

Significance
We show that in a human pancreatic β cell line the human insulin gene promoter on chromosome 11 physically contacts sites on other chromosomes. Many of these contacted sites contain type 1 or type 2 diabetes susceptibility loci. We find that insulin gene expression can affect expression of contacted genes on other chromosomes. Some of these genes, in turn, regulate insulin secretion. These results reveal physical regulatory mechanisms in which the level of insulin expression controls expression of genes involved in insulin transport and metabolism. We study the properties of one such gene, somatostatin receptor 5 antisense (SSTR5-AS1), and show that it regulates SSTR5 expression, which affects insulin secretion. Analysis of insulin contacts thus may reveal new insulin metabolic pathways.
Keywords: pancreatic beta cells, 4C-seq, insulin, gene regulation, somatostatin
Abstract
Both type 1 and type 2 diabetes involve a complex interplay between genetic, epigenetic, and environmental factors. Our laboratory has been interested in the physical interactions, in nuclei of human pancreatic β cells, between the insulin (INS) gene and other genes that are involved in insulin metabolism. We have identified, using Circularized Chromosome Conformation Capture (4C), many physical contacts in a human pancreatic β cell line between the INS promoter on chromosome 11 and sites on most other chromosomes. Many of these contacts are associated with type 1 or type 2 diabetes susceptibility loci. To determine whether physical contact is correlated with an ability of the INS locus to affect expression of these genes, we knock down INS expression by targeting the promoter; 259 genes are either up or down-regulated. Of these, 46 make physical contact with INS. We analyze a subset of the contacted genes and show that all are associated with acetylation of histone H3 lysine 27, a marker of actively expressed genes. To demonstrate the usefulness of this approach in revealing regulatory pathways, we identify from among the contacted sites the previously uncharacterized gene SSTR5-AS1 and show that it plays an important role in controlling the effect of somatostatin-28 on insulin secretion. These results are consistent with models in which clustering of genes supports transcriptional activity. This may be a particularly important mechanism in pancreatic β cells and in other cells where a small subset of genes is expressed at high levels.
It is well established that within the nucleus the genome is organized into a series of domains of varying scale that can serve to segregate active from inactive chromatin, block inappropriate interactions between regulatory sites, or in other cases, bring together regulatory elements widely separated on the genome, such as enhancers and promoters (1). Some of these interactions in vertebrates are mediated by pairs of sites separated by as much as a megabase of DNA and occupied by the protein CTCF in association with the cohesin complex. Such interactions result in formation of large loop domains within chromosomes.
Other kinds of interactions between distant sites, often on different chromosomes, arise from the clustering of transcribed genes. In an early example (2), it was shown that the Hbb-b1 β hemoglobin gene in mouse erythroid cells colocalizes at transcription factories (for a recent review on transcription factories, see ref. 3) with other expressed genes. These include the erythroid-specific gene Eraf; the Uroporphyrinogen III synthetase gene Uros, essential for heme biosynthesis (both on the same chromosome as Hbb-b1); and Hba, the gene for hemoglobin α, on a different chromosome. In this and other cases, the clusters included coregulated genes (4–7). Other transcription factory-mediated interchromosomal interactions have been reported in mouse B lymphocytes between the Myc gene on chromosome 15 and the Igh gene on chromosome 12 (4), and in human endothelial cells, which in response to TNF-α organize target genes to form “NFκB factories” (8). It has been suggested that chromosome conformation capture-based methods could detect long-range interactions within the same transcription factory (9).
Type 1 and type 2 diabetes are two distinct disease entities (10). In type 1 diabetes (T1D), the patients’ immune system attacks and destroys the insulin-producing pancreatic β cell; it comprises about 5% of all cases of diabetes. Type 2 (T2D) is the most common form of diabetes. An estimated 30.3 million people in the United States, or 9.4% of the population, have type 2 diabetes, and it remains the seventh leading cause of death in the United States (https://www.niddk.nih.gov/health-information/diabetes). It is a long-term metabolic disorder that is characterized by high blood sugar, insulin resistance, and relative lack of insulin. It has multiple causes, including both lifestyle and genetic elements.
Recent advances in genome-wide association studies (GWAS) have made it possible to identify genetic variants that are associated with T1D and T2D, and many of them are important for β cell function (11). Nevertheless, GWAS studies have only identified a small fraction of the risks attributable to genetic factors, so that diabetes is still recognized as “a geneticist’s nightmare” (12). Our laboratory has been interested in the physical interactions, in nuclei of human pancreatic β cells, between the insulin (INS) gene and other genes that are involved in insulin metabolism. In earlier work, we used 4C in human pancreatic islets to find contacts between the INS promoter and other genes on chromosome 11 (13, 14). This led to the identification of two genes, synaptotagmin 8 (SYT8) and anoctamin 1 (ANO1), ∼300 kb and 68 Mb away, respectively, from the INS locus, that we showed contacted the INS promoter. These physical contacts were correlated with expression of INS: Blocking INS promoter activity resulted in decreased contact between the promoter and both SYT8 and ANO1, as well as their deceased expression. In addition, we showed that both SYT8 and ANO1 proteins promote secretion of insulin. Thus, these interactions constitute a physical feedback transvection-like mechanism in which contacts with the highly active INS promoter stimulate expression of genes associated with insulin metabolism. It appeared that these regulatory pathways might be relevant to normal and abnormal β cell function. At least 40% of the genes associated with T1D susceptibility loci are expressed in human islets and β cells (15). Similarly, many loci known to harbor common SNPs contributing to T2D contain genes associated with β cell or islet function (11).
Here, we take advantage of the availability of a human pancreatic β cell line, EndoC-βH1 (16), to collect much higher density 4C data without the complicating presence of signals from the other kinds of cells present in islets. We show that in EndoC-βH1 cells, the INS promoter makes many strong cell type-specific contacts over large distances. The increased sequencing depth allows us not only to detect interactions within chromosome 11, on which INS resides, but also to identify many contacts of INS with sites on other chromosomes. We find that such contacts often mark sites near T1D or T2D susceptibility loci (17–22).
A large number of these sites include genes whose expression is sensitive to the level of INS transcriptional activity. Consistent with our earlier observations in human islets, many of these genes are part of regulatory mechanisms for insulin secretion. Among these we identified SSTR5-AS1, (somatostatin receptor 5 antisense) located on chromosome 16. Down-regulation of this previously uncharacterized gene results in down-regulation of the nonoverlapping gene SSTR5 (somatostatin receptor 5). Through this mechanism, SSTR5-AS1 indirectly controls the extent of SSTR5 signaling with SST28 (somatostatin-28), and this, in turn, affects the known inhibitory effect of SST on insulin secretion.
Results
Local Interactions on Chromosome 11.
