

Abstract
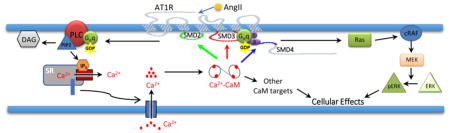
The angiotensin II receptor type 1 (AT1R) mediates many Ca2+-dependent actions of angiotensin II (AngII). Calmodulin (CaM) is a key transducer of Ca2+ signals in cells. Two locations on the receptor’s submembrane domains (SMD) 3 and 4 are known to interact with CaM. However, the binding sites for CaM, biochemical properties of the interactions, and their functional impact are not fully understood. Using a FRET-based screening method, we identified a new binding site for CaM on SMD2 (a.a. 125–141), in addition to SMD3 and the juxtamembranous region of SMD4 (SMD4JM, a.a., 309–327). Simultaneous measurements of CaM binding and free Ca2+ show that the interactions are Ca2+-dependent, with disparate Kd and EC50(Ca2+) values within the physiological range of cytoplasmic Ca2+. Full interaction between CaM and SMD3 requires the entire domain (a.a. 215–242) and has an EC50(Ca2+) value in the range of resting cytoplasmic Ca2+, suggesting AT1R-CaM interaction can occur in resting conditions in cells. AngII induces robust ERK1/2 phosphorylation in primary vascular smooth muscle cells. This effect is suppressed by AT1R inhibitor losartan and virtually abolished by CaM antagonist W-7. AngII-induced ERK1/2 phosphorylation is suppressed in cells expressing mutant AT1R with reduced CaM binding at each identified binding domain. AngII triggers transient Ca2+ signals in cells expressing wild-type AT1R. These signals are reduced in cells expressing mutant AT1R with reduced CaM binding at SMD3 or SMD4JM, but are very slow-rising, low amplitude signal in cells expressing AT1R with reduced CaM binding at SMD2. The data indicate that CaM interactions with AT1R can occur at various domains, with different affinities, at different physiological Ca2+ levels, and are important for AT1R-mediated signaling.
Keywords: Angiotensin II receptor type 1, calmodulin, calcium, FRET biosensor, angiotensin II
Graphical Abstract
1. Introduction
Angiotensin II (AngII) is among the most potent vasoactive substances produced in humans. Its effects are numerous, from vasoconstriction to control of fluid and electrolytes balances. Many of the physiological and pathological effects of AngII are mediated by the angiotensin II receptor type 1A (AT1R). Activation of AT1R triggers cardiovascular remodeling in subjects with hypertension and heart failure [19]. Pharmacological inhibition of AT1R has been exploited extensively in the treatment of hypertension and heart failure. Classically, AT1R is a 7-pass transmembrane receptor associated with Gαq/11, whose dissociation upon AngII binding to AT1R is associated with increases in intracellular free Ca2+ concentration, followed by both pre-genomic actions such as contractions of the heart and muscle cells and genomic actions that alter the expression of various genes [17]. AT1R also interacts with β-arrestin, triggering a G protein-independent cascade of downstream events, with distinct physiological outcomes [11]. Additionally, the activities of AT1R are regulated by receptor-interacting proteins [1, 6, 32]. Nevertheless, aspects of signaling via AT1R at the receptor level and its regulatory inputs remain incompletely understood.
Calmodulin is a ubiquitous transducer of intracellular Ca2+ signals. It is estimated to bind up to 300 intracellular proteins [21]. Despite its universal requirement, CaM is not expressed sufficiently for all its binding sites within the cell [10]. Shortage of CaM relative to its binding sites [12, 14, 23, 24, 31], coupled with disparate binding affinities for its target proteins from ~ 10−11 – 10−5 M, has made CaM a target for dynamic competition among its binding proteins, leading to new modes of functional coupling in cells [23, 24]. Over the last two decades, CaM has been shown to interact with several GPCRs, such as the metabotropic glutamate receptors mGluR1 and mGluR5 [18], opioid μ receptors [30], the parathyroid hormone receptor 1 [15], the 5-HT(1A) and 5-HT(2C) receptors [3, 28], the D2 dopamine receptors [7], AT1R [22, 32], and the G protein-coupled estrogen receptor 1 (GPER) [25, 27]. CaM binding to the 5-HT(1A) alters receptor phosphorylation and G protein coupling [28]; while its association with 5-HT(2C) and GPER is important receptor-mediated ERK1/2 activation [3, 25]. In 1999, a CaM-binding domain was identified at the juxtamembranous region of the 4th submembrane domain (SMD4JM) of AT1R (a.a. 305–327, rat sequence) [22]. The interaction was shown to be Ca2+-dependent. In 2013, Zhang et al. identified another CaM-binding domain on SMD3 (a.a. 215–232) and confirmed the previously identified domain on the juxtamembranous domain of the 4th submembrane domain of the receptor [32]. In that work, CaM was shown to compete for Gβγ association with both the partial SMD3 and SMD4JM peptides in vitro, indicating a role for CaM binding in Gβγ coupling.
Despite identification of two CaM-binding domains on AT1R, questions remain to be answered regarding total number of binding sites, biochemical properties and regulatory inputs of the interactions with CaM. Additionally, the functional impact of these interactions in cells are unknown. For example, are segments a.a. 305–327 (rat) in SMD4JM [22] and a.a. 215–232 (rat sequence) in SMD3 [32] the only locations where CaM interacts with AT1R? How much Ca2+ is needed for these interactions to occur? In this regard, for most known CaM-GPCR interactions, information is only available whether CaM binding to GPCR domains occurs in saturating Ca2+ or absence of Ca2+. The lack of EC50(Ca2+) values for CaM-GPCR domain interactions makes it difficult to predict when an interaction occurs in cells. Additionally, what are the functional implications of AT1R-CaM interactions? We recently used a FRET-based method to identify and characterize four distinct CaM-binding domains in the G protein-coupled estrogen receptor 1 (GPER) [27]. The method is highly specific and allows for precise determinations of apparent Kd values of CaM binding to the GPCR domains as inserts between the donor-acceptor FRET pair. These values serve as sensitive parameters to determine the binding domains, especially those that do not conform to any known binding motifs. Simultaneous measurements of biosensor-CaM interaction and responses of suitable Ca2+ indicator also enables determination of the precise EC50(Ca2+) values of the interactions. These values, coupled with measured free Ca2+ concentrations in cells, enable predictions of the physiological scenarios in which a particular CaM GPCR interaction may occur [27].
