

Abstract
Background & Aims
Myeloid cell leukemia1 (MCL1), a prosurvival member of the BCL2 protein family, plays a pivotal role in human cholangiocarcinoma (CCA) cell survival. We have previously reported that fibroblast growth factor receptor (FGFR) signaling mediates MCL1-dependent survival of CCA cells in vitro and in vivo. However, the mode and mechanisms of cell death in this model were not delineated.
Methods
Human CCA cell lines were treated with the pan-FGFR inhibitor LY2874455 and the mode of cell death examined by several complementary assays. Mitochondrial oxidative metabolism was examined using a XF24 extracellular flux analyzer. The efficiency of FGFR inhibition in patient-derived xenograft (PDX) was also assessed.
Results
CCA cells expressed two species of MCL1, a full-length form localized to the outer mitochondrial membrane, and an N-terminus truncated species compartmentalized within the mitochondrial matrix. The pan-FGFR inhibitor LY2874455 induced non-apoptotic cell death in the CCA cell lines associated with cellular depletion of both MCL1 species. The cell death was accompanied by failure of mitochondrial oxidative metabolism and was most consistent with necrosis. Enforced expression of N-terminus truncated MCL1 targeted to the mitochondrial matrix, but not full length MCL1 targeted to the outer mitochondrial membrane, rescued cell death and mitochondrial function. LY2874455 treatment of PDX-bearing mice was associated with tumor cell loss of MCL1 and cell necrosis.
Conclusions
In conclusion, FGFR inhibition induces loss of matrix MCL1 resulting in cell necrosis. These observations support a heretofore unidentified, alternative MCL1 survival function, namely prevention of cell necrosis, and have implications for treatment of human CCA.
Keywords: A-1210477, Fibroblast Growth Factor Receptor (FGFR), LY2874455, Patient derived xenograft (PDX), Seahorse extracellular flux analysis
Graphical Abstract
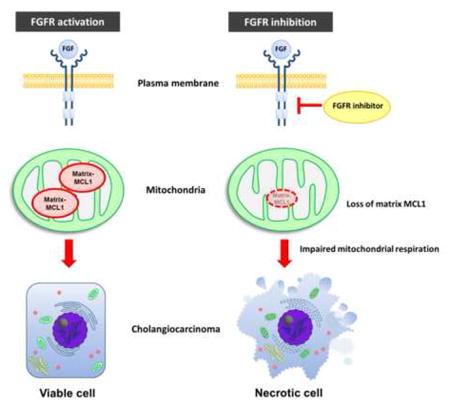
INTRODUCTION
Cholangiocarcinoma (CCA) is a lethal hepatobiliary malignancy with limited therapeutic options [1, 2]. Advances in CCA therapy will require an understanding of oncogenic signaling networks that contribute to CCA pathogenesis and can potentially be disrupted therapeutically. Like other malignancies, a cardinal feature of CCA is inhibition of cell death pathways that are engaged by oncogenic signaling pathways [3]. Members of the BCL2 gene family encode proteins that regulate the mitochondrial or intrinsic apoptotic pathway [4]. Of the BCL2 prosurvival members, MCL1 is most frequently amplified and overexpressed in CCA [5–7], and plays a pivotal role in CCA cell survival [8]. Hence, targeting MCL1 is a strategy for the treatment of CCA and other malignancies [9, 10].
Anti-apoptotic BCL2 proteins such as MCL1 block cell death by binding pro-apoptotic BCL2 effectors, such as BAX, BAK or BCL2 homology-3 (BH3) only proteins [4]. By sequestering these proteins on the outer mitochondrial membrane, MCL1 effectively blocks mitochondrial outer membrane permeabilization (MOMP) – a requisite event for the intrinsic pathway of apoptosis [4]. However, in addition to residing on the outer mitochondrial membrane, MCL1 may also undergo proteolytic processing at its N-terminus to generate a truncated species that translocates to and localizes within the mitochondrial matrix [11–14]. While full-length MCL1 on the outer mitochondrial membrane functions as an anti-apoptotic protein, N-terminal truncated MCL1 within the mitochondrial matrix regulates mitochondrial oxidative metabolism and dynamics [14].
Recently, we reported that pharmacologic inhibition of fibroblast growth factor receptor (FGFR) signaling resulted in cell death that was accompanied by loss of MCL1 mRNA and protein, and was rescued by enforced MCL1 expression [15]. However, the cell death process was not characterized by morphologic features of apoptosis, and biochemically was accompanied by reductions in cellular oxygen consumption and cellular ATP levels, observations more consistent with cell death by necrosis than apoptosis [16]. The mode of cell death is therapeutically relevant, as it provides mechanistic insight for the development of combinatorial therapy. For example, necrosis is thought to be a more immunogenic cell death than apoptosis, and combining immunotherapy with drugs inducing necrosis may be synergistic.
Herein, we explored the mode of cell death during FGFR inhibition in human CCA cells. These studies revealed that FGFR pharmacologic inhibition resulted in impairment of mitochondrial oxidative metabolism and diminished ATP levels followed by a non-apoptotic cell death that was rescued by enforced expression of an N-terminus truncated MCL1 targeted to the mitochondrial matrix. These observations suggest that, in addition to its anti-apoptotic role, MCL1 also plays an alternative survival function, namely prevention of cell necrosis.
Materials and Methods
Cell Culture
The human CCA cell lines KMCH-1 (hereafter referred to as KMCH) and KMBC-1 (hereafter referred to as KMBC), which have been previously described [17, 18], human pancreatic ductal adenocarcinoma cell lines PANC-1 and DanG were cultured in Dulbecco’s modified Eagle’s medium (DMEM). Panc04.03 and BxPC-3 cells were cultured in RPMI-1640, HepG2 cells were cultured in Eagle’s minimum essential medium. HT-29 cells were cultured in McCoy’s 5a medium. DanG was obtained from Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures. All cell lines except for KMCH, KMBC and DanG were obtained from American Type Culture Collection. All culture media were supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (100 μg/mL) and cells were cultured in a 5% CO2 incubator at 37°C. Cells with inducible MCL1 sgRNA were maintained in DMEM supplemented with 10% Tet system approved FBS (Clontech Laboratories). For authentication of the cell lines, short tandem repeat (STR) analysis was performed on KMCH and KMBC cell lines by the Genome Analysis Core of the Medical Genome Facility (Mayo Clinic, Rochester, MN). All cell lines underwent Mycoplasma contamination testing periodically using PlasmoTest™ -Mycoplasma Detection kit (InvivoGen). Experiments were performed using cells within maximum of 15 passages after thawing.
