

Abstract
Background
AMP-activated Protein Kinase (AMPK) is a stress-activated kinase that protects against cardiomyocyte injury during ischemia and reperfusion. c-Jun N-terminal kinase (JNK), a mitogen activated protein kinase, is activated by ischemia and reperfusion. NF-κB is an important transcription factor involved in ischemia and reperfusion injury.
Methods and Results
The intrinsic activation of AMPK attenuates the inflammation which occurred during ischemia/reperfusion through the modulation of the JNK mediated NF-κB signaling pathway. Rat cardiac myoblast H9c2 cells were subjected to hypoxia and/or reoxygenation to investigate the signal transduction that occurred during myocardial ischemia/reperfusion. Mitochondrial function was measured by the Seahorse XF24 V7 PS system. Hypoxia treatment triggered AMPK activation in H9c2 cells in a time dependent manner. The inhibition of hypoxic AMPK activation through a pharmacological approach (Compound C) or siRNA knockdown of AMPK α catalytic subunits caused dramatic augmentation in JNK activation, inflammatory NF-κB phosphorylation, and apoptosis during hypoxia and reoxygenation. Inhibition of AMPK activation significantly impaired mitochondrial function and increased the generation of reactive oxygen species (ROS) during hypoxia and reoxygenation. In contrast, pharmacological activation of AMPK by metformin significantly inhibited mitochondrial permeability transition pore (mPTP) opening and ROS generation. Moreover, AMPK activation significantly attenuated the JNK-NF-κB signaling cascade and inhibited mRNA and protein levels of pro-inflammatory cytokines, such as TNF-α and IL-6, during hyopoxia/reoxygenation in H9c2 cells. Intriguingly, both pharmacologic inhibition of JNK by JNK-IN-8 and siRNA knockdown of JNK signaling pathway attenuated NF-κB phosphorylation and apoptosis but did not affect AMPK activation in response to hypoxia and reoxygenation.
Conclusions
AMPK activation modulates JNK-NF-κB signaling cascade during hypoxia and reoxygenation stress conditions. Cardiac AMPK activation plays a critical role in maintaining mitochondrial function and inhibiting the inflammatory response caused by ischemic insults.
Keywords: AMP-activated protein kinase, inflammation, hypoxia and reoxygenation
1. Introduction
Cardiovascular disease, the main cause of death globally, was responsible for more than 17.3 million deaths in 2013 [1], and that number is anticipated to reach more than 23.6 million by 2030 [2]. Among these deaths, those due to ischemic heart disease increased by an approximated 41.7% from 1990 to 2013 [3]. Myocardial ischemia is the lack of blood flow to the myocardium, resulting in insufficient oxygen, glucose, and other nutrients which can support mitochondrial oxidative phosphorylation. Perfusion after ischemia is an important therapeutic strategy, but damage also occurs in the myocardium due to reperfusion, termed “reperfusion-injury” [4]. Myocardial ischemia/reperfusion (I/R) injury is an complex pathophysiological process in which calcium is overloaded, mitochondrial permeability transition pores open, and reactive oxygen species (ROS) are excessively produced, leading to mitochondrial dysfunction and cell death [5]. Many harmful inflammatory cytokines are released during I/R, such as tumor necrosis factor-α (TNF-α), IL-1β, and IL-6 [6].
AMP-activated protein kinase (AMPK) is an energy sensor that regulates the intracellular ATP to AMP ratio. It is regarded as a regulator of cardiac metabolism and is activated when the cellular energy state is low [7]. AMPK has many ways to increase ATP production and decrease ATP consumption. It can increase glucose and fatty acid uptake, enhance glycolysis and fatty acid oxidation, and inhibit glycogen and protein synthesis [8]. There is evidence that AMPK activation can modulate glucose and fatty acid metabolism via the phosphorylation of the downstream acetyl-CoA carboxylase (ACC) in order to protect the heart from ischemic damage caused by I/R [9–11]. During I/R, excessive ROS production can topple naturally occurring antioxidant systems, and excessive ROS can cause the mitochondrial permeability transition pore (mPTP) to open and activate harmful signaling [12]. Mitochondrial dysfunction during I/R is mainly caused by the opening of the mPTP [13]. Chronic energy deficiency stimulates AMPK in order to facilitate muscle mitochondrial biogenesis [14], but whether the activated AMPK can protect mitochondrial function during I/R remains uncertain.
The c-Jun N-terminal protein kinase (JNK) is a mitogen activated protein kinase (MAPK) family member that modulates multiple cellular functions, including proliferation, differentiation, and apoptosis [15]. Chronic AMPK activation decreased JNK activation via hydrogen peroxide in endothelial cells [16]. Interestingly, metformin, as an AMPK agonist used for the treatment of Diabetes has been recently recognized for protecting cardiomyocytes from high glucose and hypoxia/reoxygenation-induced injury via modulating the JNK signaling pathway [17]. The Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is a nuclear transcription factor that can regulate gene expression, which is very important to apoptosis and inflammation signaling in many diseases, including ischemic pathology [18]. Inactive NF-κB is located in the cytoplasm, where it is limited by the IκB family of proteins, including IκB-α. When NF-κB is activated, IκB-α is phosphorylated by IκB kinase (IKK) followed by degradation, leading to the translocation of NF-κB subunits from the cytoplasm to the nucleus. Previous studies have shown that the NF-κB subunit p65 is associated with I/R injury in a liver model, due to increased inflammation [19]. Moreover, the components of the mitogen-activated protein kinase (MAPK) signaling pathway, which include p38 MAPK, extracellular signal-regulated kinase (ERK) and JNK, also participate in inflammation and are considered to be upstream factors of NF-κB [20]. We hypothesize the AMPK signaling pathway could protect mitochondrial function under ischemic conditions and modulate the inflammatory response through ROS-mediated JNK signaling.
2. Methods
2.1. Cell Culture and reagents
Rat cardiac myoblast H9c2 cells (ATCC CRL-1446™) were cultured in high glucose (4.5 g/L) DMEM (Corning Cellgro) supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco. Life Technologies) and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) (ATCC). The cells were maintained in a humidified incubator with 95% air and 5% CO2 at 37°C. Cells were grown in 100 mm plates and sub-cultured when they reached 70–80% confluence. AMPK inhibitor Compound C (Dorsomorphin) and JNK inhibitor JNK-IN-8 were purchased from Sigma-Aldrich Co.; metformin was obtained from MP Biomedicals LLC.
2.2. Hypoxia/Reoxygenation
To establish an I/R model in vitro, a H/R cell model was used in our study. Before different treatments were given to H9c2 cells, the culture medium would be changed to serum-free no glucose DMEM (Gibco Life Technologies) when the H9c2 cells were grown to about 80% confluence. The cells were placed in a hypoxia incubation chamber, and normal air was replaced with 95% N2 and 5% CO2. The cells were cultured in hypoxic conditions for 15 hours. Following hypoxia, the cells were removed from the hypoxia chamber, and the medium was replaced with culture medium. Cells were kept in a normoxic incubator for 1 hour. For each evaluation, all experiments were conducted in triplicate.
2.3. Transient transfection with small interfering RNA (siRNA)
To knockdown the endogenous AMPK and JNK, H9c2 cells were transiently transfected with 100 nM mouse siRNAs targeting AMPKα1/α2, JNK1/2, or with the non-silencing control siRNA (scram) (Ambion) using Lipofectamine™ RNAiMAX (Invitrogen) in culture medium without antibiotics according to the manufacturer’s recommendations.
2.4. Mitochondrial respiration measurements
Mitochondrial oxygen consumption rate (OCR) was assessed with a Seahorse XFe24 Analyzer (Seahorse Bioscience). H9c2 cells (20,000/well) were seeded into 24 wells of Seahorse XF24 V7 PS cell culture microplates (Seahorse Bioscience). After treatment, the medium was replaced with XF Assay Medium (Seahorse Bioscience), supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM D-glucose. After placing the cell culture microplate into a 37°C non-CO2 incubator for 1 hour, OCR was measured using the Seahorse Bioscience XF24 Extracellular Flux Analyzer (Seahorse Bioscience). Measurements were taken as the cells were incubated sequentially under four conditions: (1) basal levels were measured with no additives; (2) oligomycin (1 μM) was added to reversibly inhibit ATP synthase and OXPHOS, showing glycolysis alone; (3) FCCP (2 μM), a mitochondrial uncoupler, was added to induce maximal respiration; and (4) Rotenone/antimycin A (0.5 μM), a Complex I inhibitor and mitochondrial poison, was added to end the reaction. The Seahorse software was used to plot the results. OCR was normalized to cell protein concentration per well.