The EndoC-βH1 cell line is an insulin-secreting cell line derived from human pancreatic β cells (16). To explore the interactions between the INS locus and other genomic sites, we used these cells to carry out 4C-seq experiments (23), with a site near the INS promoter as anchor (Fig. 1A). The bait sequence contains the PDX1-binding elements A3 and A5, the enhancer core element (binding region for histone H3 lysine 27 acetyltransferase p300), the T1D susceptibility-linked microsatellite (VNTR), along with some other elements (24). We focused initially on contacts between INS and sites on either side within chromosome 11. We had shown in earlier, lower-resolution studies (13, 14) in human pancreatic islets that contacts are made with many loci in these regions. This is confirmed (Fig. 1B) in EndoC-βH1 cells, where, as expected, there are strong contacts between INS and immediately adjacent sites, but also contacts with sites extending at least 700 kb in the 3′ (q-arm) direction on chromosome 11, a region containing a number of SNPs associated with both T1 and T2 diabetes susceptibility loci. The strongest contact site is 14.5 kb away from SNP rs7111341, which is associated with both T1D and T2D susceptibility (25). Many, but not all, of the contacts seen in EndoC-βH1 cells were also observed in the earlier study of pancreatic islets (Fig. 1E) (13).
Fig. 1.

The cis interactions of the INS promoter in EndoC-βH1 and HeLa cells. (A) The human INS gene locus. VNTR, variable number of tandem repeats. (B) Distance-corrected 4C-seq contact map for 1 Mbp to 4 Mbp on chromosome 11, in EndoC-βH1 cells, generated by 4Cseqpipe (58). (Upper) Normalized 4C contact profile using a 10-kb window size. (Lower) The 4C domainogram (heat map), displaying mean contact profile trend per fragment end across a range of window sizes, from 2 kb to 50 kb, shown on the y axis. Color intensity reflects normalized number of contacts (23). Red bar, viewpoint (bait). (C) Distance-corrected 4C-seq contact map for 1 Mbp to 4 Mbp on chromosome 11, in HeLa cells, generated by 4Cseqpipe. (Upper) Normalized 4C contact profile using a 10-kb window size. (Lower) The 4C domainogram as in B. Red bar, the viewpoint near INS promoter. (D) Quantitative 3C contact analysis for 1.4 Mbp to 2.5 Mbp on chromosome 11 in EndoC-βH1 cells from INS promoter. Red dash line, viewpoint. (E) Circos diagram showing 4C-seq contact map for cis interactions on chromosome 11 in EndoC-βH1 (gray links) and HeLa (red links) cells, calculated from an R script from literature (27). The red dots in the outer circle are the strong contacts detected in human pancreatic islets (13).
As a control, we carried out parallel 4C measurements in HeLa cells (Fig. 1C). There was a major shift in the pattern of INS contacts, from the extended series of 3′ interactions seen in EndoC-βH1 cells to a more limited, clustered set of surprisingly strong contacts in the opposite 5′ (q-arm) direction in HeLa cells about 500 kb from INS. This region contains a T2D susceptibility marker, rs2334499 (26). We used the chromatin conformation capture (3C) method to confirm a significantly lower level of contact frequency between sites close to rs2334499 and the INS promoter in EndoC-βH1 cells compared with HeLa (Fig. 1D, Student’s t test, P < 0.05). Although local interactions such as these are present in both EndoC-βH1 and HeLa cells, there is a dramatic difference in the abundance of long-range contacts along chromosome 11 in the two kinds of cells: many such contacts in the EndoC-βH1 cells but very few in HeLa (Fig. 1E).
Interchromosomal Interactions.
The 4C data also contain information about contacts between chromosomes. Interchromosomal interactions occur at low frequency. To identify reproducible 4C-contact regions, we applied a window-based approach to analyze the data; details of the procedure have been described in published literature (27), and further information is in Methods. This is the best-established method to identify interchromosomal interactions for our dataset (28). Only a few are detectable in the HeLa control, but they are abundant in EndoC-βH1 cells (Fig. 2). There are significant contacts with 43 individual regions in EndoC-βH1 cells (SI Appendix, Table S1A). Remarkably, a considerable number of INS 4C contact sites include T1D and T2D susceptibility loci (SI Appendix, Table S1B). Some of these loci are clustered together, as for example in the HLA loci. Several loci close to the INS gene (on chr11 p15.5) are also associated with diabetes. For clarity, we omit interactions within chromosome 11 from SI Appendix, Table S1B. The distribution of the sites in this table across the genome is shown in Fig. 3. Among the T2D loci showing interchromosomal contacts in SI Appendix, Table S1B with known function, we find only those involved in insulin secretion, but none associated with insulin resistance. Among the 34 T2D susceptibility loci listed by Bonnefond and Froguel (11) that affect insulin secretion or islet function, we find seven that contact INS, indicating that there is enrichment of those loci in the contact region (χ2 test, P = 0.003). A recent report showed that the expression levels of one of the loci, the HMG20A gene, are decreased in islets from T2D donors compared with islets from nondiabetic donors (29). The enrichment of T1D susceptibility loci is more significant, as the HLA loci resides inside the 4C contact regions. Among the 183 T1D-associated SNPs from PheGenI (20), 128 are in the 4C contact region (χ2 test, P < 0.001, 7 SNPs from the IGF2-INS-TH locus excluded from calculation). After excluding 108 SNPs at the HLA loci, the enrichment is still significant (expect number = 5, actual number = 20, χ2 test, P < 0.001, 7 SNPs from the IGF2-INS-TH locus excluded). The size of the measured contacted regions varies (SI Appendix, Fig. S1) from about 100 kb for sites on chromosome 11 to a median size of 3 Mb for other chromosomes, reflecting in part that with the reduced data density for those distant contacts, “a larger window size is required to pick up trans-interactions” (27).
Fig. 2.

The interchromosomal interactions between the INS promoter and sites on other chromosomes in EndoC-βH1 and HeLa cells. (A) 4C-seq contact map for interchromosomal interactions in EndoC-βH1 cells. Color of the links uses the default Circos setting for each chromosome. (B) 4C-seq contact map for interchromosomal interactions in HeLa cells. Color of the links as in A.
Fig. 3.

Circos diagram showing 4C contacts in EndoC-βH1 cells. Those diabetes susceptibility loci within the contact regions are also shown. The 4C contacts are represented in the inner circle. Blue line, contacts not containing diabetes susceptibility loci; red Line, contacts containing diabetes susceptibility loci. Contacts that are also present in HeLa cells are in lighter color. These diabetes susceptibility loci are represented in the outer circle: black tiles, monogenic forms of diabetes susceptibility loci; green tiles, T2D susceptibility loci; red tiles, T1D susceptibility loci. Tile thickness is larger than locus size for legibility.
Many of the contacted regions are enriched in acetylation at histone H3 lysine 27 (H3K27Ac, a marker for active enhancers and promoters; ref. 30). We carried out ChIP-seq analysis in EndoC-βH1 cells for the H3K27Ac marker. We excluded those regions that are also present in HeLa (all eight of the contacts detected in HeLa cells are also detected in EndoC-βH1 cells) and compared the remaining regions to the top 2% of H3K27Ac sites in EndoC-βH1 cells (SI Appendix, Table S2). We found that these regions are enriched 2.8-fold for H3K27Ac sites compared with the rest of the genome (χ2 test, P < 0.001). Meanwhile, we did not detect significant enrichment of the top H3K27Ac sites in those regions also detected in HeLa cells (χ2 test, P = 0.4).