In this study, we have generated new FRET reporters to screen all submembrane domains of AT1R for interactions with CaM. We identified a new CaM-binding domain on the receptor’sSMD2 (a.a. 125 – 141), confirmed interactions at SMD4JM (a.a. 309 – 327) and characterized the full interacting domain in SMD3. All three domains interact with CaM in a Ca2+-dependent fashion. EC50(Ca2+) values determined for each specific interaction by simultaneous measurements of biosensor responses and free Ca2+ indicate that AT1R-CaM binding at different SMDs can take place at drastically different physiological Ca2+ concentrations in the cell. Mutagenesis studies were used in vitro and in cells to examine the functional impact of CaM binding at the individual sites on AT1R-mediated signaling.
2. Materials and Methods
2.1. Materials
All molecular biology enzymes and competent cells were from New England Biolabs (Ipswich, MA). RNeasy kit was from Qiagen. High Capacity cDNA reverse transcription kit was from Applied Biosystems (Foster City, CA). Fetal bovine serum, isopropyl β-D-1-thiogalactopyranoside, and M-199 culture medium were obtained from Sigma-Aldrich (St. Louis, MO). HisPur cobalt resin was from Thermo Fisher (Waltham, MA). Phenyl Sepharose CL-4B resin was from GE Healthcare Life Sciences (Milwaukee, MI). DMEM culture medium was from Caisson Laboratories (Smithfield, UT). Polyethylenimine and penicillin/streptomycin were from ThermoFisher (Carlsbad, CA). Fura-2/AM was purchased from Teflabs (Austin, TX). Ionomycin, indo-1, XRhod-5F, collagenase type A, papain and dithiothreitol were obtained from ThermoFisher Scientific (Carlsbad, CA). Angiotensin II peptide, W-7 and losartan potassium were from Sigma Aldrich (St. Louis, MO). Pierce™ BCA protein assay kit, enhanced chemiluminescence detection kit, and Br2BAPTA were from ThermoScientific (Carlsbad, CA). Bovine serum albumin (BSA) was from Research Products International Corp. (Mt. Prospect, IL). Anti-β actin antibody was from ThermoScientific (RB9421P1). Anti-smooth muscle α actin antibody was from Abcam (ab5694, Cambridge, MA). Anti-ERK1/2 antibody was from Santa Cruz Biotechnology (SC514302, Santa Cruz, CA). Anti-phospho-p44/42 MAPK mouse monoclonal antibody (L34F12) and rabbit anti-hemagglutinin (HA, C29F4) antibody were obtained from Cell Signaling Technology (Danvers, MA).
2.2. Molecular Biology
Total mRNA from HEK 293 cells was isolated using the RNeasy mini kit. Full-length human AT1R was reverse transcribed from this total mRNA at 100 ng/μL RNA using the High Capacity cDNA reverse transcription kit. To clone AT1R in frame, a BamHI restriction site was incorporated into the N-terminus of the receptor, while a stop codon followed by an XhoI restriction site was incorporated into its C-terminus. Primers for this reaction are listed in Table-1. The BamHI- AT1R-XhoI sequence was then cloned into a pcDNA3.1 mammalian expression vector downstream a hemagglutinin (HA) epitope tag as previously described [27].
Table 1.
Primers
| Fragment generated | Forward primer | Reverse primer |
|---|---|---|
| BamHI-AT1R-Stop-XhoI | CTCGGGATCCATGATTCTCAACTCTTC | CTCGCTCAAGTCACTCAACCTCAAAAC |
| KpnI-mKate2-BamHI | CGACGGTACCATGTCAGAACTTATCAAGGA | CGC TGG ATC CTC TGT GCC CCA GTT T |
| KpnI-AT1R52–64-AgeI | CTCGGGTACCGTCATTTACTTTTATAT | CTCGACCGGTACTGGCCACAGTCTTCA |
| KpnI-AT1R125–141-AgeI | CTCGGGTACCGATCGATACCTGGCTAT | CTCGACCGGTTGTGCGTCGAAGGCG |
| KpnI-AT1R215–232-AgeI | CTCGGGTACCTATACTCTTATTTGG | CTCGACCGGTTTTGTTCTTCTTCTGAATTTC |
| KpnI-AT1R215–242-AgeI | CTCGGGTACCTATACTCTTATTTGG | CTCGACCGGTAATTATCTTAAAAAT |
| KpnI-AT1R309–327-AgeI | CTCGGGTACCTTTAAAAGATATTTT | CTCGACCGGTGTGGGATTTGGCTTTTGG |
| KpnI-AT1R304–323-AgeI | CTC GGG TAC CTT TCT GGG GAA AAA A | CTC GAC CGG TTT TTG GGG GAA TAT A |
| KpnI-AT1R305–327-AgeI | CTC GAC CGG TCT GGG GAA AAA ATT T | CTC GAC CGG TGT GGG ATT TGG CTT TTG G |
| KpnI-AT1R328–359-AgeI | CTCGGGTACCTCAAACCTTTCAACAAAA | CTCGACCGGTGAATTCCTCAACCTCAAAACATGGTGC |
| KpnI-AT1R125–141mut-AgeI | CTGTGGTACCGATCAAGCCCTGGC | CTGGACCGGTTGTTTGTCGAAGTTGGG |
| KpnI-AT1R215–242mut-AgeI | CAGCGGTACCTATACTCTTATTGCG | CGCCACCGGTAATTATCTTAGCAAT |
| KpnI-AT1R309–327mut-AgeI | CGCTGGTACCGCTAAAAGATATGCTC | GCTACCGGTGTGGGATTGGGCTTTT |
Constructs for FRET-based biosensors to screen for CaM binding domains in AT1R were generated as described previously for GPER [27]. Insert sequences from AT1R’s submembrane domains or fragments thereof were PCR amplified from the full-length AT1R sequence, with incorporation of the KpnI and AgeI restriction sites to the N- and C-terminal ends of the sequences. The insert sequences were then substituted in frame in between the citrine EYFP (EYFPc)-ECFP pair of previously reported pET bacterial expression vectors that encode GPER biosensors [25, 27] to generate biosensor for AT1R. As with previously described biosensors for GPER, we used the BSAT1Rx nomenclature, where x denotes the amino acid sequence (human) of the AT1R fragment used as biosensor insert. Biosensors and primers are listed in Table-1.