2.5. Reactive oxygen species (ROS) measurement
To examine the generation of ROS, the ROS-ID™ Total ROS Detection kit (Enzo Life Sciences, Inc., Plymouth Meeting, PA) was used according to the manufacturer’s instructions. H9c2 cells were seeded in a 96-well plate with a clear bottom and reached 70~80% confluence. Fluorescent signals were measured through the bottom of the cell culture plates at an excitation wavelength of 490 nm and an emission wavelength of 525 nm (Spectramax M2 plate reader)
2.6. Flow cytometric measurement of the mitochondrial permeability transition pore
The MitoProbe transition pore assay kit (Invitrogen) and the MACS flow cytometry (Miltenyi Biotec) was used to study the opening of the mitochondrial transition pore (mPTP) in H9c2 cells. This kit provides a more direct method of measuring mPTP opening than assays relying on mitochondrial membrane potential alone. In brief, 1×106 cells were loaded with the acetoxymethyl ester of calcein dye, calcein AM, which passively diffused into the cells and accumulated in cytosolic compartments, including the mitochondria. Once inside cells, intracellular esterases cleaved the acetoxymethyl esters to liberate the very polar fluorescent dye calcein. The fluorescence from cytosolic calcein was quenched by the addition of CoCl2 (cobalt chloride), while the fluorescence from the mitochondrial calcein was maintained. However, opening the mPTP instigated the release of mitochondrial calcein to the cytosol where the signal was quenched by CoCl2, leading to the dramatic reduction of calcein fluorescence.
2.7. mRNA analysis of pro-inflammatory cytokines
mRNA levels were quantified using the Real-Time PCR ΔT method with the CFX96 platform (Bio-Rad Laboratories, Inc.). Total RNA was purified using the Pure Link RNA mini kit (Ambion) according to the manufacturer’s instructions. cDNA was synthesized with the High Capacity RNA-to-cDNA Kit (Applied Biosystems). Quantitative PCR reactions were performed using the CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.). All primers were synthesized by Integrated DNA Technology (IDT, Canada). The primer sequences for mRNA analysis are: TNF-α, forward: 5′-CGTGTTCATCCGTTCTCTACC-3′; reverse: 5′-GCAATCCAGGCCACTACTT-3′; IL-6, forward: 5′-CCGTTTCTACCTGGAGTTTGT-3′; reverse: 5′-GTTTGCCGAGTAGACCTCATAG-3′; 18S rRNA: forward: 5′-TTGATTAAGTCCCTGCCCTTTGT-3′; reverse: 5′-CGATCCGAGGGCCTAACTA-3′. Expression of 18S rRNA was used to normalize gene expression. Quantification of gene expression was carried out using the CFX manager software (Bio-Rad Laboratories, Inc.).
2.8. Pro-inflammatory cytokines
Pro-inflammatory cytokines TNF-α and IL-6 were estimated using ELISA kits (R&D Systems and BD Biosciences, U.S.A. respectively) and analyzed through an iMark™ Microplate Absorbance Reader (Bio-Rad Laboratories, Inc,).
2.9. Immunoblotting
Immunoblots were performed as previously described [21, 22]. Rabbit antibodies p-AMPKα (Thr172) (#2535), AMPKα (#5831), p-Acetyl-CoA carboxylase (ACC) (Ser79) (#3661), ACC (#3676), p-JNK (Thr183/Tyr185) (#4668), JNK (#9252), p-c-Jun (Ser73) (#3270), c-Jun (#9165), p-p65 (Ser536) (#3033), p65 (#8242) and β-tubulin (#2146) were all obtained from Cell Signaling Technology (Danvers, MA, USA).
2.10. Statistical analysis
All data was acquired from independent experiments and represented as the mean ± SEM. The comparisons between two groups were analyzed using the non-parametric Mann-Whitney test using GraphPad Prism 5.0 (GraphPad Software, Inc.). A value of p < 0.05 was considered statistically significant.
3. Results
3.1. AMPK and JNK signaling in response to hypoxia and reoxygenation
In order to characterize the roles of AMPK and JNK signaling pathways in response to hypoxic stress, the H9c2 myoblast cells were treated with 95% nitrogen (N2) and 5% carbon dioxide (CO2) for different time points; the results demonstrated that hypoxia treatment can trigger AMPK activation by stimulating phosphorylation of the catalytic α subunit at Thr172 in a time dependent manner (Figure 1A). The peak activation time of AMPK due to hypoxic stress in the H9c2 cells was 15 hours. Interestingly, JNK signaling was significantly inhibited during hypoxia (Figure 1B), and was augmented by hypoxia and reoxygenation (H/R), especially at 15 hrs hypoxia followed by 1 hour reoxygenation (Figure 1B). Conversely, AMPK signaling was activated during hypoxia and attenuated during reoxygenation following hypoxia (Figures 1A and 1B).
Figure 1. The kinetics of AMPK activation and JNK activation in response to hypoxia with or without reoxygenation in H9c2 myoblast cells.
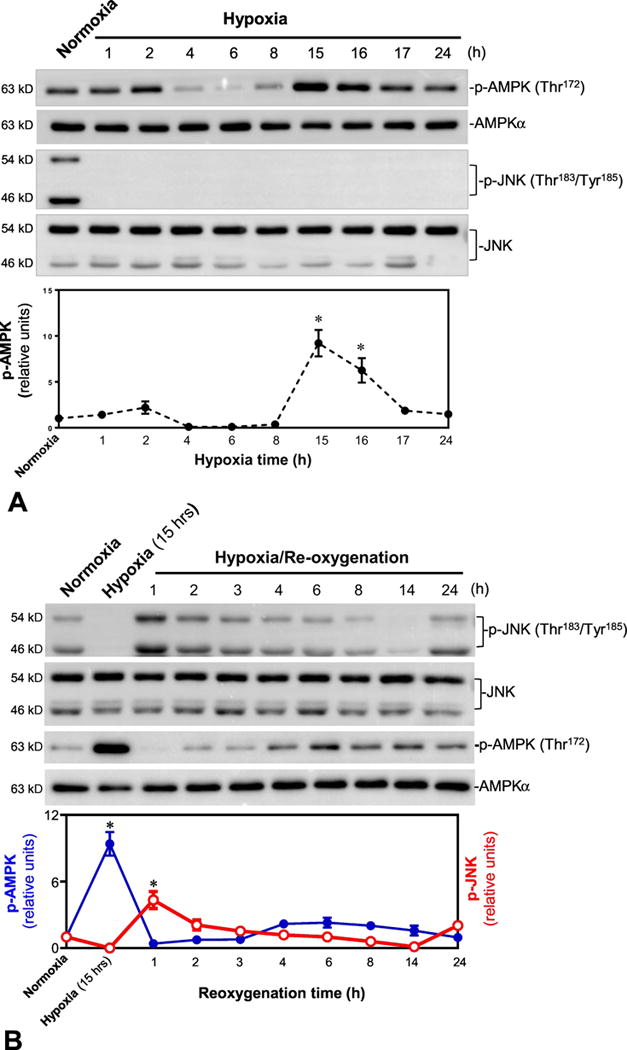
(A) Immunoblotting measured the levels of p-AMPK and AMPKα protein expression in normoxia or hypoxia conditions. Values are mean ± SEM. N=3, *p<0.05 vs. normoxia. (B) Immunoblotting measured the levels of p-AMPK, AMPKα, p-JNK, and JNK protein expression in normoxia, hypoxia or hypoxia/reoxygenation conditions. Values are mean ± SEM. N=3, *p<0.05 vs. normoxia.