Expression Levels of Contacted Genes Affected by INS Expression Levels.
In earlier studies in human pancreatic islets, we analyzed INS contacts with other sites on chromosome 11 and showed that the expression of some of these sites was correlated with the level of INS expression. We asked whether the contacted genes detected on other chromosomes in EndoC-βH1 cells behaved similarly. To inhibit INS expression, we used shRNA to target the INS promoter, a method that has been extensively described (for a recent review containing its applications and mechanisms, see ref. 31). It is important to emphasize that this strategy affects INS promoter activity and the ability of the active promoter to function in transactivation of target genes (Discussion). We find that knocking down INS expression with such an shRNA targeting the INS promoter significantly affects expression of 259 genes (Fig. 4A and SI Appendix, Table S3, α = 0.05). (Since insulin is not a transcription factor and it is the activity of its promoter in transvection that is critical, the great majority of genes are unaffected.) Of those that are affected, 45 genes lie in detected 4C contact regions; most of them show decreased expression. Thus, there is statistically significant enrichment of 4C contact regions among these 259 genes (expect number = 19, actual number = 45, χ2 test, P < 0.001).
Fig. 4.
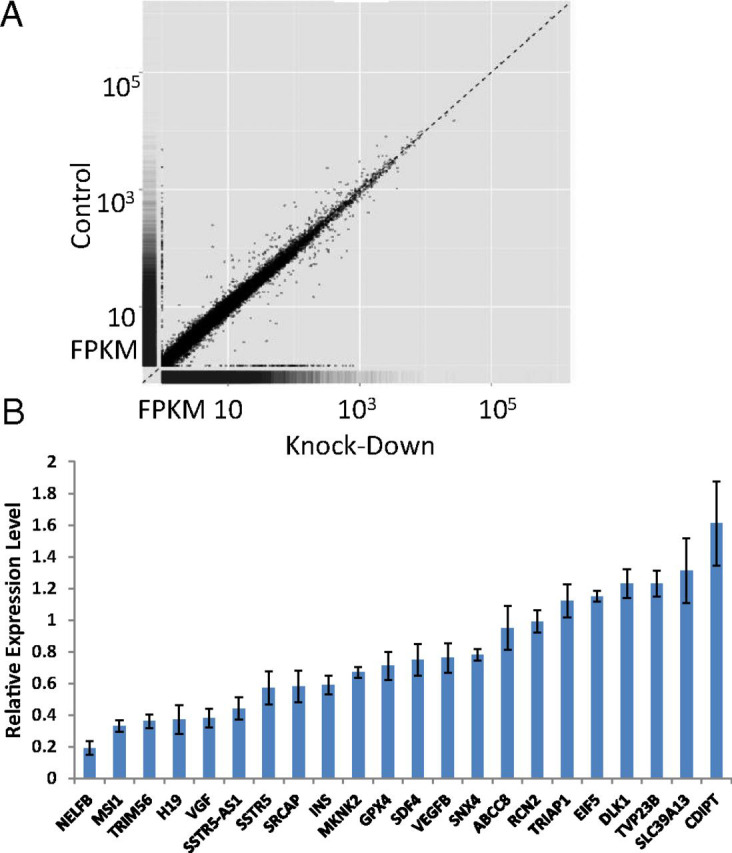
The effect of INS promoter knockdown by shRNA on gene expression in EndoC-βH1 cells. (A) Scatter plot showing the effect of INS promoter knockdown at the transcriptome level in EndoC-βH1 cells. FPKM, fragments per kilobase of transcript per million mapped reads. (B) qRT-PCR analysis of a selected subset of genes verifying expression change upon knockdown of INS promoter in EndoC-βH1 cells.
Expression of 20 selected genes with known biological functions and appreciable expression levels, both increased and decreased in expression, was verified by RT-qPCR (Fig. 4B). Not every gene associated with insulin secretion is affected by lowering INS expression; ABCC8 (ATP binding cassette subfamily C member 8) gene expression was not significantly changed, as indicated by RNA-seq and verified by RT-qPCR (Fig. 4B). These 20 genes are all associated with the H3K27Ac mark of transcriptionally active loci (SI Appendix, Fig. S2). Many of them have strong and extended H3K27Ac signals. When we examined the genomic features of these genes in the Islet Regulome Browser (32), we noticed a tendency for these down-regulated genes to associate with “enhancer clusters” (33). Such enhancer clusters “share many features and may thus represent the same phenomenon as super-enhancers” (33). Besides INS, among the genes that are down-regulated by 25% or more, seven of nine have an enhancer cluster within 100 kb of the gene (SI Appendix, Fig. S3). As a control, we chose 60 random genes and found 18 of them associated with an enhancer cluster within 100 kb of the gene (SI Appendix, Table S4). There is a significant enrichment of the enhancer clusters among these down-regulated genes (Fisher’s exact test, P < 0.01).
It is known that insulin controls gene transcription by modifying the binding of transcription factor Forkhead Box O1 (Foxo1) or insulin response element-binding protein-1 (IRE-BP1) at insulin response elements. We checked the expression of a few known target genes of Foxo1 (34) and IRE-BP1 (35) and did not find significant changes of their expression levels (SI Appendix, Fig. S4) when the INS promoter was targeted for silencing.
Contacts with Genes Affecting Insulin Metabolism: Function of SSTR5-AS1.
We searched for interactions that might be related to insulin metabolism. Among the genes shown in Fig. 4B, several are known to relate to INS regulation. MSI1 (Musashi RNA Binding Protein 1) expression in β cells coordinates INS expression in response to endoplasmic reticulum stress in diabetes (36). A peptide derived from VGF (VGF nerve growth factor inducible) helps control insulin secretion (37). DLK1 (Delta Like Non-Canonical Notch Ligand 1) (38) induces insulin synthesis and secretion, and SLC39A13 (Solute Carrier Family 39 Member 13) is a transporter protein for Zn++, which could play a role in insulin storage, secretion, and activity (39). In searching for an example of INS expression-dependent interchromosomal transvection that affected insulin metabolism, our attention was drawn to SSTR5-AS1, which codes for an antisense RNA to SSTR5. SST, produced by pancreatic δ cells, is an inhibitor of insulin secretion (40). (Newly synthesized insulin is not secreted into the extracellular space immediately. Instead, it is deposited into insulin secretion granules in the crystal form of insulin hexamer and is secreted in response to increased glucose concentration; ref. 41.) SSTRs are attractive drug targets for treating diabetes (42). Expression of SSTR5-AS1 is reduced by 30–55% when INS expression is down-regulated (Figs. 4B and 5A) and SSTR5 expression is also decreased (Fig. 4B) (Student’s t test, P < 0.05), but the SST receptor genes SSTR1 and SSTR2 are unaffected (Fig. 5A). Very little is known about the function of SSTR5-AS1, which is present in humans but has not been reported in other vertebrates such as mouse. ShRNA-mediated down-regulation of SSTR5-AS1 expression results in a decrease in SSTR5 expression (Student’s t test, P < 0.01), consistent with this antisense RNA normally acting to stimulate transcription of the SSTR5 gene (Fig. 5B).