Substitutions in the submembrane domains of AT1R to reduce CaM binding were generated by incorporating gBlock gene fragments containing the desired sequence (IDTDNA, Coraville, IA) in frame into the full-length receptor. The SMD wild-type and mutant amino acid sequences are listed in Table-2. Gblock gene fragments for the mutant sequences as well as restriction enzymes used to clone the mutant domains in frame to full-length AT1R are listed in Table-3. Mutant biosensors were subsequently generated by PCR amplifying SMD sequences from the full-length mutant AT1R, followed by incorporation of these sequences in between the EYFPc-ECFP pair as described above. Primers to generate the mutant inserts are listed in Table-1.
Table 2.
Substitutions in AT1R’s CaM-binding domains
| Domain targeted | Wild-type sequence | Mutant sequence |
|---|---|---|
| SMD2 (AT1R125–141) | DRYLAIVHPMKSRLRRT | DQALAIVHPMKSQLRQT |
| SMD3 (AT1R215–242) | YTLIWKALKKAYEIQKNKPRNDDIFKII | YTLIAKALKKAAEIQKNQPRNDDIAKII |
| SMD4JM (AT1R309–327) | FKRYFLQLLKYIPPKAKSH | AKRYALQLLKAIPPKAQSH |
Table 3.
Gblock gene fragments. Substitutions from wild-type sequenced are in bold-face fonts and italicized. Identified CaM binding domain sequences are underlined.
| AT1R Fragment |
gBlock sequence | Restriction enzymes to clone to full-length AT1R |
|---|---|---|
| 125–141 mut | CTACTCACGTGTCTCAGCATTGATCAAGCCCTGGCTATTGTTCACCCAATG AAGTCCCAACTTCGACAAACAATGCTTGTAGCCAAAGTCACCTGCATCAT CATTTGGCTGCTGGCAGGCTTGGCCAGTTTGCCAGCTATAATCCATCGAA ATGTATTTTTCATTGAGAACACCAATATTACAGTTTGTGCTTTCCATTATG AGTCCCAAAATTCAACCCTCCCGATAGGGCTGGGCCTGACCAAAAATATA CTGGGTTTCCTGTTTCCTTTTCTGATCATTCTTACAAGTTATACTCTTATTG CGAAGGCCCTAAAGAAGGCTTATGAAAT |
PmlI – EcoNI |
| 215–242 mut | CAGCATTGATCGATACCTGGCTATTGTTCACCCAATGAAGTCCCGCCTTC GACGCACAATGCTTGTAGCCAAAGTCACCTGCATCATCATTTGGCTGCTG GCAGGCTTGGCCAGTTTGCCAGCTATAATCCATCGAAATGTATTTTTCATT GAGAACACCAATATTACAGTTTGTGCTTTCCATTATGAGTCCCAAAATTC AACCCTCCCGATAGGGCTGGGCCTGACCAAAAATATACTGGGTTTCCTGT TTCCTTTTCTGATCATTCTTACAAGTTATACTCTTATTGCGAAGGCCCTAA AGAAGGCTGCTGAAATTCAGAAGAACCAACCAAGAAATGATGATATTGC TAAGATAATTATGGCAATTGTGCTTTTCTTTTTCTTTTCCTGGATTCCCCAC CAAATATTCACTTTTCTGGATGTATTGATTCAACTAGGCATCATACGTGAC TGTAGAATTGCAGATATTGTGGACACGGCCATGCCTATCACCATTTGTAT AGCTTATTTTAACAATTGCCTGAATCCTCTTTTTTATGGCTTTCTGGGGAA AAAATTTAAAAGATATTTTCTCCAGCTTCTAAAATATATTCCCCCAAAAG CCAAATCCCACTCAAACCTTTCAACAAAAATGAGCACGCTTTCCTACCGC CCCTCAGATAATGTAAGCTCATCCACCAAGAAGCCTGCACCATGTTTTGA GGTTGAGTGACTCGAGCATGCATC |
ClaI – XhoI |
| 309–327 mut | GAAGGCCCTAAAGAAGGCTTATGAAATTCAGAAGAACAAACCAAGAAAT GATGATATTTTTAAGATAATTATGGCAATTGTGCTTTTCTTTTTCTTTTCCT GGATTCCCCACCAAATATTCACTTTTCTGGATGTATTGATTCAACTAGGCA TCATACGTGACTGTAGAATTGCAGATATTGTGGACACGGCCATGCCTATC ACCATTTGTATAGCTTATTTTAACAATTGCCTGAATCCTCTTTTTTATGGC TTTCTGGGGAAAAAAGCTAAAAGATATGCTCTCCAGCTTCTAAAAGCTAT TCCCCCAAAAGCCCAATCCCACTCAAACCTTTCAACAAAAATGAGCACGC TTTCCTACCGCCCCTCAGATAATGTAAGCTCATCCACCAAGAAGCCTGCA CCATGTTTTGAGGTTGAGTGACTCGAGCATGCATC |
EcoNI – XhoI |
To generate fusions between mKate2 and wild-type or mutant AT1R, mKate2 was PCR amplified from the pDONR-P4-P1-mKate2 plasmid, a gift from Dr Planas, Institut d’Investigacions Biomediques de Barcelona (Addgene.org). The KpnI and BamHI restriction sites were incorporated into the N- and C-terminal ends of mKate2. This fragment was then substituted for the HA tag in the HA-AT1R fusions. All constructs were verified by DNA sequencing (University of Missouri).
2.3. Expression and purification of proteins
pET plasmids encoding biosensors and CaM were expressed in BL21(DE3) cells. Biosensors and CaM were purified as described in detail previously [25, 27].
2.4. Screening for CaM-binding domains in AT1R using BSAT1RX
Purified BSAT1RX were mixed at 22°C with titration buffer (25 mM Tris, 100 mM KCl, pH 7.5), 0.1 mg/mL bovine serum albumin and 1 mM CaCl2 in a quartz cuvette (Hellma Analytics, Plainview, NY). Incremental purified CaM was added to the mixture as BSAT1RX fluorescence spectra were monitored in a QuantaMaster™-40 spectrofluorometer (Photon Technology International, Inc., Birmingham, NJ). Direct interaction between Ca2+-CaM and BSAT1RX insert was defined by disruption of FRET between ECFP and EYFPc, manifested as an increase in ECFP emission, a decrease in EYFPc emission and crossing of ECFP and EYFPc spectra at the isoemissive point (~510 nm). BSAT1RX fractional responses were determined by the formula
| (1) |
where Rmin and Rmax are the ratios between emission intensities at λ475 and λ535 nm (F475/F535) when the biosensor is in unbound and maximally bound state, respectively. Dilution due to addition of CaM was minimal, yet was calculated using a detailed excel spreadsheet algorithm; however, the use of ratio in formula (1) negated the need to correct for dilution. Fractional biosensor responses were plotted against titrated CaM concentrations. Apparent Kd values of BSAT1RX-CaM interactions were obtained by fitting fractional responses as a function of CaM concentration to a hyperbolic or quadratic binding equations as described [20, 27]:
| (2) |
| (3) |
where BSfract, [BS] and [CaM] are BSAT1RX fractional response, total concentration of BSAT1RX and CaM in the reaction mix, respectively.