3.2. Inhibition of AMPK augmented JNK activation by hypoxia and reoxygenation
There is evidence that inactivation of AMPK increases JNK signaling during reperfusion after cardiac ischemia [23]. In order to determine whether there is a feedback loop relationship between AMPK signaling and JNK activation in response to hypoxic stress, AMPK specific inhibitor compound C [24] and JNK specific inhibitor JNK-IN-8 [25] were used to characterize the relationship between AMPK and JNK signaling pathways in response to hypoxia with or without reoxygenation treatments. The results showed that hypoxia triggered phosphorylation of AMPK and downstream Acetyl CoA carboxylase (ACC) but not JNK phosphorylation (Figure 2A); compound C pretreatment significantly inhibited hypoxia-triggered AMPK phosphorylation, while JNK inhibitor JNK-IN-8 did not have any effect on hypoxic AMPK activation (Figure 2A). JNK signaling pathways were activated, as shown by phosphorylation of JNK and downstream c-Jun during H/R; intriguingly, AMPK inhibitor compound C significantly augmented JNK phosphorylation during H/R (Figure 2A), and JNK-IN-8 treatment completely blocked JNK phosphorylation induced by H/R (Figure 2A).
Figure 2. AMPK inactivation augments JNK activation during hypoxia and reoxygenation.
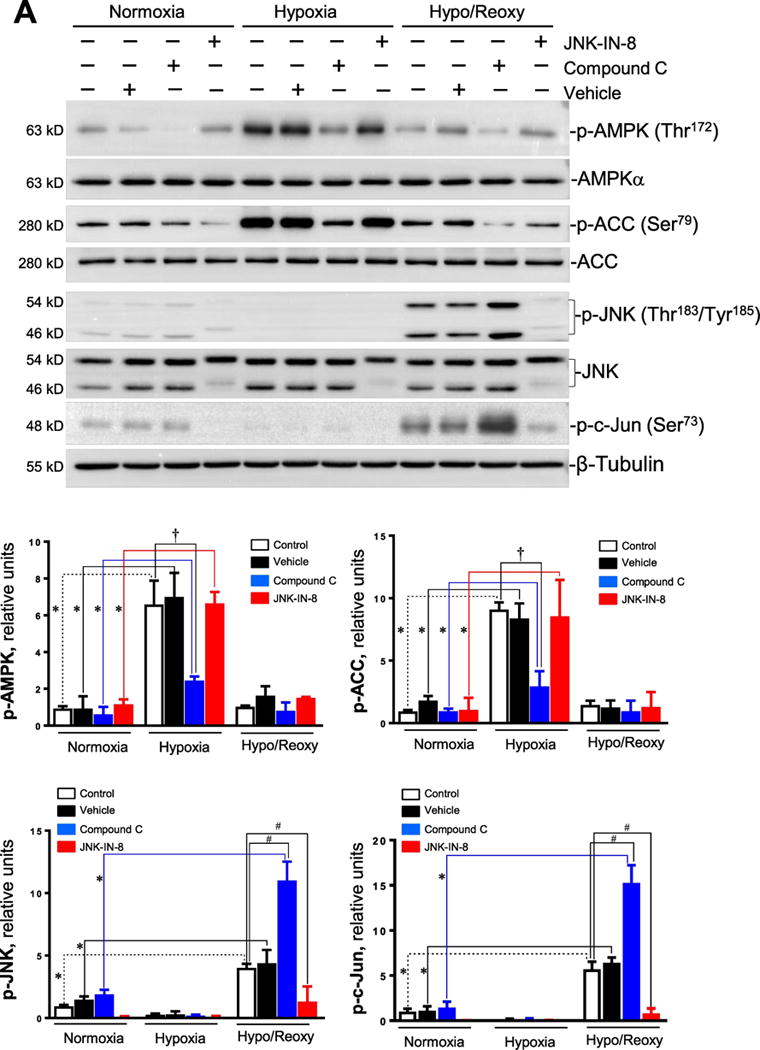

(A) H9c2 cells were treated with compound C (10 μM) and JNK-IN-8 (1 μM) under normoxia, hypoxia and hypoxia/reoxygenation. Upper: Immunoblotting showed phosphorylation of AMPK and downstream ACC, and phosphorylation of JNK and downstream c-Jun in H9c2 cells with or without AMPK inhibitor compound C and JNK inhibitor JNK-IN-8 under normoxia, hypoxia, and hypoxia/reoxygenation conditions; Lower: quantitative analysis of the relative levels of p-AMPK, p-ACC, p-JNK, and p-c-Jun in H9c2 cells with or without AMPK inhibitor or JNK inhibitor under normoxia, hypoxia, and hypoxia/reoxygenation. Values are mean ± SEM. N=4, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypoxia vehicle; #p<0.05 vs. hypo/reoxy vehicle. (B) AMPKα1/α2 siRNA knockdown AMPKα catalytic subunit and JNK1/2 siRNA knockdown JNK1/2 in the H9c2 myoblast cells under normoxia, hypoxia, and hypoxia/reoxygenation. Upper: Immunoblotting showed phosphorylation of AMPK and downstream ACC, and phosphorylation of JNK and downstream c-Jun in H9c2 cells with or without AMPKα1/α2 siRNA and JNK1/2 siRNA under normoxia, hypoxia, and hypoxia/reoxygenation conditions; Lower: quantitative analysis of the relative levels of p-AMPK, p-ACC, p-JNK, and p-c-Jun in H9c2 cells with or without AMPKα1/α2 siRNA and JNK1/2 siRNA under normoxia, hypoxia and hypoxia/reoxygenation conditions. Values are mean ± SEM. N=3, *p<0.05 vs. normoxia, repectively; †p<0.05 vs. hypoxia scram; #p<0.05 vs. hypo/reoxy scram.
Furthermore, the genetic approach with AMPKα1/α2 siRNA and JNK1/2 siRNA was used to avoid the non-specific effects of the pharmacological approach regarding the relationship between AMPK and JNK signaling pathways during H/R stress conditions. The immunoblotting results demonstrated that AMPKα1/α2 siRNA knocked down AMPKα protein levels and attenuated the phosphorylation of AMPK and downstream ACC induced by hypoxia in H9c2 cells (Figure 2B). JNK1/2 siRNA clearly knocked down JNK protein levels of H9c2 myoblast cells and attenuated the phosphorylation of JNK and downstream c-Jun by H/R (Figure 2B). Intriguingly, the AMPKα1/α2 siRNA knockdown of AMPKα significantly augmented activation of JNK caused by H/R, as shown by phosphorylation of JNK and downstream c-Jun (Figure 2B), but the JNK1/2 siRNA knockdown of JNK1/2 did not have any effect on AMPK signaling triggered by hypoxia, as shown by phosphorylation of AMPK and downstream ACC (Figure 2B).
3.3. AMPK plays a critical role in mitochondrial respirarion during hypoxic stress conditions
In order to better understand how AMPK activation modulates the JNK signaling pathway during hypoxic stress conditions, the mitochondrial oxidative capacity of H9c2 myoblast cells was evaluated by Seahorse analyzer with different treatments under normoxia or H/R conditions. The mitochondrial functions of H9c2 myoblast cells were measured with the Seahorse XF Cell Mito Stress Test under normoxia or H/R (Figure 3A). The results demonstrated that the oxygen consumption rate (OCR) of mitochondria in H9c2 cells was significantly decreased during H/R as compared to normoxia conditions (Figures 3A and 3B). Interestingly, AMPK inhibitor compound C did not affect mitochondrial functions of H9c2 under normoxia conditions, as shown by lack of changes in basal respiration, maximal respiration, spare respiratory capacity, and ATP production (Figure 3B), but compound C treatment significantly exacerbated mitochondrial dysfunctions during H/R, as exhibited by decreased basal respiration, decreased maximal respiration, decreased spare capacity, and lower ATP production (Figure 3B). However, JNK signaling inhibitor JNK-IN-8 did not show any effects on mitochondrial functions of H9c2 myoblast cells under normoxia or H/R conditions (Figures 3A and 3B). In addition, the genetic approaches with AMPKα1/α2 siRNA and JNK1/2 siRNA to treat H9c2 myoblast cells showed that both AMPK and JNK signaling pathways did not affect mitochondrial functions of H9c2 cells, as shown by unchanged basal respiration, maximal respiration, spare capacity, and ATP production under normoxia conditions (Figures 3C and 3D). However, lack of AMPK signaling significantly exacerbated mitochondrial dysfunction during H/R conditions, while JNK signaling did not make any contributions to the mitochondrial dysfunction caused by H/R (Figures 3C and 3D).
Figure 3. AMPK plays a role in maintaining mitochondrial integerity during hypoxia and reoxygenation.
