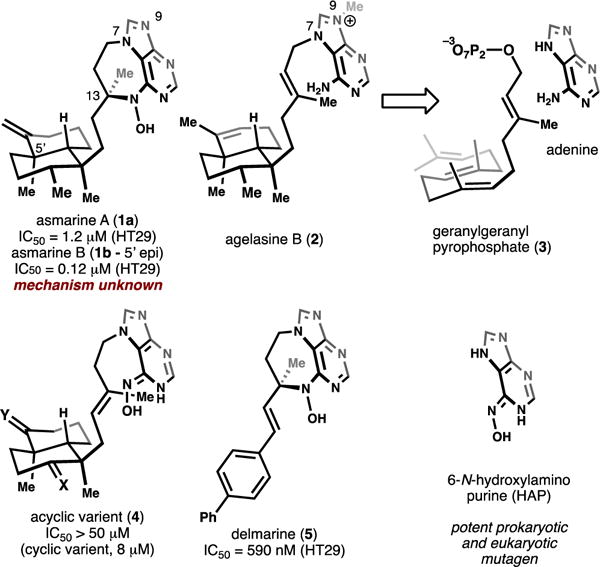
Structures of asmarine A and related compounds.
The structural complexity of the asmarines (two chiral lobes connected via an ethyl bridge) makes them some of the most challenging structures in the purine-diterpene class to prepare and has resulted in interest from chemists seeking to synthesize and understand their cytotoxicities.10–16 Unfortunately, the lack of any remaining material from the initial isolation has been a major impediment towards annotation of the biological mechanism of these compounds.17 Additionally, to this point their structural complexity has precluded their chemical synthesis. However, through the isolation literature and synthesis of asmarine-like analogs, some structure-activity-relationship (SAR) trends have been established. For instance, the N-hydroxy (N-OH) functionality is critical for cytotoxic activity as similar N-H and N-OCH3 diazepines are inactive.6, 7, 13 Synthesis of asmarine-like acyclic compounds (e.g. 4, Figure 1) has shown the necessity for the diazepine as the cyclic variants retain potency whereas acyclic compounds are inactive.15, 18 This trend suggests that the asmarines and agelasines are unrelated functionally as the agelasines inhibit various ATPases9, 19 and maintain cytotoxic activity despite the absence of the diazepine. Additionally, the parent N-hydroxypurine (HAP, Figure 1) heterocycle is a known mutagen as a base analog20: an unlikely source of cytotoxicity for the asmarines. Hydrophobic substitution of the clerodane core has been shown to be well tolerated and the stereochemistry of C13 has been shown to be unimportant for cytotoxicity.13, 15, 18 Considering this information, our lab designed a structurally simplified compound termed “delmarine” (5, Figure 1) and showed that it is a similarly potent toxin as asmarines A and B.15, 18
Here we show that delmarine inhibits DNA synthesis by sequestration of iron in mammalian cells. The SAR trends described above are fully explained by this newly identified mechanism of action. Additionally, this mechanism clarifies the unique structural features of these compounds and shows why evolution arrived at the unusual asmarine motif. With this information, we redesigned the asmarines using building blocks that are more easily assembled in a non-cellular environment in as few as two synthetic steps.
RESULTS AND DISCUSSION
Delmarine induces iron-dependent G1 arrest
Cell cycle analysis by flow cytometry can be used to ‘bin’ small molecules according to their effect on the cell state. For example, anti-mitotics such as taxol, colchicine and vinblastine all arrest the cell cycle in G2/M phase, whereas inhibitors of DNA synthesis such as thymidine and 5-fluorouracil arrest in S phase.21 Therefore, in our first interrogation of mechanism, cells treated with delmarine were subjected to cell cycle analysis by propidium iodide staining and flow cytometry. Delmarine was found to arrest cells primarily in G1 phase in both the HeLa (cervical cancer, Figure 2A,B) and HT1080 (fibrosarcoma, Supplementary Figure 1A,B) cell lines. At low concentrations of delmarine, cells passed the G1/S checkpoint and were found to arrest in S phase (Supplementary Figure 1C).
Figure 2.
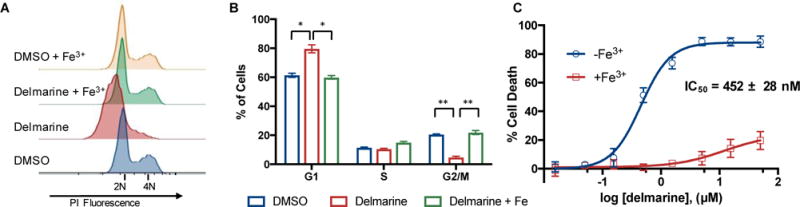
A) Delmarine induces G1 arrest as assessed by propidium iodide staining and flow cytometry in HeLa cells. Arrest can be rescued by addition of 100 μM ferric ammonium citrate. B) Quantification of data in A), N=3. C) 48 h cytotoxicity of delmarine in HT1080 cells as assessed by alamar blue. Cytotoxicity can be rescued by addition of 100 μM ferric ammonium citrate (red curve), N=3. * = p < 0.005, ** = p < 1×10−4.
