

Abstract
Nanoparticle-based systems for concurrent delivery of multiple drugs can improve outcomes of cancer treatments, but face challenges because of differential solubility and fairly low threshold for incorporation of many drugs. Here we demonstrate that this approach can be used to greatly improve the treatment outcomes of etoposide (ETO) and platinum drug combination (“EP/PE”) therapy that is the backbone for treatment of prevalent and deadly small cell lung cancer (SCLC). A polymeric micelle system based on amphiphilic block copolymer poly(2-oxazoline)s (POx) poly(2-methyl-2-oxazoline-block-2-butyl-2-oxazoline-block-2-methyl-2-oxazoline) (P(MeOx-b-BuOx-b-MeOx) is used along with an alkylated cisplatin prodrug to enable co-formulation of EP/PE in a single high-capacity vehicle. A broad range of drug mixing ratios and exceptionally high two-drug loading of over 50% wt. drug in dispersed phase is demonstrated. The highly loaded POx micelles have worm-like morphology, unprecedented for drug loaded polymeric micelles reported so far, which usually form spheres upon drug loading. The drugs co-loading in the micelles result in a slowed-down release, improved pharmacokinetics, and increased tumor distribution of both drugs. A superior antitumor activity of co-loaded EP/PE drug micelles compared to single drug micelles or their combination as well as free drug combination was demonstrated using several animal models of SCLC and non-small cell lung cancer.
Keywords: cancer, chemotherapy, cisplatin, drug delivery, etoposide, nanomedicine, polymeric micelles
Graphical Abstract
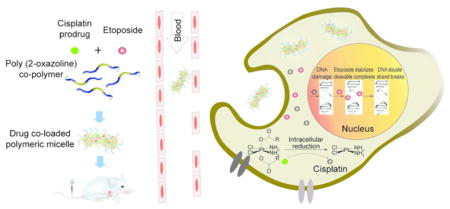
Drug combination therapy is a common way to treat cancer.1 The therapeutic outcomes can possibly be improved by using nanoparticle-based formulations that could provide a single vehicle for the concurrent delivery of multiple drugs to tumor cells to maximize therapeutic efficacy, overcome treatment resistance, and decrease adverse side effects of these drugs.2 One notable success in this area is CPX-351 (Vyxeos),3 a fixed-combination of daunorubicin and cytarabine in liposomes, recently approved by the US Food and Drug Administration for adult patients with newly diagnosed therapy-related acute myeloid leukemia (t-AML) or AML with myelodysplasia-related changes (AML-MRC), based on an improvement in overall survival in a phase III study. Here we explore the possibility of extending the nanoformulation approach to improve combination therapy to one of the most challenging malignancies, lung cancer.
Lung cancer is the most common malignancy and the leading cause of death from cancer in the world for several decades, with an estimated 1.6 million new cases and 1.38 million deaths per year.4 Of all lung cancers about 80–85% are non-small cell lung cancer (NSCLC), while 10–15% are small cell lung cancer (SCLC).5 Depending on the stages, these cancers differ in the prognosis and therapy.5 Although several new treatments have been introduced for NSCLC, the advances in chemotherapy of SCLC have been poor.4 Currently etoposide (ETO) and platinum remain (“EP/PE”) as the backbone of therapy of SCLC, and since introduction of this drug combination chemotherapy there have been no significant advances in first line therapy of SCLC for over 30 years.5 We posit that the EP/PE combination therapy outcomes can be improved by using a nanoformulation approach. The co-delivery of drugs in a single nanofomulation remains a principal challenge since poor solubility and differential pharmacokinetics (PK) severely restrict the selection of drugs that can be translated into successful combination treatments. Critical to success is optimizing the relative doses of the drugs to obtain synergistic effects on tumors.6 Most nanoparticles have a fairly low threshold for incorporation of such drugs. Previously, we have reported a nanosized polymeric micelles (PM) formulation based on highly defined amphiphilic triblock copolymers of poly(2-oxazoline)s (POx), poly(2-methyl-2-oxazoline-block-2-butyl-2-oxazoline-block-2-methyl-2-oxazoline) (P(MeOx-b-BuOx-b-MeOx)), that have greatly enhanced the solubility of single and multiple drug combinations.7,8 In particular, the POx-based PM of paclitaxel (PTX) with unprecedentedly high drug loading of nearly 45% wt. and controllable ~30–40 nm size displayed reduced toxicity and superior efficacy in early and late stage breast cancer models compared to clinically approved Taxol and Abraxane.9 Here, we propose that co-loading of ETO and platinate at synergistic drugs ratios in the POx-based drug delivery platform can safely and efficiently treat both SCLC and NSCLC lung cancers.
RESULTS
Synthesis of CPs
Cisplatin as a hydrophilic water-soluble drug would not preferentially incorporate in a hydrophobic core of PM. To enable cisplatin formulation and delivery within POx micelles, we used a prodrug strategy.10 As the synthetic approach, cisplatin derivatives with aliphatic chains of different length (n = 4, 6, 8, and 10 carbon atoms) at the axial positions have been synthesized as previously reported.14,15 The derivatives are designated as C4CP, C6CP, C8CP, and C10CP according to the length of the aliphatic chain (Figure 1a). Their chemical structures were confirmed by 1H NMR spectroscopy and mass spectrometry (Supplementary Figures S1 and S2).
Figure 1.

Preparation of binary PM containing ETO and CPs: (a) Chemical structure of the hydrophobic CPs and the scheme of release of cisplatin in the cells. (b) Schematic and chemical structures of POx triblock copolymer, ETO, and C6CP. (c) Schematic representation of the POx micelles preparation by the thin-film technique.
Preparation and Characterization of Drug-Loaded Micelles
High-Capacity ETO PM
Previously, we have reported on ETO-loaded POx-based PM but did not examine different feed concentrations of this drug and the morphology of the particles formed.8 In this study, we set the polymer concentration to a fixed value (10 g/L) and increased the ETO feeding concentration (here and below we designate the feeding ratios for single drug micelles as wt. ratios of targeted amounts “drug/polymer”). Surprisingly, even at the highest ETO concentration (feeding ratio 10/10), nearly all of the drug was incorporated into the PM (Supplementary Figure S3). Under these conditions the LE and LC were ~98% and ~50%, respectively, and the micellar solution contained ~9.7 g/L ETO, which is at least ~300-fold higher than ETO maximal solubility in pure water.16 For the entire range of ETO concentrations, the drug loaded PM solutions were transparent or slightly opaque. The DLS analysis revealed the presence of small particles with the hydrodynamic diameters ranging from ~25 nm to ~36 nm, depending on the drug feeding ratio (Supplementary Table S1). Interestingly, the ETO PM displayed a relatively broad size distribution (PDI > 0.5). Such size distribution can possibly be caused by the nonspherical worm-like morphology of these micelles observed by TEM (Figure 2e).
Figure 2.

Characterizations of C6CP/ETO PM formulations. (a) LE, (b) LC, and (c) drug concentration at various drug feeding ratios C6CP/ETO/POx: 8/4/10, 6/6/10, and 4/8/10. (d) DLS size (Deff) distribution analysis at ~1 h after preparation and (e, f) stability of the C6CP/ETO PM (4/8/10) at r.t. as determined by the particle size and PDI measurements over time. The DLS analysis was performed at a POx final concentration of 1 g/L in DI water. The Deff of the micelles at ~1 h after preparation is ~75 nm, and the PDI is ~0.220. (g) TEM images of ETO PM (ETO/POx 8/10), C6CP PM (C6CP/POx 4/10), and co-loaded POx micelle (C6CP/ETO/POx 4/8/10). Scale bar = 100 nm.
High-Capacity CPs PM
As in the previous case, the polymer concentration in the final formulation was set at 10 g/L, and the CPs concentration was varied from 2 g/L to 10 g/L. For each of the four CPs the maximal compound concentration in the micellar solution was achieved at a drug/polymer feeding ratio of 10/10 (Supplementary Figure S4). However, there were marked differences in the solubility of these compounds that were as high as ~8.4 g/L for C4CP and C6CP and relatively lower ~2.6 to 3.1 g/L for C8CP and C10CP. These differences are also reflected in the corresponding LE and LC values (LE ~82–85% and LC ~45–46% for C4CP and C6CP, and LE ~32–50% and LC ~ 23–32% for C8CP and C10CP) (Supplementary Tables S2–S5). The CPs PM solutions were transparent and remained stable with no signs of drug precipitation while stored for at least 2 weeks at 4 °C. The DLS hydrodynamic diameters of the micelles increased as the length of the aliphatic chain of the CPs increased from under 30 nm for C4CP to over 140 nm for C10CP. At high drug loading, the particle size distribution varied from being very narrow for C6CP PM (PDI < 0.1), to moderately narrow for C8CP PM and C10CP PM (PDI < 0.2), to relatively broad for C4CP PM (PDI ~ 0.3–0.4) (Supplementary Tables S2–S5). Based on the solubilization, particle size, stability, and cytotoxicity analysis (described below), we selected C6CP PM for further testing.