Fig. 5.

Expression of SSTR-related genes in EndoC-βH1 cells. (A) RNA-seq differential expression analysis for SSTR5-AS1, SSTR1, and SSTR2 in EndoC-βH1 cells upon INS promoter knockdown by shRNA. FPKM, fragments per kilobase of transcript per million mapped reads. (B) Effect of knockdown of SSTR5-AS1 on expression of SSTR5 and SSTR5-AS1. Samples without shRNA treatment were used as the control.
There are two active forms of SST, SST14 and SST28, respectively, 14 and 28 amino acids long. SST28 inhibits insulin secretion from EndoC-βH1 cells, with a maximum decrease of about 55% (under our experimental conditions) and an IC50 of 0.018 nM (Fig. 6A). When SSTR5-AS1 expression is depleted, IC50 increases to 0.43 nm (Fig. 6A). The decrease in SSTR5-AS1 decreases SSTR5 expression (Fig. 5B), and this, in turn, reduces the sensitivity of insulin secretion to inhibition by SST28. In contrast, SSTR5-AS1 has little effect on the ability of SST14 to inhibit insulin secretion (IC50 changes from 1.5 nM to 0.5 nM upon SSTR5-AS1 depletion) (Fig. 6B).
Fig. 6.

The effect of knockdown of SSTR5-AS1 on the SST-mediated inhibitory pathway of insulin secretion in EndoC-βH1 cells. (A) Insulin secretion by EndoC-βH1 cells in the presence of SST28. Blue circles and solid line (R2 = 0.89), SSTR5-AS1 knockdown cells; red circles and dashed line (R2 = 0.95), normal cells. (B) Insulin secretion by EndoC-βH1 cells in the presence of SST14. Blue circles and solid line (R2 = 0.96), SSTR5-AS1 knockdown cells; red circles and dashed line (R2 = 0.83), normal cells. Error bar: SD.
Discussion
In earlier studies in human pancreatic islets, our laboratory showed that the INS locus physically contacts other genes on chromosome 11 (13, 14). The expression level of two such genes was positively correlated with the level of INS expression. We showed that each of these genes, SYT8 and ANO1, played a role in regulation of insulin secretion. These results suggested that important aspects of insulin metabolism might be controlled by physical contact between INS and genes in its regulatory network, and that a more comprehensive study of those contacts could provide valuable information. The availability of a pancreatic β cell line, EndoC-βH1, and improved 4C analytic methods made this possible. Here, we identify INS contacts with sites distributed on most of the other chromosomes. Most of these contacts are not found in HeLa cells. Remarkably, many of the contact sites are associated with T1D or T2D susceptibility loci.
To identify genes potentially associated with insulin metabolism, we searched for those genes in contacted sites whose expression was affected by knocking down INS expression. We focused first on INS contacts within chromosome 11, which are the strongest, particularly within ∼2 Mb on either side of the INS promoter (Fig. 1B). Unexpectedly, the INS locus also makes contacts with distant chromosome 11 sites in HeLa cells (Fig. 1C). However, these sites are to a large extent distinct from those observed in EndoC-βH1 cells and upstream (p-arm) rather than downstream of INS. The major peak of interaction in HeLa is with a locus that contains the T2D risk variant rs2334499 (26); the risk allele is correlated with decreased DNA methylation. In EndoC-βH1 cells, INS promoter activation is associated with decreased contact to this T2D susceptibility locus 480 kb upstream (p-arm) of the INS gene and with increased contacts with many downstream sites.
A ChIA-PET study of CTCF-mediated contacts in K562 cells (43) is consistent with a model in which the contact between the CTCF site near the INS promoter and the region near rs2334499 is important for INS regulation. The risk is associated with paternal transmission of the allele; maternal transmission is protective. The contacts with this upstream region are present in other non-β cells such as K562 (43), but our results show that they are weaker in EndoC-βH1 than in HeLa cells (Fig. 1 B–D). It is possible that the susceptibility marker activity reflects an increased ability to compete for the INS promoter (Fig. 7A). We speculate that in β cells the T2D risk variant may strengthen loop formation between INS and the upstream sites (Fig. 1C) at the expense of the downstream contacts that are presumably important for β cell function (Fig. 1B).
Fig. 7.
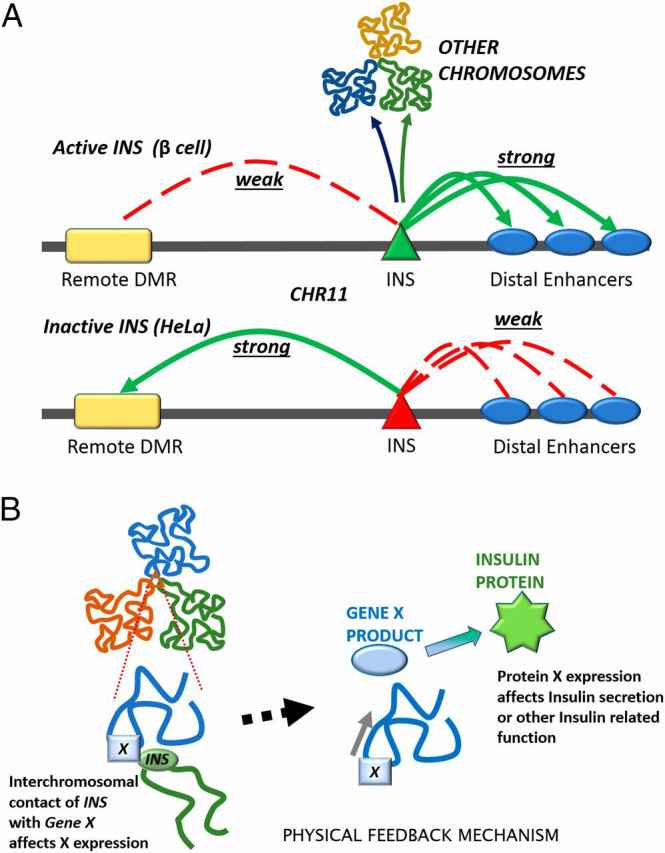
(A) Comparing intrachromosomal contacts between the INS promoter and other sites on chromosome 11 in EndoC-βH1 (Upper) and HeLa (Lower) cells (Fig. 1). The INS promoter also contacts other chromosomes (Fig. 2). (B) Schematic of the structurally based feedback mechanism, which allows INS expression to regulate its own metabolism.
The difference between HeLa and EndoC-βH1 nuclear architecture becomes evident beyond distances from INS greater than 2 Mb. There are very few longer-range interactions within chromosome 11 in HeLa cells, whereas they are abundant in EndoC-βH1 (Fig. 1E). This difference is also obvious in patterns of interaction between INS and loci on other chromosomes (Fig. 2). We have noticed that all of the interchromosomal contacts observed in HeLa cells are also present in EndoC-βH1. Since HeLa cells are well known for their remarkably high level of aneuploidy and numerous large structural variants, the lack of unique interchromosomal contacts in HeLa cells indicates that our 4C analysis is unlikely to be affected significantly by the copy number variation between the cell lines.