A number of iron chelators, such as the clinically approved agents hydroxyurea22 and deferoxamine,23 are known to arrest cells in the G1/S phase of the cell cycle. Although iron chelation affects many cellular proteins and processes, G1/S arrest induced by these agents is believed to be principally caused by sequestration of iron needed for the 8 fold upregulation of ribonucleotide reductase necessary for DNA synthesis during S phase.24, 25 Due to structural homology between the N-hydroxy purine of delmarine and the functionality present in these other molecules, we hypothesized that iron binding may be involved in the mechanism of action of delmarine. Screening of the NCI-60 with delmarine and use of the COMPARE algorithm26 returned several iron binding small molecules as top hits (Supplementary Tables 1 and 2), consistent with delmarine-induced cell death being related to iron binding.27
Therefore, the influence of iron on delmarine-induced cell cycle arrest was examined next. The addition of excess ferric ammonium citrate to the cellular growth medium rescued the G1 arrest of delmarine, but had no effect on untreated cells (Figure 2A,B and Supplementary Figure 1A,B). Similarly, addition of excess ferric ammonium citrate to the medium rescued delmarine-induced cytotoxicity in a nearly quantitative manner (Figure 1C, Supplementary Figure 1D). Taken together, this data suggested that iron binding may be involved in the mechanism of delmarine.
Delmarine binds iron with a 3:1 stoichiometry
The ability of delmarine to bind iron in aqueous buffer and its preferred stoichiometry of iron binding was next probed. Delmarine was found to directly bind to iron as assessed by UV-Visible spectroscopy (Figure 3A), and this interaction was found to occur with 3: 1 delmarine : iron stoichiometry by titration with increasing concentrations of iron (Figure 3B), as well as with the method of continuous variations (Figure 3C). The formation constant of delmarine for iron was approximated by EDTA titration (Supplementary Figure 2) and found to be 7.9×1023, a value similar in magnitude to that of other bivalent iron chelators.28
Figure 3.
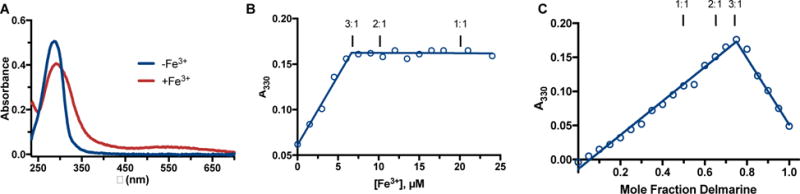
A) Delmarine binds to iron in phosphate buffered saline as assessed by UV-Vis spectroscopy. B) delmarine binds to iron with 3:1 ligand:metal stoichiometry as assessed by titration of 20 μM delmarine with increasing concentration of ferric ammonium citrate and absorbance at 330 nm. C) delmarine shows maximal absorbance at 330 nm at a ligand:metal ratio of 3:1 by the method of continuous variations.
The ability of delmarine to bind to other cellular metal cations and the ability of these complexes to modulate delmarine cytotoxicity was next examined. In bulk solvent, binding was seen with metals other than ferric iron including ferrous iron (Fe2+), copper (Cu2+), and zinc (Zn2+) (Figure 4A). Delmarine was not found to bind to magnesium, manganese or calcium (Figure 4A). Except for iron, the addition of metals involved in cellular processes was found to have little effect on cytotoxicity induced by delmarine (Figure 4B, Supplementary Figure 3). These results can likely be explained by the substantially higher concentration of labile cellular iron (10−5 to 10−6 M) relative to zinc (10−11 M) and copper (10−15 M): while the binding affinity of delmarine for copper and zinc is significant, free iron is more available for chelation in cells and is thus more relevant for cytotoxic activity.29, 30
Figure 4.
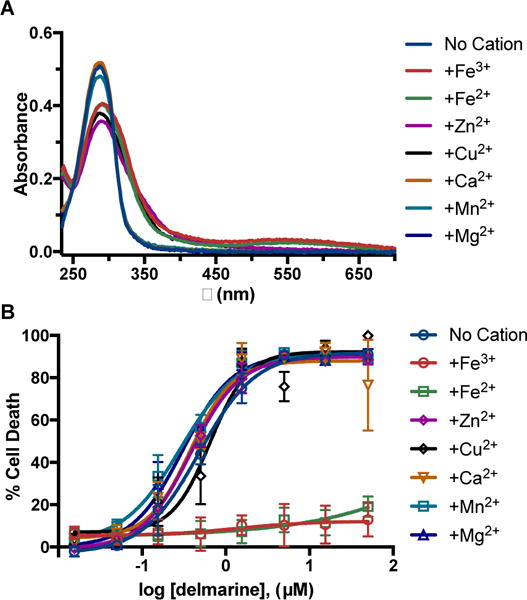
A) Delmarine binds to Fe2+, Fe3+, Zn2+, and Cu2+ in phosphate buffered saline but not to Mn2+, Mg2+, or Ca2+ as assessed by UV-Vis spectroscopy. B) Only addition of Fe3+ and Fe2+ rescues delmarine-induced cell death in HT1080 cells as assessed by alamar blue (48 h), N = 3.
Cells treated with delmarine display proteomic changes consistent with altered iron homeostasis
Having shown that delmarine binds to iron and that its cell cycle and cytotoxic effects can be rescued by exogenous addition of iron, we next asked whether cellular processes consistent with iron deprivation were observed upon treatment with delmarine. A variety of protein markers have been demonstrated to correlate with iron homeostasis in cells, including those of the iron storage protein ferritin (FTH1), the iron responsive element binding protein (IRP2) and the transferrin receptor (CD71). Treatment of HT1080 cells with delmarine and analysis of cell lysates by western blot showed a dose-dependent reduction of protein levels of FTH1, whereas levels of CD71 and IRP2 were found to be increased (Figure 5). Similar trends were seen in HeLa cells (Supplementary Fig. 4), though the effects were more bimodal in this cell line.