2.5. Measurement of Ca2+ sensitivities of AT1R-CaM interactions
Ca2+ sensitivities of the interactions between CaM and different submembrane domains of BSAT1RX were measured by simultaneously monitoring the responses of a suitable Ca2+ indicator and BSAT1RX in the presence of saturating CaM concentration (determined from Kd titrations) and incremental increases in added Ca2+. Starting reaction mixture contained 0.5 μM BSAT1RX, 2 μM Ca2+ indicator, 0.1 mg/ml BSA, 0.25 mM Br2BAPTA, and saturating CaM concentration (determined from Kd determinations), in titration buffer at 22°C. The choice of suitable Ca2+ indicators were made by initially comparing the responses of BSAT1RX and a Ca2+ indicator to achieve relatively consistent response ranges as Ca2+ was titrated in the mixture. For example, a Ca2+ indicator whose response was saturated before BSAT1RX begins to respond, or vice-versa, would be unsuitable for measurement of Ca2+ sensitivity. These initial titrations identified XRhod-5F (Kd for Ca2+ = 1.6 μM) and indo-1 (Kd for Ca2+ = 0.23 μM) as suitable indicators for BSAT1RX. For XRhod-5F, free Ca2+ concentrations were determined by its emission intensity at λ600nm with excitation at λ580 nm, measured simultaneously with BSAT1RX responses. Dilution factors, though minimal, were carefully recorded on an Excel spreadsheet for each addition of Ca2+ and used to correct for emission intensity. Free Ca2+ values were calculated using the formula
| (4) |
where, 1.6 is the in vitro Kd value of XRhod-5F for Ca2+; Fmin and Fmax are XRhod-5F emission intensities at λ600nm under nominally Ca2+-free and Ca2+-saturating conditions, respectively. For indo-1, indicator responses were obtained as ratios between emission intensities at λ405nm and λ485nm, with excitation at λ340nm. Free Ca2+ values were calculated from these ratios using the formula
| (5) |
where 0.23 is the in vitro Kd of indo-1 for Ca2+ in μM, R is the ratio between indo-1 emission intensities at λ405nm and λ485nm, Rmin and Rmax are the F405/F485 ratios obtained at nominally Ca2+-free and Ca2+-saturating conditions, respectively.
Ca2+ sensitivities of BSAT1RX-CaM interactions were determined by the EC50(Ca2+) values, derived from fits of BSAT1RX fractional responses (eq. 1) as a function of free Ca2+ using the equation:
| (6) |
where BSfract is obtained from equation (1); n is the Hill coefficient.
2.6. Cell isolation and culture
Primary VSMCs were isolated from descending section of porcine thoracic aortas based on a modification of the methods published by Bolzon, Ulrich-Merzenich and Leik as recently described [8, 13, 27, 29]. Descending porcine thoracic aortas were obtained fresh from a local slaughter house and transported in sterile phosphate buffered saline containing 3% penicillin/streptomycin. After removal of perivascular adipose tissue, the aortas were cut open and endothelial cells were removed by mechanical scraping and short incubation with 0.02% collagenase, 0.1% papain and 4 mM dithiothreitol and re-scraping. After rinsing in sterile PBS, the aortas were dissected into strips and the luminal surface was incubated with the enzyme mix for 90 minutes at 37°C. The luminal surface was subsequently scraped again, and cells from this treatment were grown in M-199 medium containing 10% fetal bovine medium and 2% penicillin/streptomycin in 90% humidified condition with 5% CO2 at 37°C for one week. This approach consistently yields highly homologous populations of VSMCs, verified by both morphological features and smooth muscle α-actin expression. VSMCs were used for 2–3 passages after isolation. The protocol was approved by Des Moines University Institutional Biosafety Committee. Immortalized human embryonic kidney (HEK293) cells were purchased from ATCC (CRL-1573, initial passage 15) and cultured in DMEM medium containing and 10% fetal bovine serum. Cells were used for up to 15 passages from the initial stock from vendor.
2.7. Transfection
HEK293 cells were transfected with plasmids encoding mKate2 in isolation or fused with wild-type or mutant full-length AT1R using polyethylenimine (PEI) based on a modification of the method published by Aricescu et al. [2]. Briefly, HEK293 cells were grown to 60% confluency by the time of transfection. Plasmid DNA and PEI were allowed to form a complex at room temperature at a PEI:DNA mass ratio of 1.5:1 for 20 minutes in serum-free, antibiotic-free culture medium (DMEM or M-199). The cells were subsequently incubated with 1:5 vol/vol DNA-PEI complex in DMEM containing 2% FBS for 6 hours before medium was replaced by regular culture medium. The cells were cultured for another 12 hrs before experimentation.
2.8. Western Blotting
All cell treatments were performed at room temperature in modified Tyrode’s buffer(composition in mM: 150 NaCl, 2.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 10 HEPES, pH 7.5) containing 1.5 mM CaCl2. Following treatment and cell lysis, the protein content was measured using BCA assay performed in triplicate for all samples. All lanes in each gel were loaded with equal amounts of total proteins, adjusted for detectability with each antibody. After detection of the levels of ERK1/2 phosphorylation, the same membrane fragments were stripped and reprobed for total ERK1/2. Following secondary incubation, membrane fragments were developed using enhanced chemiluminescence in a ChemiDoc™ XRS+ imaging system (Bio-Rad). Image Lab 5.0 software (Bio-Rad) was used for densitometry. Densitometric values of phosphorylated ERK1/2 bands were corrected for corresponding values of total ERK1/2 bands and normalized to control values.