Reagents and Antibodies
LY2874455 (ApexBio) was reconstituted with dimethyl sulfoxide (DMSO) and added to cells at a final concentration of 5 μM. Obatoclax (GlaxoSmithKline PLC) and TNF-related apoptosis-inducing ligand (TRAIL) (R&D Systems) was reconstituted with 0.1% BSA in PBS. They were added to cells at final concentrations of 5 μM and 20 ng/mL, respectively. Necrostatin-1 (Selleck Chemicals) [19] was reconstituted with DMSO and added to cells at a final concentration of 25 μM. A-1210477 (Selleck Chemicals) was reconstituted with DMSO and added to cells at a final concentration of 10 μM. Doxycycline hyclate was purchased from Sigma-Aldrich and dissolved in sterile water. The following primary antibodies were used for immunoblot analysis: phospho-FRS2 (ab195826), total-FRS2 (ab137458), phospho-MLKL (ab187091) and TIMM50 (ab23938) from Abcam; TNF-R1 (sc-8436), MCL1 (sc-819), HSP60 (sc-1052), TOM70 (sc-390545), BAX (sc-493) BAK (sc-832) and β-actin (sc-1615) from Santa Cruz Biotechnology; PARP (9542S), MLKL (14993) and RIP3 (13526) from Cell Signaling Technology; RIP1 (610458) from BD Transduction Laboratories; and GAPDH (MAB374) from Millipore.
Immunoblot analysis
Whole cell lysates were prepared as previously described in detail [20]. Proteins were resolved by 12.5% SDS-PAGE, transferred on to an Immunobilon-FL PVDF membrane (EMD Millipore), and blotted with primary antibodies. Proteins were detected by Odyssey (LI-COR) infrared scanning system or visualized using enhanced chemiluminescence reagents (ECL PLUS, Amersham GE) and ChemiDoc imaging systems (BIO-RAD) or Kodak X-OMAT film.
Cell death assays
To evaluate cytotoxicity, cells were grown in 96-well plates (5 × 103 cells per well) and assayed for survival using several different assays, each of which was performed according to the manufacturer’s instructions. These assays included: MTS [3,4-(5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt] assay (CellTiter 96 Aqueous One Solution cell proliferation assay, Promega); Sulforhodamine B (SRB) assay (In Vitro Toxicology Assay Kit Sulforhodamine B Based, SigmaAldrich), CellTiter-Blue assay (CellTiter-Blue Cell Viability Assay, Promega); and CellTiter-Glo luminescent cell viability assay (Promega). Cell death in KMCH cells was also characterized by annexin V and propidium iodide (PI) labeling using flow cytometry. KMCH cells were plated and treated with 5 μM obatoclax plus 20 ng/mL TRAIL or 5 μM LY for 24 hours. Using the Alexa Fluor® 488 annexin V/Dead cell apoptosis kit (Invitrogen), dual staining with annexin V and PI was performed according to the manufacturer’s instructions. Trypsinized cells were washed in PBS and suspended at 1 × 106/mL in annexin-binding buffer. After staining, flow-cytometric analysis was carried out on a FACS Canto (BD Biosciences).
CRISPR/Cas9 genome editing
The sequences of single guide RNA (sgRNA) for BAX and BAK were designed using online CRISPR design tool (http://crispr.mit.edu). They included a 4-bp overhang for the forward (ccgg) and complementary reverse (aaac) oligos to enable cloning into pGuide-it-ZsGreen1 (for BAX) or pGuide-it-tdTomato (for BAK) vector (Clontech). These vectors were transiently transfected into KMCH cells using FuGene HD reagent (Promega). Dual-positive ZsGreen and tdTomato cells were sorted on a single-cell basis into single wells of a 96-well plate using flow cytometry. The sgRNA sequences are as follows: BAX exon 2: 5′-CTGCAGGATGATTGCCGCCG-3′; BAK exon 4: 5′-ACGGCAGCTCGCCATCATCG-3′. FgH1tMCL1 plasmid (for inducible expression of sgRNA) was generated as described previously [21]. The sequences of sgRNAs for MCL1 (sgMCL1#1 and sgMCL1#2) have been described [21]. To produce lentiviruses, FgH1tMCL1 or FUCas9Cherry plasmids were transiently co-transfected into HEK293T cells with the packaging plasmids pMDLg/pRRE, pRSV-rev and pCMV-VSV-G (all from Addgene) using FuGene HD reagent (Promega). Harvested lentiviruses were passed through a 0.45-μm filter; KMCH cells were transduced with the lentiviral constructs in the presence of 8 μg/mL polybrene for 24 hours. After transduction, m-cherry (Cas9) and GFP (inducible-sgRNA) dual-positive cells were sorted on a single-cell basis into individual wells of a 96-well plate using flow cytometry. To induce sgRNA expression in cells, doxycycline hyclate was added to culture medium at a final concentration of 5 μg/mL. The efficiency of all CRISPR/Cas9-mediated gene deletions was examined by immunoblot analysis.
Necroptosis induction
Cells were pretreated with ZVAD-fmk (Santa Crus Biotechnology) at a final concentration of 20 μM for 1 hour. Following pretreatment, tumor necrosis factor alpha (TNF-α, R&D Systems) and TL-32711 (Birinapant, Selleck Chemicals), a SMAC mimetic, were added at a final concentration of 20 ng/mL and 1 μM, respectively. Following a 7 hr incubation period, cells were harvested for immunoblot analysis.
Quantitative real-time polymerase chain reaction (qPCR) and general PCR
Total RNA was isolated from cells using RNeasy Mini kit (QIAGEN). Reverse transcription polymerase chain reaction (RT-PCR) was performed using SuperScript First Strand Synthesis System for RT-PCR (Life Technologies). Real-time PCR was performed using SYBR Green system on a Light Cycler 480 instrument (Roche Diagnostics). Target gene expression was calculated by the ΔΔCt method and normalized to 18S ribosomal RNA expression levels. General PCR performed using PrimeSTAR Max DNA polymerase (Clontech). The following primer sequences were used: MCL1 forward; 5′-GAGGGCGACTTTTGGCTAC-3;, reverse; 5;-GTACCCGTCCAGCTCCTCTT-3;, FGFR1 forward; 5′-TGAAGATGATCGGGAAGCAT-3′, reverse; 5;-GGCCGTTGGTTGTCTTTTT-3;, FGFR2 forward; 5′-GATGGTGCGGAAGATTTTGT-3′, reverse; 5′-CCAGGTGGTACGTGTGATTG-3′, FGFR3 forward; 5′-TGGGCTTCTTCCTGTTCATC-3′, reverse; 5′-TCAGTGGCATCGTCTTTCAG-3′, FGFR4 forward; 5′-GTGCTGGTGACTGAGGACAA-3′, reverse; 5′-AGACAGAATCGCTGGAGGAG-3′, CK-19 forward; 5′-TTTGTGTCCTCGTCCTCCTC-3′, reverse; 5′-AGAGCCTGTTCCGTCTCAAA-3′
Mitochondria isolation and trypsin treatment
Mitochondria were isolated from KMCH and KMBC cells using a Mitochondria Isolation Kit for Cultured Cells (Thermo Scientific) according to the manufacturer’s instructions. Following isolation, mitochondria were incubated with 50 μg/mL trypsin for 20 minutes on ice. Trypsin was then inhibited by adding soybean trypsin inhibitor (200 μg/mL) for 5 minutes on ice.