Figure 5.
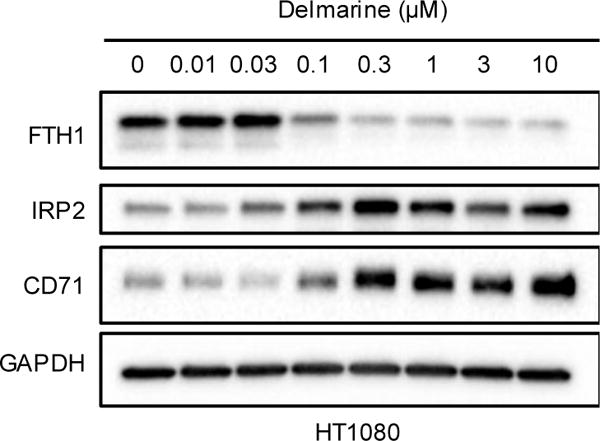
Changes of proteins associated with cellular iron levels in HT1080 cells as assessed by western blot. Representative results of one of two replicates shown.
Delmarine-induced cytotoxicity is not driven by ROS
Some iron chelators are thought to possess a cytotoxic mechanism strongly associated with radical oxygen species (ROS) production.31–33 It has been suggested that tri- and bidentate chelators can cause increases in cellular ROS through redox cycling whereas hexadentate chelators exhibit no such phenotype.34 The ability of delmarine (bidentate) as well as the small molecule chelators triapine (tridentate) and deferoxamine (DFO, hexadentate) to affect the generation of cellular ROS was examined. Preloading of HT1080 cells with the ROS-responsive probe H2-DCFDA followed by incubation with delmarine and the small molecule chelators showed a slight but not statistically significant increase in ROS by flow cytometry, with no significant difference between the different iron chelators (Figure 6A,B). Further, while EDTA showed activity in an in vitro ascorbate oxidation ROS generation assay,31 no such activity was seen for delmarine, triapine or deferoxamine (Figure 6C). Finally, delmarine-induced cell death was not protected by co-treatment with the ROS scavenger N-acetylcysteine (NAC) (Figure 6D).35 Taken together, this data suggests that delmarine does not induce substantial ROS as part of its cytotoxic mechanism. The presence or lack of ROS induction by triapine has been debated in the literature;33, 36 and ROS generation often occurs more generally in concert with cell death.37, 38
Figure 6.
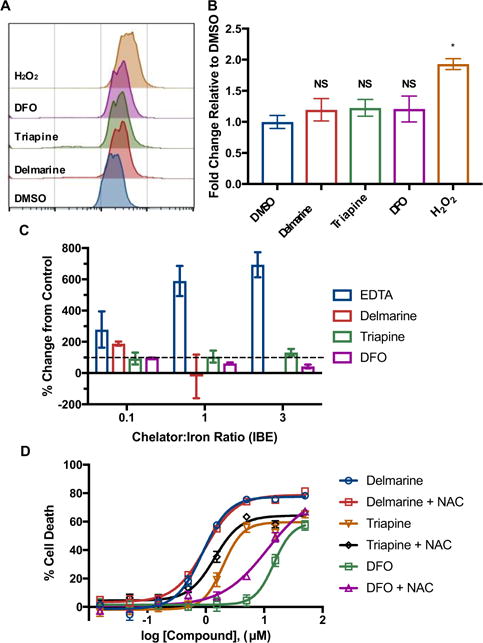
A) ROS induction as assessed by the ROS-responsive dye H2-DCFDA by flow cytometry. B) Quantification of data in A), N = 3, * = p < 0.001. C) Significant ROS production is not seen in vitro by the ascorbate oxidation assay, N = 3. D) The antioxidant N-acetyl cysteine (NAC) does not protect from cell death induced by iron chelators as assessed in HeLa cells at 48 h by alamar blue, N = 3.
Dissection of the asmarine scaffold
The determination that the asmarines induce cytotoxicity primarily through iron chelation allowed for the further dissection of the functionality present in these natural products. We reasoned that in all likelihood the nitrogen atom of N-1 and the N-6 hydroxylamine comprise the motif that is principally responsible for iron binding and that large changes could be made to the rest of the asmarine structure without sacrificing activity so long as the iron binding motif was retained. An effort to chemically synthesize simplified analogs with asmarine-like function was therefore undertaken (Figure 7).
Figure 7.