2.9. Measurement of intracellular Ca2+ concentration in HEK293 cells expressing mKate2-AT1R
Intracellular Ca2+ concentration was measured in HEK293 cells expressing mKate2 only or mKate2 in fusion with wild-type or mutant full-length AT1R as described previously [25, 26]. HEK293 cells were loaded with 4 μM fura-2/AM for 30 min in culture medium at 37°C. After removal of fura-2/AM, the cells were equilibrated in modified Tyrode’s buffer (composition in mM: 150 NaCl, 2.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 10 HEPES, 1 CaCl2, pH 7.4) for 15 min at room temperature. mKate2 fluorescence was first identified and selected in RFP channel. Filter cube was then switched to fura-2 channel without changing the microscopic field. Excitation of fura-2 was alternated between λ340 nm and λ380 nm for 100 ms per 1-s cycle from an ultra-high speed wavelength switcher (Lambda DG-4, Sutters Instruments) at 1-ms switching interval. Emission at λ510 nm was collected via an EMCCD camera (DU-885, Andor Technology). At the end of the agonist-induced Ca2+ signal time course, Rmax values of fura-2 were obtained in all cells by adding 10 μM ionomycin and 10 mM CaCl2. Rmin values were determined in all cells using a previously developed equation [26]. Free Ca2+ concentrations were calculated using the standard equation
| (7) |
Where the Kd value is 224 nM for fura-2; R is the observed fura-2 ratio during the experiments. Sf and Sb represent the emission intensities at λ510 nm corresponding to the Ca2+-free and Ca2+-bound states of fura-2. Sb values were readily measured in all cells together with Rmax values. Sf values were determined in each cell using a previously developed equation [26].
2.10. Statistical Analysis
Data were normally distributed and are expressed as means ± SD. Statistical analysis was performed using one-way ANOVA, followed by a Tukey post-hoc test. Statistical significance was set at P < 0.05.
3. Results
3.1. Screening for all CaM-binding domains in AT1R
The two known CaM-binding domains in AT1R include a.a. 215–232 in SMD3 and a.a. 305–327 in SMD4JM [22, 32]. To confirm and screen for all CaM-binding sites on AT1R, we used a FRET-based approach to screen for CaM-binding sites in the submembrane domains of human AT1R. This method has allowed us to identify four distinct CaM-binding sites on the four submembrane domains of GPER, some of which do not conform to any known CaM-binding motifs [27]. We generated a host of FRET biosensors that consist of partial or full-length submembrane domains of AT1R flanked by the FRET donor ECFP and acceptor citrine EYFP (EYFPc). These included BSAT1R52–64 (covering SMD1), BSAT1R125–141, (covering SMD2), BSAT1R 215–232 (covering previously identified CaM-binding domain in SMD3 [32]), BSAT1R215–242 (covering the entire SMD3), and a series of BSAT1RX to scan SMD4 due to the length of this domain (Table-1). Figures 1A–B recapitulate the principle of the biosensors for screening of CaM-binding sequences. A domain to be screened for CaM binding is inserted between EYFPc-ECFP (see also Materials and Methods). In the absence of CaM interaction with the insert sequence, proximity between EYFPc and ECFP allows for robust FRET between them upon excitation of the ECFP moiety (430 nm) (Fig. 1A). Specific interaction between CaM and the insert domain will disrupt FRET (Fig. 1B), causing an increase in ECFP emission, a decrease in EYFP emission, and crossing of the emission spectra at the isoemissive point (~510 nm) [27]. In the absence of specific binding with the insert, up to 700 μM CaM caused no emission spectral changes of the FRET pair [27]. With high sensitivity of FRET change in response to conformational changes of the insert upon CaM binding, it is easy to determine with high precision the apparent binding affinities between CaM and the insert domain in biosensor format. The length of the insert domain can be varied until the highest binding affinity is achieved, which identifies the correct domain required for full interaction with CaM. Figure 1C shows a Coomassie gel of the biosensors characterized. Figure 1D shows Coomassie staining of three separate preparations of CaM used to screen CaM binding. All proteins showed as single bands on the gels, demonstrating high purity.
Fig. 1.

Method to screen for CaM binding domains in AT1R. (A & B) Design of FRET biosensors to screen for CaM interaction (see text for explanation), depicted in unbound (A) and CaM-bound (B) conditions. EYFPc, citrine enhanced yellow fluorescence protein; ECFP, enhanced cyan fluorescence protein; N and C, N-terminal and C-terminal ends, respectively, of fluorophores. (C) Representative Coomassie gel of the biosensors generated. (D) Coomassie gel of three separate CaM preps.
Using these biosensors, our first goal was to confirm the reported interactions at a.a. 305–327 on SMD4JM and a.a. 215–232 on SMD3 and to screen for any other sites of interactions on the submembrane domains. As reported interactions were Ca2+-dependent, all screens were first done with Ca2+-liganded purified CaM. BSAT1R52–64 (covering SMD1) did not interact with CaM; its emission spectra virtually overlapped in response to buffer alone or high concentration of CaM (Fig. 2A). Surprisingly, BSAT1R125–141, which covers SMD2, demonstrated clear signature spectral changes upon Ca2+-CaM titration (Fig. 2B). Initial screenings for the entire SMD4 sequence using BSAT1RX spanning a.a. 304–323, 305–327, 309–327, and 328–359 indicated that BSAT1R328–359 does not interact with CaM (not shown), while BSAT1R309–327 possesses the highest affinity for interaction with CaM of all BSAT1RX covering the SMD4JM region. Figure 2C shows the signature spectral changes for positive interaction upon titration of Ca2+-liganded CaM into BSAT1R309–327. BSAT1R215–232 also showed positive responses to Ca2+-liganded CaM, confirming previously reported results on this segment [32] (Fig. 2D). Since a.a. 215–232 only represents a portion of SMD3, we questioned if this represents the entire CaM-binding domain here. BSAT1R215–242, which covers the entire SMD3, clearly binds CaM (Fig. 2E). However, BSAT1R215–242 has a 3-fold higher affinity for Ca2+-CaM than BSAT1R215–232, indicating that full interaction with CaM requires the entire SMD3 (Fig. 2F). Figure 2G shows fractional saturation of BSAT1R125–141, BSAT1R215–242, and BSAT1R309–327 as a function of Ca2+-liganded CaM. The apparent Kd values for these interactions are shown in Table-4.
Fig. 2.