Combination C6CP/ETO PM Have Increased Drug Loading and Worm-like Shape
The preparation of PM containing two drugs is as straightforward and simple as the procedure to solubilize a single drug loaded POx micelle. The drug(s) and the polymer are solubilized in a common solvent (ethanol), the solution mixed with a desired drug(s) polymer ratio, the solvent removed to obtain a thin solid film, and the film is finally hydrated with buffer or water to give a micellar solution (Figure 1b,c). To prepare C6CP/ETO PM, we selected three different C6CP/ETO wt. ratios of 8/4, 6/6, and 4/8 (g/g) and, as in the cases of single drugs, kept the POx concentration constant (10 g/L) to examine the drug loading. (Consequently, for the co-loaded drug micelles, the feeding ratios were defined as wt. ratios “drug1/drug2/polymer”.) In this case, however, each drug contents in the micellar solution were measured separately, to give two sets of values for LE and LC. With any of the chosen ETO/C6CP ratios, the LE of C6CP was above 85%, and the LE of ETO was nearly quantitative (Figure 2a, see also Supplementary Table S6). The LC values for each drug varied with the drug feed, but the net LC for both drugs always exceeded 50%. Remarkably, the total concentration of two drugs in the micellar solution was nearly 11 g/L (Figure 2b,c) with only 10 g/L polymer used. Thus, the total drug solubilization is higher for the co-loading of both drugs compared to the solubilization of the respective single drugs.
The particle size and polydispersity were strongly affected by the drug ratio (Figure 2d–f, see also Supplementary Table S6). At similar overall drug loading, the micelles with the highest ETO content had the smallest size ~75 nm, while the micelles with highest C6CP content were considerably larger ~103 nm. Interestingly, the PDI changed in the opposite direction. The co-loaded PM with the highest C6CP content were close to uniform (PDI 0.08), while the co-loaded PM with the highest ETO content were more heterogeneous (PDI 0.22). Nevertheless the co-loading of the drugs greatly decreased the polydispersity of C6CP/ETO PM as compared to ETO single loaded micelles (PDI > 0.5), indicating that upon co-loading both drugs strongly interact with each other and/or the block copolymer. The co-loaded micelles remained stable in solution, and no change in size or PDI was detectable for at least 2 weeks of storage (Figure 2e,f). Notably, although their PDI decreased and the size increased, the co-loaded micelles still retained the elongated worm-like shape characteristic of single ETO PM, in contrast to CP PM loaded with only one drug or empty micelles as observed by TEM (Figure 2g).
The changes in the morphology of the PM upon co-loading of the drugs were further examined by the AFM. Based on this study, the majority of empty micelles adopted a spherical shape, however, there were few exceptions where elongated particles formed, attributable to micelle aggregation (Figure 3a). A size histogram of dry empty micelles is presented in Figure 3c with analysis provided in Supplementary Figure S5. Micelle height is uniform (4 nm) and shows insignificant signs of spreading upon deposition (“Mexican hat”). Assuming a spherical shape, we estimate the radius of a dry micelle to be R = 9 nm. Drug loading had a major impact on the resulting micelle shape. In particular, dry C6CP/ETO PM displayed two distinct populations of spherical and elongated, worm-like particles (Figure 3b). These populations were separated based on the particle circularity, and their dimensions were measured independently as shown in histograms (Figure 3d and Supplementary Figure S6). In addition, the spherical and worm-like particles differed in their average aspect ratios, which in the case of elongated particles (circularity from 0.5 to 0.9) varied from ~1.3 to ~3.5 (for details see Supplementary Figures S7–S8). We then assessed the effect of concentration of the drug loaded C6CP/ETO PM micelles by varying the POx in solution from 5 to 500 μg/mL, while keeping the drug ratio the same. At the lower concentrations, the micelles were predominantly spheres, but as concentration increased, the micelles became elongated and mostly worm-like (Supplementary Figure S9). Further increase in concentration leads to limitations in AFM imaging, where rods begin to assemble upon drying on the substrate and ultimately form a grid of mass where individual particles are indistinguishable. The attempt to examine the effect of the FBS using AMF was not successful and resulted in the images with poor resolutions of the morphology, possibly due to adsorption and aggregation of the protein on the mica.
Figure 3.

(a, b) Typical AFM images and (c, d) particle area histograms for (a, c) empty POx PM and (b, d) drug loaded micelles, C6CP/ETO PM (4/8/10). (a, b) Red circles indicate elongated, worm-like particles. The area histograms present (c) the entire population of particles for empty POx PM and (d) separately, elongated (red, circularity 0.5–0.9) and spherical (blue, circularity >0.9) for C6CP/ETO PM (4/8/10). Sample solutions contain 20 μg/mL POx in H2O.
Drug Release and Serum Binding Are Decreased in Co-Loaded C6CP/ETO PM
The rate of drug release from a nanoparticle carrier can significantly affect the therapeutic outcomes. This makes a time-controlled drug release a key consideration in the micelle design. As PM are dynamic structures that can exchange both the macromolecules and the drugs with the environment, we determined the release of C6CP and ETO from the single and co-loaded drug micelles in the presence of serum protein using two distinct techniques. First, the drug release was studied by the dialysis method under sink conditions using PBS with 40%w/w BSA as the release media. The C6CP was released faster than ETO from the single drug as well as co-loaded micelles. Surprisingly, the release rates of both C6CP and ETO from the co-loaded micelles were lower as compared to the corresponding single drug micelles (Figure 4). Specifically, the time of 50% drug release from C6CP/ETO PM was prolonged from ~2 to ~3 h for C6CP and from ~4 h to ~6 h for ETO.
Figure 4.
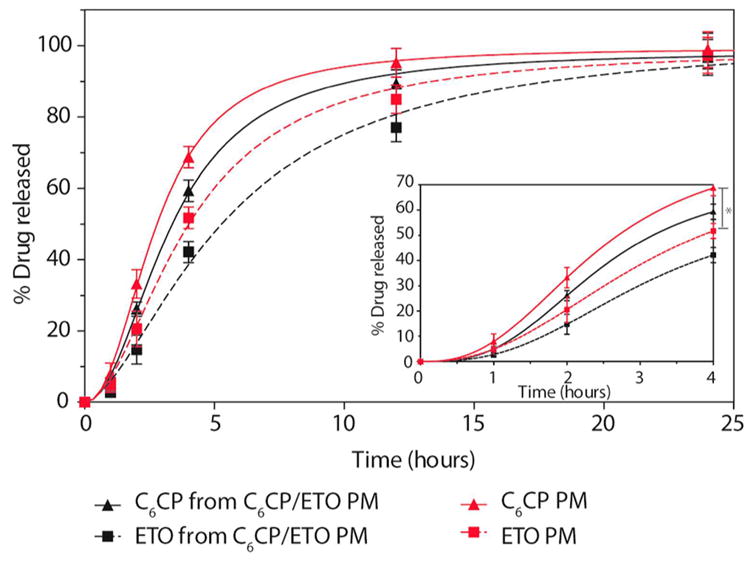
Drug release profiles for single and co-loaded drug PM. Micelles were prepared at following feeding ratios: C6CP/ETO PM (4/8/10), C6CP PM (4/10) and ETO PM (8/10). The drug release study was performed at a drug concentration of 0.1 mg/mL in PBS, pH7.4 at 37 °C under sink conditions (against 200 × volume of 40 g/L BSA in PBS). The data are mean ± SD, n = 3, * p < 0.05.
Second, the binding of micellar drugs with serum proteins was examined. In this experiment, the PM containing single and co-loaded drugs as well as the mixture of PM each containing different single drugs were incubated for either 1 h or 4 h with 2 mL of FBS to mimic the dilution of the formulations in the blood. The distribution of the drug between the micelles and serum was determined using a solid-phase extraction (SPE) column (SOLA HRP), which selectively retains free and protein-bound drugs, but not the drug incorporated in the micelles. The results suggest that the co-loaded drug micelles retained higher amounts of both drugs as compared to single drug loaded micelles of their mixture (Supplementary Table S7). For example, after 1 h of incubation time, ~79% of ETO remained encapsulated in the C6CP/ETO PM compared to ~71% for the mixture of C6CP PM and ETO PM and ~70% for the single drug loaded ETO PM. Likewise, ~78% of C6CP remained encapsulated in the C6CP/ETO PM but only ~70% for the mixture of C6CP PM and ETO PM and ~63% for the single drug loaded C6CP PM. The differences between the single drug micelles and C6CP/ETO PM somewhat diminished after 4 h incubation time, especially for the mixture group but remained considerable in comparison with either C6CP PM or ETO PM groups. Probably, a slow intermicellar drug exchange occurs in the mixture of single loaded micelles upon incubation produces some co-loaded micelles. In conclusion, coformulation of the two drugs in the same PM increased the retention of these drugs in the micelles by slowing down the drug release and exchange with the serum proteins.
Combination C6CP/ETO PM Display High Activity and Drug Synergy in Cancer Cells
We evaluated the in vitro drug cytotoxicity of each of the CPs PM in H69AR human SCLC cells and 344SQ/Luc. murine NSCLC cells using the MTT assay (Supplementary Figure S10 and Table S8). In H69AR cell line, the cytotoxicity profiles of the CPs PM strongly depended on the length of CPs aliphatic chain. For these micelle-incorporated prodrugs, the cytotoxicity increased almost 50-fold as the length of the aliphatic chain increased (IC50 2.86 μg/mL for C4CP, 0.31 μg/mL for C6CP, 0.06 μg/mL for C8CP and 0.06 μg/mL for C10CP). However, each prodrug in PM was substantially more active in this cell line as compared to free cisplatin (IC50 3.13 μg/mL). The cytotoxicity differences between various CPs PM were only about 4-fold in the 344SQ/Luc. cells (IC50 4.01, 0.65, 0.47, and 0.38 μg/mL for C4CP, C6CP, C8CP, and C10CP, respectively). In this case, the prodrug with the shortest aliphatic chain was less toxic, and the three other prodrugs were more toxic than cisplatin (IC50 2.01 μg/mL).