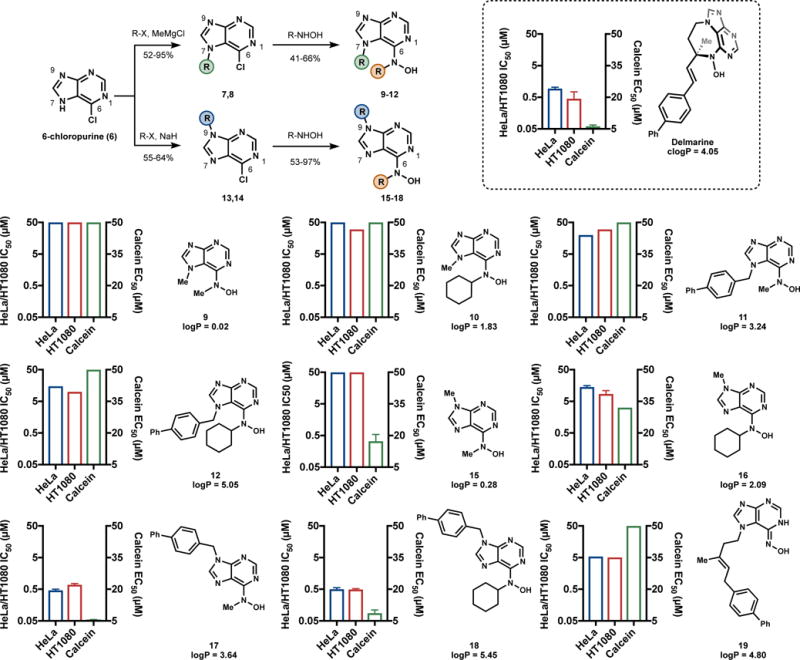
Synthesis and evaluation of delmarine analogs. IC50 values represent 48 h values assessed by alamar blue and are reported in μM. Calcein EC50 values represent the concentration of compound required to increase fluorescence of 1 μM calcein in the presence of 3 μM ferric ammonium citrate to 50% of iron-free 1 μM calcein.
To retain similarity to the asmarines (N-6 and N-7 substituted), a series of analogs substituted at the N-6 and N-7 positions were first made. A regioselective N-7 alkylation39 of 6-chloropurine (6) followed by displacement of the chlorides of compounds 7 and 8 with various hydroxylamines provided compounds 9-12. Compounds 9-12 were designed to have varying values of logP as we had previously found this to be an important parameter.15, 18 Also, increased potency of iron binders with increased logP is a known phenomenon linked to the ability of more hydrophobic chelators being better able to transport iron across cellular membranes.40, 41 Interestingly, all these N-7 substituted analogs were found to have little activity in HeLa or HT1080 cells (Figure 7), even when their logP matched or exceeded that of delmarine. The reason for this inactivity soon became clear, as these compounds were shown to be unable to bind iron in aqueous buffer with anywhere near the affinity of delmarine as determined by a calcein competition assay (Figure 7). It was reasoned that steric interactions between substituents at the N-6 and N-7 positions that do not exist in the diazepine structure force the NO bond out of plane in the simplified analogs (Figure 8). We reasoned that analogs substituted at N-7 lost the ability to bind iron efficiently as a result of this loss of planarity.
Figure 8.
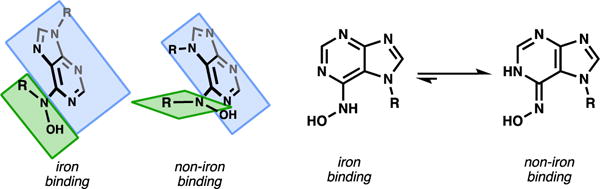
Iron binding requirements for the asmarines and asmarine analogs.
Compounds with N-9 substituents were synthesized next (Figure 7) using standard conditions42, 43 to provide regioselective N-9 substitution. Analogous to the N-7 series, displacement of the chlorides in compounds 13 and 14 with hydroxylamines provided analogs 15-18. The iron binding ability of the N-9 series was improved (Figure 7) and the potency of these compounds in HeLa and HT1080 cells was proportional to their logP. Compounds with higher logP values such as 17 and 18 were found to have cytotoxic activity on par with or exceeding that of delmarine (Figure 7).
The large differences in iron binding and cytotoxic activity between N-7 analogs versus N-9 analogs assigned an important role to the asmarine diazepine: the ring allows N6 and N7 alkyl groups to occupy the same plane by alleviating steric repulsion through covalent bond formation (Figure 8). Although the same effects can be achieved by appendage of lipophilic groups to N9, available biosynthetic intermediates guide the evolution of function. No similar purine metabolites have been isolated that contain an N9-terpenyl linkage, whereas N7 connections are common.
While the necessity of the diazepine to enforce a proper iron-binding conformation on the asmarines provided some clarity with respect to its cytotoxicity, it did not necessarily explain the inactivity of acyclic agelasine-like intermediates (e.g. 4, Figure 1). To better understand such compounds, we assessed compound 19 (Figure 7), the immediate synthetic precursor to delmarine15 in both the calcein iron-binding competition assay as well as for cytotoxicity. Compound 19 was found to possess diminished cytotoxicity in cells relative to delmarine and compounds 17 and 18 and was found to not bind iron in the calcein assay (Figure 7). The lack of iron binding of compounds such as 19 which lack N-substitution can be explained by the preference of such compounds for oxime tautomers over N-hydroxy aminopyridine tautomers in solution (Figure 8). As a result, if iron binding does not offset the energy of tautomerization, these linear purines do not bind iron and are not cytotoxic. This result suggests a second role for the asmarine diazepine: to provide N6-substitution and force the iron-binding N-hydroxy aminopyridine tautomer in solution (Figure 8).
