

Abstract
The aryl hydrocarbon receptor (AHR) is a ligand-activated signaling molecule which involves in diverse biological functions ranging from cancer metastasis to immune regulation. This receptor forms a cytoplasmic complex with Hsp90, p23, and XAP2. We have previously reported that down-regulation of p23 triggers degradation of the AHR protein, uncovering a potentially dynamic event which controls the cellular AHR levels without ligand treatment. Here we investigate the underlying mechanisms for this p23 effect using wild-type HeLa and the p23 knockdown HeLa cells. Reduction of the Hsp90 and XAP2 contents, however, did not affect the AHR protein levels, implying that this p23 effect on AHR is more than just alteration of the cytoplasmic complex dynamics. Association of p23 with Hsp90 is not important for the modulation of the AHR levels since exogenous expression of p23 mutants with modest Hsp90-binding affinity effectively restored the AHR message and protein levels. The protein folding property of p23 which resides at the terminal 50-amino acid region is not involved for this p23 effect. Results from our interaction study using the affinity purified thioredoxin fusion proteins and GST fusion proteins showed that p23 directly interacts with AHR and the interaction surface lies within AHR amino acid 1 to 216 and p23 amino acid 1 to 110. Down-regulation of the p23 protein content promotes the ubiquitination of AHR, indicating that p23 protects AHR from the ubiquitin-meditated protein degradation.
Keywords: aryl hydrocarbon receptor, AHR, p23, protein degradation, Hsp90, ARNT
Graphical Abstract
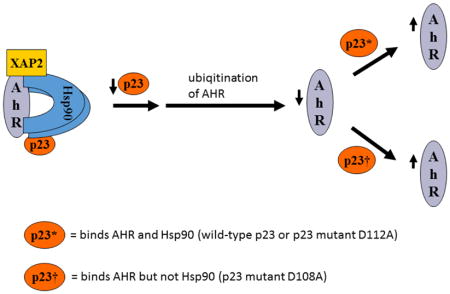
1. Introduction
The aryl hydrocarbon receptor (AHR) is best known as a ligand-activated transcription factor: Upon ligand binding, the AHR protein complex, which is consisted of AHR, Hsp90, XAP2, and p23 in the cytoplasm, translocates into the nucleus and then dissociated upon formation of the AHR/ARNT heterodimer. This heterodimer is responsible for recruitment of coactivators locally to remodel chromatin structure, allowing transcription of its target genes, notably the xenobiotic metabolizing enzymes [1, 2]. In addition to regulation of drug metabolism, AHR is involved in diverse cellular events – organ development [3, 4], glucose homeostasis [5], immune responses [6], stem cell differentiation [7], and cancer cell invasion and metastasis [8]. Although endogenous ligands of AHR have been identified, which are FICZ, kynurenine, and other tryptophan metabolites [9, 10], the precise mechanism to explain many AHR functions may very well go beyond its well established cell signaling mechanism and are largely unknown. Some of its action is peculiar and complex: different ligands of AHR can cause completely opposite immune responses [6] and AHR may promote or suppress cancer cell invasion and metastasis, depending on the cell types [11–13].
In addition to be part of the AHR cytoplasmic complex, p23 is present as a co-chaperone in a number of Hsp90 client complexes – namely progesterone receptor [14], estrogen receptor [15], androgen receptor [16], and telomerase [17]. Interference of the Hsp90 and p23 interaction has been reported to be a viable strategy to suppress cancer cell growth and metastasis [18, 19]. And it appears that most, if not all, of the p23 function involves Hsp90.
Many aggressive tumors appear to overexpress AHR [10, 20] whereas increased AHR levels in lung tumor, on the other hand, suppresses epidermal to mesenchymal transition [8]. It would be beneficial to understand how the AHR protein levels can be maintained from a perspective of honing in AHR as a potential drug target. However, we currently have very limited knowledge with regards to mechanisms which regulate the cellular AHR levels. The best known mechanisms that trigger the AHR proteasomal degradation are mediated through binding of a ligand [21] and treatment with an Hsp90 inhibitor such as geldanamycin [22]. In addition, we have reported that down-regulation of p23 causes degradation of the AHR protein in various mouse and human cell lines [23]. Here we present data supporting that this p23 effect is not merely changing the dynamics of the AHR cytoplasmic complex. This p23-mediated AHR protein degradation is not Hsp90-dependent and appears to be mediated through ubiquitination.
2. Materials and methods
2.1. Reagents
3-MC and cell culture media were purchased from Sigma (St. Louis, MO). MG132 was purchased from Cayman Chemical (Ann Arbor, MI). Cell culture reagents, if not specified, were purchased from Invitrogen (Carlsbad, CA). All other chemicals, if not specified, were purchased from Sigma (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA). Oligonucleotides were purchased from Invitrogen (Carlsbad, CA). The DRE oligonucleotides conjugated with IRDye 680 (OL439, IRD680-TCGAGTAGATCACGCAATGGGCCCAGC; OL440, IRD680-TCGAGCTGGGCCCATTGCGTGATCTAC) were purchased from Integrated DNA Technologies (San Diego, CA). Fetal bovine serum was purchased from Tissue Culture Biologicals (Tulare, CA) or Thermo Fisher Scientific (Rockford, IL). HeLa (ATCC, Manassas, Virginia) and the stable cells derived in our laboratory were grown in DMEM supplemented with 10% fetal bovine serum, 2 mM GlutaMAX-I, 10 U/ml of penicillin, and 10 mg/ml of streptomycin. All cells were maintained at 37°C and 5% CO2. EndoFectin transfection reagent was purchased from GeneCopeia (Rockville, MD). Anti-AHR rabbit IgG (SA210) was purchased from Enzo Life Sciences (Farmingdale, NY). Anti-p23 mouse IgG (JJ3) was purchased from Thermo Fisher Scientific (Rockford, IL). Anti-GAPDH rabbit IgG (G9545) and anti-thioredoxin rabbit IgG (T0803) were purchased from Sigma (St. Louis, MO). Anti-GFP rabbit IgG (sc-8334), anti-Hsp90 goat IgG (N-17), and anti-ubiquitin P4D1 mouse IgG (sc-8017) were purchased from Santa Cruz Biotechnology (Dallas, TX). Anti-β-actin mouse IgG was purchased from Ambion (Austin, TX). Anti-XAP2 (ARA9) rabbit IgG was purchased from Novus Biologicals (Littleton, CO). All secondary IgGs conjugated with IRDye 800CW or 680 were purchased from LI-COR Bioscience (Lincoln, NE). The Hsp90AA1 (Hsp90 alpha)-specific (NM_001017963) and XAP2 (AIP)-specific (NM_001302959.1) shRNA pLKO.1 plasmids were purchased from Dharmacon (Lafayette, CO). psPAX2 was a gift from Didier Trono (Addgene plasmid # 12260), pMD2.G was a gift from Didier Trono (Addgene plasmid # 12259), scramble shRNA was a gift from David Sabatini (Addgene plasmid # 1864), and pET LIC cloning vector (2A-T) was a gift from Scott Gradia (Addgene plasmid # 29665) (Addgene, Cambridge, MA). The codon humanized pGFP2-N2 plasmid was purchased from BioSignal Packard (Montreal, Canada). The pET-42a plasmid was purchased from MilliporeSigma (Burlington, MA). The pThioHis plasmid was purchased from Invitrogen (Carlsbad, CA). The full-length human Hsp90 cDNA was purchased from Sino Biological (Beijing, China). Direct-zol RNA kit was purchased from Zymo Research (Irvine, CA). MMLV high performance reverse transcriptase was purchased from Epicentre (Madison, WI). Cobalt agarose beads and IPTG were purchased from Gold Biotechnology (St. Louis, MO). Gibson assembly kit was purchased from New England BioLabs (Ipswich, MA). QuikChange Lightning site-directed mutagenesis kit was purchased from Agilent (San Diego, CA). Millipore Amicon Ultra-4 device was purchased from Thermo Fisher Scientific (Grand Island, NY). Nitrocellulose membrane for LI-COR western and 10X orange loading dye for gel shift assay were purchased from LI-COR Bioscience (Lincoln, NE). Bovine serum albumin and cycloheximide were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Glutathione magnetic beads and Protein G Dynabeads were purchased from Thermo Fisher Scientific (Rockford, IL).