To identify genes with expression levels coupled to that of INS, we used shRNA to inhibit INS expression. As noted above, this strategy targets the INS promoter (13, 44). A genome-wide survey (Fig. 4A) revealed that expression of 259 genes was affected. Of this group, 104 (40%) are associated with metabolic pathways out of a total of 6,090 genes associated with that category genome wide (P = 2.96e-5). Also, 126 of these genes are associated with breast neoplasms (P = 3.1e-4), perhaps related to the recently reported connection between breast cancer and T2D (45). We selected from this list the 45 genes that reside in INS contact regions detected by our 4C analysis and verified changed expression for 20 of these by RT-qPCR (Fig. 4B). ChIP-seq analysis of histone H3K27Ac showed that all these genes carry this modification at or near their promoters, consistent with their actively transcribed status (46). It is conceivable that some genes affected by levels of INS expression are not regulated by physical contact with the INS gene; one could imagine, for example, pathways that respond to nascent INS RNA. However, targeting the INS promoter, as we have done here, only slightly reduced the basal level of secretion of insulin, of which there is a large store in β cells. We found that shRNA knockdown cells secreted 91 ± 4% of insulin in resting conditions compared with the control samples. It should be noticed that this basal level of insulin secretion under low glucose cell growth condition is distinct from the glucose-stimulated insulin secretion under high glucose condition. We did not observe significant change of SSTR5-AS1 level in INS promoter knockdown cells when we restored the extracellular insulin level by changing the medium from the nonknockdown samples (SI Appendix, Fig. S5). For these reasons, it is unlikely that the changes we observe in gene expression are caused by the observed small changes in the secreted insulin protein abundance. It is likely that changes in expression levels of some genes result from indirect effects of changes in regulatory gene expression. We also cannot eliminate the possibility that the INS promoter-targeting strategy we used might have some off-target effects on expression of other genes.
Although plausible connections can be made between the factors regulated by these contact-dependent mechanisms and susceptibility to T1D or T2D, the contact with the HLA loci is more difficult to explain. As shown in SI Appendix, Fig. S6, HLA-A expression decreases with decreasing INS expression; expression of other HLA genes is not significantly altered. We suggest that there is a coupling between the expression of INS and HLA-A. The greater the expression of the HLA-A presenting antigen, the more likely the β cell is to present a complete representative set of noninsulin protein fragments on its surface. Given the large amounts of insulin protein in β cells, it may make regulatory sense that increased expression of insulin should stimulate HLA-A production.
We reasoned that there might be uncharacterized diabetes-associated genes among those with expression coupled to that of insulin. Indeed, this set of 20 genes includes four that are already known to be important for insulin metabolism. We further investigated a possible feedback regulation mechanism by focusing on another of these target genes, SSTR5-AS1, which had not previously been well characterized. Early studies showed that in rodent islets, SSTR2 and SSTR5 principally mediate the suppression of glucagon and insulin release respectively (reviewed in ref. 40). The studies in human islets and human β cells indicated that the situation differs from that in rodent β cells (40, 47, 48). These data suggest that SST effects are most significantly mediated by SSTR2, but SSTR5 and SSTR1 are also involved. In EndoC-βH1 cells, the short isoform of SSTR5 is expressed; transcription is in the opposite direction from SSTR5-AS1, so that transcripts of the gene and its antisense do not overlap. We showed that SSTR5-AS1 expression stimulates expression of SSTR5 (Fig. 5B). It should be noted that earlier work identified SSTR2 as the functionally dominant receptor in human pancreatic β cells (47); however, these observations were made with SST14. Our work shows that SST28 function principally involves SSTR5 (Fig. 6A). The analysis of long-range contacts thus leads to discovery of a negative feedback system for controlling insulin secretion, in which lower levels of INS expression result in decreased expression of SSTR5-AS1 to stimulate insulin export (SI Appendix, Fig. S7). Perhaps the reason that this significant role for SSTR5-AS1 has been overlooked is that it is not present in most vertebrates and is found largely in a subset of primates.
It will be obvious that the gene SSTR5 is a principal active component in this regulatory system. It behaves identically to the antisense gene, although its mRNA levels are much lower. It too is stimulated by INS expression, makes the same physical contacts with the INS gene, and is directly responsible for binding to SST28. SSTR5 thus acts in concert with SSTR5-AS1 to serve in the INS regulatory feedback mechanism. Our results indicate that SSTR5 is responsible only for SST28 signaling. The principal receptor for SST14 in β cells is presumably SSTR2. Thus, there seem to be two parallel signaling pathways in β-cells: SST14–SSTR2 and SST28–SSTR5. SST28 is at least 80-fold more potent than SST14 in terms of IC50. The SST28 inhibitor curve is a classic simple receptor binding curve. The SST14 inhibitor curve has a much narrower dynamic range, mimicking an “on-and-off” switch. Future drug designs should consider the properties of these two signaling pathways. It is possible that a combination of both SSTR2 and SSTR5 agonists also could be useful in diabetes therapy.
An important question raised by these results is how such long-range interactions are maintained between genes on different chromosomes. Some shorter range intrachromosomal contacts are at least in part stabilized by CTCF-mediated loop formation, in most cases associated with cohesin binding (49–52). Such a role for CTCF was shown earlier to be associated with the formation of contacts between INS and SYT8 in human pancreatic islets (13). This seems an unlikely mechanism to explain the results in this paper. As pointed out in the Introduction, there are now many examples of contacts between genes on different chromosomes, in which the genes can share common transcription factors as well as access to RNA polymerase II. The interchromosomal contacts we identified are associated with high levels of histone H3K27 acetylation, a hallmark of very active or “super” enhancers. Recent studies have detected such contacts between sites on different chromosomes. These are associated with high densities of actively transcribed genes and with histone modifications connected with transcriptional activity (53, 54).
It seems likely that the interactions we observe between the INS gene on chromosome 11 and loci on most of the other chromosomes involve the formation of transient transcription factories (Fig. 7B). Pancreatic β cells are committed largely to the production of insulin, and we estimate that there are 30,000 copies of INS mRNA per EndoC-βH1 cell from the RNA-seq data. There are multiple binding sites for transcription factors upstream of the INS gene, marked by the highest level of histone H3K27 acetylation we measure in EndoC-βH1 cells (SI Appendix, Table S2). Consistent with the idea that the interactions we observe involve clusters of active genes, all of the target genes shown in Fig. 4B carry this modification. A recent study in an engineered system showed that chromatin segments ranging in size from 0.6 to 3 Mb cluster with segments of the same chromatin class (55). These segments acquire “corresponding positions” in the nucleus irrespective of their chromosomal context. The size range of the segments matches well with that of the interchromosomal contacts we identified.