By poisoning the DNA topoisomerase II, ETO suppresses the repair of cisplatin-induced DNA lesions, which results in the synergy of these drugs.17,18 Based on the cytotoxicity results for the C6CP prodrug as well as the data on the co-formulation loading and stability, we further examined the cytotoxicity of the co-loaded C6CP/ETO PM for different drug ratios. In both cell lines the cytotoxicity of the co-loaded drug micelles increases as the fraction of ETO in the drug mixture increases (Figure 5a,c, Table 1). The lowest IC50 value of the three drug ratios was observed for C6CP/ETO PM (4/8/10). Interestingly, the mixture of C6CP PM (4/10) and ETO PM (8/10) with the same drug ratio as C6CP/ETO PM (4/8/10) was less active in both cell lines. To assess drugs synergy, the CI was calculated using the isobologram equation of Chou–Talalay.19 In H69AR cells, the drug synergy was observed for all tested C6CP/ETO PM formulations (Figure 5b). In 344SQ/Luc. cells, the C6CP/ETO PM (4/8/10) formulation displayed a clear synergistic effect between the drugs (CI < 0.5) (Figure 5d). However, the two other formulations with lower ETO content displayed drug antagonism (CI > 1.0). To further validate the observed trend where the synergy increases as the C6CP:ETO ratio decreases, we examined one more formulation, C6CP/ETO PM (2/6/10). Indeed, for this additional formulation in both cell lines, the IC50 values were lower and the drug synergy same or greater than for C6CP/ETO PM (4/8/10) (Supplementary Table S9 and Figure S11). Based on the in vitro cell studies, we selected the C6CP/ETO PM (4/8/10) system for further animal studies.
Figure 5.

Cytotoxicity, synergy and drug uptake for single and co-loaded drug PM in (a, b) SCLC H69AR and (c–f) NSCLC 344SQ/Luc. cell lines: (a, c) Cell inhibition curves, (b, d); CI as a function of cell inhibition Fa (CI < 1 indicates synergistic effect); (e, f) Kinetics of intracellular accumulation of C6CP and ETO in cells exposed to various drug formulations. Drug formulations were (a–d) free cisplatin, free ETO, C6CP/ETO PM (6/6/10), and C6CP/ETO PM (8/4/10), (a–f) C6CP PM (4/10), ETO PM (8/10), C6CP/ETO PM (4/8/10), and mixture C6CP PM (4/10) and ETO PM (8/10). Drug concentrations were varied (a–d) or IC50 of C6CP/ETO PM (4/8/10) (e, f) in each experiment. (a, c, e, f) Data points are means ± SD, n = 6 (a, c) or n = 3 (e, f) (* p < 0.05, ** p < 0.01).
Table 1.
Cytotoxicity of the Single Drug and Co-Loaded Drug PM in SCLC H69AR and NSCLC 344SQ/Luc. Cell Lines
| cell lines | IC50, μg/mL (μM)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| POx | ETO | cisplatin | cisplatin and ETO (1/4) | ETO PM (8/10) | C6CP PM (4/10) | C6CP/ETO PM | mixture C6CP PM (4/10) and ETO PM (8/10) | |||
|
| ||||||||||
| (8/4/10) | (6/6/10) | (4/8/10) | ||||||||
| H69AR | >100 (>100) | 19.21 (32.64) | 3.13 (10.43) | 4.87 (9.89) | 12.2 (20.69) | 0.35 (0.60) | 0.53 (0.97) | 0.35 (0.63) | 0.26 (0.46) | 0.68 (1.20) |
| 344SQ/Luc | >100 (>10) | 3.01 (5.11) | 2.01 (6.70) | 2.24 (4.55) | 2.2 (3.70) | 0.64 (1.21) | 0.61 (1.11) | 0.46 (0.82) | 0.37 (0.65) | 0.76 (1.34) |
The cytotoxicity data are expressed as IC50 values, μg/mL or molar concentrations presented in the brackets (counting per single drug or both drugs for co-loaded drug PM and mixture of single drug PM). Based on data in Figure 5a,c.
Combination C6CP/ETO PM Increase Drug Accumulation in Cancer Cells
The intracellular accumulation of ETO and C6CP in PM was examined in 344SQ/Luc. cells. In this study, the cells were incubated for up to 50 h with either co-loaded C 6CP/ETO PM (4/8/10) or single drug PM or the mixture of respective single drug loaded PM. As can be seen in Figure 5e,f, the uptake of both drugs is enhanced when the drugs are co-formulated C6CP/ETO PM (4/8/10) compared to the mixture of C6CP PM (4/10) and ETO PM (8/10) with the same drugs ratio. Although the difference with the single drug micelles C6CP PM (4/10) and ETO PM (8/10) was not so dramatic, the co-loaded drug micelles appear to be superior to these formulations as well.
Toxicology Studies MTD and Toxicology Profiles of Co-Loaded C6CP/ETO PM
The MTD for the co-loaded drug micelles C6CP/ETO PM (4/8/10) was determined in healthy 6–8 week old female nude mice using a q4d × 4 regimen. The MTD was determined as 15 mg/kg for C6CP and 30 mg/kg for ETO. Therefore, we further used this dose for animal treatments with C6CP/ETO PM (4/8/10) as well as all other micellar formulations in this study. When converted to a human (60 kg body weight), this dose is equivalent to ~90 mg/kg ETO and ~45 mg/kg C6CP (or ~25.5 mg/kg of cisplatin assuming its complete release from the prodrug). This is comparable or less than the doses during EP/PE regimen in clinics that contain 60 mg/m2 cisplatin i.v. on day 1 plus 120 mg/m2 ETO i.v. on days 1–3 every 21 days for 4 cycles.20 POx micelle formulations are potentially safer because they use much less excipient.9 For example, the single drug ETO PM (8/10) in the present study contains 8 mg/mL drug and 10 mg/mL of POx in aqueous saline. There are no added alcohols and >35 times less excipients per gram of the drug compared to the current ETO injection (Toposar), USP for i.v. use, which contains 20 mg/mL ETO USP, 2 mg citric acid, 30 mg benzyl alcohol, 80 mg modified polysorbate 80/Tween 80, 650 mg polyethylene glycol 300, and 30.5% (v/v) alcohol.
In vivo safety of drugs was also evaluated in the H69AR tumor bearing nude mice by examining clinical chemistry parameters for kidney and liver function after administering MTDs of free or micelle incorporated drugs. The MTD of the free cisplatin was 2.5 mg/kg, and the MTD of the mixture of free drugs was 2 mg/kg cisplatin and 4 mg/kg ETO (Toposar) mg/kg body weight. There were no significant changes in ALT, BUN, and albumin levels that were in the normal range (Supplementary Table S10). Albeit the levels of creatinine seemed to be below the normal range, they were similar to the levels measured in mice treated with saline (Supplementary Table S10).
Mild renal toxicity was observed in animals treated with the combination of free cisplatin and ETO (Supplementary Figure S12). The samples from these animals revealed mild to moderately atrophied tubules having thinner and outstretched cell lining. Rare individual sloughed (dead) cells were also seen in the lumen of the tubules. Few scattered tubules have proteinaceous fluid or casts (Supplementary Figure S12 arrow in kidney samples). The renal toxicity appeared to be less in mice treated with the C6CP/ETO PM (4/8/10) and the mixture of C6CP PM (4/10) and ETO PM (8/10), where considerably lower scattered tubular damage was observed.
Combination C6CP/ETO PM Have Improved PK in Tumor-Bearing Mice
The drug PK and distribution profiles for C6CP/ETO PM (4/8/10) at MTD were compared to those for the mixture C6CP PM (4/10) and ETO PM (8/10) as well as the single drug micelles and commercial ETO Toposar in H69AR tumor bearing mice (Figure 6a,b). The PK parameters calculated using a noncompartment model are presented in Supplementary Table S11. It is apparent that the co-loaded drug micelles considerably increased the plasma half-life of each drug as compared to the single drug micelles and their mixture ( t1/2,α 6.78 h vs 5.38 and 5.10 h for C6CP and 7.18 h vs 4.04 and 5.30 h for ETO). Moreover, there was a ~1.5-fold increase in the plasma AUC (~1.5-fold decrease in CL) and ~1.2-fold decrease Vdobs for C6CP in the co-loaded drug micelles compared to single drug micelles. There were little if any changes in the corresponding parameters for ETO. The Cmax in the plasma did not seem to change much for any of the drugs in different formulations. However, the tumor Cmax was considerably increased for both drugs in the co-loaded drug micelles when compared to the single micelles or their mixtures (Cmax 3.88 μg/g vs 1.91 μg/g and 2.60 μg/g for C6CP and 12.90 μg/g vs 8.29 μg/g and 8.72 μg/g for ETO). The peak concentrations of both drugs in the tumors were reached at ~1 h post-injection for all investigated formulations, however, for at least as long as 6 and 24 h, the tumor concentrations of both C6CP and ETO for the co-loaded drug micelles were significantly higher than these concentrations for the single drug micelles and their mixture (Figure 6c,d). Notably, the tumor AUCs were increased by ~2.0- to 2.2-fold for C6CP and ~1.7- to 2.7-fold for ETO when these drugs were administered in the C6CP/ETO PM (4/8/10) format compared to either single micelles or their mixture. Further, analysis suggests that the tumor to plasma AUC ratios for C6CP in the co-loaded drug micelles, C6CP/ETO PM (4/8/10), exceeded these ratios for C6CP in the single drug micelles, C6CP PM (4/10), or mixture of the micelles, C6CP PM (4/10) and ETO PM (8/10), by ~1.4 and ~1.7 times, respectively (Supplementary Figure S13). The similar AUC ratio for ETO in co-loaded drug micelles exceeded these ratios for ETO in the single drug micelles and the mixture of micelles by ~1.3 and ~2.1 times, respectively. Overall the co-loaded drug micelles displayed a considerably improved PK and biodistribution in the blood and tumor as compared to the single drug micelles or their mixture.