2.2. Generation of Hsp90 and XAP2 stable knockdown cells
Protocols for generation of shRNA-containing lentiviruses and lentiviral infection were previously described [23]. Hsp90 knockdown HeLa cells were generated using the Hsp90 alpha-specific shRNA (#1, 3299; #2, 3300; #3, 3301; #5, 2153; #6, 8563) whereas XAP2 knockdown HeLa cells were generated using the XAP2-specific shRNA #2 (3923) or #3 (3924).
2.3. Cloning of recombinant plasmids
The GST fusion of p23 with the GST tag at the N-terminus was generated by cloning the full-length human p23 cDNA into the Nco I/PST I sites of the pET-42a plasmid. The GST fusion plasmids carrying a p23 mutant cDNA were generated by using the sense and antisense sequences (Fig. 1) to perform QuikChange site-directed mutagenesis. The GFP fusion plasmids carrying a p23 cDNA were generated by cloning the corresponding cDNA with a Kozak sequence (ACCATGGGT) into the Eco RV site of the pGFP2-N2 plasmid using Gibson assembly to generate GFP at the C-terminus of the fusion protein. The pThioHis plasmid was used to generate the thioredoxin (TH) fusions of an AHR construct. The TH553 expressing plasmid carrying the aa1-295 human AHR cDNA was generated previously [24]. The TH217-549 (aa217-549) and TH515 (aa516-849) expressing plasmids were generated by cloning the corresponding cDNA into the Stu I site of the pThioHisA plasmid using Gibson assembly. The full-length human Hsp90 cDNA was amplified and cloned into the Ssp I site of the pET-His6 plasmid using Gibson assembly. All cloned plasmids were sequenced to confirm the identity (Functional Biosciences, Madison, WI). DNA was purified for transfection using Zymopure maxiprep kit (Zymo Research, Irvine, CA) and stored at −20°C.
Fig. 1.

Wild-type human p23 coding sequence from nucleotide 301 to 360 is shown, with translation start site as +1. Amino acid (aa) 106 to 112 represents the region where point mutants W106A, W109A, D108A, D111A, and D112A were generated. Sense mutagenic primers are shown with the mutated codons bolded and the nucleotide numbers. Lines represents the length of the primers. The antisense mutagenic primers are the reverse complement of the sense primers.
2.4. Bacterial expression of GST fusions of p23, thioredoxin fusions of AHR, and histidine fusion of Hsp90
Metal-affinity purification was performed as previously described [25]. In brief, 600 ml of culture was induced with 1 mM IPTG for 5–6 h at 37°C. Metal-affinity purification using cobalt resin was performed and the affinity purified fractions were eluted with 0.5 M imidazole. All fractions were concentrated, when necessary, using the Amicon Ultra-4 device and then stored at −80°C.
2.5. Western analysis
Protocols for LI-COR Western analysis was previously described [25]. The transferred nitrocellulose membrane was blocked in 5% BSA for 30 min, followed by the primary antibody incubation in the same blocking buffer overnight. Dilutions for antibodies were as follows: 1:1,000 for p23 (JJ3), 1:5,000 for AHR (SA210), 1:5,000 for β-actin and GAPDH, 1:200 for Hsp90 (N-17), 1:5,000 for XAP2, 1:5,000 for thioredoxin (T0803), and 1:1,000 for ubiquitin (P4D1).
2.6. RT-qPCR using RNA from transiently transfected p23KD HeLa cells
When the p23KD HeLa cells were about 90% confluence in a 12-well plate, transient transfection was initiated using the EndoFectin reagent as follows: plasmid (6 μg, pGFP2-N2 empty plasmid or cloned pGFP2-N2 plasmid carrying a wild-type p23 or p23 mutant cDNA) and EndoFectin reagent (12 μL) were incubated in a final volume of 100 μL of OPTI-MEM for 20 min before being added onto a well containing 1 mL of fresh media. Media was exchanged after 24 h and cells were harvested 52 h after transfection. Direct-zol kit was used to isolate RNA and reverse transcription was performed with 1 μg of RNA, as estimated by the Fisher Nanodrop Lite (Rockford, IL), using Epicentre MMLV reverse transcriptase. Quantitative PCR was performed with 1 μL of solution after reverse transcription, 10 μL of Bio-Rad iTaq SYBR green supermix (Hercules, CA), and 1 μL (0.8 pmol) of sequence-specific primers (AHR primers are OL615, ACATCACCTACGCCAGTCGC; OL616, TCTATGCCGCTTGGAAGGAT whereas β-actin primers are OL101, CCACACTGTGCCCATCTAGG; OL102, AGGATCTTCATGAGGTAGTCAGTCAG) using a Bio-Rad CFX Connect real-time PCR machine with the following protocol: 40 cycles of 90°C for 10 s/60°C for 1 min with fluorescence readings taken at 60°C. The 2−ΔΔCq method was used to present the normalized values [26].
2.7. GST pulldown assay
For the Hsp90:p23 interaction study, a normalized amount (4 μg) of GST, GST-p23, or GST-p23 mutants was incubated with 5 μL of the affinity purified Hsp90 (20 μg) in 1 mL of the assay buffer (25 mM HEPES, pH 7.4, 100 mM KCl, 10% glycerol, 5 mM ATP, 0.01% NP-40, 20 mM sodium molybdate) at 30°C for 20 min. The sample solution was then incubated with 20 μL of the pre-equilibrated glutathione magnetic beads at 4°C for 2 h while mixing at a rotation of 42 rpm. The pellet was washed four times with the wash buffer (assay buffer plus 0.4% Tween-20) and then eluted with 30 μL of the SDS–PAGE sample buffer. The samples were subjected to 12% SDS–PAGE, followed by LI-COR Western analysis using the anti-GST or anti-Hsp90 IgG for signal detection. For the p23:AHR interaction study, a normalized amount (100 μg) of a thioredoxin fusion of an AHR construct (TH553, TH217-549 or TH515) was incubated with 3 μL of GST-p23 (7.5 μg) or GST (3.5 μg) in 200 μL of the assay buffer (25 mM HEPES, pH 7.4, 100 mM KCl, 10% glycerol, 1 mg/ml of BSA, 0.01% NP-40, 10 mM DTT) at room temperature for 30 min. BSA was included in the assay buffer to minimize non-specific protein interactions. The sample solution was then incubated with 3 μL of the pre-equilibrated glutathione magnetic beads at 4°C overnight at a rotation of 42 rpm. The pellet was washed four times with the wash buffer (assay buffer plus 0.2% Tween-20) and then eluted with 20 μL of the SDS–PAGE sample buffer. The samples were subjected to 15% SDS–PAGE, followed by Western analysis with a LI-COR CLx Odyssey system (Lincoln, NE) using the anti-thioredoxin IgG for signal detection.