Plasmids, expression constructs and generation of mutants
The plasmid encoding S peptide-tagged MCL1 was subcloned into pcDNA as previously described [22]. To generate outer mitochondrial membrane MCL1 (MCL1OM), arginine 6 was substituted with alanine utilizing Q5 site-directed Mutagenesis kit (New England BioLabs). The N-terminal 86 amino acids of MCL1 were deleted by PCR using PrimeSTAR Max DNA polymerase (Clontech), and fused to the N-terminal 58 amino acids Neurospora crassa ATP-synthase encoded gBlocks (Integrated DNA Technologies) using Gibson Assembly Master Mix (New England BioLabs) to generate matrix MCL1 (MCL1Matrix) as described by Perciavalle et al. [14]. All MCL1 mutants were subcloned into pLenti plasmid (OriGene). To generate stable transfectants, HEK 293T cells were transfected with pCMV-VSV-G (Addgene), pCMV-dR8.2 dvpr (Addgene), and the lentiviral MCL1OM, MCL1Matrix or empty vector, respectively, using FuGene HD reagent (Promega). After 48 and 72 hours, supernatant was passed through a 0.45-μm pore filter, and polybrene (Sigma Aldrich) was added to a final concentration of 8 μg/mL. Target KMCH cells were incubated with lentivirus-containing medium from the HEK 293T cells for 24 hours before medium was replaced with fresh complete medium. Cells were cultured for an additional 3 days in lentivirus-free media, and then placed into selection medium containing 5 μg/mL puromycin. Expression of MCL1 mutants was confirmed by immunoblot analysis.
ATP assay
Cells were plated in 24-well plates (2 × 104 cells per well) and LY2874455 was added for 6 hours. Cellular ATP levels in KMCH cells were measured by a commercial fluorometric assay (BioVision, Milpitas, CA) according to manufacturer’s instructions. Fluorescence was measured using a multi-detection microplate reader (BioTek). ATP concentration was determined by comparing values to a standard curve.
Extracellular flux analysis of overall oxygen consumption rates (OCR)
Mitochondrial respiration was assessed using an XF24 extracellular flux analyzer (Seahorse Biosciences) as described by Wu et al. [23]. Briefly, MCL1OM and MCL1Matrix expressing cells were seeded in XF-24 cell culture plates (Seahorse Bioscience) at 7 × 104 cells/well and incubated under standard conditions for 24 hours. Beginning 6 hours before the measurement, cells were treated with or without LY2874455. Thereafter, the cells were washed with XF Base DMEM (Seahorse Bioscience) containing 25 mM glucose and 10 mM pyruvate. The overall oxygen consumption rate was measured sequentially after addition of 1 μM oligomycin, 0.2 μM FCCP and 1 μM rotenone plus 1 μM antimycin A. Experiments were conducted using five replicates for each condition and repeated in three independent experiments. At the end of the experiments, cells were harvested and protein measured by the Bradford protein assay. OCR values were normalized to the protein content of each well.
Mitochondrial membrane potential Δψm) measurements
Cells were plated on BIOCOAT 96 well plates (Falcon) and treated with vehicle or 5 μM LY2874455 for 6 hours. After tetramethylrhodamine methyl ester (TMRM) (Invitrogen) was added to cells at a final concentration of 100 nM, incubation was continued for 20 minutes. Fluorescence was measured at Excitation/Emission wave lengths of 560 nm/590 nm on a BIOTEK Synergy fluorescent plate reader. Then, 50 μM of 1799 (Dupont), a non-fluorescent mitochondrial uncoupler, was added and incubation was continued for an additional 20 minutes. Mitochondrial membrane (Δψm) values were expressed as a percentage of the same time vehicle control as follows: Δψm = [(experimental fluorescence - uncoupled fluorescence)/(control fluorescence - uncoupled fluorescence)] × 100.
Reactive oxygen species (ROS) detection assay
Cells were plated in 96-well black plates with optically clear bottom (Perkin Elmer) and 5 μM LY and 10 μM dihydroethidium were added for 6 hours at 37°C. Following this incubation period, cells were washed with Fluorobrite DMEM (Gibco) and fluorescence intensity was assessed using Celigo Imaging Cytometer (Nexcelom Bioscience, Lawrence, MA).
Patient-derived xenograft (PDX) model
This study was approved by the Mayo Clinic Ethics Committee (IRB#66-06) and the Institutional Animal Care and Use Committee, and all human subjects gave written informed consent for participation in medical research. Patient-derived xenografts, from an individual patient with intrahepatic CCA, were implanted in 6- to 8-week old male NOD/SCID mice (Charles River, strain code: 394) as previously described [15, 24]. Briefly, after mice were anesthetized with 1.5–3 % isoflurane, an incision was made in the flank area in the middle of the thigh line and enlarged to 0.5 cm. Following implantation of a tissue fragment (1 mm × 1 mm × 1 mm) into a pocket created under the flank fat pad, the incision was sealed with Vetbond™ (3M). Once the diameter of the tumors reached 1 cm, mice were randomly divided into 2 groups (Vehicle: n=5, LY: n=7) and treated with either vehicle (2% sodium carboxymethyl cellulose) or LY2874455 (3 mg/kg) via oral gavage twice daily for 7 days as described previously [25]. At the end of the treatment, all mice were sacrificed and tumor tissue was obtained for further studies. Mice were housed on a 12 hours light-dark cycle and fed a normal chow diet. All animal experiments were performed in accordance with a protocol approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Biochemical analysis
Serum albumin, alkaline phosphatase (ALP), alanine aminotransferase (ALT) and total bilirubin were measured by a commercially available veterinary chemistry analyzer (VetScan2, Abaxis Veterinary Diagnostics).
Immunohistochemistry
PDX tumors were embedded in paraffin or Tissue-Tek® O.C.T. Compound (Sakura® Finetek). Frozen sections were fixed in 3.2% paraformaldehyde for 20 minutes and endogenous peroxidase was quenched using 0.3% H2O2 in methanol. After permeabilization using Trion X-100, sections were subsequently blocked for 1 hour at room temperature with phosphate-buffered saline (PBS) containing 5% bovine serum albumin (BSA) and incubated with primary antibody in 5% BSA overnight at 4°C. Bound antibody was detected using a Dako EnVision+ system containing horseradish peroxidase conjugated secondary antibody and diaminobenzidine (Vector Laboratories). Sections were counterstained with hematoxylin prior to mounting. Primary antibodies and their dilutions were as follows: pFRS2 (AF5126, 1:50) from R&D Systems; and SOX9 (82630S, 1:600) and MCL1 (39224S, 1:150) from Cell Signaling Technology. Quantification of positive cells was performed using ImageJ software.