The approach described here is particularly useful in cell types such as pancreatic β cells or cells producing hemoglobin, where strong enhancers or superenhancers (56) may serve as foci for organization of active transcription centers. It allows us to identify genomic regions and individual genes that are important for INS regulation and that may not be easy to detect by other methods. The exploration of some of these contacts in detail has already led to identification of novel pathways for regulation of insulin secretion. In depth examination of each of the many contacts is likely to reveal other previously unrecognized regulatory mechanisms. It would not be surprising that future studies using this approach would identify more diabetes associated genes.
Materials and Methods
Culture of EndoC-βH1 Cells.
EndoC-βH1 cells (16) were cultured in low-glucose DMEM (catalog no. 11885; Life Technology) with 2% BSA (fatty acid free and heat shocked, catalog no. catBAH66; Equitech), 50 μM 2-mercaptoethanol, 10 mM nicotinamide, 5.5 mg/mL human transferrin (catalog no.T8158; Sigma–Aldrich), 6.7 ng/mL sodium selenite, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. Cells were seeded at a density of 6 × 105/cm2 on ECM-gel (1%) (catalog no. E1270; Sigma-Aldrich)/fibronectin (2 μg/mL, catalog no. F1141; Sigma–Aldrich) coated plates and cultured at 37 °C and 5% CO2.
4C-Seq Procedure.
The 4C-seq experiment was conducted according to published procedures (57). Ten million EndoC-βH1 or HeLa cells were fixed by formaldehyde. The cross-linked chromatin was digested by BglII (New England Biolabs) and religated by T4 DNA ligase (New England Biolabs). After reversal of cross-links, DNA was digested by NlaIII (New England Biolabs) and religated by T4 DNA ligase (New England Biolabs). The DNA product served as template for 4C-PCR. The 4C-PCR primers (SI Appendix, Table S5) were designed according to published procedures (23). The PCR product was sequenced by the Illumina HiSeq 2500 system in the NIH NIDDK Genomic Core Facility. The samples were prepared as three biological replicates in each cell line.
4C-Seq Data Analysis.
The reads mapping to the bait itself were filtered out. The short-range intrachromosomal contacts were analyzed using the 4Cseqpipe program (58). The “stat_type” parameter was set to the default “median” option. The “trend_resolution” parameter was set to 10 kb. The “interval_type” parameter was set to the default value. The long-range intrachromosomal contacts and interchromosomal contacts were analyzed using the protocol developed by de Laat (23, 27). The data are transformed to unique coverage (>1 reads per fragment end is set to 1) to avoid possible PCR artifacts. To determine the contact map on chromosome 11, a window with a size from 1 to 100 runs along the chromosome, accompanied by a background window around the other window of 3,000 fragment ends. The false discover rate (FDR) limit was set to 0.01 in 1,000 random permutations. Windows with a score above the FDR are scored as “interacting region.” For interchromosomal interactions, an FDR threshold of 0.01 is determined based on 100 or 1,000 random permutations of the data for each chromosome. A window size of 500 unique fragment ends is used; windows that exceed the threshold are scored as interchromosomal interactions. The contact regions in SI Appendix, Table S1A were from the interchromosomal interactions determined after 1,000 random permutations. Almost identical interactions were determined after 100 random permutations. Only the edges of a few contact regions shift slightly. Only the contacts detected in all three biological replicates were considered as reproducible. The interchromosomal contacts maps were generated by Circos (59).
Quantitative 3C Analysis.
Fifty nanograms of 4C ligation product were used as input in the RT-PCR assay on CFX96 Touch Real-Time PCR Detection System (Bio-Rad). The contact frequency was quantified with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) with the primers listed in SI Appendix, Table S5. The strong contact 45-kb 3′ to the INS promoter was chosen as the normalization point.
Viral shRNA Knockdown of INS Promoter Activity in EndoC-βH1 Cells.
The shRNA sequence (SI Appendix, Table S5) used to knock down the activity of the INS promoter was designed based on previous work (13). The shRNA sequence is cloned into Tet-pLKO-puro vector (Addgene) using the indicated primers (SI Appendix, Table S5). The cloned plasmid containing the desired shRNA sequence was cotransfected with the helper plasmids pDM2.G and psPAX2 into 293T cells using X-tremeGENE HP DNA Transfection Reagent (Roche). The lentiviral particles were collected after 48 h. The viral particles were used to transfect 1 × 108 EndoC-βH1 cells. The cells were selected against puromycin at 1 μg/mL for 2 wk and then maintained at 0.5 μg/mL subsequently. To performing knockdown experiments, the viral-transduced cells were treated with 1 μg/mL doxycycline for 7 d (one cell cycle). The media was changed every 48 h. No doxycycline was added in the negative control samples.
ChIP-Seq.
The ChIP-seq experiments were performed with three biological replicates. The chromatin immune-precipitation was performed with 50,000 EndoC-βH1 cells using True MicroChIP Kit (Diagenode catalog no. C01010130). The chromatin was sheared by a Bioruptor Plus sonication device. The sequencing library was generated using MicroPlex Library Preparation Kit v2 (Diagenode catalog no. C05010012) on an IP-Star Compact Automated System (Diagenode catalog no. B03000002) at the NIDDK Genomics Core Facility. The sequencing was performed on an Illumina HiSeq 2500 system at the NIDDK Genomics Core Facility. The anti-Histone H3 (acetyl K27) antibody was purchased from Abcam (catalog no. ab4729). The regions enriched with the relevant histone mark were analyzed using CCAT3.0 (60). The input was used as the negative control. The data are visualized in UCSC Genome Browser (https://genome.ucsc.edu/) with assembly GRCh37/hg19 (61).
RNA Isolation, Reverse Transcription, RNA-Seq, and Quantitative RT-PCR.
The total RNA from 5 × 105 EndoC-βH1 cells that were induced with shRNA was isolated with a NucleoSpin RNA XS kit (Clontech). The c-DNA library was prepared with a SMART-Seq v4 Ultra Low Input RNA Kit (catalog no. 634888; Clontech) with 10 ng of input RNA. The full-length cDNA output of the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing was processed with the Nextera XT DNA Library Preparation Kit (catalog no. FC-131-1024; Illumina). The sequencing library was sequenced by an Illumina HiSeq 2500 system. The shRNA-induced samples were prepared as three biological replicates, and three biological replicates without shRNA induction served as negative control. More than 90% of the reads for each library were effectively mapped to the hg19 human genome assembly using TopHat2 (62). Subsequently, both quantification and differential analysis of the reads were performed using Cufflinks (63). The gene expression data were visualized by Bioconductor package CummeRbund (63). The mapping parameters are largely unchanged from the Cufflinks reference above. We carried out similar experiments with a scramble shRNA (Plasmid 1864; Addgene), and removed the genes significantly affected by the scramble shRNA from our list. Primers in SI Appendix, Table S5 were used to confirm the gene differential expression data from RNA-seq. The c-DNA library prepared by the SMART-Seq v4 Ultra Low Input RNA Kit were used as input to be quantified with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) on CFX96 Touch Real-Time PCR Detection System (Bio-Rad). GAPDH (Glyceraldehyde-3-Phosphate Dehydrogenase) endogenous control gene was used to normalize gene expression by the ΔΔCt method (Applied Biosciences).