2.8. Electrophoretic mobility shift assay
Samples containing AHR and ARNT (2 μL/1 μL (4 μg/1 μg) of affinity purified Pichia AHR/ARNT or 0.01 μL/1 μL (7 μg/1 μg) of TH553/affinity purified Pichia ARNT) were incubated with 5 mg of Pichia ySMD1163 extract in the presence or absence of a ligand (10 μM βNF or DMSO) at 30°C for 10 min. The final volume was adjusted to 11 μL with the HEDG buffer (25 mM HEPES, 10 mM EDTA, 1 mM DTT, 10% glycerol). In some cases, Pichia extract was replaced by p23 (1 μL of affinity purified GST-p23 (40 μg) or 6His-p23 (40 μg)). Calf thymus DNA (0.3 μg) and poly-dIdC (1.5 μg) were then added for another incubation for 10 min at room temperature. After that, samples were incubated with the IRDye 680 conjugated DRE (0.5 pmol) for 10 min at room temperature. 10X orange loading dye was then added to each sample, followed by electrophoresis on a native 5% polyacrylamide TBE gel and resolved at 185 volts for 2 h at 4°C. The sandwiched gel was analyzed directly using a LI-COR CLx Odyssey near-infrared imaging system (Lincoln, NE).
2.9. Transient Transfection for whole cell extract generation
When the p23KD HeLa cells were about 90% confluence in a 6-well plate, transient transfection was initiated using the EndoFectin reagent as follows: plasmid (4 μg, pGFP2-N2 empty plasmid or cloned pGFP2-N2 plasmid carrying the p23 or p23 mutant cDNA) and EndoFectin reagent (8 μL) were incubated in a final volume of 200 μL of complete media for 20 min before being added onto a well containing 2 mL of fresh media. Media was exchanged after 24 h and cells were harvested 72 h after transfection. The lysis buffer (25 mM HEPES, pH 7.4, 0.4 M KCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 1 mM PMSF, and 2 μg/ml of leupeptin) was used to harvest cells from a 75 cm2 flask (300 μL) or wells from a 6-well plate (50 μL/well). After three cycles of freeze/thaw, lysates were kept on ice for 30 min and then centrifuged at 14,000 g for 10 min at 4°C. The supernatants were defined as whole cell extract and were subjected to LI-COR Western analysis.
2.10. Ubiquitinated protein analysis
Wild-type HeLa and the p23KD HeLa cells were grown to 90% confluence in 100 mm culture dishes. HeLa cells were treated with DMSO, 10 μM MG132, or co-treated with 1 μM 3-MC and 10 μM MG132 for 2 h. The p23KD HeLa cells were treated with DMSO or 10 μM MG132 for 2 h. Cells were harvested and lysates were prepared using the lysis buffer (25 mM HEPES, pH 7.4, 0.15 M KCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 1 mM PMSF, 2 μg/ml of leupeptin, and 10mM N-ethylmaleimide). Samples (2 mg, from four 100 mm dishes) were subjected to immunoprecipation with the anti-AHR SA210 IgG (2 μL) for 30 min at room temperature. The pre-equilibrated Protein G Dynabeads (3 μL) were added to each sample with the assay buffer (25 mM HEPES, pH 7.4, 0.15 M NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 0.1% Tween-20, and 1 mg/mL of BSA) and the samples were incubated with a rotation of 45 rpm overnight at 4°C. The beads were then washed three times with the assay buffer plus 0.1% Tween-20, followed by LI-COR Western analysis to detect the ubiquitinated AHR using the anti-ubiquitin IgG.
2.11. Statistical analysis
GraphPad Prism 7.04 software (La Jolla, CA) was utilized for statistical analysis. The significant difference was determined by two-tailed unpaired t-test to determine the statistical significance with 95% confidence intervals with *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns, not significant (p > 0.05). In some cases, unpaired t-test using the Holm-Sidak method was used to determine statistical significance (Fig. 2B and 3B).
Fig. 2.


Down-regulation of the p23 protein decreased the AHR protein and message levels in HeLa cells. (A) Western and qPCR results showing that AHR protein and message levels were reduced in p23 knockdown (p23KD) stable HeLa cells when normalized to the wild-type (WT) HeLa cells (arbitrarily set as 1). 1 μg of RNA from HeLa or p23KD cells was used to follow the qPCR protocol described under 2.6. The percentages represent the % content remaining. (B) Western results showing the amount of AHR after cycloheximide (40 μg/ml) treatment was plotted with zero time point arbitrarily set as one. All plots (A and B) are quantified data (means ± SD, n = 3 for Western data whereas n = 5 for qPCR data). The images are representative of replicate data. Each Western lane contained 40 μg of protein.
Fig. 3.

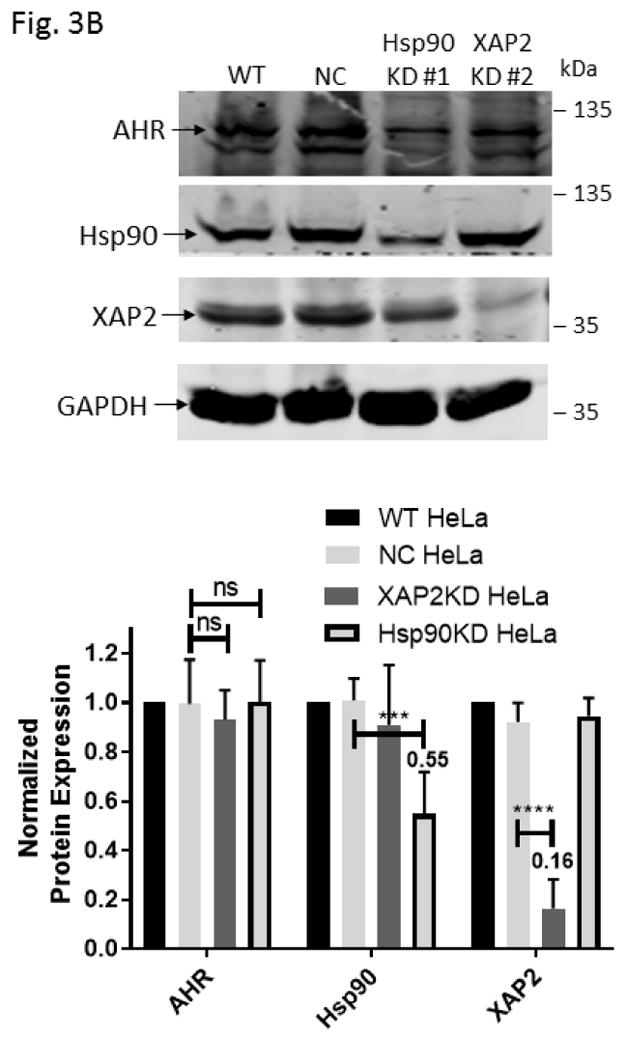
Down-regulation of the Hsp90 and XAP2 proteins did not decrease the AHR protein levels in HeLa cells. (A) AHR protein levels were determined by Western analysis in Hsp90- (left panel) and XAP2- (right panel) knockdown stable HeLa cells. Eight cell lines were examined: wild type (WT), negative control knockdown stable (NC), Hsp90 knockdown (KD) #1–3, 5, 6, and XAP2 knockdown (KD) #3. The images in left panel were performed once whereas images in right panel were repeated with different samples twice with similar results (n=3). (B) Western analysis of AHR, Hsp90, and XAP2 expression in wild-type HeLa cells (WT), scramble shRNA-treated stable HeLa cells (NC), Hsp90-knockdown stable HeLa cells using #1 shRNA (Hsp90KD), and XAP2-knockdown stable HeLa cells using #2 shRNA (XAP2KD). Images on top are representative of the replicates and the graph was plotted below (n = 6, means ± SD). Each lane of A and B contained 40 μg of whole cell extract and was normalized by GAPDH.