Figure 6.

PK and tumor accumulation of C6CP and ETO in H69AR SCLC bearing mice. (a, b) Plasma (a) and tumor (b) biodistribution of both C6CP (black) and ETO (red) after single injection of various POx micelle formulations: C6CP/ETO PM (4/8/10), C6CP PM (4/10), and ETO PM (8/10) mixture, ETO PM (8/10), C6CP PM (4/10), and free ETO (Toposar). (c) C6CP and (d) ETO levels in accumulated in tumor 6, 24, and 48 h post-injection. Data are mean ± SD, * p < 0.05 (n = 3).
Comparison of the co-loaded drug micelles with the free ETO (Toposar) administered at MTD suggests that the PM formulation displays ~1.4 times higher half-life and 1.3 times lower Vdobs in plasma and ~20.7 times higher tumor AUC, which after normalization to the administered drug dose translates to ~2.76 increase in the dose-normalized AUC. An increase in the plasma half-life of ETO in co-loaded PM by about 2 h when compared with the free ETO (~7.2 h for co-loaded PM vs ~5 h for free ETO) is generally consistent with the relatively fast kinetics of the drug release from the micelles observed in this study (around 40% in 4 h). After release from the micelles, a hydrophobic drug is likely to be bound to the serum proteins and in this format circulate, distribute to the tumor and organs and ultimately cleared. The free drug also binds to the serum proteins and is distributed in a serum-bound format.21
Analysis of the tumor PK parameters also allows computing the C6CP/ETO ratios actually delivered to the tumor. Specifically, the tumor Cmax ratios were ~0.30 for both the co-loaded micelles and the micelle mixture. Likewise, the tumor AUC ratios were ~0.34 and ~0.42 for the co-loaded micelles and the micelle mixture, respectively. Therefore, the C6CP/ETO ratio at the tumor seems to be less than the C6CP/ETO ratio of 0.5 in the initial formulations. However, based on the in vitro cytotoxicity study for C6CP/ETO PM (2/6/10) that has C6CP/ETO ratio ~0.33, and assuming that the drugs are released and C6CP is converted to cisplatin at tumor sites at the same rate as in cell culture study, one could expect that the drugs actually delivered to the tumor would exhibit a synergistic anticancer effect.
Combination C6CP/ETO PM Have Superior Anti-Tumor Activity in Vivo
The in vivo antitumor activity of the PM formulations was evaluated in three models of lung cancer: (1) A549 NSCLC, (2) H69AR SCLC, and (3) 344SQ NSCLC. The animals were treated with the co-loaded drug micelles, C6CP/ETO PM (4/8/10), the single drug micelles, or their mixture. For some experiments, the treatment groups also included free cisplatin, free ETO, or the mixture (4/8) of the free drugs. All drugs formulated in PM were administered at the MTD of C6CP/ETO PM. The free drugs were administered at the MTD of cisplatin and ETO mixture (2 mg/kg cisplatin and 4 mg/kg ETO).
A549 Xenograft Model of NSCLC
The tumor growth curves after various treatments are presented in Figure 7a. The control groups treated with saline or PM alone displayed similar growth rates with the tumor volume increasing from ca. 100 to 1500 mm3 during 40 days. The groups treated with single drug micelles, ETO PM or C6CP PM, displayed slight, albeit significant, decrease in the tumor volumes compared to the saline controls. Considerable tumor inhibition was observed for animals treated with the mixture of the single drug micelles, C6CP PM and ETO PM, where the final tumor volume was ca. 600 mm3. The co-loaded drug micelles, C6CP/ETO PM, exhibited the most pronounced antitumor effect and decreased the final tumor volume to only ca. 350 mm3 (Figure 7a). The mixture of C6CP PM and ETO PM and the co-loaded C6CP/ETO PM extended the animals median survival from 45 days (saline control) to 67 and 69 days, respectively (Supplementary Figure S14a). Notably, while all animals in the C6CP PM and ETO PM mixture group reached the end point by day 80, 2/6 of mice treated with the co-loaded drug micelles survived beyond day 90.
Figure 7.

Antitumor effects of the single and co-loaded drug PM in NCSLC and SCLC animal models: (a) A549 and (b–f) H69AR. (a, b) Tumor volume changes, (c, d) histology, and (e, f) quantitation of % Pt-DNA adduct (red) and Caspase-3 (red) in H69AR tumor sections 2 days after the last treatment (DAPI staining (blue) indicates nucleus, the scale bar is 20 μm). The treatments regimen was q4d × 4. Drug injection doses were: 30 mg ETO/kg and 15 mg C6CP/kg for C6CP/ETO PM (4/8/10) and mixture of C6CP PM (4/10) and ETO PM (8/10); 30 mg ETO/kg for ETO PM (8/10), 15 C6CPs mg/kg for C6CP PM (4/10); 2 mg/kg cisplatin and 4 mg/kg ETO for free drugs mixture. Empty PM were injected at the polymer dose equivalent to that in the co-loaded micelle formulation. (a, b) Data are mean ± SD, n = 6, * p < 0.05 (vs C6CP PM and ETO PM mixture group (a) or vs C6CP PM group (b)), * p < 0.05 (vs C6CP PM and ETO PM mixture group (b)). For detailed statistical comparisons see Supplementary Table S12. (e, f) Five randomly selected microscopic fields were analyzed on ImageJ. * p < 0.05; ** p < 0.01.
H69AR Xenograft Model of SCLC
Similar promising efficacies of PM formulations were observed in a more aggressive H69AR SCLC xenograft model that rapidly grew from ca. 100 to 2400 mm3 during 31 days (Figure 7b). In this case, however, single drug micelles, C6CP PM, caused modest tumor inhibition, while ETO PM had a slight but statistically significant effect on the tumor volume. The mixture of the single drug micelles, C6CP PM and ETO PM, inhibited tumor growth considerably and extended the mean survival time from 32 to 55 days, albeit all mice reached the end point before day 60 (Supplementary Figure S14b). Like in the previous model case, the co-loaded drug micelles, C6CP/ETO PM, produced the strongest tumor growth inhibition, with 2/6 long-term survivors recorded beyond day 60.
344SQ Orthotopic Model of NSCLC
We further evaluated various formulations in the orthotopic 344SQ NSCLC model. The 344SQ/Luc. cancer cells used in this experiment were transfected with luciferase, allowing a continuous monitoring of the tumor growth by IVIS imaging (Supplementary Figure S15) as well as direct observation of the animals lifespan (Figure 8). The free drugs as well as their combination had little if any antitumor effect in this model with no animals surviving by day 29. In contrast, both single drug micelles produced some extension of the lifespan. Specifically, 2/5 and 3/5 mice survived at day 29 in the groups treated with C6CP PM and ETO PM, respectively. The mixture of the single drug micelles, C6CP PM and ETO PM, significantly extended the lifespan as compared to the controls, with 4/5 mice surviving on day 29. But still, the longest lifespan was observed with the co-loaded drug micelles, C6CP/ETO PM with all (5/5) mice surviving on day 29. The IVIS images clearly show that the tumor burden in this group was much less as compared to the group treated with the mixture of the single drug micelles as well as in all other groups (Supplementary Figure S15).
Figure 8.
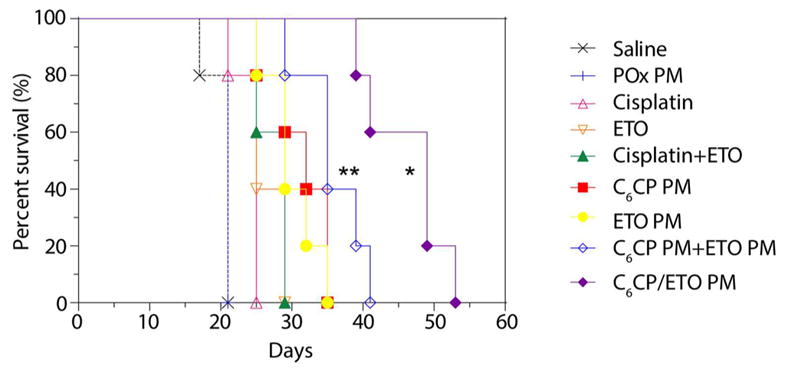
Kaplan–Meier survival plot showing antitumor effects of the single and co-loaded drug PM in 344SQ/Luc. NCSLC animal model. The treatments regimen was q4d × 4. Drug injection doses were: 30 mg ETO/kg and 15 mg C6CP/kg for C6CP/ETO PM (4/8/10) and mixture of C6CP PM (4/10) and ETO PM (8/10); 30 mg ETO/kg for ETO PM (8/10), 15 C6CPs mg/kg for C6CP PM (4/10); 2 mg/kg cisplatin or 4 mg/kg ETO for free drugs; 2 mg/kg cisplatin and 4 mg/kg ETO for free drugs mixture. Empty PM were injected at the polymer dose equivalent to that in the co-loaded micelle formulation. * p < 0.05 (vs C6CP PM and ETO PM mixture group), ** p < 0.01 (vs C6CP PM group). For detailed statistical comparisons see Supplementary Table S13.