Finally, the logP trends associated with the synthetic compounds (Figure 7) provide a rationale for the existence of the clerodane core in the asmarine structure. The clerodane core imparts substantial hydrophobicity on the asmarine system (logP of asmarine A = 5.79, logP of delmarine = 4.05). As we and others have already shown that this motif can be substituted without loss of cytotoxic activity,13, 15, 18 it is likely that the clerodane is present for its hydrophobic nature to enable iron movement across cellular membranes. In this context, the purported ten-fold potency increase from asmarine A to B is curious. In this context, the purported ten-fold potency increase from asmarine A to B is curious. If real, the difference may result from altered transport across membranes, or may point to a minor, secondary pharmacology. Secondary effects might result from the unusual reactivity of the asmarine phramacohophore5,15 or metalloprotein binding.
CONCLUSION
Here we have shown that the asmarine alkaloids are optimized for cytotoxic iron binding through three main structural features. First, appendage of a terpenyl substituent is necessary to increase hydrophobicity and is likely present to enhance membrane permeability. Second, N6 alkylation enforces an iron-binding ‘hydroxy-aniline’ tautomer, whereas non-alkylated analogs populate an ‘oxime’ tautomer that binds poorly. Third, the diazepine ring of the asmarines allows the hydroxyl-aniline group to rotate into the plane of the purine without steric repulsion by the N7 substituent. It appears that evolution of cytotoxicity requires these three related structural features to be present to accommodate the restrictions of biosynthetic starting materials. Now that the iron-binding function of the asmarines has been discovered, a function-oriented remodeling of the asmarine structure is possible using a greater breadth of building blocks than is available within cellular environments. We have shown that the hydrophobic chain can be rearranged to N9 which allows the N6-alkylated hydroxyl-amine tautomer to remain in plane, retaining iron binding capacity, as demonstrated by competition with calcein and mammalian cytotoxicity. Not only can chemical synthesis in the modern era deliver large quantities of complex molecules, but it also can fully annotate every substructure in a complex metabolite and simplify such molecules to smaller fragments.44 This exploration has uncovered the detailed structural requirements for asmarine cytotoxicity; other natural products undoubtedly hold their own trove of secrets.
METHODS
Cell Lines and Reagents
HeLa and HT1080 cells were cultured in RPMI 1640 or MEM, respectively. Media was supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C and 5% CO2 and MEM was additionally supplemented with 2 mM glutamax.
Cell Death Assessment by Alamar Blue
HeLa cells or HT1080 cells were seeded in 96-well plates in 50 μL of the appropriate medium at cell numbers of 2,500 cells/well for HeLa or 5,000 cells/well for HT1080. Cells were allowed to adhere for 6-8 h and compounds were added in 50 μL of medium from DMSO stocks to final concentrations of 50-0.016 μM with a final DMSO concentration of 0.5% (v/v). Cells were incubated for 48 h and resazurin (10 μL of a 1 mg/mL solution in water) was added. Staurosporine (10 μM) was used as the positive control for 100% cell death. For experiments involving supplementation of media, appropriate metal salts were added directly to the media to final concentrations of 100 μM. For these experiments, media was removed and replaced with fresh media containing resauzrin after 48 h. For NAC protection, N-acetyl cysteine was added to a final concentration of 5 mM. Data was analyzed in Prism and IC50 values were estimated in cases where dose response curves failed to converge.
Cell Cycle Analysis by Flow Cytometry
HeLa or HT1080 cells (200,000) were plated in 6-well plates in a volume of 2 mL and allowed to adhere 4-8 h. Prior to treatment, the media was removed and replaced and delmarine was added to the appropriate concentration (10 μM unless otherwise specified). The compounds were incubated for 18 h at which point the media was collected and the cells were trypsinized and harvested. The cells were centrifuged at 1000 × g for 5 min, washed with 3 mL of PBS, resuspended in 500 μL of PBS, and fixed by dropwise addition to 3 mL of 70% EtOH at −20°C. The fixed cells were centrifuged, washed once with 3 mL PBS, 500 μL of FxCycle PI/RNAse solution (ThermoFisher) was added, and the cells were incubated at room temperature for 30 min. The cells were analyzed by flow cytometry using a NovoCyte flow cytometer and 15,000 events were collected.
Detection of ROS by Flow Cytometry
HT1080 cells (100,000) were plated in 2 mL of MEM in wells of a six-well plate. The cells were allowed to adhere for 6 h and were treated with 20 μM H2-DCFDA 1 h. The media was then removed and replaced with media containing delmarine (10 μM), Triapine (10 μM) or DFO (100 μM). Cells were incubated 16 h, the media collected, the cells trypsinized, and centrifuged at 1000 × g for 5 min. The cell pellet was washed with 1 mL HBSS, centrifuged again, resuspended in 450 μL HBSS and analyzed by flow cytometry.
UV Titration of Delmarine with Fe3+
Delmarine was diluted to a final concentration of 20 μM in 1 mL of PBS with final DMSO concentration = 1% (v/v). Ferric ammonium citrate was added from a concentrated stock of 1 mM to final concentrations of 1.5-24 μM. Additionally, delmarine-free solutions with corresponding amounts of iron were prepared in PBS to allow for blanking of the absorbance of iron. Samples were incubated for 30 min at room temperature and a spectrum of each solution was collected in a quartz cuvette.
UV Titration of Delmarine Complex with EDTA
EDTA was added to concentrations of 10-0.0001 μM from 50× stocks to solutions of delmarine (20 μM) and ferric ammonium citrate (6.67 μM). The solutions were incubated for 30 min and analyzed by UV-Vis spectroscopy. Maximal binding was defined as no added EDTA and minimal binding was determined by addition of no ferric ammonium citrate to the ligand.