3. Results
3.1. Stable knockdown of p23 in HeLa cells decreases the ahr message content and increases the AHR protein degradation
Our p23 knockdown (p23KD) stable HeLa cells has 58% and 51% of the AHR message and protein contents, respectively, when the p23 protein was reduced to 52% of the wild-type content (Fig. 2A). Down-regulation of the ahr message content suggested that the reduction of the AHR protein levels could partially be mediated through pre-translational control. But more definitively, reduction of the AHR protein levels was caused by increased degradation of the AHR protein in the p23KD HeLa cells when compared to the wild-type control: the AHR protein was degraded at a faster rate in the p23 knockdown cells when degradation was measured up to 6 h after protein synthesis was inhibited by 40 μg/ml of cycloheximide (Fig. 2B). This observation of the human AHR is consistent with our published results of the mouse AHR that down-regulation of p23 facilitates the AHR protein degradation [23].
3.2. Down-regulation of the Hsp90 and XAP2 proteins does not reduce the AHR protein levels
Five Hsp90 alpha-specific shRNA plasmids were able to down-regulate Hsp90 alpha in HeLa cells to some extent (Fig. 3A, left panel). Less efficient down-regulation of Hsp90 was expected due to the importance of heat shock proteins for cell viability. Nonetheless, we observed that the Hsp90 protein levels could be reduced to as low as 55 % with Hsp90-specific shRNA #1 when compared to the HeLa cells treated with the scramble shRNA. These Hsp90 down-regulated cells exhibited no significant change of the AHR protein levels (Fig. 3B). More effective down-regulation of XAP2 was observed when compared to the Hsp90 down-regulation (Fig. 3A, right panel and Fig. 3B). Both XAP2-specific shRNAs (#2 and 3) effectively down-regulated the XAP2 cellular content. XAP2-specific shRNA #2 down-regulated 84 % of XAP2 expression while the AHR protein levels were essentially unchanged. Collectively, down-regulation of XAP2 and Hsp90 clearly caused no reduction of the AHR protein levels, suggesting that p23 has a unique mechanism of modulating the AHR protein levels, which cannot be explained by merely alteration of the AHR cytoplasmic complex.
3.3. Bacterially expressed GST-p23 promotes the formation of the AHR gel shift complex in vitro
In an effort to examine how p23 controls the AHR protein levels, we generated various GST fusions of p23 to study the p23:AHR and p23:Hsp90 interactions. To ensure that GST-p23 is functional, we tested whether it can promote the formation of the AHR/ARNT/DRE complex in vitro since our previous data clearly showed that the bacterially expressed histidine fusion of p23 (6His-p23) can effectively promote this formation [27]. Results from our gel shift assay showed that GST-p23 promoted the ligand-dependent formation of the Pichia expressed human AHR gel shift complex with 6His-p23 as the positive control for comparison (Fig. 4A, left panel). Next, we explored whether GST-p23 could promote the formation of the AHR gel shift complex caused by a truncated human AHR lacking the ligand binding domain (CΔ553, aa1-295) fused to thioredoxin (TH553). Both of the p23 fusions also formed the ligand-independent TH553/ARNT/DRE gel shift complex, further validating that GST-p23 is functional (Fig. 4A, right panel). These gel shift experiments were repeated so that the gel shift complexes could be confirmed and quantified by their band intensities (Fig. 4B). Formation of the AHR gel shift complex requires exogenous proteins [28] and thus either Pichia extract or p23 was required to form the gel shift complex.
Fig. 4.


GST-p23 promoted the formation of the AHR gel shift complex in vitro. (A) Gel shift assay images showing that GST fusion of p23 (GST-p23) and histidine fusion of p23 (6His-p23) promoted formation of the βNF-dependent AHR/ARNT/DRE complex formation (left panel, lanes 1–6). AHR and ARNT are full length human proteins expressed in Pichia. Both p23 fusions also promoted the formation of the ligand-independent AHR/ARNT/DRE complex with TH553 as the thioredoxin fusion of the human AHR aa1-295, a constitutively active AHR construct (right panel, lanes 7–15). Samples with 6His-p23 and Pichia extract were controls. Arrows indicate the two different AHR gel shift complexes. The experiment was repeated and the gel shift band intensity was quantified using a LI-COR CLx Odyssey imager with either AHR/ARNT/βNF/Pichia extract/DRE or TH553/ARNT/Pichia extract to be arbitrarily set as one. The graph was plotted in B (n = 4, means ± SD).
3.4. Control of the cellular AHR protein levels by p23 does not require Hsp90 or the p23-mediated protein folding property
We examined the requirement for the p23 control of the AHR protein levels. We suspected that this p23 effect on AHR might be mediated through its interaction with Hsp90. Therefore p23 mutants were generated to address this hypothesis. Specifically we generated five p23 point mutants as GST fusions – W106A, W109A, D108A, D111A, and D112A – where a tryptophan or aspartate residue which is potentially important for the p23:Hsp90 interaction was changed to an alanine. Results from our GST pulldown experiments revealed that all five mutants had less affinity to Hsp90 when compared to the wild-type p23 (Fig. 5A and B). Among them, D108A has the least affinity with Hsp90, which was 12% of the wild-type p23 and was statistically similar to the GST negative control (Fig. 5B and C). On the contrary, D112A has the highest affinity with Hsp90 among the five mutants, which was 41% of the wild-type p23. Next, we selected D108A and D112A with the lowest and the highest Hsp90 affinity, respectively, to restore the AHR protein levels in the p23KD HeLa cells via transient transfection as GFP fusions. We observed that both D108A and D112A could restore the AHR protein (Fig. 6A and B) and message (Fig. 6C) levels to a similar extent as the wild-type p23, suggesting that interaction with Hsp90 is not necessary for p23 to modulate the AHR levels. Although GFP-p23 appeared to be nuclear, GFP-p23 D108A and GFP-p23 D112A were both cytoplasmic and nuclear (Fig. 6D), showing that subcellular localization of p23 might not be important for the control of the AHR protein levels. In addition, a p23 construct lacking the terminal 50 amino acids (CΔ50) which are thought to be essential for protein folding could equally be effective in restoring the AHR protein levels (Fig. 6A and B), indicating that protein folding property of p23 is not required for controlling the AHR levels.
Fig. 5.