Statistical analysis
Data are represented as mean ± S.E. from at least three independent experiments. To assess statistical significance of differences, the two tailed t-test (for two groups) or one-way analysis of variance (ANOVA) followed by Dunnett’s test (for multiple groups) was performed.
RESULTS
The pan-FGFR inhibitor, LY2874455, induces necrosis in human cholangiocarcinoma cells
The pan-FGFR inhibitor LY2874455 (hereafter referred to as LY) [25] was employed for these studies. To confirm that LY inhibits FGFR signaling, we examined phosphorylation of the common FGFR receptor substrate, FRS2 (fibroblast growth factor receptor substrate 2), in KMCH and KMBC cells. As indicated in Fig. 1A, FRS2, was phosphorylated in both CCA cell lines at baseline; and incubation with LY blocked this phosphorylation, indicating inhibition of FGFR signaling cascades.
Fig. 1. Fibroblast growth factor receptor (FGFR) inhibition induces necrosis in CCA cells.

(A) KMCH and KMBC cells were treated with vehicle or FGFR inhibitor LY28744055 (LY, 5 μM) for 24 hours in DMEM with 2% FBS. Cell lysates were subjected to immunoblot analysis of p-FRS2. (B) Cell death rate of KMCH and KMBC cells assessed by MTS and SRB assay after a 24 h treatment with LY (5 μM). (C) KMCH and KMBC cells were treated for 24 h as indicated and cell death rate was assessed by SRB assay. (D) Immunoblot analysis for PARP in cells treated with LY (5 μM) or obatoclax (5 μM) plus TRAIL (20 ng/mL) for 24 h. (E) Cell death rate of wild type (WT) and BAX−/−/BAK−/− cells treated with LY (5 μM) or obatoclax (5 μM) plus TRAIL (20 ng/mL) for 24 h. (F) Flow cytometry analysis of propidium iodide (PI) and annexin V staining in KMCH cells treated with obatoclax (5 μM) plus TRAIL (20 ng/mL) or LY (5 μM). Numbers indicate the percentage of cells (mean ± SEM, n=3) in each fraction. Bar graphs represent mean ± SEM for at least three independent experiments. ns, non-significant. ***p < 0.001 or as indicated. Statistical significance was determined using the two-tailed t test or one-way ANOVA followed by Dunnett’s test. (G) Necroptosis induction in KMCH and KMBC cells. Cells were treated with ZVAD-fmk (20 μM), TNF-α (20 ng/mL) and TL-32711 (1 μM) with or without necrostatin-1 (20 μM). HT29 cells were used as a positive control for necroptosis signaling.
Further studies demonstrated that LY also induces cell death in both the KMCH and KMBC cell lines (Fig. 1B). Interestingly, this LY-induced death process was not blocked by necrostatin-1, a receptor-interacting protein (RIP) 1 kinase inhibitor, which blocks necroptosis [19], nor by the potent caspase inhibitor Q-VD-OPh [26] (Fig. 1C).
To further characterize the LY-induced death process, we compared it to cell death induced by obatoclax plus TRAIL, a combination that induces apoptosis in CCA cells as previously reported by us [20]. Cleavage of poly (ADP-ribose) polymerase (PARP), which is mediated by active caspases [27, 28], was detected in the presence of obatoclax plus TRAIL but not LY (Fig. 1D). Likewise, deletion of genes encoding BAX and BAK, pro-apoptotic effector proteins necessary for activation of the mitochondrial apoptotic pathway, using CRISPR/Cas9 [29] diminished cell death induced by obatoclax plus TRAIL but not LY-induced cell death (Fig. 1E). Flow cytometry after staining with annexin V plus PI revealed that obatoclax plus TRAIL treatment increased annexin V single positive population (Fig. 1F), indicative of apoptosis, whereas LY treatment significantly increased the annexin V and PI dual positive cell population, suggesting plasma membrane rupture.
Given that a necrotic mode of cell death was occurring in LY-treated CCA cells, we next sought to examine our model for potential necroptosis signaling processes. In necroptosis, formation of the necrosome, a complex consisting of RIP1, RIP3 and Fas-associated protein, results in phosphorylation of mixed lineage kinase domain-like protein (MLKL) resulting in cell membrane permeabilization [30, 31]. To induce necroptosis, cells were treated with ZVAD-fmk, TNF-α, and TL-32711 with or without necrostatin-1. However, expression of MLKL and RIP3 was absent in the KMCH and KMBC cells whereas both proteins were readily identified in the HT29 cells, a positive control for necroptosis (Fig. 1G). These results suggest that necroptosis was not a mode of cell death in LY-treated CCA cells. Taken together, these data suggest FGFR inhibition mediates predominantly a non-apoptotic cell death most compatible with necrosis in human CCA cells. Finally, LY induced cell death was also assessed in a malignant hepatocyte derived cell line, HepG2 cells, and pancreatic ductal adenocarcinoma cells (PDAC). While HepG2 cells were resistant to LY-mediated cell death, the PDAC cell lines, PANC1, BxPC3, DanG and Panc 04.03 cells were susceptible to LY triggered cell death (Supplemental Fig. 1A).
FGFR inhibition depletes both outer mitochondrial membrane and matrix MCL1 species in human CCA cells
Consistent with our earlier report that the pan-FGFR inhibitor BGJ398 resulted in depletion of total cellular MCL1 in human CCA cells due to downregulation of MCL1 mRNA [15], we observed that LY treatment also resulted in loss of MCL1 mRNA and protein (Fig. 2). Importantly, when we examined both the full length and truncated MCL1 species, both of these proteins decreased in a dose-(Fig. 2A) and time-dependent (Fig. 2B) manner.
Fig. 2. FGFR inhibition depletes MCL1 expression in a dose- and time-dependent manner leading to cell death.

(A) After KMCH and KMBC cells were treated with increasing concentrations of LY2874455 (LY) for 24 h, MCL1 mRNA and mitochondrial protein levels were assessed by qPCR and western blotting, respectively. HSP60 was used as a loading control for mitochondrial lysate. (B) KMCH and KMBC cells were treated with LY (5μM) for 4, 8, and 24 hours and MCL1 mRNA and mitochondrial protein levels were examined. Bar graphs represent mean ± SEM for at least three independent experiments. ***p < 0.001 compared to vehicle treatment. (C) KMCH cells were stably transfected with Cas9 and doxycycline-inducible sgRNA targeting MCL1 gene (isgMCL1). Cells were treated with doxycycline (5 μg/mL) for several time points and cell death rate was evaluated by CellTiter-Glo luminescent assay. ***p < 0.001, **p < 0.01. Statistical significance was determined using the two-tailed t test or one-way ANOVA followed by Dunnett’s test.