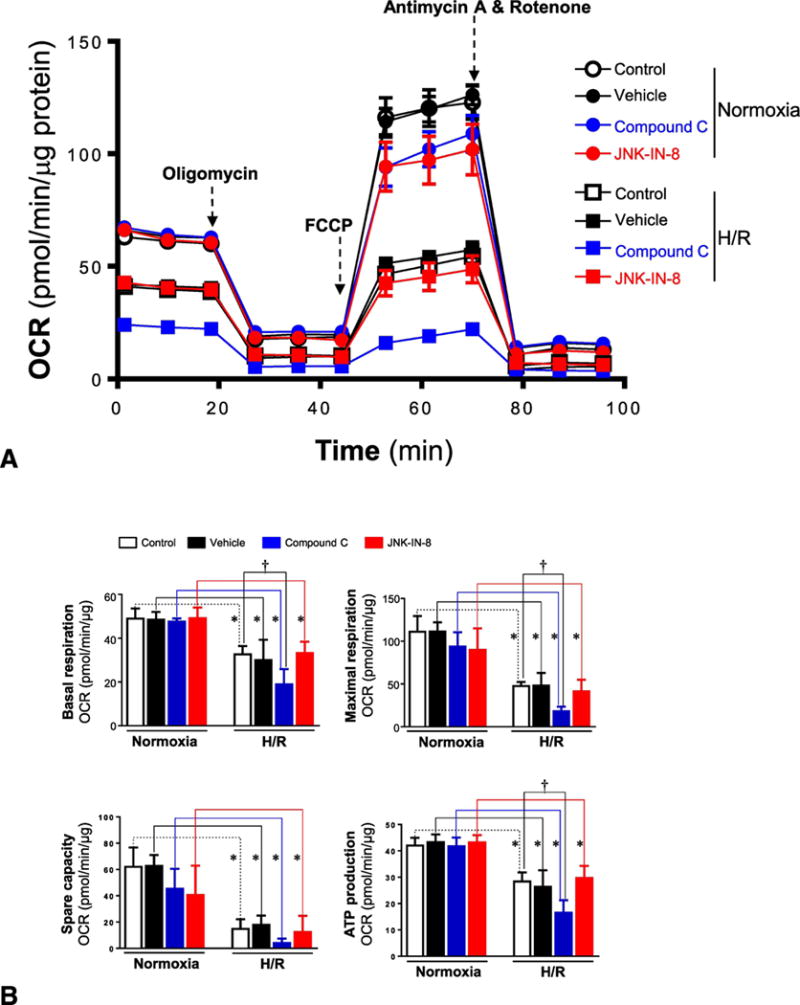
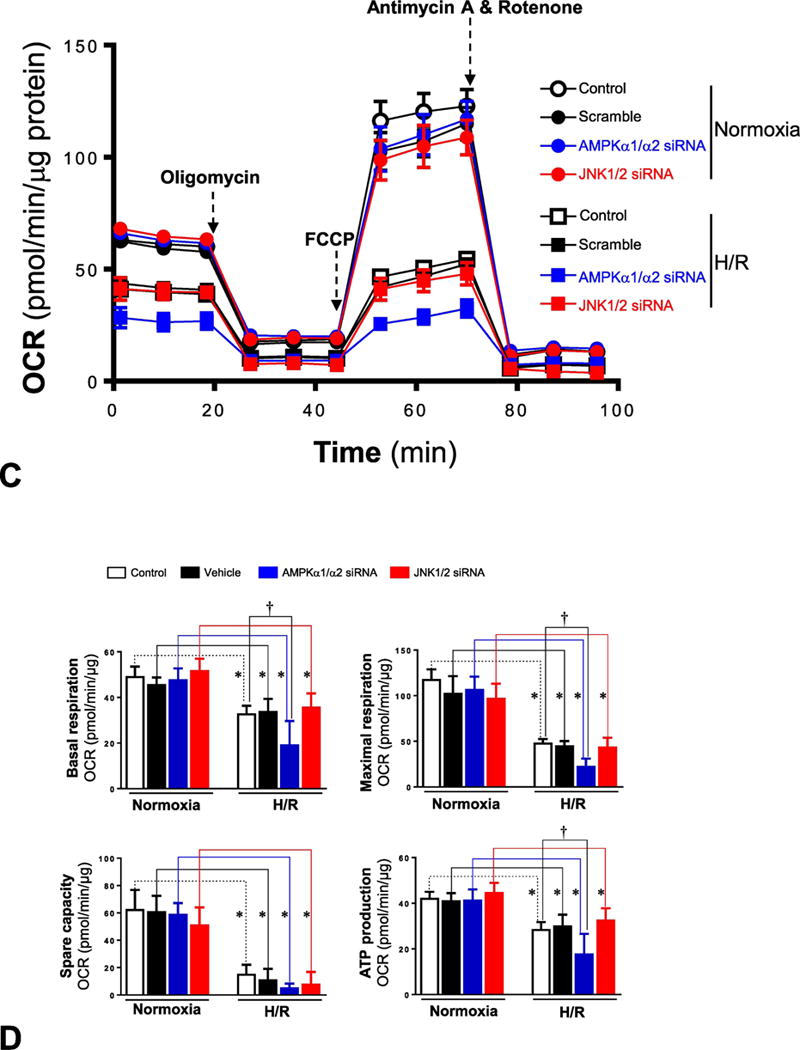
(A) Mitochondrial respiration measurements of oxygen consumption rate (OCR) were performed with a Seahorse metabolic analyzer. Oligomycin (1 μM), FCCP (2 μM), and rotenone (0.5 μM) combined with antimycin (0.5 μM) were added sequentially to H9c2 cells treated with or without compound C (10 μM) and JNK-IN-8 (1 μM) under normoxia and hypoxia/reoxygenation conditions. (B) Quantitative analysis of mitochondrial function parameters (basal respiration, maximal respiration, spare capacity, and ATP production) are shown in the bar charts. Values are mean ± SEM. N=5, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy vehicle. (C) Mitochondrial respiration measurements of OCR were performed with a Seahorse metabolic analyzer. Oligomycin (1 μM), FCCP (2 μM), and rotenone (0.5 μM) combined with antimycin (0.5 μM) were added sequentially to H9c2 cells treated with AMPKα1/α2 siRNA or JNK1/2 siRNA under normoxia and hypoxia/reoxygenation conditions. (D) Quantitative analysis of mitochondrial function parameters (basal respiration, maximal respiration, spare capacity, and ATP production) are shown in the bar charts. Values are mean ± SEM. N=5, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy scram.
3.4. AMPK modulates hypoxic stress-mediated mitochondrial reactive oxygen species (ROS) production
The mitochondria are a major source for generation of ROS, especially under stress conditions [26]. In order to characterize the role of AMPK signaling in hypoxic stress induced ROS generation, the amount of ROS generated under different conditions was measured with the ROS-ID™ Total ROS Detection kit. The results demonstrated that AMPK inhibition by compound C significantly augmented ROS production of H9c2 cells during H/R (Figure 4A), while JNK signaling blocked by JNK-IN-8 appears to not affect ROS generation under both normoxia and H/R conditions (Figure 4A). The genetic approaches with AMPKα1/α2 siRNA and JNK1/2 siRNA also showed that the AMPK signaling pathway plays a critical role in hypoxic stress mediated ROS production of H9c2 myoblast cells, as AMPKα knockdown by AMPKα1/α2 siRNA augmented ROS production by H/R (Figure 4B), while JNK signaling did not have any effect on mitochondrial ROS generation under both normoxia and H/R conditions, as JNK1/2 knockdown by JNK1/2 siRNA did not affect ROS production of H9c2 cells (Figures 4A and 4B).
Figure 4. AMPK rgulates opening of mitochondrial permeability transition pore (mPTP) and production of reactive oxygen species (ROS) during hypoxia and reoxygenation.