Delmarine-Fe3+ Assessment by the Method of Continuous Variations
Delmarine was added from DMSO stocks to final concentrations of 0-20 μM with DMSO = 2% (v/v). Ferric ammonium citrate was added so that the sum of the concentration of delmarine and ferric ammonium citrate was always 20 μM. Samples were incubated for 30 min and analyzed by UV-visible spectroscopy.
Binding of Delmarine to Other Metals
Delmarine was added to a final concentration of 20 μM in PBS with final DMSO = 2% (v/v). Solutions of the appropriate cation were added from 10 mM stocks to final concentrations of 20 μM. Samples were incubated for 30 min and analyzed by UV-visible spectroscopy.
Calcein Competition Assay
Solutions of analogs were added to a 96 well plate at concentrations of 50-0.39 μM from DMSO stocks (final DMSO = 5% v/v). A solution of calcein (1 μM) and ferric ammonium citrate (3 μM) in PBS was added and the solutions were incubated for 1 h in the dark. Calcein fluorescence was read on a plate reader and the percentage of ligand bound was determined by normalizing to ligand free and iron free solutions.
LogP Calculation
LogP values were calculated using the property calculation software FAFDrugs4: (http://fafdrugs3.mti.univ-paris-diderot.fr/index.html).45
Ascorbate Oxidation Assay
The assay was performed as described previously.31 Briefly, ferric ammonium citrate was dissolved to a final concentration of 30 μM. Ligands were added to final concentrations of iron binding equivalents of 0, 0.1, 1, or 3:1. Ascorbate was added to a final concentration of 100 μM and A260 was measured at 0 and 30 min. Change in A260 between the 0 and 30 min time points was measured and plotted for each point relative to control (0 iron binding equivalents).
Immunoblotting
HeLa or HT1080 cells (200,000 or 350,000 cells, respectively) were added to a 6 well plate and allowed to adhere for 4-8 h. Cells were treated with the indicated concentrations of compound (final DMSO = 1% v/v) and incubated for 18 h. After 18 h the media was removed, the cells were washed once with cold PBS, trypsinized and harvested. The cells were lysed in 50 μL of ice cold RIPA buffer for 30 min. After 30 min, the cells were centrifuged at 14,000 × g and the supernatant was resolved by SDS-PAGE. After transfer to a membrane, the blots were blocked in 5% milk in TBST for 1 h and incubated with primary antibody (1:500 dilution in TBST with 5% BSA for FTH1, IRP2, and CD71 or 1:5,000 dilution in 2.5% milk for GAPDH) overnight at 4 °C. The blots were washed with TBST, incubated with anti-rabbit HRP secondary antibody (1:5,000 for FTH1, IRP2, and CD71, 1:10,000 for GAPDH) for 1 h, washed with TBST, treated with luminol/peroxide, and imaged.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the NIH (1R35GM122606-01 to R.A.S.) and the Arnold and Mabel Beckman Foundation (Postdoctoral Fellowship to M.J.L.). We thank C. Fearns for critical review of the manuscript.
ABBREVIATIONS
- SAR
Structure-Activity Relationship
- ROS
Radical Oxygen Species
- NAC
N-Acetyl Cysteine
- DFO
Deferoxamine
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supporting Figures. Chemical and biological procedures. Characterization data for new compounds. (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
References
- 1.Dewick PM. Medicinal Natural Products A Biosynthetic Approach. 3. John Wiley & Sons; West Sussex, United Kingdom: 2009. [Google Scholar]
- 2.Lachance H, Wetzel S, Kumar K, Waldmann H. Charting, Navigating, and Populating Natural Product Chemical Space for Drug Discovery. J Med Chem. 2012;55:5989–6001. doi: 10.1021/jm300288g. [DOI] [PubMed] [Google Scholar]
- 3.Gerry CJ, Hua BK, Wawer MJ, Knowles JP, Nelson SD, Jr, Verho O, Dandapani S, Wagner BK, Clemons PA, Booker-Milburn KI, Boskovic ZV, Schreiber SL. Real-Time Biological Annotation of Synthetic Compounds. J Am Chem Soc. 2016;138:8920–8927. doi: 10.1021/jacs.6b04614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yosief T, Rudi A, Stein Z, Goldberg I, Gravalos GMD, Schleyer M, Kashman Y. Asmarines A-C; Three novel cytotoxic metabolites from the marine sponge Raspailia sp. Tetrahedron Lett. 1998;39:3323–3326. [Google Scholar]
- 5.Yosief T, Rudi A, Kashman Y. Asmarines A−F, Novel Cytotoxic Compounds from the Marine Sponge Raspailia Species. J Nat Prod. 2000;63:299–304. doi: 10.1021/np9902690. [DOI] [PubMed] [Google Scholar]
- 6.Rudi A, Shalom H, Schleyer M, Benayahu Y, Kashman Y. Asmarines G and H and Barekol, Three New Compounds from the Marine Sponge Raspailia sp. J Nat Prod. 2004;67:106–109. doi: 10.1021/np030261x. [DOI] [PubMed] [Google Scholar]