To assess the impact of the decreased MCL1 species, we genetically deleted MCL1 in KMCH cells using a doxycycline-inducible sgRNA expression system [21]. Following doxycycline treatment, both MCL1 species were deleted in 24 hours (Fig. 2C, right panel). MCL1 deleted KMCH cells underwent cell death in a time-dependent manner (Fig. 2C, left panel), suggesting that loss of MCL1 species is sufficient to induce cell death.
In further experiments, we assessed the submitochondrial distribution of the two MCL1 species. Mitochondria were isolated from KMCH cells and treated with trypsin, reasoning that outer mitochondrial membrane proteins would undergo proteolytic degradation while the matrix proteins would be protected. Using the mitochondrial translocases TOM70 and TIMM50 as markers of the outer and inner mitochondrial membranes, respectively [32], this analysis demonstrated that TOM70, but not TIMM50, was susceptible to trypsin proteolysis. Consistent with a location on the outer mitochondrial membrane, the larger MCL1 species also underwent efficient proteolytic degradation, whereas the lower molecular weight species was more resistant to trypsin digestion (Fig. 3A).
Fig. 3. Identification of MCL1 species in CCA cells.

(A) Isolated mitochondria from KMCH cells were digested with 50 μg/mL trypsin for 20 min and subjected to SDS-PAGE. HSP60 was used as a loading control, and TOM70 and TIMM50 were used as markers for the mitochondrial outer and inner membrane, respectively. (B) KMCH cells were treated with cycloheximide (CHX, 100 μg/mL) for 2 and 4 h. Expression of MCL1 species was examined by western blotting.
Prior studies have suggested that full length MCL1 also undergoes more rapid cellular depletion than the matrix species in the presence of cycloheximide (i.e., the half-life of the full length protein is shorter than that of the truncated protein) [11, 12]. Consistent with these prior studies, incubation of KMCH cells with cycloheximide resulted in more rapid loss of full length MCL1 compared to the truncated species (Fig. 3B).
Collectively, these results are consistent with the presence of full length MCL1 on the outer mitochondrial membrane and a truncated MCL1 species within the mitochondrial matrix in human CCA cells. Both MCL1 species are decreased following incubation with LY, likely reflecting diminished MCL1 transcription as represented by rapid depletion of cellular MCL1 mRNA [9].
Matrix MCL1 regulates CCA cell death by modulating mitochondrial oxidative metabolism
To assess whether loss of mitochondrial outer membrane MCL1 versus matrix MCL1 contributed preferentially to LY-induced cell death, we expressed MCL1 targeted to the mitochondrial outer membrane or matrix in KMCH cells from plasmids employing the CMV promoter (Fig. 4A) and assessed the ability of the constructs to rescue the cells. MCL1 targeted to the mitochondrial matrix (MCL1Matrix) was generated by fusing the mitochondrial targeting sequence of matrix-localized ATP synthase to N-truncated MCL1, producing a mutant that resides solely within the mitochondrial matrix and is incapable of blocking apoptosis [14]. Mutagenesis of the highly conserved arginine residue of MCL1 at position 6 to alanine generated MCL1OM, which maintains its localization on the outer mitochondrial membrane [14]. LY significantly reduced endogenous MCL1 expression in mock transfected cells, but the two exogenous constructs continued to be expressed (Fig. 4B). Importantly, KMCH cells stably transfected with MCL1Matrix were resistant to cell death during LY incubation, whereas cells stably transfected with MCL1OM were not protected from cell death (Fig. 4C). In an effort to complement these data, we attempted to demonstrate MCL1 in the mitochondrial outer membrane and mitoplasts using mitochondrial subfractionation. However, after extensive attempts using a multitude of published approaches, we were unable to isolate mitoplasts without outer membrane contamination. One possible explanation for this is that unlike non-malignant cells, cancer cell mitochondria may not demonstrate high amplitude swelling in the presence of hypotonic buffers or permit separation of the two membranes. Presumably, the contact sites between the outer and inner membranes are quite abundant and “tight” in these mitochondria precluding complete separation of the two membranes.
Fig. 4. Loss of matrix MCL1 induces cell death by causing failure of mitochondrial oxidative metabolism.

(A) Schematic illustration of matrix and outer membrane (OM) MCL1 mutants. N. crassa ATP-synthase mitochondrial targeting sequence (MTS) is indicated in black. Point mutation (R6A) is indicated in white. MCL1 mutants were stably expressed in KMCH cells and used for experiments in B–F. (B) KMCH cells expressing MCL1 mutants were treated with vehicle or LY2874455 (LY, 5 μM) for 24 h. Lysates of isolated mitochondria were subjected to immunoblot analysis of MCL1. (C) Cell death rate of KMCH cells expressing MCL1 mutants treated with vehicle or LY (5 μM) for 24 h was assessed by SRB assay. (D) KMCH cells expressing MCL1 mutants were treated with vehicle or LY (5 μM) for 6 h, and cellular ATP levels were measured. (E) KMCH cells expressing MCL1 mutants were treated with vehicle or LY (5 μM) for 6 h, and oxygen consumption rate (OCR) and ATP production rate were measured using the Seahorse XF24 extracellular flux analyzer. (F) WT, BAX−/−/BAK−/−, and MCL1 mutant cells were treated with vehicle or LY (5 μM) for 6 h, and mitochondrial membrane potential (Δψm) was evaluated by TMRM (100 nM) staining. (G) Cells were treated with vehicle or LY (5 μM) for 6 hours. Reactive oxygen species (ROS) levels were assessed by dihydroethidium fluorescence intensity using Celigo Imaging Cytometer. Bar graphs represent mean ± SEM for at least three independent experiments. ns, non-significant. ***p < 0.001, **p < 0.01. Statistical significance was determined using the two-tailed t test or one-way ANOVA followed by Dunnett’s test.
Further experiments designed to investigate the mechanistic basis for this protection evaluated the impact of MCL1OM and MCL1Matrix on mitochondrial function. Cellular ATP levels, oxygen consumption rate (OCR) and ATP production rate all decreased with FGFR inhibition; however, these mitochondrial functional deficits were largely restored by expression of MCL1Matrix (Fig. 4D, 4E). In contrast, enforced expression of MCL1OM had no effect on FGFR-inhibitor induced impairment of mitochondrial oxidative function (Fig. 4D, 4E).