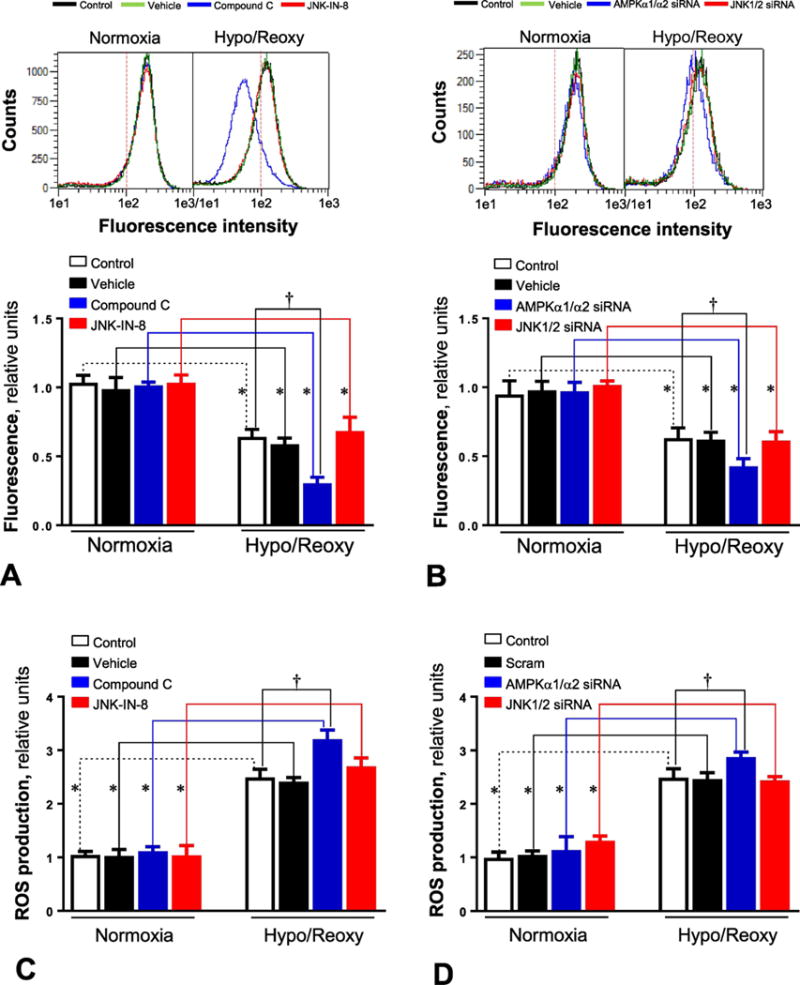
(A) H9c2 myoblast cells were treated with AMPK inhibitor compound C (10 μM) or JNK inhibitor JNK-IN-8 (1 μM) under normoxia and hypoxia/reoxygenation conditions. The opening of mitochondrial permeability transition pore (mPTP) was measured with the MitoProbe transition pore assay kit. Values are mean ± SEM. N=5, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy vehicle. (B) The mPTP opening in H9c2 myoblast cells treated with AMPKα1/α2 siRNA or JNK1/2 siRNA under normoxia and hypoxia/reoxygenation conditions. Values are mean ± SEM. N=5, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy scram. (C) The levels of reactive oxygen species (ROS) in H9c2 cells treated with compound C (10 μM) or JNK-IN-8 (1 μM) under normoxia or hypoxia and reoxygenation conditions. Values are mean ± SEM. N=6, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy vehicle. (D) The ROS production in H9c2 cells treated with AMPKα1/α2 siRNA or JNK1/2 siRNA under normoxia and hypoxia/reoxygenation conditions. Values are mean ± SEM. N=6, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy scram.
3.5. AMPK limits mitochondrial permeability transition pore opening by hypoxic insults
There is evidence that myocardial ischemia and reperfusion induces opening of the mitochondrial permeability transition pore (mPTP), which can decrease mitochondrial respiration and result in cardiomyocyte death [27]. The opening of mPTP in H9c2 myoblast cells was measured with the MitoProbe transition pore assay kit. The results demonstrated that mPTP opening in H9c2 cells was increased in response to H/R as compared to normoxia conditions (Figure 4C). AMPK signaling inhibitor compound C augmented the mPTP opening by H/R, while JNK inhibitor JNK-IN-8 did not show this augmentation effect in H9c2 cells under H/R conditions (Figure 4C). The genetic approach with AMPKα1/α2 siRNA and JNK1/2 siRNA also demonstrated that AMPKα knockdown enhanced the hypoxic stress-induced mPTP opening, while JNK1/2 knockdown did not show significant effect on mPTP opening of H9c2 cells under both normoxia and hypoxic stress conditions (Figure 4D).
3.6. AMPK inhibits hypoxic stress-mediated inflammatory response via modulating NF-κB activity
There are several reports that intracellular redox alterations leading to ROS generation could trigger inflammation [26, 28]. In order to determine whether AMPK and JNK signaling pathways play a role in the hypoxic stress-mediated inflammatory response, the inflammation-related signaling pathway NF-κB was observed in H9c2 myoblast cells with or without AMPK/JNK antagonist under normoxia and H/R conditions. The immunoblotting data showed that H/R stimulated NF-κB signaling, demonstrated by an increased phosphorylation of p65 at Ser276 (Figure 5A), and AMPK inhibitor compound C treatment significantly augmented NF-κB activation by H/R (Figure 5A). Intriguingly, JNK inhibitor JNK-IN-8 can attenuate the hypoxic stress induced NF-κB activation (Figure 5A). The genetic approach with AMPKα1/α2 siRNA and JNK1/2 siRNA also showed that AMPKα knockdown can enhance hypoxic stress-triggered NF-κB activation, but JNK1/2 knockdown abolished the NF-κB activation caused by H/R (Figure 5B). We further measured the mRNA and protein levels of two pro-inflammatory cytokines, TNF-α and IL-6, by performing real-time RT-PCR and ELISA measurements, respectively. The results demonstrated that H/R induced up-regulation of TNF-α and IL-6 mRNA and increased the secretion of TNF-α and IL-6 to the medium (Figures 5C and 5D). AMPK inhibitor treatment significantly augmented this up-regulation, while JNK inhibitor JNK-IN-8 significantly inhibited the up-regulation of both TNFα and IL-6 by H/R (Figure 5C). The genetic approach with AMPKα1/α2 siRNA and JNK1/2 siRNA showed that AMPKα knockdown enhanced hypoxic stress-mediated up-regulation of TNFα and IL-6 while JNK1/2 knockdown inhibited the up-regulation by H/R (Figure 5D).
Figure 5. AMPK modulates inflammatory NF-κB signaling activation and proinlammatory cyokine expression during hypoxia and reoxygenation.