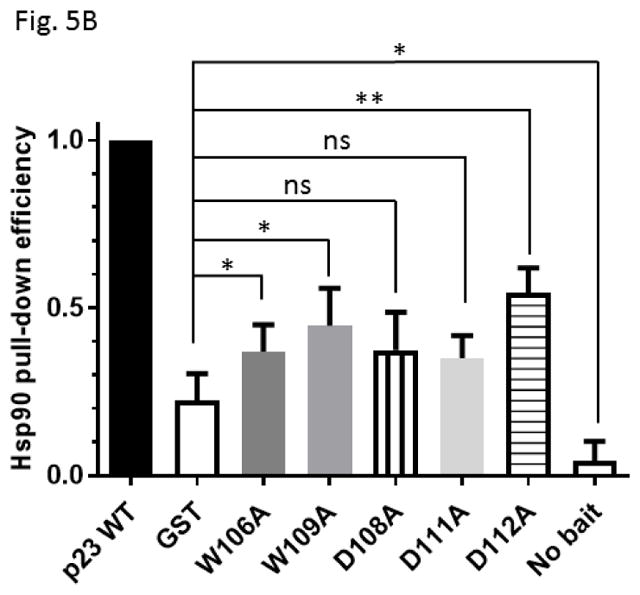
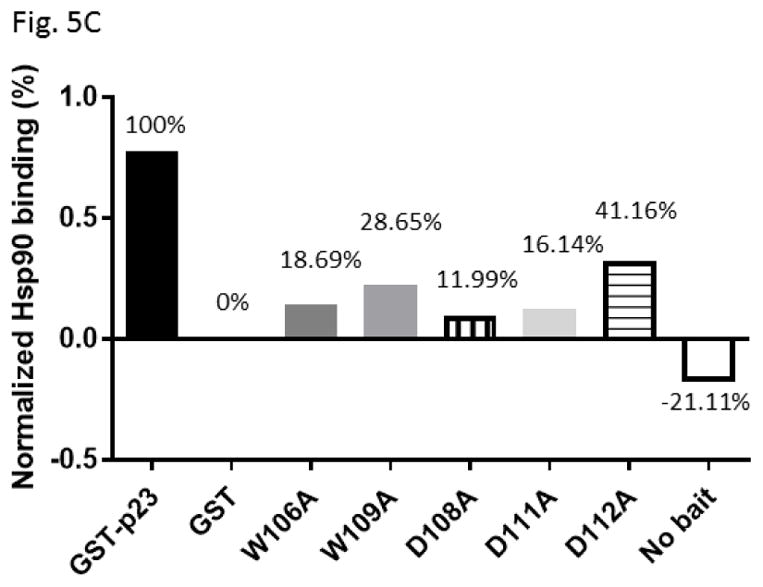
Hsp90 interacted with p23 and its mutants. (A) Western blot comparing Hsp90 co-precipitation of GST-wild-type (GST-p23) to GST and GST-p23 mutants W106A, W109A, D108A, D111A and D112A. 100% input represents the total amount of the test protein added to the sample to start the GST pulldown experiment. Unlabeled arrows within the images indicate the Hsp90 band that was co-precipitated by GST-p23 as the positive control. The image is representative of replicate data. (B) Quantitative analysis of Hsp90 co-precipitation with p23 mutants normalized to GST-p23 which was arbitrarily set as one. The plot is quantified data showing the means with error bars (mean ± SD, n=4). Statistical significance was calculated when compared to the negative control GST. (C) Normalized co-precipitation of Hsp90 by GST-p23 and mutants when the GST value was subtracted from all conditions. This plot was generated using the average values in B.
Fig. 6.

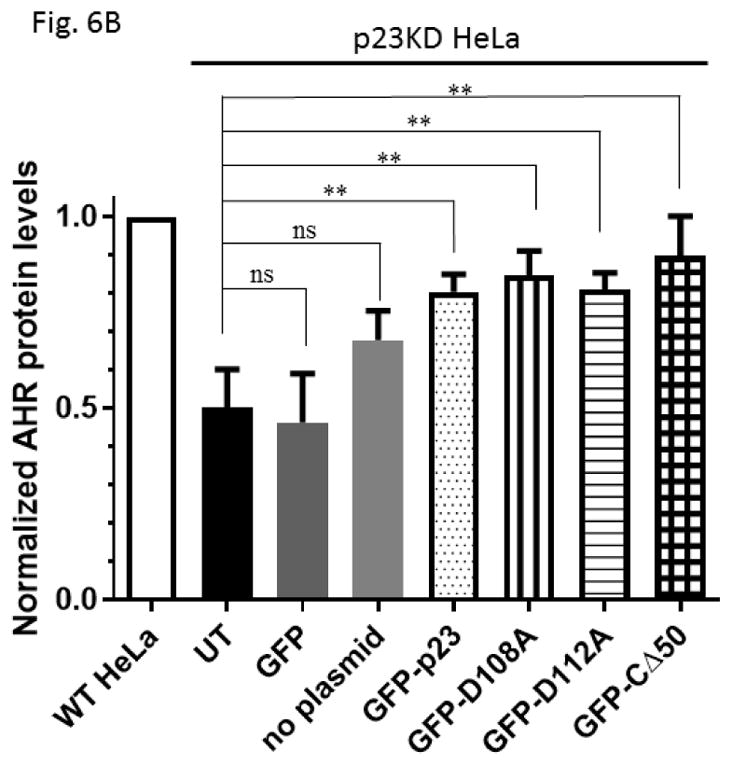
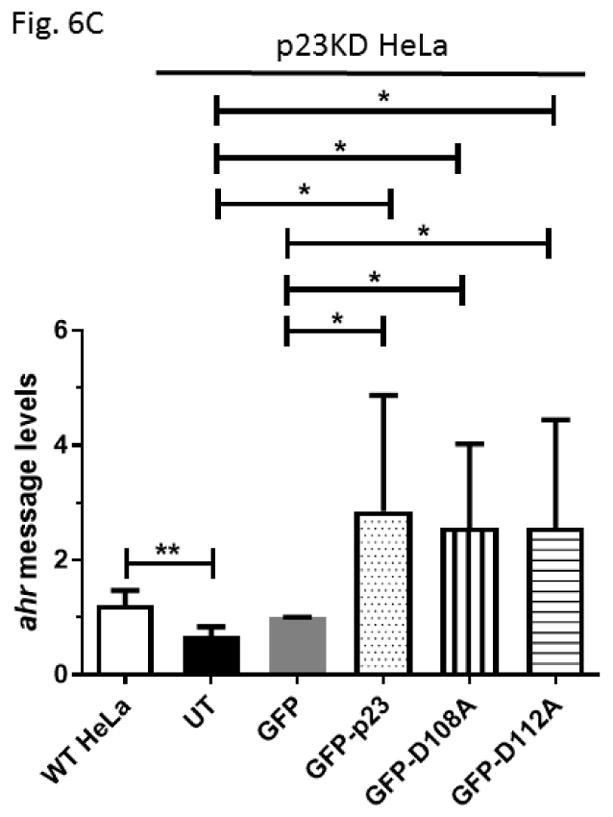

GFP fusion of wild type p23 and p23 mutants restored the AHR protein and message levels in p23 knockdown (p23KD) stable HeLa cells. (A) Western results showing GFP-p23 wild type (WT), GFP-p23 D108A, GFP-p23 D112A, and GFP-p23 CΔ50 restored the AHR protein levels in p23KD HeLa cells. UT, untransfected p23KD cells; no plasmid, p23KD cells had undergone transfection with no plasmid; GFP, p23KD transfected with the GFP control plasmid. The image is representative of triplicate data. Each lane contained 40μg of protein. (B) Quantitative analysis of western blot in A. (C) Exogenous p23 treatment also restored the ahr message levels in p23KD HeLa cells. The plots are quantified qPCR data showing the means with error bars (mean ± SD, n=5 for all except n=7 for GFP and GFP-p23 D112A groups). Statistical significance were calculated when compared to untransfected (UT) p23KD cells or p23KD cells transfected with the GFP control plasmid. (D) Florescence microscopy using a Keyence microscope showing that GFP-p23 WT was nuclear whereas p23 mutants (D108A and D112A) were cytoplasmic and nuclear.