- 7.Rudi A, Aknin M, Gaydou E, Kashman Y. Asmarines I, J, and K and Nosyberkol: Four New Compounds from the Marine Sponge Raspailia sp. J Nat Prod. 2004;67:1932–1935. doi: 10.1021/np049834b. [DOI] [PubMed] [Google Scholar]
- 8.Hertiani T, Edrada-Ebel R, Ortlepp S, van Soest RWM, de Voogd NJ, Wray V, Hentschel U, Kozytska S, Müller WEG, Proksch P. From anti-fouling to biofilm inhibition: New cytotoxic secondary metabolites from two Indonesian Agelas sponges. Bioorg Med Chem. 2010;18:1297–1311. doi: 10.1016/j.bmc.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura H, Wu H, Ohizumi Y, Hirata Y. Agelasine-A, -B, -C and -D, novel bicyclic diterpenoids with a 9-methyladeninium unit possessing inhibitory effects on Na,K-ATPase from the Okinawa sea sponge Agelas sp. Tetrahedron Lett. 1984;25:2989–2992. [Google Scholar]
- 10.Pappo D, Rudi A, Kashman Y. A synthetic approach towards the synthesis of asmarine analogues. Tetrahedron Lett. 2001;42:5941–5943. [Google Scholar]
- 11.Ohba M, Tashiro T. Preparatory Study for the Synthesis of the Marine Sponge Alkaloids Asmarines A-F: Synthesis of Their Heterocyclic Portions. Heterocycles. 2002;57:1235. [Google Scholar]
- 12.Pappo D, Kashman Y. Synthesis of 9-substituted tetrahydrodiazepinopurines—asmarine A analogues. Tetrahedron. 2003;59:6493–6501. doi: 10.1021/jo048622g. [DOI] [PubMed] [Google Scholar]
- 13.Pappo D, Shimony S, Kashman Y. Synthesis of 9-Substituted Tetrahydrodiazepinopurines: Studies toward the Total Synthesis of Asmarines. J Org Chem. 2005;70:199–206. doi: 10.1021/jo048622g. [DOI] [PubMed] [Google Scholar]
- 14.Vik A, Gundersen LL. Synthetic studies directed towards asmarines; construction of the tetrahydrodiazepinopurine moiety by ring closing metathesis. Tetrahedron Lett. 2007;48:1931–1934. [Google Scholar]
- 15.Wan KK, Iwasaki K, Umotoy JC, Wolan DW, Shenvi RA. Nitrosopurines en route to potently cytotoxic asmarines. Angew Chem Int Ed Engl. 2015;54:2410–2415. doi: 10.1002/anie.201411493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodgen SA, Schaus SE. Efficient Construction of the Clerodane Decalin Core by an Asymmetric Morita–Baylis–Hillman Reaction/Lewis Acid Promoted Annulation Strategy. Angew Chem Int Ed Engl. 2006;45:4929–4932. doi: 10.1002/anie.200601076. [DOI] [PubMed] [Google Scholar]
- 17.Kashman Y. Personal Communication.
- 18.Wan KK, Shenvi RA. Conjuring a Supernatural Product – DelMarine. Synlett. 2016;27:1145–1164. [Google Scholar]
- 19.Pimentel AA, Felibertt P, Sojo F, Colman L, Mayora A, Silva ML, Rojas H, Dipolo R, Suarez AI, Compagnone RS, Arvelo F, Galindo-Castro I, De Sanctis JB, Chirino P, Benaim G. The marine sponge toxin agelasine B increases the intracellular Ca2+ concentration and induces apoptosis in human breast cancer cells (MCF-7) Cancer Chemother Pharmacol. 2012;69:71–83. doi: 10.1007/s00280-011-1677-x. [DOI] [PubMed] [Google Scholar]
- 20.Kozmin SG, Schaaper RM, Shcherbakova PV, Kulikov VN, Noskov VN, Guetsova ML, Alenin VV, Rogozin IB, Makarova KS, Pavlov YI. Multiple antimutagenesis mechanisms affect mutagenic activity and specificity of the base analog 6-N-hydroxylaminopurine in bacteria and yeast. Mutat Res Fund Mol Mech Mut. 1998;402:41–50. doi: 10.1016/s0027-5107(97)00280-7. [DOI] [PubMed] [Google Scholar]
- 21.Hung DT, Jamison TF, Schreiber SL. Understanding and controlling the cell cycle with natural products. Chem Biol. 1996;3:623–639. doi: 10.1016/s1074-5521(96)90129-5. [DOI] [PubMed] [Google Scholar]
- 22.Adams RLP, Lindsay JG. Hydroxyurea: Reversal of Inhibition and Use as a Cell-Synchronizing Agent. J Biol Chem. 1967;242:1314–1317. [PubMed] [Google Scholar]
- 23.Brodie C, Siriwardana G, Lucas J, Schleicher R, Terada N, Szepesi A, Gelfand E, Seligman P. Neuroblastoma Sensitivity to Growth Inhibition by Deferrioxamine: Evidence for a Block in G1 Phase of the Cell Cycle. Cancer Res. 1993;53:3968–3975. [PubMed] [Google Scholar]
- 24.Eriksson S, Gräslund A, Skog S, Thelander L, Tribukait B. Cell cycle-dependent regulation of mammalian ribonucleotide reductase. The S phase-correlated increase in subunit M2 is regulated by de novo protein synthesis. J Biol Chem. 1984;259:11695–11700. [PubMed] [Google Scholar]
- 25.Engström Y, Eriksson S, Jildevik I, Skog S, Thelander L, Tribukait B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J Biol Chem. 1985;260:9114–9116. [PubMed] [Google Scholar]