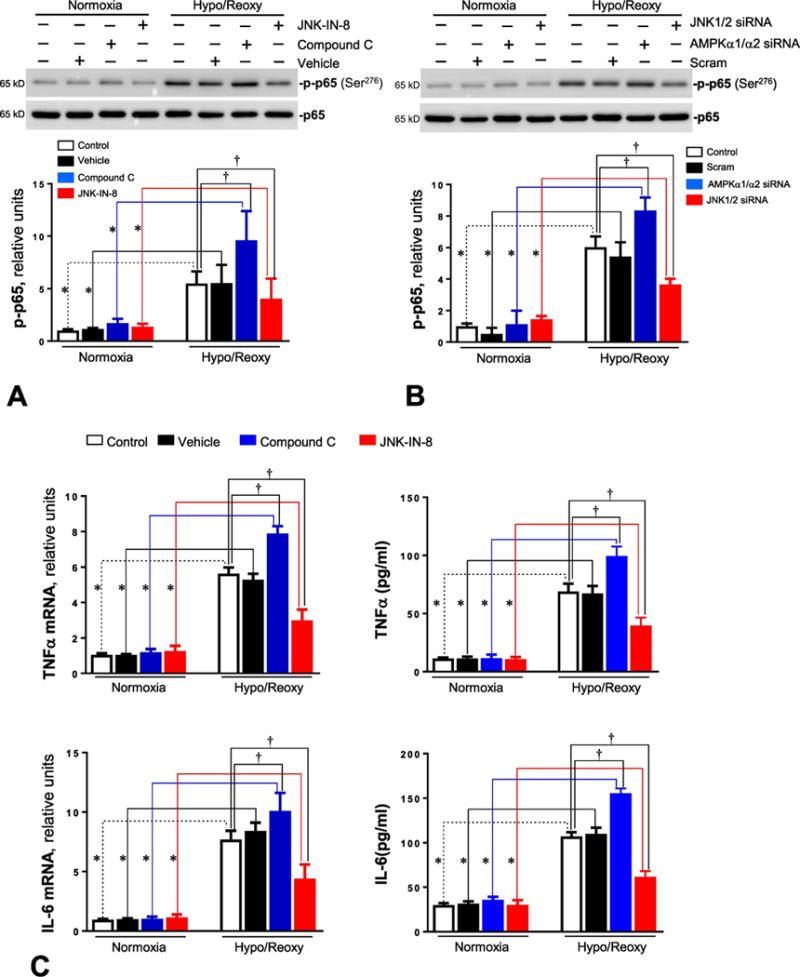
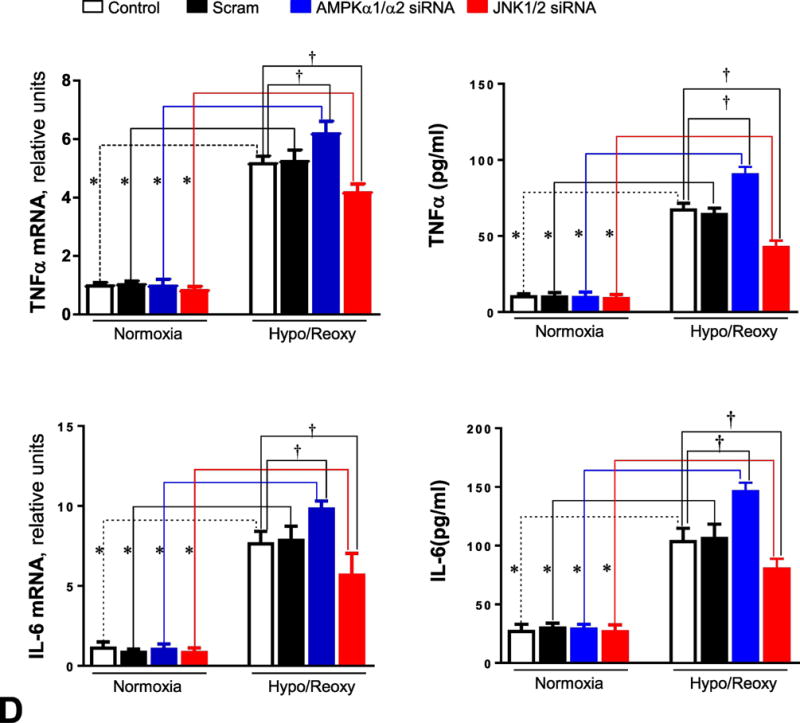
(A) Immunoblotting measured the levels of p-p65 and p65 protein expression in H9c2 myoblast cells treated with compound C (10 μM) or JNK-IN-8 (1 μM) under normoxia and hypoxia/reoxygenation conditions. Values are mean ± SEM. N=3, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy vehicle. (B) Immunoblotting measured the levels of p-p65 and p65 protein expression in H9c2 myoblast cells treated with AMPKα1/α2 siRNA or JNK1/2 siRNA under normoxia and hypoxia/reoxygenation conditions. Values are mean ± SEM. N=3, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy scram. (C) The total RNA was isolated and subjected to real time RT-PCR to analyze the products of pro-inflammatory cytokines, TNF-α and IL-6. 18S rRNA was used as an internal control. Real-time mRNA levels of TNFα and IL-6 were measured in H9c2 cells treated with compound C (10 μM) or JNK-IN-8 (1 μM) under normoxia and hypoxia/reoxygenation conditions. The secreted TNF-α and IL-6 in the culture medium were measured with ELISA kit. Values are mean ± SEM. N=5, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy vehicle. (D) Real-time RT-PCR measured the mRNA levels of TNFα and IL-6 in H9c2 cells treated with AMPKα1/α2 siRNA or JNK1/2 siRNA under normoxia and hypoxia/reoxygenation conditions. The secreted TNF-α and IL-6 in the culture medium were measured with ELISA kit. Values are mean ± SEM. N=5, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy scram.
3.7. AMPK protects H9c2 cells from apoptosis induced by hypoxia and reoxygenation
In order to determine whether the AMPK regulated inflammatory response is involved in apoptosis during H/R, H9c2 myoblast cells were treated with AMPK inhibitor compound C under normoxia or H/R conditions. The fluorescence imaging data showed that H/R caused an upregulation of apoptosis in H9c2 cells as compared to normoxia conditions (Figures 6). AMPK inhibitor compound C treatment can significantly augment apoptosis of H9c2 by H/R, but JNK inhibitor JNK-IN-8 attenuated this apoptosis (Figures 6A and 6C). The genetic approach with AMPKα1/α2 siRNA and JNK1/2 siRNA showed that AMPKα knockdown enhanced apoptosis of H9c2 cells by H/R, but JNK1/2 knockdown inhibited the hypoxic stress-triggered apoptosis of H9c2 cells (Figures 6B and 6D).
Figure 6. AMPK inhibits while JNK promotes apoptosis of H9c2 cells during hypoxia and reoxygenation.
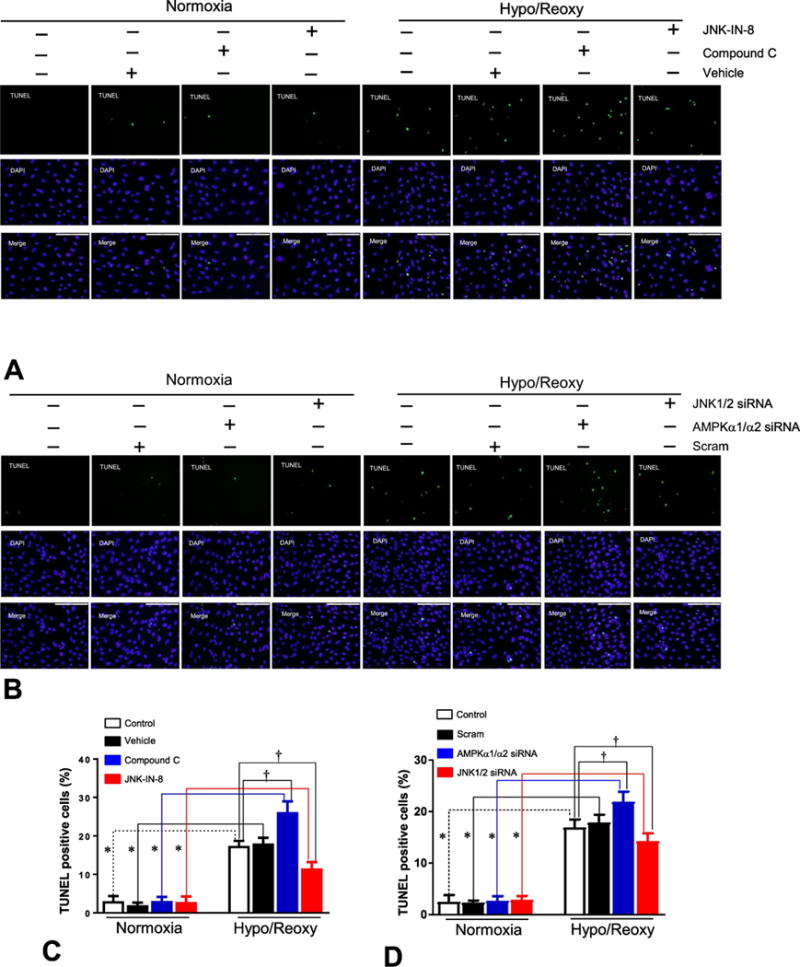
(A) Apoptotic cells were detected by staining with TUNEL and DAPI, as described in Methods. Apoptosis in H9c2 myoblast cells treated with compound C (10 μM) or JNK-IN-8 (1 μM) under normoxia and hypoxia/reoxygenation conditions. (B) Apoptosis in H9c2 myoblast cells treated with AMPKα1/α2 siRNA or JNK1/2 siRNA under normoxia and hypoxia/reoxygenation conditions. (C) Quantitative analysis of apoptosis in H9c2 cells treated with compound C or JNK-IN-8 under normoxia and hypoxia/reoxygenation conditions. Value are mean ± SEM. N=3, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy vehicle. (D) Quantitative analysis of apoptosis in H9c2 cells treated with AMPKα1/α2 siRNA or JNK1/2 siRNA under normoxia and hypoxia/reoxygenation conditions. Values are mean ± SEM. N=3, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy scram.