3.5. p23 interacts with the N-terminal 216 amino acids of AHR and this interaction cannot be inhibited in the presence of ARNT
We generated various thioredoxin fusions of AHR, namely TH553 (aa1-295), TH217-549 (aa217-549), and TH515 (aa516-848), to address whether p23 would directly interact with AHR. These fusions were expressed in BL21 DE3 and then enriched using metal-affinity purification prior to the GST pulldown experiments. We observed that only TH553 was co-precipitated with GST-p23, but not when GST was used as the bait (Fig. 7A), suggesting that p23 binds to the N-terminal 216 amino acids of AHR. GST-p23 D108A, GST-p23 D112A, and GST-p23 CΔ50 all interacted with TH553 but not GST (Fig. 7B), further validating that p23, more specifically the aa1-110 p23 region (CΔ50), interacts with the N-terminal region of AHR. This aa1-110 p23 construct was sufficient to restore the AHR protein levels in the p23KD cells (Fig. 6B). Collectively, these results are consistent with the possibility that direct interaction between p23 and AHR is essential for p23 to control the AHR protein levels. Although this N-terminal AHR region overlaps with the region where AHR interacts with ARNT, five-fold increase of the Pichia ARNT amount used for gel shift assay did not seem to affect the interaction between GST-p23 and TH553 (Fig. 7C). Double that amount of Pichia ARNT to 10 μL still did not affect the p23:AHR interaction (data not shown).
Fig. 7.


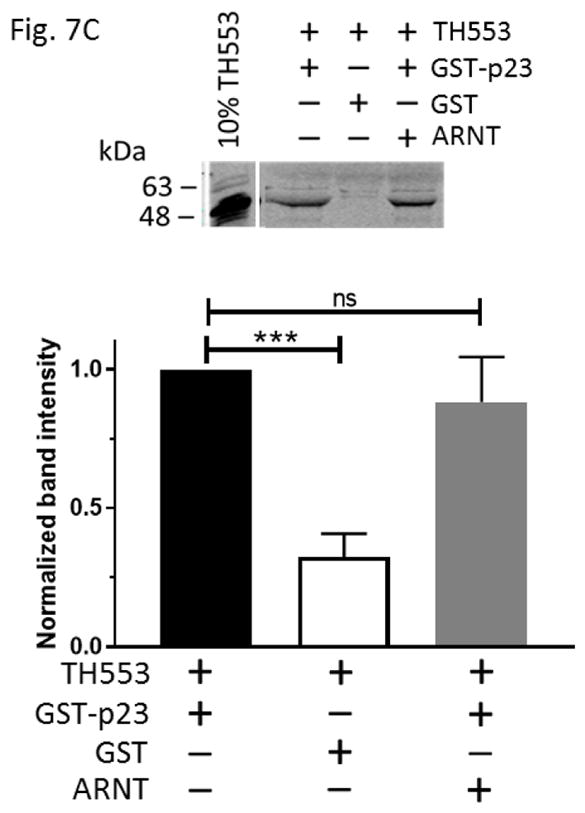
p23 interacted with AHR at the N-terminal 216 amino acids of AHR. (A) GST-p23 wild type (WT) (left panel) but not GST (right panel) co-precipitated AHR aa1-295 (TH553) but not AHR aa217-549 (TH217-549) or aa516-848 (TH515). TH, thioredoxin. Arrows indicate the location of the expected band. (B) All transfected p23 mutants were able to interact with TH553 similar to GST-p23 WT. All images are representative of triplicate data that are plotted with means ± SD (n = 3). (C) The above Western images showing that addition of affinity purified Pichia ARNT (5μL) did not interfere with the interaction between affinity purified GST-p23 (0.35 μl) and TH553 (5 μl). All images are representative of triplicate data that are plotted below with means ± SD (n=3).
3.6. Reduced p23 content causes ubiquitination of AHR without ligand treatment
We examined whether the increase of the AHR protein degradation in the p23KD HeLa cells is related to the ubiquitination of AHR. It is well established that upon ligand binding, AHR undergoes ubiquitination, which leads to degradation of the receptor via 26S proteasome. In HeLa cells, we could observe the formation of the ubiquitinated AHR only when cells were treated with 3-MC for 2 h in the presence of MG132, a proteasomal degradation inhibitor (Fig. 7). Without ligand treatment, the ubiquitinated AHR was detectable in the p23KD HeLa cells, but not in HeLa cells, suggesting that the unliganded AHR in the p23KD HeLa cells undergoes ubiquitination, leading to protein degradation. The modest ubiquitin signals observed in the DMSO-treated HeLa cells versus the p23KD cells (Fig. 8, lanes 4 and 6) were likely nonspecific background signals independent of AHR since MG132 was not added for accumulation of the ubiquitinated proteins.
Fig. 8.

Down-regulation of p23 increased ubiquitination of AHR in HeLa cells. Western data showing that ubiquitinated AHR (AHR-Ub) could be observed when wild-type HeLa (WT) cells were treated with 3-MC for 2 h in the presence of 10 μM MG132 (lane 2) and also when the p23KD HeLa cells were treated with 10 μM MG132 without ligand treatment (lanes 3 and 7). Lanes 1–3 and 4–7 are images from two separate Western experiments. Anti-ubiquitin IgG was used to detect the signals. Western of lanes 1–3 was repeated once with similar results (n = 2). Experiment to detect the ubiquitinated AHR in HeLa (WT) and p23 knockdown stable HeLa cells (p23KD) ± 10 μM MG132 (lanes 4–7) was repeated twice and was plotted below the representative images (n = 3, means ± SD).
4. Discussion
We previously reported that both the message and protein levels of the mouse AHR are reduced by approximately 50% in a p23 knockdown stable Hepa1c1c7 cells [23]. This extent of the AHR down-regulation is functionally significant: both the ligand-induced, DRE-driven EROD and reporter luciferase activities are significantly reduced in p23 knockdown Hepa1c1c7 cells when compared to the wild-type cells. In an effort to address the human relevance on this regard, we conducted experiments and proved that the same down-regulation can be observed in human (HeLa) cells. The p23 co-chaperone appears to be important in maintaining the human AHR protein levels by modulating the AHR protein degradation. This p23 effect is not caused by unrelated cellular events during stable cell generation since this reduction of the message and protein levels of AHR can be reversed with exogenous wild-type p23, confirming that this AHR reduction in human cells is p23-specific. At present, we cannot rule out the involvement of pre-translational control since the amount of the ahr transcript is consistently reduced when p23 is down-regulated.
It is conceivable that down-regulation of p23 might affect the formation or the structural dynamic of the AHR cytoplasmic complex, thereby leading to the fate of receptor degradation. This argument is supported by the observation that geldanamycin disrupts the p23:Hsp90 interaction and releases AHR from the complex for proteasomal degradation [29]. Moreover, reduction of the Hsp90 client proteins ErbB2 and HIF-1α by docosahexaenoic acid in an A549 xenograft model is thought to be mediated through disruption of the Hsp90:p23 interaction [30]. However, decreased levels of the other components of the AHR complex – namely Hsp90 and XAP2 – does not cause reduction of the AHR protein. Although it has been reported that down-regulation of XAP2 in Hepa1c1c7 cells reduced up to 30% of the protein levels of the mouse AHR (Ahb-1 allele) [31], XAP2 apparently has different roles in affecting the human AHR signaling when compared to the Ahb-1 receptor [32]. Our knockdown results with XAP2 are in line with the observation that down-regulation of XAP2 does not affect the Ahb-2 allele of AHR in C2C12 myoblasts [31]. Collectively, we believe that this p23 effect on AHR is not merely affecting the turnover of the cytoplasmic complex. However, we cannot rule out the possibility that the assembly of the AHR cytoplasmic complex is more sensitive to p23 than to Hsp90 and XAP2, since both complex disruption and p23 down-regulation cause increased in AHR ubiquitination. In addition, down-regulation of p23 might trigger degradation of the AHR protein by altering the ATP binding status of Hsp90, as in the case of geldanamycin treatment. This mechanism would favor the argument that 55 % of the wild-type Hsp90 content, as we have observed, might be sufficient to form the p23-bound AHR complex to stabilize AHR. However, the p23 mutant D108A that binds Hsp90 minimally can effectively restore the AHR protein levels, strongly supporting the irrelevance of the Hsp90 involvement in this p23-mediated AHR protein degradation. Moreover. geldanamycin causes 70–80 % degradation of the AHR protein as early as two hours after treatment [22, 29]. This time course of pronounce degradation is very similar to the ligand-triggered degradation of AHR via 26S proteasome, but is rather different from the sustained reduction of the AHR levels mediated by p23.