To provide an explanation for these findings, we also examined mitochondrial membrane potential (Δψm). Opening of the inner mitochondrial membrane permeability transition pore results in a collapse of ion gradients across this membrane, thereby impairing oxidative phosphorylation, and is a potential explanation for our observations [33]. Loss of Δψm is employed to assess opening of this pore [34]. Therefore, we initially examined Δψm during FGFR inhibition in wild type and BAX−/−BAK−/− cells, which are protected from BH3-only protein-induced mitochondrial outer membrane permeabilization, to assess if this was the cause of impaired oxidative phosphorylation. During FGFR inhibition, MCL1Matrix enforced expression conserved Δψm, while MCL1OM transfected cells underwent a reduction of Δψm (Fig. 4F). Impairment of the electron transport chain during oxidative phosphorylation results in enhanced generation of reactive oxygen species (ROS) [35, 36]. Accordingly, we observed that LY treatment resulted in a significant increase in ROS generation in CCA cells (Fig. 4G). These observations are most compatible with cell death by necrosis due to inhibition of oxidative phosphorylation, a function apparently regulated by mitochondrial matrix levels of MCL1.
FGFR plus selective MCL1 inhibition is neither antagonistic nor synergistic in regards to CCA cell death
Having gained insight into mechanisms of CCA cell death by FGFR inhibition, we next examined the possibility that inhibiting the anti-apoptotic function of MCL1 may also modulate LY cytotoxicity. In particular, we sought to determine if combining LY with an MCL1-targeting BH3 mimetic would be antagonistic, as MCL1-targeting BH3 mimetics increase MCL1 protein levels [37], or synergistic by promoting both apoptosis and necrosis. For these studies, we employed A-1210477, a selective MCL1 inhibitor that binds with high affinity to the BH3 groove on the MCL1 protein [37]. As reported previously [37], A-1210477 resulted in an increase in cellular MCL1 protein levels by stabilizing the protein at a post-translational level (Fig. 5A). However, A-1210477 did not prevent cellular loss of MCL1 by LY, consistent with the effect of LY on inhibiting MCL1 transcription (Fig. 5A). There was modest CCA cell death with A-1210477 treatment alone in KMCH and KMBC cells (Fig. 5B). In comparison, LY resulted in greater cytotoxicity. The combination of LY and A-1210477 was neither antagonistic nor synergistic in inducing CCA cell death (Fig. 5B). Hence, LY is more cytotoxic than A-1210477, suggesting that cellular loss of MCL1 is more deleterious than inhibiting its anti-apoptotic function.
Fig. 5. FGFR inhibition plus an MCL1-selective BH3 mimetic is neither antagonistic nor synergistic in regards to CCA cell death.

KMCH and KMBC cells were treated with MCL1 inhibitor A-1210477 (10 μM) and/or LY2874455 (LY, 5 μM) for 24 h. (A) Mitochondrial lysates were subjected to immunoblot analysis of MCL1 and HSP60 (loading control). (B) Cell death rate was assessed by SRB assay. Bar graphs represent mean ± SEM for at least three independent experiments. ns, non-significant. ***p < 0.001, **p < 0.01. Statistical significance was determined using one-way ANOVA followed by Dunnett’s test.
FGFR inhibition reduces tumor burden in a patient-derived xenograft (PDX) model of CCA
After CCA PDXs were implanted heterotopically and allowed to grow to ~1 cm in diameter, mice were treated for 1 week with LY. This PDX constitutively expressed FGFR 1–4 mRNA (Fig. 6A); and FRS2 (a proximate substrate of FGFR tyrosine kinase activity) was phosphorylated in the PDX (Fig. 6B), indicating active FGFR expression and signaling. Also, the dose of LY was sufficient to reduce FRS2 phosphorylation in the PDX, indicating pharmacologic FGFR inhibition at achievable drug concentrations (Fig. 6B). LY treatment was tumor suppressive with this short duration of treatment, as the tumors from LY-treated mice were smaller by weight vs. those from vehicle-treated animals (Fig. 6C). The LY-treated tumors also displayed areas of cellular necrosis by H&E staining (Fig. 6D). Moreover, tumor area, as examined by morphometric analysis of SOX9 immunohistochemistry, was correspondingly reduced in the LY-treated PDX (Fig. 6E). MCL1 expression by immunohistochemistry, albeit variable, was also decreased in the LY-vs. vehicle-treated PDXs (Fig. 6F). LY treatment was not associated with hepatobiliary toxicity as indicated by normal serum levels of aminotransferases and absence of histological damage in tissue including specific examination of cholangiocytes (Supplemental Fig. 2A and 2B). Thus, FGFR inhibition by LY was cytotoxic for CCA cells, but hepatocytes nor cholangiocytes, in vivo apparently by a process apparently similar to the in vitro observations.
Fig. 6. FGFR inhibition reduces patient-derived xenograft (PDX) tumor weight by inducing necrosis.

CCA PDX was implanted heterotopically in NOD/SCID mice. When the tumor diameter reached 1 cm, mice were randomized to receive either vehicle (Veh, n=5) or LY2874455 (LY, 3 mg/kg body weight, n=7) via oral gavage for 1 week. (A) At the end of the study, tumor RNA was extracted and RT-PCR was performed for FGFR1, 2, 3, 4 and CK19 (loading control). (B) Immunohistochemistry for p-FRS2 in tumors of vehicle and LY-treated mice. Top panel: 20× objective; bottom panel: 40× objective. Scale bar: 20 μm. (C) Tumor weight in vehicle and LY-treated mice. *p < 0.05. (D) Hematoxylin and eosin-stained tumors in vehicle and LY-treated mice. Scale bar: 20 μm. (E) Immunohistochemistry for CCA marker SOX9 in vehicle and LY-treated mice. Scale bar: 20 μm. SOX9-positive area was assessed by morphometry (20× objective). ***p < 0.001. (F) Immunohistochemistry for MCL1 in vehicle and LY-treated mice. Top panel: 20× objective; bottom panel: 40× objective. Scale bar: 20 μm. MCL1-positive area was assessed by morphometry (20× objective). ***p < 0.001. All statistical significance was determined using the two-tailed t test.
DISCUSSION
The principal findings of this study provide new mechanistic insight regarding the contribution of matrix MCL1 to cell survival in human CCA cells. Our results indicate the following when cell death is induced by FGFR inhibition: (i) the model of cell death is predominantly necrotic rather than apoptotic, and accompanied by a reduction in mitochondrial oxidative metabolism; (ii) mitochondrial oxidative metabolism and cell death are rescued by targeting MCL1 to the mitochondrial matrix, but not by targeting it to the outer mitochondrial membrane; and (iii) FGFR inhibition induces necrosis in human CCA PDX models. These observations are discussed in greater detail below.