3.8. Activation of AMPK by metformin protects cells from hypoxic insults
Lastly, we tested the protective effects of AMPK agonist metformin on hypoxic injury in H9c2 myoblast cells. Metformin (5 mM) treatment triggered AMPK activation as shown by phosphorylation of the AMPK α subunit at Thr172 under normoxia and H/R conditions (Figure 7A). Interestingly, metformin treatment can attenuate JNK activation by H/R, as demonstrated by the phosphorylation of JNK (Figure 7B). Additionally, metformin inhibited hypoxic stress-triggered NF-κB activation, as shown by phosphorylation of p65 at Ser276 (Figure 7B). The level of mPTP opening was decreased by metformin treatment during H/R (Figure 7C). Moreover, metformin treatment significantly inhibited ROS production in H9c2 cells by H/R (Figure 7D), and the up-regulation of levels of pro-inflammatory cytokines TNF-α and IL-6 caused by H/R was inhibited (Figures 7E).
Figure 7. AMPK activator inhibits JNK activation by hypoxia and reoxygenation and modulates inflammaroty response during hypoxia and reoxygenation.


(A) Immunoblotting showed the phosphoryaltion of AMPK α catalytic subunit at Thr172 site in H9c2 cells treated with AMPK activator metformin (5 mM) under normoxia, hypoxia, and hypoxia/reoxygenation conditions. Values are mean ± SEM. N=3, *p<0.05 vs. vehicle, respectively; †p<0.05 vs. normoxia vehicle. #p<0.05 vs. normoxia metformin. (B) Immunoblotting showed the phosphoryaltion of JNK and p65 in H9c2 cells treated with AMPK activator metformin (5 mM) under normoxia, hypoxia, and hypoxia/reoxygenation conditions. Values are mean ± SEM. N=3, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy vehicle. (C) The mPTP opening in H9c2 myoblast cells treated with or without metformin. Values are mean ± SEM. N=5, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy vehicle. (D) The ROS production in H9c2 myoblast cells treated with or without metformin. Values are mean ± SEM. N=6, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy vehicle. (E) The total RNA was isolated and subjected to real time RT-PCR to analyze the products of pro-inflammatory cytokines, TNF-α and IL-6 in H9c2 myoblast cells treated with or without metformin. 18S rRNA was used as an internal control. The secreted TNF-α and IL-6 in the culture medium were measured with ELISA kit. Values are mean ± SEM. N=5, *p<0.05 vs. normoxia, respectively; †p<0.05 vs. hypo/reoxy vehicle.
4. Discussion
There is evidence suggesting a potential link between metabolic disorders and inflammation, both of which are associated with abnormal cytokine production [29]. AMPK is a heterotrimeric enzyme composed of a catalytic α subunit tethered to the regulatory β and γ subunits and is an important regulator of cardiac energy homeostasis [11]. This serine/threonine kinase is activated by ATP depletion through phosphorylation at its Thr172 residue by upstream kinases such as liver kinase B1 (LKB1) [26]. AMPK responds to increases in the AMP/ATP ratio by switching off energy-consuming pathways and stimulating ATP-producing pathways such as fatty acid β-oxidation and glycolysis [26]. A principal event of AMPK activity is the phosphorylation-mediated inhibition of acetyl-CoA carboxylase (ACC), resulting in the down-regulation of malonyl-CoA levels. Since malonyl-CoA is an inhibitor of carnitine palmitoyltransferase I (CPT-1), which is a mitochondrial enzyme responsible for the formation of acyl carnitine by catalyzing the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to I-carnitine, AMPK activation will result in an increase in energy production [11]. AMPK activation has been reported to protect the heart against ischemic injury [9], and these cardioprotective effects stem from its capacity to improve both glucose and fatty acid metabolism [21, 30].
The ischemia and reperfusion (I/R) injury in the heart has been recognized to induce the release of many cytokines and chemokines, resulting in cell apoptosis and death [31, 32]. There is accumulating evidence that an inflammatory response occurs during human myocardial ischemia/reperfusion injury. Moreover, AMPK activation has been shown as a promising pharmacologic approach to limit organ injury caused by ischemia/reperfusion before transplantation [33]. In order to investigate the signaling mechanisms associated with the inflammatory response of ischemic stress, the H9c2 cardiomyoblast cell culture model is used to determine the signaling events which occur during hypoxia/reoxygenation and reveal potential molecular targets to limit cardiac injury during ischemia/reperfusion. These reports were checked in our study, during which we found that hypoxia/reoxygenation, a model of I/R injury, induced cardiac myoblast H9c2 cell apoptosis and ROS production. Our study also showed that the levels of pro-inflammation cytokines IL-6 and TNF-α were increased in H/R. The AMPK signaling pathway plays a critical role in controlling mPTP opening of H9c2 cell mitochondria under H/R, since AMPK inhibitor and AMPKα1/α2 siRNA knockdown both augment mPTP opening and ROS production in H9c2 myoblast cells under H/R. However, the JNK signaling pathway did not make contributions to hypoxic stress-mediated mPTP opening and ROS generation in the H9c2 cells, Although, JNK signaling is involved in mediating the inflammatory response during H/R, as both JNK inhibitor and JNK1/2 siRNA knockdown JNK1/2 treatment reduced NF-κB activation and mRNA levels of proinflammatory cytokines TNFα and IL-6.
Interestingly, AMPK signaling appears to modulate JNK activation in response to H/R, since both AMPK inhibitor and AMPKα1/α2 siRNA knockdown AMPKα significantly augmented JNK activation while AMPK agonist metformin can reduce the hypoxic stress-induced JNK activation and downstream NF-κB activation and inflammatory response by up-regulation of TNFα and IL-6 mRNA. NF-κB is an important transcription factor. It has function in the processes of inflammation and apoptosis induced by I/R injury [34, 35]. It was verified that the NF-κB subunit p65 is activated during I/R injury in the rat steatotic liver, resulting in inflammation and necrosis [19]. Accordingly, the suppression of NF-κB by its inhibitor BAY 11-7802 or the inhibition of IκB phosphorylation has been proven to reduce the inflammation and apoptosis induced by myocardial or cerebral I/R injury [36]. Our results showed that the inflammatory response-related NF-κB signaling was activated during H/R in H9c2 myoblast cells, which leads to the upregulation of mRNA levels of the targets of NF-κB inflammation-related genes such as TNF-α and IL-6. It is well known that c-Jun N-terminal protein kinase (JNK) induces NF-κB activation in response to a variety of stress stimuli [35, 37]. The response of AMPK, JNK and NF-κB signaling pathways to ischemia and/or reperfusion was also observed in the in vivo regional myocardial ischemia and reperfusion in mouse hearts by ligation of the left anterior descending coronary artery (Suppl. Fig. 1). Our study also suggests that AMPK plays a critical role in limiting H9c2 cell apoptosis caused by H/R, since both AMPK inhibitor compound C and AMPKα1/α2 siRNA knockdown AMPKα can significantly enhance the apoptosis occurring under H/R conditions. In contrast, both JNK inhibitor JNK-IN-8 and JNK1/2 siRNA knockdown JNK1/2 can reduce activation of the NF-κB and apoptosis in H9c2 cells under H/R conditions. The study demonstrated that hypoxic AMPK activation controls mPTP opening and ROS generation that triggers the JNK signaling pathway; in other words, JNK activation is a downstream event of mPTP opening and ROS generation by hypoxic stress. Therefore, the inhibition of the JNK signaling pathway did not affect mPTP opening and ROS generation in the H9c2 cells. AMPK regulation of the JNK signaling cascade could make a critical contribution to limiting cell damage by hypoxic stress via modulating mitochondrial homeostasis and inflammatory response.
Finally, a well-known AMPK activator, metformin, was used to test whether pharmacological intervention could be applied to prevent H9c2 myoblast cells from hypoxic injury. Metformin can activate the AMPK signaling pathway in H9c2 cells under both normoxia and H/R conditions. Metformin inhibited mPTP opening and ROS generation caused by H/R. Moreover, metformin treatment attenuates JNK-NF-κB signaling and inhibits mRNA levels of pro-inflammatory cytokines TNF-α and IL-6 during H/R.
In conclusion, the results indicate that intrinsic AMPK plays a critical role in maintaining mitochondrial integrity during hypoxic stress conditions; thus, AMPK can control ROS production and the JNK-NF-κB signaling cascade for inflammatory response and can limit cell damage under H/R conditions.
Supplementary Material
Highlights.