Combination C6CP/ETO PMs Increase DNA Damage and Apoptosis in Tumors
The inhibition of DNA repair by topoisomerase II upon treatment with ETO slows down the removal of Pt-DNA adducts formed during treatment with cisplatin and results in the synergistic effects of these drugs.17,18 In this work, we evaluated these effects in the H69AR tumor model, by staining the tumor sections with the Alexa-555-labeled anti Pt-DNA adduct antibody on day 2 after the last drug treatment (Figure 7c). As seen from quantifications in Figure 7e, a significant increase in the amount of Pt-DNA adducts was observed in groups treated with the co-loaded drug micelles, C6CP/ETO PM, compared to both the single drug C6CP PM and mixture of C6CP PM and ETO PM treatment groups. Further, to elucidate the level of apoptosis following various treatments, the apoptosis marker Caspase-32 was quantified in the tumor sections on day 2 days after the last treatment (Figure 7d). Consistent with the data on the Pt-DNA adducts formation, Caspase-3 was significantly elevated in the C6CP/ETO PM treatment groups compared to all other groups (Figure 7f).
DISCUSSION
Development of nanoparticle drug carriers that can simultaneously encapsulate several drugs that differ in physicochemical and pharmacological properties is extremely important for combination chemotherapy of malignant diseases.2 Critical to the success of such a strategy are efficient drug loading and ability to precisely control the ratio of the drugs to be administered. We report on PM based on POx block copolymers that can simultaneously incorporate two important chemotherapeutic drugs, ETO and cisplatin, in a form of a prodrug, in a single, stable, and injectable solution and with minimal amount of the polymer excipient. This study demonstrates that these two drugs can be combined in a broad range of drug ratios, with unparalleled high net drug loadings over 50 wt % as stable micellar formulations. The herein reported drug loadings are, to the best of our knowledge, unprecedented for other PM containing two different drugs.22
The ETO and cisplatin combination chemotherapy is a well-known chemotherapy treatment used to treat several cancers.23 ETO induces DNA strand breaks by forming a tertiary complex with DNA and DNA topoisomerase II. When used together with cisplatin, ETO suppresses the DNA repair, leading to more efficient DNA damage.24 Due to the pharmacological synergy between these two drugs, ETO and cisplatin combination has shown cumulative effects against many forms of cancer, but in particular against NSCLC and SCLC. Based on the clinical successes of this combination therapy, we posited that co-encapsulation within POx PM could even further improve the therapeutic index. As expected, the co-loaded drug micelles displayed greatly improved in vitro cytotoxicity compared to the single drug micelles or even the mixture of the two single drug micelles. A profound synergistic effect has been observed in CI studies in the lung cancer cells for the co-loaded drug micelles. However, in addition to the pharmacological synergy of the drugs, the co-loaded drug micelles displayed several unexpected and highly beneficial properties.
These properties are beneficial for therapy as have been evident by the increase of the loading capacity as well as the reduced release rates and improved retention of drugs in the co-loaded micelles. This phenomenon probably reflects an increased miscibility of the specific drug pair ETO and C6CP within the BuOx core of the micelles. Surprisingly, the uptake of each of the drugs in the co-loaded drug micelles was also improved as compared to the mixture of the single drug micelles. We posit that the co-loaded drug micelles are more efficiently delivered into the cells due to a smaller number of particles, in which both drugs are residing and which are taken up into the cells by endocytosis. In this regard we would also like to point out that improved in vitro antitumor effect of 2 drugs in 1 nanoparticle when compared to the mixture of free drugs or drugs in separate nanoparticles was explained by deterministic spatially constrained delivery of both drugs as opposed to their stochastic distribution in the target cells.25 It is possible that a similar effect contributes to the improved cytotoxicity of the co-loaded PM.
Perhaps, most notably we observed the elongated, worm-like morphology of the co-loaded drug micelles. It has been reported that nonspherical particles, such as gold rods or polymer brushes, have altered cellular uptake and PK as compared to respective spheres.26,27 Of relevance to our work, Discher et al. has suggested potential benefits of worm-like vs spherical micelles in terms of PK for drug delivery.28 Yet their study neither used drug loaded micelles nor characterized drug delivery to tumors. To the best of our knowledge, herein we describe the highly loaded PM compositions with elongated morphology (short worms) that is highly unusual and very rare for drug loaded micelles. In previous reports, addition of the drug disrupted worm-like morphology. For example, empty PM represented a heterogeneous mixture of spheres and filaments/worms in the absence of the drug, but became spherical and uniform upon addition of PTX.29 The complete transition from worms to spheres occurred at the drug loadings <3% for P(MeOx-b-(2-nonyl-2-oxazoline)-b-MeOx) and <10% for the high capacity P(MeOx-b-BuOx-b-MeOx) triblock.29 The recently reported poly(ethylene glycol-b-ε-caprolactone) and poly(ethylene glycol-b-α-benzyl carboxylate ε-caprolactone) filomicelles containing PTX have very low drug loadings of only ~2.5 to ~3.2%.30 Such low drug loadings are very unfavorable for drug delivery of chemotherapeutic agents to treat cancer.9 In the present study, however, the highly loaded ETO-containing POx micelles displayed elongated morphology, which was sustained for the ETO/C6CP PM even though the single drug micelles of C6CP were spherical. It is amazing that the elongated morphology was observed at drug loadings as high as ~43% for single drug ETO PM and ~52% for co-loaded C6CP/ETO PM. When compared with the literature29 for the same POx polymer, we observed the worm-like micelles with least 10 times higher ETO loading as compared to PTX. This suggests that the filament morphology is governed by specific drug–polymer interactions and is characteristic for a given drug structure, in our case ETO. Moreover, drug–drug interactions in the micelle core can result in the morphology switch, specifically, transition from spheres for single C6CP micelles to worms upon addition of ETO in the co-loaded drug micelles. It is not surprising that the formation of the worm-like micelles was concentration-dependent, with the worms being more prevalent at high concentrations and the spheres dominating at low concentrations. Moreover, since in our case the loading of the drug (ETO) into the micelles promotes the worm-like morphology, one would expect that the drug release to the serum proteins in the blood should favor the transition of the remaining micelles from worms to spheres. Notably, the worm-like micelles were observed at the sample concentration at least 500 times less than the concentration of the solutions injected to animals (10 mg/mL). Therefore, one would expect that the worms are initially present in the circulation and then, consistent with the previous report by Discher et al.,28 transform into spheres. In view of the transient character of the worm-like micelles, it is hard to speculate at this point on their specific benefits for drug delivery. However, the discovery of this morphology for highly loaded PM is unusual and significant from the scientific standpoint and enables further assessment of the role of the shape of the micelles on their capture by the mononuclear cells, clearance, circulation, and distribution into tumors.
The PK and biodistribution data of the co-loaded drug micelles for both delivered drugs are highly encouraging. In particular, co-loaded drug micelles considerably increased the plasma half-life of each drug compared to the single drug micelles and their mixture. Moreover, the tumor Cmax and the tumor AUCs of each drug in the co-loaded drug micelles were also greatly increased, with the overall result being a drastic improvement of the PK and tumor distribution of the co-loaded formulation compared to the single drug micelles or their mixture. The superior PK and tumor distribution of the drugs in the co-loaded drug micelles may be due to several factors including (1) improved circulation of the drugs in the blood due to shape differences, (2) decreased drug release rates and better retention of the drugs in the micelles in the presence of serum, and (3) perhaps improved drug internalization in the cells as observed in the drug uptake studies. Noteworthy, consistent with the results of our study, Shin et al. demonstrated improvement of the blood AUC of the 2-in-1 and 3-in-1 co-loaded drugs in poly(ethylene glycol)-blockpoly( D,L-lactic acid) PM when compared to the single drug PM.31 Interestingly, for the 3-in-1 PM, the improvement was only observed with the drugs administered at high doses but not at the modest doses, which may underscore additional advantage of our high loaded PM approach.
The major and most impressive manifestation of the drug co-loading effect is a considerably increased antitumor activity of co-loaded drug micelles, compared not only with the individual single drug micelles but also with the mixture of the two single drug micelles administered at the same dose. This has been shown using three different tumor models of the NSCLC and SCLC along with the observed great improvement of the co-loaded drug micelle therapy compared to the free drug combinations. Overall, the highly loaded, worm-like micelles carrying ETO and C6CP have shown high promise in treatment of lung cancer. The ability of controlling the nanoparticle morphology, drug retention, PK, and therapeutic efficacy by blending multiple drugs in a single particle is of both basic and practical significance.
MATERIALS AND METHODS
Materials
Amphiphilic triblock copolymers P(MeOx37-b-BuOx21-b-MeOx36), Mn = 10.0 kg/mol, Đ (Mw/Mn) = 1.14 were synthesized as described in the previous studies by means of living cationic ring-opening polymerization and characterized by 1H NMR spectroscopy and gel permeation chromatography.8,9 ETO was purchased from LC Laboratories (Woburn, MA). Cisplatin was purchased from Sigma, and Toposar was purchased from UNC Hospital Pharmacy. All other materials were from Fisher Scientific Inc. (Fairlawn, NJ), and all reagents were HPLC grade. The A549 NSCLC cell and H69AR SCLC cell lines were originally obtained from Sigma-Aldrich (St. Louis, MO). Cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) (Gibco 11965-092) supplemented with 10% fetal bovine serum (FBS) and 1% pen-strep. The 344SQ/Luc. NSCLC cell line (expressing luciferase) was originally provided by Dr. John Kurie (MD Anderson Cancer Center, Houston, TX). The 344SQ/Luc. cells were cultured in DMEM medium with 10% FBS and 5 μg/mL puromycin.