Viral shRNA Knockdown of lnc_RNA SSTR5-AS1 in EndoC-βH1 Cells.
The shRNA sequence (SI Appendix, Table S5) used to knock down the lnc-RNA SSTR5-AS1 was designed based on Silencer Select siRNA (siRNA ID no. n269672; Ambion). The inducible shRNA was introduced into EndoC-βH1 cells via lentiviral particles as described above. The shRNA-induced samples were prepared as three biological replicates, and three biological replicates without shRNA induction served as negative control. To perform knockdown experiments, the viral transduced cells were treated with 2 μg/mL doxycycline for 7 d. The media was changed every 48 h. No doxycycline was added in the negative control samples. To assess the knockdown efficiency and the effect of knockdown of SSTR5-AS1 on SSTR5 expression, total RNA was isolated from the cells by CellAmp Direct RNA Prep Kit for RT-PCR (catalog no. 3732; TaKaRa). The c-DNA was synthesized from the RNA by PrimeScript RT Reagent Kit (catalog no. RR037A; TaKaRa). The c-DNA generated were used as input to be quantified with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). GAPDH endogenous control gene was used to normalize gene expression by the ΔΔCt method. Primers in SI Appendix, Table S5 were used.
Assessment of Insulin Secretion from EndoC-βH1 Cells.
EndoC-βH1 cells expressing shRNA targeting SSTR5-AS1 were seeded onto ECM gel fibronectin-coated 96-well plates at 5 × 104 cells per well. Two days after cell seeding, the medium was changed to a glucose starvation medium (same components as growth media without glucose). After incubation at 37 °C in a CO2 incubator for 18 h, the medium was changed to Krebs-Ringer solution supplemented with 0.5 mM glucose (64) for 1 h. Then using the Krebs-Ringer solution, with 15 mM glucose and 0.5 mM IBMX (3-isobutyl-1-methylxanthine), the cells were incubated for 1 h at 37 °C in a CO2 incubator. At this stage, varying concentrations of somatostatin-14 (catalog no. S1763; Sigma-Aldrich) or somatostatin-28 (catalog no. S6135; Sigma-Aldrich) were added as well. Insulin concentration in the supernatants was measured by ELISA according to manufacturer’s instructions using an Insulin ELISA Kit (catalog no. IS130D; Calbiotech). The wells without somatostatin added were used for normalization. EndoC-βH1 cells containing the integrated shRNA targeting SSTR5-AS1 but without doxycycline treatment were used as negative controls. There were four biological replicates for each data point in Fig. 6. The nonlinear regression was to fit the data to either a simple isotherm equation or a Hill equation with the Hill constant at 3. The coefficient of determination, R2, was reported in the main text.
Supplementary Material
Acknowledgments
We thank Inserm Transfert, CNRS, and Endocells for providing the pancreatic β cell line, EndoC-βH1; Harold Smith for the help on 4C-seq experimental design; and Lynn Young and Gregory Riddick for their help on bioinformatics. This work utilized the computational resources of the NIH High-Performance Computing Biowulf cluster (https://hpc.nih.gov). This work was supported by the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases, NIH.
Footnotes
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, https://www.ncbi.nlm.nih.gov/geo (accession no. GSE112346).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1803146115/-/DCSupplemental.
References
- 1.Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet. 2016;17:661–678, and erratum (2016) 17:772. doi: 10.1038/nrg.2016.112. [DOI] [PubMed] [Google Scholar]
- 2.Osborne CS, et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet. 2004;36:1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- 3.Buckley MS, Lis JT. Imaging RNA polymerase II transcription sites in living cells. Curr Opin Genet Dev. 2014;25:126–130. doi: 10.1016/j.gde.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osborne CS, et al. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schoenfelder S, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet. 2010;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li G, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fanucchi S, Shibayama Y, Burd S, Weinberg MS, Mhlanga MM. Chromosomal contact permits transcription between coregulated genes. Cell. 2013;155:606–620. doi: 10.1016/j.cell.2013.09.051. [DOI] [PubMed] [Google Scholar]
- 8.Papantonis A, et al. TNFα signals through specialized factories where responsive coding and miRNA genes are transcribed. EMBO J. 2012;31:4404–4414. doi: 10.1038/emboj.2012.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dekker J. The three ‘C’ s of chromosome conformation capture: Controls, controls, controls. Nat Methods. 2006;3:17–21. doi: 10.1038/nmeth823. [DOI] [PubMed] [Google Scholar]
- 10.Polonsky KS. The past 200 years in diabetes. N Engl J Med. 2012;367:1332–1340. doi: 10.1056/NEJMra1110560. [DOI] [PubMed] [Google Scholar]
- 11.Bonnefond A, Froguel P. Rare and common genetic events in type 2 diabetes: What should biologists know? Cell Metab. 2015;21:357–368. doi: 10.1016/j.cmet.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 12.Rich SS. Diabetes: Still a geneticist’s nightmare. Nature. 2016;536:37–38. doi: 10.1038/nature18906. [DOI] [PubMed] [Google Scholar]
- 13.Xu Z, Wei G, Chepelev I, Zhao K, Felsenfeld G. Mapping of INS promoter interactions reveals its role in long-range regulation of SYT8 transcription. Nat Struct Mol Biol. 2011;18:372–378. doi: 10.1038/nsmb.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Z, et al. Mapping of long-range INS promoter interactions reveals a role for calcium-activated chloride channel ANO1 in insulin secretion. Proc Natl Acad Sci USA. 2014;111:16760–16765. doi: 10.1073/pnas.1419240111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pociot F. Type 1 diabetes genome-wide association studies: Not to be lost in translation. Clin Transl Immunology. 2017;6:e162. doi: 10.1038/cti.2017.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ravassard P, et al. A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J Clin Invest. 2011;121:3589–3597. doi: 10.1172/JCI58447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohlke KL, Boehnke M. Recent advances in understanding the genetic architecture of type 2 diabetes. Hum Mol Genet. 2015;24:R85–R92. doi: 10.1093/hmg/ddv264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott RA, et al. Diabetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes. 2017;66:2888–2902. doi: 10.2337/db16-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fløyel T, Kaur S, Pociot F. Genes affecting β-cell function in type 1 diabetes. Curr Diab Rep. 2015;15:97. doi: 10.1007/s11892-015-0655-9. [DOI] [PubMed] [Google Scholar]
- 20.Qiu YH, Deng FY, Tang ZX, Jiang ZH, Lei SF. Functional relevance for type 1 diabetes mellitus-associated genetic variants by using integrative analyses. Hum Immunol. 2015;76:753–758. doi: 10.1016/j.humimm.2015.09.033. [DOI] [PubMed] [Google Scholar]
- 21.Zhao W, et al. CHD Exome+ Consortium; EPIC-CVD Consortium; EPIC-Interact Consortium; Michigan Biobank Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat Genet. 2017;49:1450–1457. doi: 10.1038/ng.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M, et al. Three single nucleotide polymorphisms associated with type 2 diabetes mellitus in a Chinese population. Exp Ther Med. 2017;13:121–126. doi: 10.3892/etm.2016.3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van de Werken HJ, et al. 4C technology: Protocols and data analysis. Methods Enzymol. 2012;513:89–112. doi: 10.1016/B978-0-12-391938-0.00004-5. [DOI] [PubMed] [Google Scholar]