AMPK activation modulates the c-Jun N protein kinase (JNK) signaling pathway under hypoxic stress conditions.
The opening of the mitochondrial permeability transition pore (mPTP) is controlled by AMPK in response to hypoxia and reoxygenation.
Pharmacological AMPK activation could inhibit the inflammatory response during hypoxia and reoxygenation.
Acknowledgments
Funding
These studies were supported by American Diabetes Association 1-17-IBS-296, NIH R21AG044820, R01AG049835, P01HL051971, and P20GM104357, the Taishan Scholar Program of Shandong Province (tshw201502046), and 2016 Yantai Shuangbai Scholar Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Design and conduct of the study: XC, XL, WZ, JH, BX, BL, WZ, CC, TR, and JL. Data collection and analysis: XC, XL, CC, TR, and JL. Data interpretation: XC, XL, ZW, CC, TR, and JL. Manuscript writing: XC, CC, TR, and JL.
Conflict of Interest
None.
References
- 1.Kovell LC, Juraschek SP, Russell SD. Stage A Heart Failure Is Not Adequately Recognized in US Adults: Analysis of the National Health and Nutrition Examination Surveys, 2007–2010. PloS one. 2015;10:e0132228. doi: 10.1371/journal.pone.0132228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Djousse L, Driver JA, Gaziano JM. Relation between modifiable lifestyle factors and lifetime risk of heart failure. Jama. 2009;302:394–400. doi: 10.1001/jama.2009.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gopal DM, Kalogeropoulos AP, Georgiopoulou VV, Smith AL, Bauer DC, Newman AB, et al. Cigarette smoking exposure and heart failure risk in older adults: the Health, Aging, and Body Composition Study. American Heart Journal. 2012;164:236–42. doi: 10.1016/j.ahj.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418–58. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- 5.Raedschelders K, Ansley DM, Chen DD. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacology & Therapeutics. 2012;133:230–55. doi: 10.1016/j.pharmthera.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Ishii D, Schenk AD, Baba S, Fairchild RL. Role of TNFalpha in early chemokine production and leukocyte infiltration into heart allografts. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2010;10:59–68. doi: 10.1111/j.1600-6143.2009.02921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–20. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 8.Qi D, Young LH. AMPK: energy sensor and survival mechanism in the ischemic heart. Trends in Endocrinology and Metabolism. 2015;26:422–9. doi: 10.1016/j.tem.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Tong C, Yan X, Yeung E, Gandavadi S, Hare AA, et al. Limiting cardiac ischemic injury by pharmacological augmentation of macrophage migration inhibitory factor-AMP-activated protein kinase signal transduction. Circulation. 2013;128:225–36. doi: 10.1161/CIRCULATIONAHA.112.000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrison A, Li J. PPAR-gamma and AMPK–advantageous targets for myocardial ischemia/reperfusion therapy. Biochemical Pharmacology. 2011;82:195–200. doi: 10.1016/j.bcp.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Annals of the New York Academy of Sciences. 2008;1147:37–52. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petronilli V, Cola C, Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. II. The minimal requirements for pore induction underscore a key role for transmembrane electrical potential, matrix pH, and matrix Ca2+ The Journal of Biological Chemistry. 1993;268:1011–6. [PubMed] [Google Scholar]
- 14.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:15983–7. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weston CR, Davis RJ. The JNK signal transduction pathway. Current Opinion in Cell Biology. 2007;19:142–9. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Schulz E, Dopheide J, Schuhmacher S, Thomas SR, Chen K, Daiber A, et al. Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation. 2008;118:1347–57. doi: 10.1161/CIRCULATIONAHA.108.784298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu M, Ye P, Liao H, Chen M, Yang F. Metformin Protects H9C2 Cardiomyocytes from High-Glucose and Hypoxia/Reoxygenation Injury via Inhibition of Reactive Oxygen Species Generation and Inflammatory Responses: Role of AMPK and JNK. Journal of Diabetes Research. 2016;2016:2961954. doi: 10.1155/2016/2961954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. Journal of Neuroimmunology. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramachandran S, Liaw JM, Jia J, Glasgow SC, Liu W, Csontos K, et al. Ischemia-reperfusion injury in rat steatotic liver is dependent on NFkappaB P65 activation. Transplant Immunology. 2012;26:201–6. doi: 10.1016/j.trim.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Himaya SW, Ryu B, Qian ZJ, Kim SK. Paeonol from Hippocampus kuda Bleeler suppressed the neuro-inflammatory responses in vitro via NF-kappaB and MAPK signaling pathways. Toxicology in Vitro. 2012;26:878–87. doi: 10.1016/j.tiv.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 21.Quan N, Sun W, Wang L, Chen X, Bogan JS, Zhou X, et al. Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. FASEB J. 2017;31:4153–67. doi: 10.1096/fj.201700063R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang H, Sun W, Quan N, Wang L, Chu D, Cates C, et al. Cardioprotective actions of Notch1 against myocardial infarction via LKB1-dependent AMPK signaling pathway. Biochemical pharmacology. 2016;108:47–57. doi: 10.1016/j.bcp.2016.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaha VG, Qi D, Su KN, Palmeri M, Lee HY, Hu X, et al. AMPK is critical for mitochondrial function during reperfusion after myocardial ischemia. J Mol Cell Cardiol. 2016;91:104–13. doi: 10.1016/j.yjmcc.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, et al. Discovery of potent and selective covalent inhibitors of JNK. Chemistry & Biology. 2012;19:140–54. doi: 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Y, Li J. Metabolic shifts during aging and pathology. Compr Physiol. 2015;5:667–86. doi: 10.1002/cphy.c140041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 28.Moussa A, Li J. AMPK in myocardial infarction and diabetes: the yin/yang effect. Acta Pharmaceutica Sinica B. 2012;2:368–78. doi: 10.1016/j.apsb.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palomer X, Salvado L, Barroso E, Vazquez-Carrera M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int J Cardiol. 2013;168:3160–72. doi: 10.1016/j.ijcard.2013.07.150. [DOI] [PubMed] [Google Scholar]
- 30.Ma Y, Wang J, Gao J, Yang H, Wang Y, Manithody C, et al. Antithrombin up-regulates AMP-activated protein kinase signalling during myocardial ischaemia/reperfusion injury. Thromb Haemost. 2015;113:338–49. doi: 10.1160/TH14-04-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura M, Wang NP, Zhao ZQ, Wilcox JN, Thourani V, Guyton RA, et al. Preconditioning decreases Bax expression, PMN accumulation and apoptosis in reperfused rat heart. Cardiovascular Research. 2000;45:661–70. doi: 10.1016/s0008-6363(99)00393-4. [DOI] [PubMed] [Google Scholar]
- 32.Ekshyyan O, Aw TY. Apoptosis in acute and chronic neurological disorders. Frontiers in Bioscience. 2004;9:1567–76. doi: 10.2741/1357. [DOI] [PubMed] [Google Scholar]
- 33.Bouma HR, Ketelaar ME, Yard BA, Ploeg RJ, Henning RH. AMP-activated protein kinase as a target for preconditioning in transplantation medicine. Transplantation. 2010;90:353–8. doi: 10.1097/TP.0b013e3181e7a3aa. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Yang L, Rezaie AR, Li J. Activated protein C protects against myocardial ischemic/reperfusion injury through AMP-activated protein kinase signaling. J Thromb Haemost. 2011;9:1308–17. doi: 10.1111/j.1538-7836.2011.04331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Wang Y, Gao J, Tong C, Manithody C, Li J, et al. Antithrombin is protective against myocardial ischemia and reperfusion injury. J Thromb Haemost. 2013;11:1020–8. doi: 10.1111/jth.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onai Y, Suzuki J, Kakuta T, Maejima Y, Haraguchi G, Fukasawa H, et al. Inhibition of IkappaB phosphorylation in cardiomyocytes attenuates myocardial ischemia/reperfusion injury. Cardiovascular Research. 2004;63:51–9. doi: 10.1016/j.cardiores.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Pan Y, Zhang X, Wang Y, Cai L, Ren L, Tang L, et al. Targeting JNK by a new curcumin analog to inhibit NF-kB-mediated expression of cell adhesion molecules attenuates renal macrophage infiltration and injury in diabetic mice. PloS one. 2013;8:e79084. doi: 10.1371/journal.pone.0079084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.