CaM-binding domains in AT1R. Incremental purified CaM was titrated as the emission spectra of BSAT1R54–66 (A), BSAT1R125–141 (B), BSAT1R309–327 (C), BSAT1R215–232 (D), and BSAT1R215–242 (E) were monitored. Initial reaction mix contained 0.5 μM BSAT1RX, 0.1mg/ml BSA and 1 mM Ca2+ in titration buffer (F) Composite plot showing difference in binding affinity for CaM between BSAT1R215–232 (open triangles) and BSAT1RX (closed circles). (G) Composite plot of average (n = 6 independent experiments) fractional saturations of BSAT1R125–141, BSAT1R215–242, and BSAT1R309–327 as a function of Ca2+-liganded CaM.
Table 4.
Apparent Kd, EC50(Ca2+) and DR values for CaM interactions with BSAT1RX
| BSAT1RX | Apparent Kd (μM) | EC50(Ca2+) (μM) | Dynamic Range | |
|---|---|---|---|---|
| BSAT1R125–141 | 13.9 ± 0.39* | 4.10 ± 0.11* | 1.58 ± 0.01* | |
| BSAT1R125–141mut | > 318 ± 13.0† | 17.6 ± 0.78† | 1.34 ± 0.01* | |
| BSAT1R215–232 | 1.05 ± 0.04* | 0.30 ± 0.03* | 2.41 ± 0.03 | |
| BSAT1R215–242 | 0.36 ± 0.05 | 0.15 ± 0.01 | 2.06 ± 0.03 | |
| BSAT1R215–242mut | 4.52 ± 0.25† | 1.49 ± 0.03† | 1.55 ± 0.01† | |
| BSAT1R309–327 | 0.51 ± 0.01 | Sp1 | Sp2 | 1.92 ± 0.04 |
| 0.31 ± 0.02* | 3.17 ± 0.2* | |||
| BSAT1R309–327mut | 6.60 ± 0.18† | Sp1mut | Sp2mut | 1.47 ± 0.01† |
| 0.77 ± 0.1† | 9.6 ± 0.49† | |||
p < 0.05 vs corresponding value of BSAT1R215–242;
p < 0.05 vs corresponding value of wild-type BSAT1RX;
We previously described the parameter dynamic range (DR), determined as the maximal change in FRET ratio of the biosensors [27]. CaM causes significant conformational changes in its target proteins upon binding, a property important for its role in promoting target activities. The dynamic range of a BSAT1RX reflects the conformational changes of the insert domain that could occur upon interaction with CaM and provides a prediction of the potential conformational change that could take place in the holoreceptor upon CaM binding. Figure 3A shows relative FRET ratios of the BSAT1RX as a function of Ca2+-CaM; Fig. 3B shows the increases in dynamic range of the BSAT1RX from unbound state (ΔDR). Full interaction with Ca2+-CaM at a.a. 215–242 (SMD3) is apparently associated with the largest dynamic range, followed by that of a.a. 309–327 (SMD4JM) and a.a. 125–141 (SMD2). This is similar to the order of affinity of these domains for Ca2+-CaM (Fig. 2G, Table-4).
Fig. 3.

Dynamic ranges of BSAT1RX. (A) Relative average ratios of BSAT1R125–141, BSAT1R215–242, and BSAT1R309–327 in response to Ca2+-liganded CaM titration. (B) Average per cent increase from unbound state in ratio of BSAT1RX. n = 6 independent experiments. *, p<0.05 vs value for BSAT1R215–242
3.2. Ca2+ dependencies and sensitivities of the interactions between AT1R and CaM
We first examined the Ca2+ dependency of the interactions between CaM and the domains identified. Previous studies have demonstrated Ca2+ dependency for the interaction at a.a. 305–317 in SMD4JM and a.a. 215–232 in SMD3 by examining binding in the presence of saturating Ca2+ or nominal absence of Ca2+ using the Ca2+ chelator EGTA [22, 32]. Although our initial biosensor screen showed that BSAT1R125–141, BSAT1R215–242, and BSAT1R309–327 all interacted with Ca2+-liganded CaM, it was necessary to examine whether any interactions take place in the absence of Ca2+, since both Ca2+-dependent and independent interactions between CaM and the same binding domain have been shown [4, 5]. Figure 4 shows the spectral changes of BSAT1R125–141, BSAT1R215–242, and BSAT1R309–327 in response to Ca2+-saturated CaM (A, C, E) or apoCaM (B, D, F), at the concentrations that demonstrated maximal binding from the Kd titrations (Fig. 2). At CaM concentrations that produced maximal biosensor response in the presence of Ca2+, no response was observed in the absence of Ca2+ (Fig. 4).
Fig. 4.

Ca2+ dependency of interactions between BSAT1RX and CaM. Specified concentrations of CaM (saturating concentrations determined from Kd titrations) were added to a mixture of 0.5 μM BSAT1R125–141, BSAT1R215–242, BSAT1R309–327 in titration buffer containing 0.1 mg/ml BSA and 1 mM Ca2+ (A, C, E, respectively) or absence of Ca2+ (apoCaM) and presence of 0.25 mM BAPTA (B, D, F, respectively). Dotted and solid spectra, BSAT1RX response before and after the addition of CaM, respectively. Data are representative of 6 independent experiments for each paradigm.