Although most p23 function involves Hsp90, p23 does not require Hsp90 for modulation of the AHR protein degradation. Such conclusion is supported by the findings using p23 mutants which have noticeably less binding to Hsp90. We generated a number of p23 point mutants at the region that is known to be important for Hsp90 binding. Among them, we chose the mutant D108A with the least Hsp90 binding (12%) and another mutant D112A which retained >3-fold more Hsp90 binding (41%) to study whether this interaction is important to affect the AHR protein levels. Although W106A has been reported by another laboratory to have negligible Hsp90 binding [33], we did not use this mutant since it exhibited somewhat significant binding (19%) when compared to our GST control. Both D108A and D112A mutants, however, were able to restore the AHR protein levels similar to the wild-type p23, showing that interaction with Hsp90 is not a prerequisite for p23 to affect AHR levels. This is not entirely surprising since other researchers reported that p23 stabilizes citrate synthase against the thermal-mediated aggregation in an Hsp90-independent manner [34]. From the context of what we know about p23 and AHR, it is somewhat challenging to comprehend how p23 can affect AHR without Hsp90 since p23 forms the AHR complex by association with Hsp90. Thus far, there is no definitive data to prove that AHR binds p23 independent of Hsp90, realizing that Hsp90 is ubiquitous and is clearly abundant in the rabbit reticulocyte lysates that were used for co-immunoprecipitation studies to prove the p23:AHR interaction [35]. In an effort to better address whether p23 binds AHR directly, we generated bacterial reagents – AHR constructs and p23 – which had undergone metal-affinity purification so that any bacterial homologs of heat shock proteins should be minimized. Our data proved that p23 can bind AHR directly, which unveils the possibility that binding of p23 to AHR protects AHR from degradation. This p23:AHR interaction requires the N-terminal 216 amino acids of AHR and the N-terminal 110 amino acids of p23. One might speculate that since this N-terminal AHR region binds DNA and ARNT, there should be some competition between p23 and ARNT in binding at that region. Surprisingly we failed to observe any competitive binding at this AHR region: formation of the AHR/ARNT/DRE gel shift complex is not suppressed by p23 and the p23:AHR interaction is unaffected by ARNT, suggesting that the p23:AHR interaction is rather complex and should be further investigated.
Interestingly, AHR in the p23 knockdown HeLa cells appears to be ubiquitinated without ligand treatment, explaining in part why the AHR protein degradation is enhanced when the p23 content is reduced. It is well known that the consequence of AHR ubiquitination, followed by ligand activation, is degradation via the 26S proteasome since MG132 treatment can attenuate the reduction of the AHR protein [21]. Without ligand treatment, AHR has been reported to undergo ubiquitination which is mediated by C-terminal hsp70-interaction protein (CHIP) – an ubiquitin E3 ligase [36]. The endogenous significance of this CHIP-mediated AHR degradation, however, is unclear since CHIP did not alter the fate of the AHR protein upon TCDD or geldanamycin treatment when comparing the transformed lung fibroblasts derived from CHIP knockout to wild-type mice [31]. Although CHIP may not be involved in the normal disposition of AHR, it is tempting to ponder whether this CHIP-mediated degradation of AHR may become important when the cellular p23 content is reduced. Another interesting observation is that the tau protein can be ubiquitinated by CHIP and this ubiquitination can be reversed by up-regulation of Hsp70 [37], implying that the folding status of tau, which is likely affected by the cellular abundance of Hsp70, may control the CHIP-mediated ubiquitination. However, the ubiquitinated tau protein appears to accumulate as insoluble aggregates rather than be efficiently degraded by the 26S proteasome [38]. This is strikingly similar to our observation of the AHR degradation when p23 is down-regulated: Although we could show 3MC-triggered degradation of AHR when p23 was down-regulated, the AHR protein levels could not be restored with MG132 treatment in p23 knockdown stable Hepa1c1c7 cells [23] and p23 knockdown HeLa cells via transient transfection (data not shown). Rather, the AHR degradation was more pronounced in the presence of MG132. This observation indicated that proteasomal degradation per se cannot fully explain how the AHR protein is degraded when p23 is down-regulated. In the case of the tau protein, it has been postulated that the ubiquitinated tau aggregates can eventually be degraded via autophagy [39]. In addition, it has been reported that MG132 can induce autophagic degradation of the progerin protein in dermal fibroblasts derived from individuals with Hutchinson-Gilford progeria syndrome [40] and expression of LC3-II, which indicates autophagic induction, can be promoted in MG132-treated MCF-7 cells [41]. Autophagy could be a mechanism for p23-mediated AHR protein degradation which should be further explored.
Acknowledgments
This work is supported by the National Institutes of Health grants R15ES023104 (W.K.C.).
ABBREVIATIONS
- AHR
aryl hydrocarbon receptor
- ARNT
AHR nuclear transclocator
- p23
p23 co-chaperone
- Hsp90
heat shock protein 90
- XAP2
hepatitis virus B X-associated protein 2
- 3-MC
3-methylcholanthrene
- βNF
β-napthoflavone
- DRE
dioxin response element
- GST
glutathione S-transferase
- TH
thioredoxin
- KD
knockdown
- WT
wild-type
Footnotes
CONFLICT OF INTEREST
On behalf of all the authors, the corresponding author confirmed that there are no known conflicts of interest associated with this publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Poland A, Glover E, Robinson JR, Nebert DW. Genetic expression of aryl hydrocarbon hydroxylase activity: Induction of monooxygenase activities and cytochrome p1–450 formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice genetically “nonresponsive” to other aromatic hydrocarbons. J Biol Chem. 1974;249:5599–5606. [PubMed] [Google Scholar]
- 2.Nebert DW, Puga A, Vasiliou V. Role of the Ah receptor and the dioxin-inducible [Ah] gene battery in toxicity, cancer, and signal transduction. Ann N Y Acad Sci. 1993;685:624–640. doi: 10.1111/j.1749-6632.1993.tb35928.x. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, et al. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci USA. 1996;93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novelli M, Piaggi S, De Tata V. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced impairment of glucose-stimulated insulin secretion in isolated rat pancreatic islets. Toxicol Lett. 2005;156:307–314. doi: 10.1016/j.toxlet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 7.Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–1348. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li CH, Liu CW, Tsai CH, Peng YJ, Yang YH, Liao PL, et al. Cytoplasmic aryl hydrocarbon receptor regulates glycogen synthase kinase 3 beta, accelerates vimentin degradation, and suppresses epithelial-mesenchymal transition in non-small cell lung cancer cells. Arch Toxicol. 2017;91:2165–2178. doi: 10.1007/s00204-016-1870-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci. 2010;115:89–97. doi: 10.1093/toxsci/kfq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 11.Contador-Troca M, Alvarez-Barrientos A, Barrasa E, Rico-Leo EM, Catalina-Fernández I, Menacho-Márquez M, et al. The dioxin receptor has tumor suppressor activity in melanoma growth and metastasis. Carcinogenesis. 2013;34:2683–2693. doi: 10.1093/carcin/bgt248. [DOI] [PubMed] [Google Scholar]