- 24.Hay CW, Docherty K. Comparative analysis of insulin gene promoters: Implications for diabetes research. Diabetes. 2006;55:3201–3213. doi: 10.2337/db06-0788. [DOI] [PubMed] [Google Scholar]
- 25.Imamura M, et al. Genome-wide association studies in the Japanese population identify seven novel loci for type 2 diabetes. Nat Commun. 2016;7:10531. doi: 10.1038/ncomms10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong A, et al. DIAGRAM Consortium Parental origin of sequence variants associated with complex diseases. Nature. 2009;462:868–874. doi: 10.1038/nature08625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Splinter E, de Wit E, van de Werken HJ, Klous P, de Laat W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: From fixation to computation. Methods. 2012;58:221–230. doi: 10.1016/j.ymeth.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 28.Raviram R, et al. 4C-ker: A method to reproducibly identify genome-wide interactions captured by 4C-seq experiments. PLOS Comput Biol. 2016;12:e1004780. doi: 10.1371/journal.pcbi.1004780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mellado-Gil JM, et al. The type 2 diabetes-associated HMG20A gene is mandatory for islet beta cell functional maturity. Cell Death Dis. 2018;9:279. doi: 10.1038/s41419-018-0272-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinberg MS, Morris KV. Transcriptional gene silencing in humans. Nucleic Acids Res. 2016;44:6505–6517. doi: 10.1093/nar/gkw139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mularoni L, Ramos-Rodríguez M, Pasquali L. The pancreatic islet regulome browser. Front Genet. 2017;8:13. doi: 10.3389/fgene.2017.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasquali L, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014;46:136–143. doi: 10.1038/ng.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang T, et al. FoxO1 plays an important role in regulating β-cell compensation for insulin resistance in male mice. Endocrinology. 2016;157:1055–1070. doi: 10.1210/en.2015-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villafuerte BC, et al. Over-expression of insulin-response element binding protein-1 (IRE-BP1) in mouse pancreatic islets increases expression of RACK1 and TCTP: Beta cell markers of high glucose sensitivity. Biochim Biophys Acta. 2017;1865:186–194. doi: 10.1016/j.bbapap.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szabat M, et al. Musashi expression in β-cells coordinates insulin expression, apoptosis and proliferation in response to endoplasmic reticulum stress in diabetes. Cell Death Dis. 2011;2:e232. doi: 10.1038/cddis.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrocchi-Passeri P, et al. The VGF-derived peptide TLQP-62 modulates insulin secretion and glucose homeostasis. J Mol Endocrinol. 2015;54:227–239. doi: 10.1530/JME-14-0313. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, et al. Overexpression of Pref-1 in pancreatic islet β-cells in mice causes hyperinsulinemia with increased islet mass and insulin secretion. Biochem Biophys Res Commun. 2015;461:630–635. doi: 10.1016/j.bbrc.2015.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukunaka A, et al. Zinc transporter ZIP13 suppresses beige adipocyte biogenesis and energy expenditure by regulating C/EBP-β expression. PLoS Genet. 2017;13:e1006950. doi: 10.1371/journal.pgen.1006950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braun M. The somatostatin receptor in human pancreatic β-cells. Vitam Horm. 2014;95:165–193. doi: 10.1016/B978-0-12-800174-5.00007-7. [DOI] [PubMed] [Google Scholar]
- 41.Guest PC. 2D gel electrophoresis of insulin secretory granule proteins from biosynthetically labelled pancreatic islets. Adv Exp Med Biol. 2017;974:167–174. doi: 10.1007/978-3-319-52479-5_12. [DOI] [PubMed] [Google Scholar]
- 42.Rai U, Thrimawithana TR, Valery C, Young SA. Therapeutic uses of somatostatin and its analogues: Current view and potential applications. Pharmacol Ther. 2015;152:98–110. doi: 10.1016/j.pharmthera.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Tang Z, et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell. 2015;163:1611–1627. doi: 10.1016/j.cell.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janowski BA, Hu J, Corey DR. Silencing gene expression by targeting chromosomal DNA with antigene peptide nucleic acids and duplex RNAs. Nat Protoc. 2006;1:436–443. doi: 10.1038/nprot.2006.64. [DOI] [PubMed] [Google Scholar]
- 45.Boyle P, et al. Diabetes and breast cancer risk: A meta-analysis. Br J Cancer. 2012;107:1608–1617. doi: 10.1038/bjc.2012.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kailey B, et al. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic β- and α-cells. Am J Physiol Endocrinol Metab. 2012;303:E1107–E1116. doi: 10.1152/ajpendo.00207.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strowski MZ, et al. Somatostatin receptor subtype 5 regulates insulin secretion and glucose homeostasis. Mol Endocrinol. 2003;17:93–106. doi: 10.1210/me.2001-0035. [DOI] [PubMed] [Google Scholar]
- 49.Parelho V, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 50.Wendt KS, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 51.Rubio ED, et al. CTCF physically links cohesin to chromatin. Proc Natl Acad Sci USA. 2008;105:8309–8314. doi: 10.1073/pnas.0801273105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stedman W, et al. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 2008;27:654–666. doi: 10.1038/emboj.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalhor R, Tjong H, Jayathilaka N, Alber F, Chen L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat Biotechnol. 2011;30:90–98. doi: 10.1038/nbt.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jost D, Carrivain P, Cavalli G, Vaillant C. Modeling epigenome folding: Formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res. 2014;42:9553–9561. doi: 10.1093/nar/gku698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van de Werken HJG, et al. Small chromosomal regions position themselves autonomously according to their chromatin class. Genome Res. 2017;27:922–933. doi: 10.1101/gr.213751.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gheldof N, Leleu M, Noordermeer D, Rougemont J, Reymond A. Detecting long-range chromatin interactions using the chromosome conformation capture sequencing (4C-seq) method. Methods Mol Biol. 2012;786:211–225. doi: 10.1007/978-1-61779-292-2_13. [DOI] [PubMed] [Google Scholar]
- 58.van de Werken HJ, et al. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat Methods. 2012;9:969–972. doi: 10.1038/nmeth.2173. [DOI] [PubMed] [Google Scholar]
- 59.Krzywinski M, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu H, et al. A signal-noise model for significance analysis of ChIP-seq with negative control. Bioinformatics. 2010;26:1199–1204. doi: 10.1093/bioinformatics/btq128. [DOI] [PubMed] [Google Scholar]
- 61.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim D, et al. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ndiaye FK, et al. Expression and functional assessment of candidate type 2 diabetes susceptibility genes identify four new genes contributing to human insulin secretion. Mol Metab. 2017;6:459–470. doi: 10.1016/j.molmet.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.