Synthesis and Characterization of Hydrophobic Platinum-(IV) Prodrugs (CPs)
Ten mL of c,c,t-[Pt(NH3)2Cl2(OH)2] (0.69 g, 2.05 mmol) solution in DMSO was mixed with 10 mL of the solution in dimethylformamide of a respective aliphatic anhydride: C4CP, butyric anhydride (0.40 g, 3.9 mmol), C6CP, hexanoic anhydride (0.90 g, 4.2 mmol), C8CP, octanoic anhydride (2.18 g, 8.2 mmol) or C10CP, decanoic anhydride (2.53 g, 7.8 mmol), and the reaction mixture was stirred at room temperature (r.t.) for 48 h. Water (approximately 20 mL) was added to the mixture to precipitate light yellow solids, followed by filtration and isolation. The yellow solids were washed several times with diethyl ether and dried.10 The yields of C4CP, C6CP, C8CP, and C10CP were all approximately 40 wt %. The synthesized prodrugs were soluble in ethanol (5, 5, 2, and 2 g/L, respectfully).
Preparation and Characterization of Drug Loaded POx PM
Drug loaded PM were prepared by the thin-film method as previously described.8,9,11 Briefly, predetermined amounts of POx, ETO, and CPs were dissolved in ethanol (5–10 g/L) and mixed, followed by complete removal of ethanol. Appropriate amounts of normal saline were used to rehydrate the dried thin film under mild heating at 50–60 °C (or r.t. for CPs PM) for up to 15 min in order to obtain drug loaded PM. The excess of non-incorporated drug was removed by centrifugation from the otherwise stable micellar solution. The resulting micelle formulation was stored as aqueous solution in a refrigerator for up to 2 weeks or as lyophilized powder for long-term storage.
The drug concentrations in PM were determined by reverse-phase high-performance liquid chromatography (HPLC) method using an Eclipse XDB-C18–5 μm column (150 mm × 4.6 mm) and Agilent 1200 HPLC. Each sample was diluted 20 times in acetonitrile/water (50/50, v/v), and 20 μL was injected into the HPLC. The gradient elution started with 50% acetonitrile for 3 min, decreased to 20% acetonitrile over 4 min and kept for additional 3 min, then increased back to 50% acetonitrile over 2 min and kept for additional 3 min. The retention times of C4CP, C6CP, C8CP, C10CP, and ETO were approximately 2.9, 6.9, 9.7, 12.6, and 3.7 min, and the detection wavelength was 245 nm, while the flow rate was 1.0 mL/min and column temperature was 30 °C. A standard curve range from 5 μg/mL to 1000 μg/mL was used to calibrate the quantity of each drug.
The drug loading capacity (LC) and loading efficiency (LE) were calculated using eqs 1 and 2, where Mdrug and Mexcipient are the weight amounts of the loaded (solubilized) drug and polymer excipient in the dispersion, while Mdrug added is the weight amount of the drug initially added to the dispersion.
| (1) |
| (2) |
A Nano-ZS (Malvern Instruments Inc., UK) dynamic light scattering (DLS) equipment was used to measure size distribution of POx micelles. Briefly, each sample was diluted 10 times with deionized water (DI H2O) to yield 1 g/L final polymer concentration before the measurement. The intensity-mean z-averaged particle size (effective diameter) and the polydispersity index (PDI) of PM were determined by cumulate analysis. Results are the average of three independent micelle samples measurements.
The morphology of PM was studied using a LEO EM910 TEM operating at 80 kV (Carl Zeiss SMT Inc., Peabody, MA). Digital images were obtained using a Gatan Orius SC1000 CCD Digital Camera in combination with Digital Micrograph 3.11.0 software (Gatan Inc., Pleasanton, CA). One drop of each PM solution (diluted 200 times using DI H2O) was deposited on a copper grid/carbon film for 5 min, and excess solution was wicked off using fine filter paper. Then one drop of staining solution (1% uranyl acetate) was added and allowed to dry for 10 s prior to the TEM imaging.
Atomic force microscopy (AFM) was used to visualize the drug loading effect on micelle shape and size. Solutions of empty and drug loaded micelles (20, 30, 100, 500 μg/mL POx in H2O) were deposited onto freshly cleaved mica substrates by spin-casting. The imaging was performed in PeakForce QNM mode using a multimode AFM (Brüker) with a NanoScope V controller and silicon probes (resonance frequency of 70 Hz and spring constant of 0.4 N/m). Both Nanoscope Analysis (Brüker) and ImageJ (NIH) softwares were used to characterize micelle dimensions.
In Vitro Drug Release
The drug release studies were performed by membrane dialysis method against phosphate buffered saline (PBS), pH 7.4 at 37 °C. Briefly, drug loaded PMs were diluted in PBS containing 40 g/L BSA to achieve ~0.1 g/L of total drug concentration. The 100 μL of the diluted micelle solutions was added into floatable Slide-A-Lyzer MINI dialysis devices (100 μL capacity, 20 kDa MWCO; Thermo Scientific) and suspended in 20 mL PBS supplemented with 40 g/L BSA to ensure the sink conditions. Three devices were used for every time point. At each time point the samples were withdrawn from dialysis device and quantified by HPLC to obtain remaining drug amount of sample. Drug release profiles were constructed by plotting the % release over time.
Serum Binding Studies
Reverse-phase Thermo Scientific SOLA HRP solid-phase extraction (SPE) cartridges were used for separation and determination of micellar and protein bound forms of ETO and C6CP in serum. The micellar drugs did not retain on the stationary phase (fractions A3 and A4), while the protein bound drug was retained and eluted only with acidified methanol (fraction A6). The sample preparation was performed as follows: The formulations (100 μL of C6CP/ETO PM (4/8/10), mixture of C6CP PM (4/10) and ETO PM (8/10), C6CP PM (4/10), and ETO PM (8/10) comprising 3H-labeled ETO (3H-ETO, 2.5 μCi/mg ETO) were added to 2 mL of 100% FBS, incubated at 37 °C, and 200 μL samples were collected from the mixture solution at 1 and 4 h. Each 200 μL serum sample was mixed with 200 μL of PM solution (2 mg/mL) in PBS, pH 7.4. The following fractionation procedure was used: (A1) Column conditioning: add 0.5 mL methanol (discard to waste); (A2) equilibrate: add 0.5 mL water (discard to waste); (A3) application: load pretreated sample (encapsulated drug, collect); (A4) wash 1:2 × 0.25 mL POx in phosphate buffered saline (2 mg/mL), pH 7.4 added sequentially (encapsulated drug, collect); (A5) wash 2:2 × 1 mL water/methanol (90:10 v/v) added sequentially (discard to waste); (A6) elution: 0.5 mL methanol and 0.1% formic acid (albumin bound drug, collect).
In Vitro Cytotoxicity and Combination Index (CI) Analysis
In vitro cytotoxicity of drug loaded PM was evaluated in A549, H69AR, and 344SQ/Luc. lung cancer cell lines using [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) assay. Briefly, cells were seeded in 96-well plates at a density of 3000 cells/well 24 h prior to drug treatment. Subsequently, cells were treated with drug loaded PM or free drugs in full medium. Following 72 h, the incubation medium was removed, and 100 μL of fresh medium with MTT (100 μg/well) was added and incubated for 4 h at 37 °C. The medium was discarded, the formed formazan salt was dissolved in 100 μL of DMSO, and absorbance was read at 562 nm using a plate reader (SpectraMax M5, Molecular Devices). Cell survival rates were calculated as normalized to control untreated wells. Data represent average of hexaplicate in means ± standard deviation (SD). The mean drug concentration required for 50% growth inhibition (IC50) was determined using Graphpad Prism 5 software. The CI analysis based on the Chou and Talalay method was performed using CompuSyn software.12 Briefly, for each level of growth inhibition Fa, the CI values for binary drug combinations are calculated according to the following equation: CI = (D)1/(Dx)1 + (D)2/(Dx)2, where (D)1 and (D)2 are the concentrations of each drug in the combination resulting in Fa × 100% growth inhibition, and (Dx)1 and (Dx)2 are the concentrations of the drugs alone resulting in Fa × 100% growth inhibition. CI values for drug combinations are plotted as a function of Fa. Generally, the CI values between Fa = 0.2 and 0.8 are considered valid. The best-fit CI value at IC50 is used to show and compare the synergistic effects of drug combinations with different drug ratios or for different cell lines. CI values less than 1 or more than 1 demonstrate synergism or antagonism of drug combinations, respectively.
In Vitro Cell Uptake
344SQ/Luc. cells (5 × 103/well) were seeded in 24-well plates. After 24 h the medium was replaced (200 μL) and exposed to (1) C6CP PM, (2) ETO PM, (3) C6CP/ETO PM, or (4) a mixture of C6CP PM and ETO PM in fresh medium for 30 min, 1, 4, 8, 24, and 48 h. The concentrations of each drug during the exposure were equal to their concentrations at IC50 of C6CP/ETO PM. All ETO test articles contained 3H-ETO (2.5 μCi/mg ETO). After that, cells were washed three times with PBS, harvested with 200 μL trypsin containing ethylenediaminetetraacetic acid (EDTA), and lysed with 100 mM Tris buffer containing 5 mM EDTA, 200 mM NaCl, 0.2% sodium dodecyl sulfate, pH 8.0 for 1 h at 37 °C. The homogenates were divided into two vials, one of which was analyzed for 3H-ETO using a Tricarb 4000 (Packard, Meriden). To the other vial ca. 100 μL of concentrated HNO3 was added, and the samples were incubated at 60 °C overnight, followed by dissolving in DI water (5 mL). Platinum concentration was measured by inductively coupled plasma mass spectrometry (ICP-MS). All experiments were repeated three times, and the average platinum and ETO contents (ng/5 × 103 cells) were calculated.13
Maximum Tolerable Dose (MTD) Determination
All animal procedures were in compliance with the United States federal animal welfare regulations and approved by the UNC Institutional Animal Care and Use Committee. MTD evaluation for C6CP/ETO PM was performed in dose escalation study in healthy 6–8 week old female nude mice. Animals (three mice per group) received i.v. injections (tail vein) of 10/20, 12.5/25, 15/30, and 20/40 C6CP/ETO mg/kg of C6CP/ETO PM using a q4d × 4 regimen (total 4 times repeated dosing, every fourth day with saline as a control). Mice survival and changes in the body weight were observed daily over 2 weeks in all groups following the last injection. The highest dose that did not cause noticeable toxicity (as defined by a median body weight loss of 15% of the control or abnormal behavior including hunched posture and rough coat) was used as MTD for efficacy experiments.