In these studies, we employed pharmacologic FGFR inhibition as a highly relevant model to induce CCA cell death. Oncogenic FGFR signaling has been strongly implicated in CCA biology. For example, FGFR2 fusion genes have been reported by several investigators in human CCA and mediate oncogenic behavior (e.g., cell proliferation, invasion, etc.); FGFR2 inhibition appears to be therapeutic for these cancers [38, 39]. We have also reported that an FGFR and Hippo autocrine, feed-forward, oncogenic signaling pathway participates in CCA pathogenesis in the absence of the fusion aberrations [15]. FGFR inhibition is therapeutic in PDX models characterized by this feed-forward signaling pathway [15]. Given these preclinical and clinical observations, a number of FGFR inhibitors are also being developed for the treatment of human cancers, including CCA [40, 41]. Hence, examining the pathways and mode of cell death by FGFR inhibition is quite pertinent to the therapy of human CCA.
We previously reported that FGFR pharmacologic inhibition induced CCA cell death through interruption of an MCL1 regulated survival pathway [15]. Cell death was accompanied by loss of MCL1 mRNA and protein, and rescued by enforced expression of MCL1. However, neither the mode nor mechanism(s) of cell death was robustly delineated. Herein, we explored the mechanism(s) of this MCL1 regulated cell death in the context of FGFR inhibition to gain further therapeutic insight. Unexpectedly, cell death in human CCA cells by FGFR inhibition was not apoptotic despite cellular loss of MCL1. Evidence for a non-apoptotic mode of cell death included the inability to identify caspase activation and lack of cell death inhibition by a caspase inhibitor or genetically depletion of BAX and BAK. Moreover, A-1210477, a pharmacologic agent that blocks MCL1 anti-apoptotic function without decreasing protein levels, caused only modest cytotoxicity. Necroptosis is a mode of cell death mediated by RIP 1 and 3 kinases independent of caspase activation [42], and is blocked by pharmacologic inhibition of RIP 1 kinase with necrostatin-1 [19]. However, in our model, necrostatin-1 also did not prevent CCA death by FGFR inhibition. Moreover, key mediators of necroptosis, MLKL and RIP3, were not expressed in the CCA cells lines, excluding necroptosis as a mediator of cell death during LY treatment. In contrast, cell death was characterized by reductions in mitochondrial respiration, loss of cellular ATP, a decrease in mitochondrial membrane potential (ΔΨm), and increased generation of reactive oxygen species. The loss of ΔΨm occurred despite genetic deletion of BAX and BAK, indicating it is unlikely to be secondary to outer mitochondrial membrane permeabilization. The loss of ΔΨm, decrease in cellular ATP, and impaired mitochondrial respiration observed in the current studies are most consistent with cell death following formation of the inner mitochondrial membrane permeabilization transition pore [33, 43]. Opening of this transition pore is a common mechanism of cell death by necrosis. Collectively, these observations suggest a defect in mitochondrial matrix function as a cause of the cell death in CCA cells after LY treatment, likely by necrosis.
Our previous observations, coupled with the results presented above, suggest that MCL1 loss may be disrupting mitochondrial oxidative metabolism in the matrix leading to the mitochondrial permeability transition pore opening. On the other hand, MCL1 is typically viewed as a regulator of apoptosis. To reconcile these findings, we postulated that loss of the previously described N-truncated matrix species of MCL1 may play a role in LY-induced cell death. Indeed, enforced expression of full length MCL1 targeted to the outer mitochondrial membrane failed to block LY-induced repression of mitochondrial oxidative metabolism and cell death. In contrast, enforced expression of a N-terminus truncated form of MCL1 to the mitochondrial matrix rescued adverse effects of LY on mitochondrial oxidative metabolism and cell death. These observations are consistent with the known effects of reducing matrix MCL1 species causing reduced protein expression of Cox 1 and Cox 2 subunits, alterations in the large supercomplexes containing complexes I, III and IV, and efficient assembly of F1F0-ATP synthase oligomers [14]. These results suggest that MCL1 has multiple survival functions and can prevent both apoptosis and necrosis, albeit by distinct mechanisms.
Previous studies have demonstrated that BCL2, which is widely viewed as a regulator of apoptosis, can regulate necrosis under certain circumstances [44, 45]. To our knowledge, the present study is the first to implicate MCL1 in regulation of anticancer drug-induced necrotic cell death. The current results cannot be simply attributed to inhibition of MCL1 expression by LY2874455, as similar effects were observed with BGJ398 in our prior study [15], namely cellular depletion of MCL1, inhibition of cellular oxygen consumption rates and loss of cellular ATP. This mode and mechanism of cell death secondary to MCL1 depletion is potentially important. Cell death by necrosis would not only bypass intrinsic anti-apoptotic cellular resistance mechanisms, but may also induce a strong immunologic anti-tumor response, a desired effect of anti-cancer therapy [46]. Indeed, necrosis elicits an inflammatory response and can lead to an immunogenic cell death, and as such may be synergistic with immunotherapy paradigms [46, 47].
Supplementary Material
Lay summary.
Herein, we report that therapeutic inhibition of a cell receptor expressed by bile duct cancer cells results in loss of a critical survival protein termed MCL1. Cellular depletion of MCL1 results in cell death of the cancer cells by a process characterized by cell rupture. Cell death by this process can stimulate the immune system and has implications for combination therapy employing receptor inhibition with immunotherapy.
Highlights.
The FGFR inhibitor, LY2874455, unexpectedly causes cell necrosis and not apoptosis in human cholangiocarcinoma cells.
LY2874455 treatment down-regulates expression of the BCL-2 family member MCL1
Cellular depletion of MCL1 within the mitochondrial matrix impairs mitochondrial respiration and mitochondrial membrane potential causing necrosis.
In a patient-derived xenograft model, LY2874455 treatment induces tumor reduction associated with loss of MCL1 expression and cell necrosis.
These data inform use of FGFR inhibitors for the treatment of human cholangiocarcinoma.
Acknowledgments
Financial support: This work was supported by NIH grants DK59427 (to G.J.G.), CA166741 (to S.H.K.), the NIDDK-funded Optical Microscopy Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), and the Mayo Clinic. Support was also provided to P. Hirsova by MH CZ - DRO (UHHK, 00179906) and Edward C. Kendall Research Fellowship Award, and to S. Rizvi by the Cholangiocarcinoma Foundation and a Pilot-Feasibility Award by the Center for Cell Signaling in Gastroenterology (P30DK084567).
We would like to thank Dr. Marco Herold (The Walter and Eliza Hall Institute of Medical Research, Victoria, Australia) for providing FgH1tMCL1 plasmids. We also thank Ms. Courtney Hoover for excellent secretarial support.
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest with the contents of this article.