- 26.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. Display and Analysis of Patterns of Differential Activity of Drugs Against Human Tumor Cell Lines: Development of Mean Graph and COMPARE Algorithm. J Natl Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 27.Sun D, Melman G, LeTourneau NJ, Hays AM, Melman A. Synthesis and antiproliferating activity of iron chelators of hydroxyamino-1,3,5-triazine family. Bioorg Med Chem Lett. 2010;20:458–460. doi: 10.1016/j.bmcl.2009.11.130. [DOI] [PubMed] [Google Scholar]
- 28.Grillo AS, SantaMaria AM, Kafina MD, Cioffi AG, Huston NC, Han M, Seo YA, Yien YY, Nardone C, Menon AV, Fan J, Svoboda DC, Anderson JB, Hong JD, Nicolau BG, Subedi K, Gewirth AA, Wessling-Resnick M, Kim J, Paw BH, Burke MD. Restored iron transport by a small molecule promotes absorption and hemoglobinization in animals. Science. 2017;356:608–616. doi: 10.1126/science.aah3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ba LA, Doering M, Burkholz T, Jacob C. Metal trafficking: from maintaining the metal homeostasis to future drug design. Metallomics. 2009;1:292–311. doi: 10.1039/b904533c. [DOI] [PubMed] [Google Scholar]
- 30.Finney LA, O’Halloran TV. Transition Metal Speciation in the Cell: Insights from the Chemistry of Metal Ion Receptors. Science. 2003;300:931–936. doi: 10.1126/science.1085049. [DOI] [PubMed] [Google Scholar]
- 31.Dean RT, Nicholson P. The Action of Nine Chelators on Iron-Dependent Radical Damage. Free Rad Res. 1994;20:83–101. doi: 10.3109/10715769409147506. [DOI] [PubMed] [Google Scholar]
- 32.Chaston TB, Lovejoy DB, Watts RN, Richardson DR. Examination of the Antiproliferative Activity of Iron Chelators. Clin Cancer Res. 2003;9:402–414. [PubMed] [Google Scholar]
- 33.Shao J, Zhou B, Di Bilio AJ, Zhu L, Wang T, Qi C, Shih J, Yen Y. A Ferrous-triapine complex mediates formation of reactive oxygen species that inactivate human ribonucleotide reductase. Mol Cancer Ther. 2006;5:586–592. doi: 10.1158/1535-7163.MCT-05-0384. [DOI] [PubMed] [Google Scholar]
- 34.Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem Sci. 2016;41:274–286. doi: 10.1016/j.tibs.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bair JS, Palchaudhuri R, Hergenrother PJ. Chemistry and Biology of Deoxynyboquinone, a Potent Inducer of Cancer Cell Death. J Am Chem Soc. 2010;132:5469–5478. doi: 10.1021/ja100610m. [DOI] [PubMed] [Google Scholar]
- 36.Trondl R, Flocke LS, Kowol CR, Heffeter P, Jungwirth U, Mair GE, Steinborn R, Enyedy EA, Jakupec MA, Berger W, Keppler BK. Triapine and a More Potent Dimethyl Derivative Induce ER Stress in Cancer Cells. Mol Pharmacol. 2013;85:451–459. doi: 10.1124/mol.113.090605. [DOI] [PubMed] [Google Scholar]
- 37.Cai J, Jones DP. Superoxide in Apoptosis: Mitochondrial Generation Triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 38.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 39.Chen S, Graceffa RF, Boezio AA. Direct, Regioselective N-Alkylation of 1,3-Azoles. Org Lett. 2016;18:16–19. doi: 10.1021/acs.orglett.5b02994. [DOI] [PubMed] [Google Scholar]
- 40.Baker E, Vitolo ML, Webb J. Iron chelation by pyridoxal isonicotinoyl hydrazone and analogues in hepatocytes in culture. Biochem Pharmacol. 1985;34:3011–3017. doi: 10.1016/0006-2952(85)90142-x. [DOI] [PubMed] [Google Scholar]
- 41.Richardson D, Tran E, Ponka P. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood. 1995;86:4295–4306. [PubMed] [Google Scholar]
- 42.de Ligt RAF, van der Klein PAM, von Frijtag Drabbe Künzel JK, Lorenzen A, Ait El Maate F, Fujikawa S, van Westhoven R, van den Hoven T, Brussee J, Ijzerman AP. Synthesis and biological evaluation of disubstituted N6-cyclopentyladenine analogues: the search for a neutral antagonist with high affinity for the adenosine A1 receptor. Bioorg Med Chem. 2004;12:139–149. doi: 10.1016/j.bmc.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 43.Yang W, Chen Y, Zhou X, Gu Y, Qian W, Zhang F, Han W, Lu T, Tang W. Design, synthesis and biological evaluation of bis-aryl ureas and amides based on 2-amino-3-purinylpyridine scaffold as DFG-out B-Raf kinase inhibitors. Eur J Med Chem. 2015;89:581–596. doi: 10.1016/j.ejmech.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 44.Crane Erika A, Gademann K. Capturing Biological Activity in Natural Product Fragments by Chemical Synthesis. Angew Chem Int Ed Engl. 2016;55:3882–3902. doi: 10.1002/anie.201505863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Néron B, Ménager H, Maufrais C, Joly N, Maupetit J, Letort S, Carrere S, Tuffery P, Letondal C. Mobyle: a new full web bioinformatics framework. Bioinformatics. 2009;25:3005–3011. doi: 10.1093/bioinformatics/btp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.