Toxicology Studies
Female athymic nude mice (6–8 weeks) with H69AR xenograft tumors were administered with saline or respective MTDs of free cisplatin and ETO (Toposar) mixture, C6CP PM (4/10) and ETO PM (8/10) mixture, or C6CP/ETO PM (4/8/10) (at MTD) using a q4d × 4 regimen. Here and below the numbers in the parentheses refer to the mass ratios C6CP/POx, ETO/POx or C6CP/ETO/POx in the formulation. Two days after the last injection, mice were euthanized and blood was collected and centrifuged at 7500 rpm for 5 min to obtain the serum. Blood urea nitrogen (BUN), creatinine, and alanine aminotransferase (ALT) levels were assayed as indicators of renal and hepatic function. Whole blood collected from animals was used for the plasma albumin level test. To evaluate the organ-specific toxicity, major organs (brain, spleen, liver, lung, kidney, heart, and tumor) were harvested, fixed in formalin, and subjected to pathological analysis by hemotoxylin and eosin (H&E) staining.
PK and Tumor Accumulation
Female athymic nude mice (6–8 weeks) with well-developed 100 mm3 H69AR xenograft tumors were administered a single dose of following formulations via tail vein: (1) C6CP/ETO PM (15 mg/kg C6CP and 30 mg/kg ETO mouse body weight, the MTD for this formulation), (2) C6CP PM and ETO PM mixture (15 mg/kg C6CP and 30 mg/kg ETO), (3) C6CP PM (15 mg/kg C6CP), (4) ETO PM (30 mg/kg ETO), and (5) “Free” ETO (Toposar) (4 mg/kg ETO). Each mL prior to injection of Toposar (contains 20 mg ETO, 2 mg citric acid, 30 mg benzyl alcohol, 80 mg Tween-80, 650 mg PEG-300, and 30.5% (v/v) alcohol) was diluted 25-times in saline. All ETO injections contained 3H-ETO (5 μCi/mouse). At several time points 0.083, 0.5, 1, 2, 6, 24, and 48 h post-injection, three animals from every treatment group were euthanized, the blood was collected by cardiac puncture, and the organs (spleen, liver, kidney) and the tumor were removed, washed in ice-cold saline, weighted, and homogenized in a glass tissue homogenizer (Tearor, BioSpec Products, Inc.). For drug concentrations in plasma, organs and tumors were measured by radioactivity counts using a Tricarb 4000 for 3H-ETO or by platinum content using ICP-MS for C6CP. PK parameters were determined with Phoenix WinNonlin (version 6.0) software using noncompartmental analysis.
In Vivo Tumor Growth Inhibition
A549 and H69AR Xenograft Models
Female athymic nude mice (6–8 weeks) were subcutaneously inoculated in the right flank with 1 × 106 human A549 cells or 3 × 106 human H69AR cells, each resuspended in 50% growth medium and 50% Matrigel. For each tumor model, the animal treatments started when tumor sizes reached ca. 100 mm3. Animals were randomized into seven groups of six mice each with similar mean tumor volumes between groups and then treated with the following formulations: (1) C6CP/ETO PM (15 mg/kg C6CP and 30 mg/kg ETO mouse body weight), (2) C6CP PM and ETO PM mixture (15 mg/kg C6CP and 30 mg/kg ETO), (3) C6CP PM (15 mg/kg C6CP), (4) ETO PM (30 mg/kg ETO), (5) free drug mixture (2 mg/kg cisplatin and 4 mg/kg ETO as Toposar), (6) PM alone (at the POx dose equivalent to that in the C6CP/ETO PM group), and (7) saline alone. The formulations were administered via tail vein using q4d × 4 regimen (on the days 0, 4, 8, 12). Survival and body weight were monitored daily. Tumor length (L) and width (W) were measured, and tumor volume (TV) was calculated as TV = 1/2 × L × W2. Tumor growth was monitored twice weekly for 13 weeks or earlier end-points defined by tumor volume (>2000 mm3), animal weight loss (>15%), or animals becoming moribund. Tumors were removed at the end of the observation and subjected to histopathological examination.
344SQ Orthotopic Model
The orthotopic model was created by directly transplanting 344SQ/Luc. cells into female mice lung by an intrapulmonary technique (5 × 103 cells/mouse in 40 μL 3:1 mixture of Hanks’ balanced salt solution, and BD Matrigel (BD Biosciences)). Mice are anesthetized using 3% isoflurane in O2 and then kept under 1.5–2% isoflurane in O2. A ~1 cm incision was made in the left chest wall. Chest muscles were separated by sharp dissection, and costal and intercostal muscles were exposed. A 27-gauge needle was inserted through the intercostal space between the third and fourth ribs. Cells were slowly injected into the left lung over 30 s. The skin was closed using tissue adhesive and surgical suture in an interrupted pattern. The animals were randomized and treated via tail vein following q4d × 4 regimen the next day after cells injection. Prior to imaging, mice were injected 10 μL per gram of body weight of intraperitoneal (i.p.) luciferin solution at a concentration of 15 mg/mL. Mice were anesthetized with isoflurane prior to imaging. IVIS (in vivo imaging system) imaging were taken every 2 days to monitor tumor growth: (1) C6CP/ETO PM (15 mg/kg C6CP and 30 mg/kg ETO mouse body weight), (2) C6CP PM and ETO PM mixture (15 mg/kg C6CP and 30 mg/kg ETO), (3) C6CP PM (15 mg/kg C6CP), (4) ETO PM (30 mg/kg ETO), (5) free cisplatin (2 mg/kg), (6) free ETO as Toposar (4 mg/kg), (7) free drug mixture (2 mg/kg cisplatin and 4 mg/kg ETO), (8) PM alone (at the POx dose equivalent to that in the C6CP/ETO PM group), and (9) saline alone (n = 5). For in vivo imaging, mice were placed onto the warmed stage inside the camera box and received continuous exposure to 2.5% isoflurane to sustain sedation during imaging. Every group of mice was imaged for 30 s. The light emitted from the mice was detected by the IVIS-100 camera system, integrated, digitized, and displayed.
Tumor Sections Histology
Pt-DNA Adducts Staining of Tumor Sections
The Pt-DNA adducts were detected using anticisplatin-modified DNA antibody [CP9/19] (Abcam, Cambridge, MA). The tumor sections were collected during efficacy experiments (the second day after the last treatment), fixed in neutral buffered formalin (4% v/v) at 4 °C for another 24 h, deparaffinized, antigen recovered, blocked with BSA (1% w/v) containing 0.1% triton X-100 in PBS for 1 h at r.t., incubated with a 1:250 dilution of [CP9/19] antibody at 4 °C overnight, and then incubated with Alexa 555-labeled goat antirat IgG antibody (1:200, Santa Cruz, CA). The sections were also counter-stained with VECTASHIELD mounting media with DAPI (Vector laboratories, Burlingame, CA). The tumor sections were analyzed, and the staining was quantified using a Zeiss LSM 700 confocal laser scanning microscope (Zeiss, USA) in the Microscopy Services Laboratory at the UNC Medical School.
Caspase-3 Levels Staining of Tumor Sections
The apoptosis was detected using Caspase-3 (Cleaved) polyclonal antibody [CP229 A] (Biocare Medical). The tumor sections were collected and processed as described above but incubated with a 1:200 dilution of [CP229 A] antibody at 4 °C overnight and then incubated with Alexa 555-labeled goat antirabbit IgG antibody (1:250, Santa Cruz, CA). The sections were also counter-stained and analyzed as described above.
Statistical Analysis
Quantitative results were expressed as mean ± SD. Statistical comparisons for drug release, cellular uptake, PK, tumor accumulation, histology quantitation images, and tumor inhibition data were done using one-way analysis of variance (ANOVA) with Holm–Sidak posthoc test for multiple comparisons. Statistical analysis was performed using Prism 7.03 software. Statistical comparison of animal survival was done by log-rank test. Differences were considered to be statistically significant if the p value was <0.05.
Supplementary Material
Acknowledgments
This work was supported by the National Cancer Institute (NCI) Cancer Nanotechnology Platform Partnership grant (U01 CA116591) and the Carolina Center of Cancer Nanotechnology Excellence Project 4 (U54 CA198999). A.Z.W. was also supported by the NIH/NCI (R21 CA182322). X.W. is grateful to the China Scholarship Council (CSC) for a predoctoral fellowship (2011601254). We are also grateful to Chad Pecot who provided 344SQ/Luc. lung cancer cell line, Charlene Santos at MP1U of UNC for helping the development of the orthotopic lung cancer model, and William Zamboni for providing advice in the PK analysis.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.7b07878.