- 12.Bogoevska V, Wolters-Eisfeld G, Hofmann BT, El Gammal AT, Mercanoglu B, Gebauer F, et al. HRG/HER2/HER3 signaling promotes AhR-mediated Memo-1 expression and migration in colorectal cancer. Oncogene. 2017;36:2394–2404. doi: 10.1038/onc.2016.390. [DOI] [PubMed] [Google Scholar]
- 13.Al-Dhfyan A, Alhoshani A, Korashy HM. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and beta-Catenin and Akt activation. Mol Cancer. 2017;16:14–31. doi: 10.1186/s12943-016-0570-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nair SC, Toran EJ, Rimerman RA, Hjermstad S, Smithgall TE, Smith DF. A pathway of multi-chaperone interactions common to diverse regulatory proteins: estrogen receptor, Fes tyrosine kinase, heat shock transcription factor Hsf1, and the aryl hydrocarbon receptor. Cell Stress Chaperones. 1996;1:237–250. doi: 10.1379/1466-1268(1996)001<0237:apomci>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knoblauch R, Garabedian MJ. Role for Hsp90-associated cochaperone p23 in estrogen receptor signal transduction. Mol Cell Biol. 1999;19:3748–5379. doi: 10.1128/mcb.19.5.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas M, Harrell JM, Morishima Y, Peng HM, Pratt WB, Lieberman AP. Pharmacologic and genetic inhibition of hsp90-dependent trafficking reduces aggregation and promotes degradation of the expanded glutamine androgen receptor without stress protein induction. Human Mol Genet. 2006;15:1876–1883. doi: 10.1093/hmg/ddl110. [DOI] [PubMed] [Google Scholar]
- 17.Holt SE, Aisner DL, Baur J, Tesmer VM, Dy M, Ouellette M, et al. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 1999;13:817–826. doi: 10.1101/gad.13.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patwardhan CA, Fauq A, Peterson LB, Miller C, Blagg BS, Chadli A. Gedunin inactivates the co-chaperone p23 protein causing cancer cell death by apoptosis. J Biol Chem. 2013;288:7313–7325. doi: 10.1074/jbc.M112.427328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cano LQ, Lavery DN, Sin S, Spanjaard E, Brooke GN, Tilman JD, et al. The co-chaperone p23 promotes prostate cancer motility and metastasis. Mol Oncol. 2015;9:295–308. doi: 10.1016/j.molonc.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novikov O, Wang Z, Stanford EA, Parks AJ, Ramirez-Cardenas A, Landesman E, et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in ER-/PR-/Her2-human breast cancer cells. Mol Pharmacol. 2016;90:674–688. doi: 10.1124/mol.116.105361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davarinos NA, Pollenz RS. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J Biol Chem. 1999;274:28708–28715. doi: 10.1074/jbc.274.40.28708. [DOI] [PubMed] [Google Scholar]
- 22.Chen HS, Singh SS, Perdew GH. The Ah receptor is a sensitive target of geldanamycin-induced protein turnover. Arch Biochem Biophys. 1997;348:190–8. doi: 10.1006/abbi.1997.0398. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen PM, Wang D, Wang Y, Li Y, Uchizono JA, Chan WK. p23 co-chaperone protects the aryl hydrocarbon receptor from degradation in mouse and human cell lines. Biochem Pharmacol. 2012;84:838–850. doi: 10.1016/j.bcp.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delucchi AB, Jensen KA, Chan WK. Synthesis of 32P-labelled protein probes using a modified thioredoxin fusion protein expression system in Escherichia coli. Biomol Eng. 2003;20:1–5. doi: 10.1016/s1389-0344(02)00050-3. [DOI] [PubMed] [Google Scholar]
- 25.Xie J, Huang X, Park MS, Pham HM, Chan WK. Differential suppression of the aryl hydrocarbon receptor nuclear translocator-dependent function by an aryl hydrocarbon receptor PAS-A-derived inhibitory molecule. Biochem Pharmacol. 2014;88:253–265. doi: 10.1016/j.bcp.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Shetty PV, Bhagwat BY, Chan WK. p23 enhances the formation of the aryl hydrocarbon receptor-DNA complex. Biochem Pharmacol. 2003;65:941–948. doi: 10.1016/s0006-2952(02)01650-7. [DOI] [PubMed] [Google Scholar]
- 28.Chan WK, Chu R, Jain S, Reddy JK, Bradfield CA. Baculovirus expression of the Ah receptor and Ah receptor nuclear translocator: evidence for additional dioxin responsive element-binding species and factors required for signaling. J Biol Chem. 1994;269:26464–26471. [PubMed] [Google Scholar]
- 29.Pollenz RS, Buggy C. Ligand dependent and independent degradation of the human aryl hydrocarbon receptor in cell culture models. Chem -Biol Int. 2006;164:49–59. doi: 10.1016/j.cbi.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 30.Mouradian M, Ma IV, Vicente ED, Kikawa KD, Pardini RS. Docosahexaenoic acid-mediated inhibition of heat shock protein 90-p23 chaperone complex and downstream client proteins in lung and breast cancer. Nutrition Cancer. 2017;69:92–104. doi: 10.1080/01635581.2017.1247886. [DOI] [PubMed] [Google Scholar]
- 31.Pollenz RS, Dougherty EJ. Redefining the role of the endogenous XAP2 and C-terminal hsp70-interacting protein on the endogenous Ah receptors expressed in mouse and rat cell lines. J Biol Chem. 2005;280:33346–33356. doi: 10.1074/jbc.M506619200. [DOI] [PubMed] [Google Scholar]
- 32.Ramadoss P, Petrulis JR, Hollingshead BD, Kusnadi A, Perdew GH. Divergent roles of hepatitis B virus X-associated protein 2 in human versus mouse Ah receptor complexes. Biochemistry. 2004;43:700–709. doi: 10.1021/bi035827v. [DOI] [PubMed] [Google Scholar]
- 33.Reebye V, Querol Cano L, Lavery DN, Brooke GN, Powell SM, Chotai D, et al. Role of the HSP90-associated cochaperone p23 in enhancing activity of the androgen receptor and significance for prostate cancer. Mol Endocrinol. 2012;26:1694–1706. doi: 10.1210/me.2012-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bose S, Weikl T, Bugl H, Buchner J. Chaperone function of Hsp90-associated proteins. Science. 1996;274:1715–1717. doi: 10.1126/science.274.5293.1715. [DOI] [PubMed] [Google Scholar]
- 35.Kazlauskas A, Sundstrom S, Poellinger L, Pongratz I. The hsp90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol Cell Biol. 2001;21:2594–2607. doi: 10.1128/MCB.21.7.2594-2607.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lees MJ, Peet DJ, Whitelaw ML. Defining the role for XAP2 in stabilization of the dioxin receptor. J Biol Chem. 2003;278:35878–35888. doi: 10.1074/jbc.M302430200. [DOI] [PubMed] [Google Scholar]
- 37.Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Human Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- 38.Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci USA. 1987;84:3033–3036. doi: 10.1073/pnas.84.9.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013;105:49–59. doi: 10.1016/j.pneurobio.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 40.Harhouri K, Navarro C, Depetris D, Mattei MG, Nissan X, Cau P, et al. MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol Med. 2017;9:1294–1313. doi: 10.15252/emmm.201607315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bao W, Gu Y, Ta L, Wang K, Xu Z. Induction of autophagy by the MG132 proteasome inhibitor is associated with endoplasmic reticulum stress in MCF7 cells. Mol Med Reports. 2016;13:796–804. doi: 10.3892/mmr.2015.4599. [DOI] [PubMed] [Google Scholar]