Author Contributions: AK and GJG coordinated and designed the study and prepared manuscript and figures. AK designed, performed and analyzed experiments in Fig. 1, 2, 3, 4, 5 and 6. PH designed, performed, and analyzed experiments in Fig. 4 and 6. PH also contributed to proofreading. SFB performed and analyzed experiments in Fig. 1, 4, 5 and 6. SR assisted in the experimental design, and writing and revising the manuscript. MCH and MJT assisted and performed experiments in Fig. 6 involving the PDX model. SHK assisted in designing the experiments in Fig. 1 and 5 and contributed to the writing of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383:2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. 2013;145:1215–1229. doi: 10.1053/j.gastro.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 5.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Churi CR, Shroff R, Wang Y, Rashid A, Kang HC, Weatherly J, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One. 2014;9:e115383. doi: 10.1371/journal.pone.0115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ross JS, Wang K, Gay L, Al-Rohil R, Rand JV, Jones DM, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist. 2014;19:235–242. doi: 10.1634/theoncologist.2013-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, Farrugia DJ, et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer research. 2004;64:3517–3524. doi: 10.1158/0008-5472.CAN-03-2770. [DOI] [PubMed] [Google Scholar]
- 9.Gores GJ, Kaufmann SH. Selectively targeting Mcl-1 for the treatment of acute myelogenous leukemia and solid tumors. Genes Dev. 2012;26:305–311. doi: 10.1101/gad.186189.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Juin P, Geneste O, Gautier F, Depil S, Campone M. Decoding and unlocking the BCL-2 dependency of cancer cells. Nature reviews Cancer. 2013;13:455–465. doi: 10.1038/nrc3538. [DOI] [PubMed] [Google Scholar]
- 11.De Biasio A, Vrana JA, Zhou P, Qian L, Bieszczad CK, Braley KE, et al. N-terminal truncation of antiapoptotic MCL1, but not G2/M-induced phosphorylation, is associated with stabilization and abundant expression in tumor cells. The Journal of biological chemistry. 2007;282:23919–23936. doi: 10.1074/jbc.M700938200. [DOI] [PubMed] [Google Scholar]
- 12.Warr MR, Mills JR, Nguyen M, Lemaire-Ewing S, Baardsnes J, Sun KL, et al. Mitochondriondependent N-terminal processing of outer membrane Mcl-1 protein removes an essential Mule/Lasu1 protein-binding site. The Journal of biological chemistry. 2011;286:25098–25107. doi: 10.1074/jbc.M111.218321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang CR, Yang-Yen HF. The fast-mobility isoform of mouse Mcl-1 is a mitochondrial matrixlocalized protein with attenuated anti-apoptotic activity. FEBS letters. 2010;584:3323–3330. doi: 10.1016/j.febslet.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nature cell biology. 2012;14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rizvi S, Yamada D, Hirsova P, Bronk SF, Werneburg NW, Krishnan A, et al. A Hippo and Fibroblast Growth Factor Receptor Autocrine Pathway in Cholangiocarcinoma. The Journal of biological chemistry. 2016;291:8031–8047. doi: 10.1074/jbc.M115.698472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43:S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 17.Yano H, Maruiwa M, Iemura A, Mizoguchi A, Kojiro M. Establishment and characterization of a new human extrahepatic bile duct carcinoma cell line (KMBC) Cancer. 1992;69:1664–1673. doi: 10.1002/1097-0142(19920401)69:7<1664::aid-cncr2820690705>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 18.Murakami T, Yano H, Maruiwa M, Sugihara S, Kojiro M. Establishment and characterization of a human combined hepatocholangiocarcinoma cell line and its heterologous transplantation in nude mice. Hepatology. 1987;7:551–556. doi: 10.1002/hep.1840070322. [DOI] [PubMed] [Google Scholar]
- 19.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature chemical biology. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 20.Mott JL, Bronk SF, Mesa RA, Kaufmann SH, Gores GJ. BH3-only protein mimetic obatoclax sensitizes cholangiocarcinoma cells to Apo2L/TRAIL-induced apoptosis. Mol Cancer Ther. 2008;7:2339–2347. doi: 10.1158/1535-7163.MCT-08-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, et al. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell reports. 2015;10:1422–1432. doi: 10.1016/j.celrep.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi S, Lee SH, Meng XW, Mott JL, Bronk SF, Werneburg NW, et al. Serine 64 phosphorylation enhances the antiapoptotic function of Mcl-1. The Journal of biological chemistry. 2007;282:18407–18417. doi: 10.1074/jbc.M610010200. [DOI] [PubMed] [Google Scholar]
- 23.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. American journal of physiology Cell physiology. 2007;292:C125–136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 24.Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc. 2009;4:1670–1680. doi: 10.1038/nprot.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao G, Li WY, Chen D, Henry JR, Li HY, Chen Z, et al. A novel, selective inhibitor of fibroblast growth factor receptors that shows a potent broad spectrum of antitumor activity in several tumor xenograft models. Mol Cancer Ther. 2011;10:2200–2210. doi: 10.1158/1535-7163.MCT-11-0306. [DOI] [PubMed] [Google Scholar]
- 26.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8:345–352. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- 27.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADPribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 28.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 29.O’Neill KL, Huang K, Zhang J, Chen Y, Luo X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes & development. 2016;30:973–988. doi: 10.1101/gad.276725.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Molecular cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- 33.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiological reviews. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 34.Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. Journal of molecular and cellular cardiology. 2015;78:100–106. doi: 10.1016/j.yjmcc.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan MJ, Kim YS, Liu ZG. TNFalpha and reactive oxygen species in necrotic cell death. Cell Res. 2008;18:343–349. doi: 10.1038/cr.2008.31. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. doi: 10.1038/ncomms14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, et al. Potent and selective smallmolecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax) Cell death & disease. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer discovery. 2013;3:636–647. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sia D, Losic B, Moeini A, Cabellos L, Hao K, Revill K, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nature communications. 2015;6:6087. doi: 10.1038/ncomms7087. [DOI] [PubMed] [Google Scholar]
- 40.Rizvi S, Gores GJ. Emerging Molecular Therapeutic Targets for Cholangiocarcinoma. Journal of hepatology. 2017 doi: 10.1016/j.jhep.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katoh M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review) International journal of molecular medicine. 2016;38:3–15. doi: 10.3892/ijmm.2016.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nature reviews Molecular cell biology. 2017;18:127–136. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 43.Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochemical Society transactions. 2006;34:232–237. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- 44.Kane DJ, Ord T, Anton R, Bredesen DE. Expression of bcl-2 inhibits necrotic neural cell death. J Neurosci Res. 1995;40:269–275. doi: 10.1002/jnr.490400216. [DOI] [PubMed] [Google Scholar]
- 45.Tsujimoto Y, Shimizu S, Eguchi Y, Kamiike W, Matsuda H. Bcl-2 and Bcl-xL block apoptosis as well as necrosis: possible involvement of common mediators in apoptotic and necrotic signal transduction pathways. Leukemia. 1997;11(Suppl 3):380–382. [PubMed] [Google Scholar]
- 46.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 47.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annual review of immunology. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.