Ca2+ dependency determined in the presence or absence of saturating concentrations of Ca2+ does not allow for predicting when AT1R-CaM interactions can occur in different physiological scenarios at different free Ca2+ levels. Currently, no information exists with respect to the Ca2+ sensitivities of these interactions. To measure these parameters, we mixed BSAT1RX and CaM in the absence of Ca2+ and simultaneously measured responses from both the biosensor and a suitable Ca2+ indicator as Ca2+ was titrated in the reaction mixture. This approach enables precise determinations of both fractional binding and the free Ca2+ concentration at which the binding occurs. The choice of a suitable Ca2+ indicator for these titrations was made easy by the commercial availability of many indicators with different Kd values and fluorescent properties. Ideally, a suitable Ca2+ indicator will have the same response range with that of BSAT1RX to Ca2+-liganded CaM. This can easily be estimated by testing a few Ca2+ indicators with different Kd values. Figure 5A shows an example of these measurements. Ca2+ was titrated in a mixture of BSAT1RX, respective saturating CaM (determined from Fig. 2), 0.25 mM BAPTA (to ensure starting condition had essentially no Ca2+), and a Ca2+ indicator, in this case indo-1 (Kd for Ca2+ =0.23 μM). BSAT1RX and Indo-1 responses were simultaneously monitored (Fig. 5A). Free Ca2+ was then calculated based on indo-1 response (Eq. (5)) and biosensor fractional saturation was calculated using Eq. (1) and plotted as a function of the corresponding free Ca2+ values. A fit from this relationship to Eq. (6) would yield the EC50(Ca2+) for the interaction. Indo-1 was used for BSAT1R215–232 and BSAT1R215–242 since initial testing showed the interactions between CaM and these biosensors have the same range of Ca2+ sensitivity with indo-1. XRhod-5F (Kd for Ca2+ = 1.6 μM) was used for BSAT1R125–141 and BSAT1R309–327 since its response range was the same with those of the biosensors over the entire Ca2+ titration. In these measurements, bleed-through artifacts were avoided by 1) alternate excitation of BSAT1RX and Ca2+ indicator and 2) well separated excitation wavelengths of indo-1 (340 nm) or XRhod-5F (590 nm) from that of BSAT1RX (435 nm). To confirm absence of bleed-through artifacts using this approach for indo-1 and BSAT1RX, reactions were started with a mixture of indo-1 and BSAT1R215–242 and 0.25 mM BAPTA in the absence of CaM. As incremental Ca2+ was added, indo-1 ratio increased until saturation of the signal (Fig. 5B, upper panel) without any alteration in BSAT1R215–242 ratio, which only increased upon addition of purified CaM (Fig. 5B, lower panel). Similarly, absence of bleed-through artifacts was confirmed for XRhod-5F and BSAT1RX using the example of BSAT1R125–141 (Fig. 5C). Aggregate plot of BSAT1RX fractional saturations as a function of free Ca2+ is shown in Fig. 5D. EC50(Ca2+) values for the interactions are listed in Table-4. The order of Ca2+ sensitivity these domains for interaction with CaM is a.a. 215–242 (SMD3) > a.a. 309–327 (SMD4JM) > a.a. 125–141 (SMD2), similar to the order of binding affinity and dynamic range. Interestingly, EC50(Ca2+) for CaM-BSAT1R215–242 interaction is 150 nM, within the resting Ca2+ range in VSMCs. The Ca2+ titration curve for CaM-BSAT1R309–327 interaction exhibits a biphasic behavior. Complexes between Ca2+-CaM and a.a. 309–327 (SMD4JM) thus apparently consist of two species, CaM-SMD4JMSp1, with an EC50(Ca2+) value of 0.31 ± 0.02 μM, and CaM-SMD4JMSp2, with and EC50(Ca2+) of 3.17 ± 0.2 μM (Table-4).
Fig. 5.
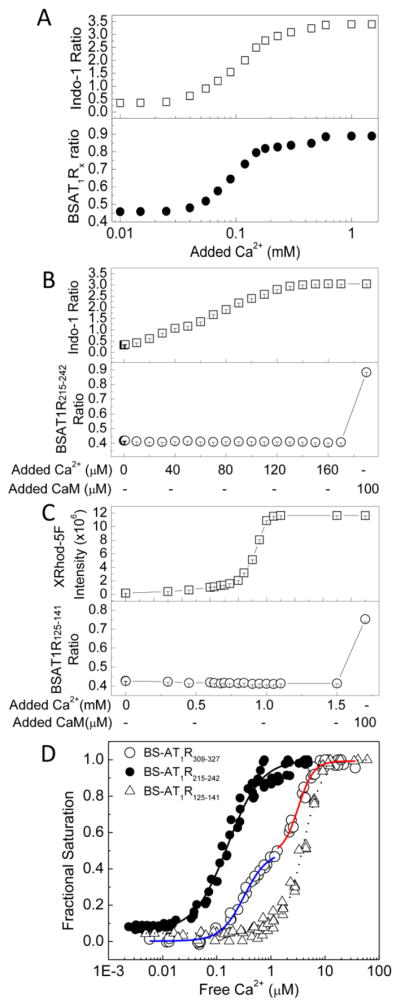
Determinations of the Ca2+ sensitivities of AT1R-CaM interactions. (A) Typical experiment to determine EC50(Ca2+) value for BSAT1RX-CaM interaction. Reactions contained 0.5 μM BSAT1RX, 2 μM Indo-1 (or XRhod-5F), 0.25 mM BAPTA, and saturating CaM concentration (obtained from Kd titrations). Incremental aliquots of Ca2+ were added as the emission intensities of Indo-1 (upper panel) and BSAT1RX responses (lower panel) were monitored. (B and C) Controls for bleed-through artifact between indo-1 or XRhod-5F and BSAT1Rx. Reactions contained 0.1 μM BSAT1R215–242 and 2 μM indo-1 (B) or 0.1 μM BSAT1R125–141 and 2 μM XRhod-5F (C) in the presence of 0.25 mM BAPTA. Incremental aliquots of Ca2+ were added as indo-1, XRhod-5F and biosensor fluorescence responses were monitored. 100 μM purified CaM was added after indo1 and XRhod-5F signals had been saturated. (D) Aggregate (n = 3) plots showing Ca2+ sensitivity of the interactions between CaM BSAT1R125–141 (open triangles), BSAT1R215–242 (closed circles), and BSAT1R309–327 (open circles). Fractional BSAT1RX response and free Ca2+ were calculated as described under Materials and Methods. Fits were performed on aggregate data from three independent experiments. Blue and red fits, CaM-BSAT1R309–327Sp1 and CaM-BSAT1R309–327Sp2, respectively (see text for explanation). Data were from 3 independent experiments for each biosensor.
3.3. CaM-AT1R interactions are required for AT1R-mediated ERK1/2 phosphorylation
The data presented so far show that AT1R possesses three distinct CaM-binding domains with disparate affinities and Ca2+ sensitivities for their interactions with CaM. To initially assess the influence of CaM binding on AT1R-mediated signaling, we tested the effect of inhibiting CaM on AngII-induced ERK1/2 phosphorylation in primary VSMCs. We have routinely isolated primary vascular endothelial cells (PAECs) and then primary VSMCs from the same porcine aortas [25–27]. Under bright-field microscopy, isolated primary PAECs demonstrated typical cobble-stone morphology (Fig. 5A) while the VSMCs demonstrated elongated morphology (Fig. 5B). To confirm the smooth muscle nature of the VSMCs isolated, lysates from PAECs and VSMCs were immunoblotted for smooth muscle α-actin and β-actin. While β-actin was expressed in both lysates, smooth muscle α-actin was only detected in lysate from VSMCs (Fig. 5C). Sub-confluent primary VSMCs were then pretreated for 30 min with vehicle, AT1R antagonist losartan (1 μM; IC50 ~ 5.5 nM), or the CaM antagonist W-7 (100 μM; Ki ~ 20 μM), followed by stimulation with 100 nM AngII for 5 min. AngII triggered robust increases in ERK1/2 phosphorylation. This effect was suppressed by losartan and abolished by W-7 pretreatment (Fig. 6).