Additional DLS characterization and stability studies, release profile, cisplatin prodrug characterization (1H-NMR and mass spectra analysis), cytotoxicity, additional PK and biodistribution data, drug serum binding experiments, histological examination, clinical chemistry, and PK parameters are described (PDF)
References
- 1.Greco F, Vicent MJ. Combination Therapy: Opportunities and Challenges for Polymer-Drug Conjugates as Anticancer Nanomedicines. Adv Drug Delivery Rev. 2009;61:1203–1213. doi: 10.1016/j.addr.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Miao L, Guo S, Zhang J, Kim WY, Huang L. Nanoparticles with Precise Ratiometric Co-Loading and Co-Delivery of Gemcitabine Monophosphate and Cisplatin for Treatment of Bladder Cancer. Adv Funct Mater. 2014;24:6601–6611. doi: 10.1002/adfm.201401076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, Stuart RK, Strickland SA, Hogge D, Solomon SR. Final Results of a Phase III Randomized Trial of Cpx-351 versus 7+ 3 in Older Patients with Newly Diagnosed High Risk (Secondary) AML. J Clin Oncol. 2016;34:7000. [Google Scholar]
- 4.Kalemkerian GP, Akerley W, Bogner P, Borghaei H, Chow LQ, Downey RJ, Gandhi L, Ganti AK, Govindan R, Grecula JC, Hayman J, Heist RS, Horn L, Jahan T, Koczywas M, Loo BW, Jr, Merritt RE, Moran CA, Niell HB, O’Malley J, et al. Small Cell Lung Cancer. J Natl Compr Cancer Network. 2013;11:78–98. doi: 10.6004/jnccn.2013.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan BA, Coward JIG. Chemotherapy Advances in Small-Cell Lung Cancer. J Thorac Dis. 2013;5:S565–S578. doi: 10.3978/j.issn.2072-1439.2013.07.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tardi PG, Dos Santos N, Harasym TO, Johnstone SA, Zisman N, Tsang AW, Bermudes DG, Mayer LD. Drug Ratio-Dependent Antitumor Activity of Irinotecan and Cisplatin Combinations in Vitro and in Vivo. Mol Cancer Ther. 2009;8:2266–2275. doi: 10.1158/1535-7163.MCT-09-0243. [DOI] [PubMed] [Google Scholar]
- 7.Luxenhofer R, Schulz A, Roques C, Li S, Bronich TK, Batrakova EV, Jordan R, Kabanov AV. Doubly Amphiphilic Poly(2-Oxazoline)S as High-Capacity Delivery Systems for Hydrophobic Drugs. Biomaterials. 2010;31:4972–4979. doi: 10.1016/j.biomaterials.2010.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han Y, He Z, Schulz A, Bronich TK, Jordan R, Luxenhofer R, Kabanov AV. Synergistic Combinations of Multiple Chemotherapeutic Agents in High Capacity Poly(2-Oxazoline) Micelles. Mol Pharmaceutics. 2012;9:2302–2313. doi: 10.1021/mp300159u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He Z, Wan X, Schulz A, Bludau H, Dobrovolskaia MA, Stern ST, Montgomery SA, Yuan H, Li Z, Alakhova D, Sokolsky M, Darr DB, Perou CM, Jordan R, Luxenhofer R, Kabanov AV. A High Capacity Polymeric Micelle of Paclitaxel: Implication of High Dose Drug Therapy to Safety and in Vivo Anti-Cancer Activity. Biomaterials. 2016;101:296–309. doi: 10.1016/j.biomaterials.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted Delivery of Cisplatin to Prostate Cancer Cells by Aptamer Functionalized Pt(Iv) Prodrug-Plga-Peg Nanoparticles. Proc Natl Acad Sci U S A. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He Z, Schulz A, Wan X, Seitz J, Bludau H, Alakhova DY, Darr DB, Perou CM, Jordan R, Ojima I, Kabanov AV, Luxenhofer R. Poly(2-Oxazoline) Based Micelles with High Capacity for 3rd Generation Taxoids: Preparation, in Vitro and in Vivo Evaluation. J Controlled Release. 2015;208:67–75. doi: 10.1016/j.jconrel.2015.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chou TC. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 13.Miura Y, Takenaka T, Toh K, Wu S, Nishihara H, Kano MR, Ino Y, Nomoto T, Matsumoto Y, Koyama H, Cabral H, Nishiyama N, Kataoka K. Cyclic Rgd-Linked Polymeric Micelles for Targeted Delivery of Platinum Anticancer Drugs to Glioblastoma through the Blood-Brain Tumor Barrier. ACS Nano. 2013;7:8583–8592. doi: 10.1021/nn402662d. [DOI] [PubMed] [Google Scholar]
- 14.Giandomenico CM, Abrams MJ, Murrer BA, Vollano JF, Rheinheimer MI, Wyer SB, Bossard GE, Higgins JD. Carboxylation of Kinetically Inert Platinum(Iv) Hydroxy Complexes. An Entr.Acte.Ee into Orally Active Platinum(Iv) Antitumor Agents. Inorg Chem. 1995;34:1015–1021. doi: 10.1021/ic00109a004. [DOI] [PubMed] [Google Scholar]
- 15.Barnes KR, Kutikov A, Lippard SJ. Synthesis, Characterization, and Cytotoxicity of a Series of Estrogen-Tethered Platinum-(Iv) Complexes. Chem Biol. 2004;11:557–564. doi: 10.1016/j.chembiol.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 16.McEvoy GKe. American Hospital Formulary Service - Drug Information 92. American Society of Hospital Pharmacists, Inc; Bethesda, MD: 1992. p. 524. [Google Scholar]
- 17.Tee AR, Proud CG. DNA-Damaging Agents Cause Inactivation of Translational Regulators Linked to Mtor Signalling. Oncogene. 2000;19:3021–3031. doi: 10.1038/sj.onc.1203622. [DOI] [PubMed] [Google Scholar]
- 18.Johnson SW, Laub PB, Beesley JS, Ozols RF, Hamilton TC. Increased Platinum-DNA Damage Tolerance Is Associated with Cisplatin Resistance and Cross-Resistance to Various Chemotherapeutic Agents in Unrelated Human Ovarian Cancer Cell Lines. Cancer Res. 1997;57:850–856. [PubMed] [Google Scholar]
- 19.Chou TC, Talalay P. Quantitative Analysis of Dose-Effect Relationships: The Combined Effects of Multiple Drugs or Enzyme Inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 20.Turrisi AT, 3rd, Kim K, Blum R, Sause WT, Livingston RB, Komaki R, Wagner H, Aisner S, Johnson DH. Twice-Daily Compared with Once-Daily Thoracic Radiotherapy in Limited Small-Cell Lung Cancer Treated Concurrently with Cisplatin and Etoposide. N Engl J Med. 1999;340:265–271. doi: 10.1056/NEJM199901283400403. [DOI] [PubMed] [Google Scholar]
- 21.Paal K, Muller J, Hegedus L. High Affinity Binding of Paclitaxel to Human Serum Albumin. Eur J Biochem. 2001;268:2187–2191. doi: 10.1046/j.1432-1327.2001.02107.x. [DOI] [PubMed] [Google Scholar]
- 22.Tian J, Min Y, Rodgers Z, Wan X, Qiu H, Mi Y, Tian X, Wagner KT, Caster JM, Qi Y, Roche K, Zhang T, Cheng J, Wang AZ. Nanoparticle Delivery of Chemotherapy Combination Regimen Improves the Therapeutic Efficacy in Mouse Models of Lung Cancer. Nanomedicine. 2017;13:1301–1307. doi: 10.1016/j.nano.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pillai RN, Owonikoko TK. Small Cell Lung Cancer: Therapies and Targets. Semin Oncol. 2014;41:133–142. doi: 10.1053/j.seminoncol.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montecucco A, Zanetta F, Biamonti G. Molecular Mechanisms of Etoposide. EXCLI Journal. 2015;14:95–108. doi: 10.17179/excli2015-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldman A, Kulkarni A, Kohandel M, Pandey P, Rao P, Natarajan SK, Sabbisetti V, Sengupta S. Rationally Designed 2-in-1 Nanoparticles Can Overcome Adaptive Resistance in Cancer. ACS Nano. 2016;10:5823–5834. doi: 10.1021/acsnano.6b00320. [DOI] [PubMed] [Google Scholar]
- 26.Arnida, Janat-Amsbury MM, Ray A, Peterson CM, Ghandehari H. Geometry and Surface Characteristics of Gold Nanoparticles Influence Their Biodistribution and Uptake by Macrophages. Eur J Pharm Biopharm. 2011;77:417–423. doi: 10.1016/j.ejpb.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mullner M, Dodds SJ, Nguyen TH, Senyschyn D, Porter CJ, Boyd BJ, Caruso F. Size and Rigidity of Cylindrical Polymer Brushes Dictate Long Circulating Properties in Vivo. ACS Nano. 2015;9:1294–1304. doi: 10.1021/nn505125f. [DOI] [PubMed] [Google Scholar]
- 28.Geng YAN, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Shape Effects of Filaments Versus Spherical Particles in Flow and Drug Delivery. Nat Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulz A, Jaksch S, Schubel R, Wegener E, Di Z, Han Y, Meister A, Kressler J, Kabanov AV, Luxenhofer R, Papadakis CM, Jordan R. Drug-Induced Morphology Switch in Drug Delivery Systems Based on Poly(2-Oxazoline)S. ACS Nano. 2014;8:2686–2696. doi: 10.1021/nn406388t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nair PR, Karthick SA, Spinler KR, Vakili MR, Lavasanifar A, Discher DE. Filomicelles from Aromatic Diblock Copolymers Increase Paclitaxel-Induced Tumor Cell Death and Aneuploidy Compared with Aliphatic Copolymers. Nanomedicine. 2016;11:1551–1569. doi: 10.2217/nnm-2016-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin HC, Alani AW, Cho H, Bae Y, Kolesar JM, Kwon GS. A 3-in-1 Polymeric Micelle Nanocontainer for Poorly Water-Soluble Drugs. Mol Pharmaceutics. 2011;8:1257–1265. doi: 10.1021/mp2000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.