Fig. 6.

AngII-induced ERK1/2 phosphorylation in primary VSMCs and effects of CaM antagonism. (A & B) Bright-field images of primary porcine endothelial cells (PAECs, A) and VSMCs (B) isolated from the same porcine aortas cultured in phenol red-containing medium for visualization purposes. Images were taken using a Moticam CMOS camera (Motic Microscopes). (C) Immunoblots for smooth muscle α-actin (lower) and β-actin (upper) from lysates of the PAECs and VSMCs isolated. (D) Primary VSMCs were serum starved for 4 hrs prior to pretreatment with or without losartan (1 μM) or W-7 (100 μM) for 30 minutes, followed by treatment for 10 minutes with vehicle or 100 nM AngII. Upper immunoblot, representative changes in phosphorylated ERK1/2; lower immunoblot, corresponding total ERK1/2 expression, reprobed from stripped upper membrane. Histogram, average (n = 6 independent experiments) relative AT1R-mediated ERK1/2 phosphorylation in response to specified treatment. Densitometric values of the phosphorylated ERK1/2 bands were divided by corresponding values for total ERK1/2 bands and normalized.
The observed effect of W-7, while consistent with a role for CaM binding on AT1R-mediated signaling, can be non-specific. To assess the role of AT1R-CaM interaction at individual SMDs, we generated several substitutions in each domain, aiming to reduce CaM binding affinities and Ca2+ sensitivities, verified changes in CaM binding affinity and Ca2+ sensitivity using biosensor approach, then tested AngII-induced ERK1/2 phosphorylation in cells expressing full-length AT1R containing wild-type or mutant sequences. The substitutions introduced in the identified CaM-binding domains are shown in Table-2. BSAT1R125–141mut, BSAT1R215–242mut, BSAT1R309–327mut all still interact with CaM in a Ca2+-dependent fashion. Figures 7A–C show plots of Ca2+-CaM binding to wild-type vs mutant BSAT1R125–141mut, BSAT1R215–242mut, BSAT1R309–327mut, respectively, demonstrating reductions in CaM binding affinities of all the mutant biosensors. Due to difficulty to saturate binding for BSAT1R125–141mut, Fig. 7A is presented as relative FRET ratio rather than fractional saturation as in Fig. 7B and 7C. Nevertheless, the available titration data clearly shows that BSAT1R125–141mut has substantially reduced affinity for CaM compared to BSAT1R125–141. Figures 7D–F show fractional responses of the wild-type vs mutant biosensors as a function of free Ca2+ determined as described in Fig. 5A. All mutant biosensors showed reduced Ca2+ sensitivities for interactions with CaM. The mutant SMD4JM again exhibited a biphasic Ca2+ response curve, similar to the wild-type version (Fig. 7F). The apparent Kd and EC50(Ca2+) values for CaM interaction with the mutant biosensors are listed in Table-4. Having confirmed the effects of the substitutions to decrease interactions with CaM at each domain, full-length AT1Rs with wild-type or mutant sequences at each SMD were tagged with a hemagglutinin (HA) epitope tag at their N termini and expressed in HEK293 cells. After 24 hrs, cells were serum starved for 4 hours prior to treatment and lysis. Immunoblotting of samples with the anti-HA antibody showed equal expression levels of the expressed receptors and no bands recognized in the mock-transfected samples (lower immunoblots, Fig. 7G(i), (ii), (iii)). Treatment of mock-transfected HEK293 cells with 100 nM AngII mildly increased ERK1/2 phosphorylation due to the activity of endogenous AT1R. Heterologous expression of wild-type AT1R substantially increased this effect. However, in cells expressing AT1R with the substitutions that were verified to reduce CaM-binding affinity and Ca2+ sensitivities (Fig. 7A–F) in SMD2, SMD3, or SMD4JM (upper blots, Fig. 7G, (i), (ii), or (iii), respectively) the effect of AngII was significantly reduced (Fig. 7G, iv). Total ERK1/2 expression levels probed from the same membranes as for ERK1/2 phosphorylation were similar, indicating equal loading (middle blots, Fig. 7G(i), (ii), (iii)).
Fig. 7.

Effects of CaM binding-reducing mutations on AT1R-mediated signaling. (A, B, C) Average (n = 6 independent experiments) responses of the specified BSAT1RX with wild-type (open symbols) or mutant (closed symbols) sequences at the identified CaM binding domain as a function of free CaM showing reduction in binding affinities. Conditions for titration reactions were as described for Fig. 2. Plot data were fit to Eq. 2 or 3 to yield Kd values (Table-4). (D, E, F) Average (n = 6 independent experiments) fractional saturations of the specified BSAT1RX with wild-type (open symbols) or mutant (closed symbols) sequences in the identified CaM-binding domains in the presence of saturating CaM and incremental free Ca2+ measured as described in Fig. 5. Data were fit to Eq. 6 to yield EC50(Ca2+) values (Table-4). Blue and red fits correspond to wild-type or mutant CaM-BSAT1R309–327Sp1 and CaM-BSAT1R309–327Sp2, respectively. (G) HEK293 cells were transfected as indicated with mock materials or plasmids encoding hemagglutinin (HA) epitope tagged to the N termini of full-length AT1R with wild-type or mutant sequences (Table-2) in SMD2 (i), SMD3 (ii), or SMD4JM (iii). 24 hrs post-transfection, cells were serum starved for 4 hrs, followed by treatment with vehicle or 100 nM AngII for 10 minutes. Upper and middle immunoblots, representative phosphorylated ERK1/2 and total ERK1/2 from the same SDS-PAGE membranes; lower immunoblots, expression of HA-AT1R across corresponding samples. (iv) Average (n = 6 independent experiments) relative phosphorylated ERK1/2. Relative densitometric values of phosphorylated ERK1/2 bands were divided by corresponding values for the total ERK1/2 bands and normalized to corresponding mock-untreated values. *, p<0.05 vs corresponding untreated conditions; †, p<0.05 vs valuesfrom cells transfected with full-length wild-type AT1R and treated with AngII.
3.4. CaM-AT1R interactions are important for AngII-induced Ca2+ signals
To corroborate the results with AT1R-mediated ERK1/2 phosphorylation, we examined the effects of the CaM binding-reducing mutations in the individual SMDs on AngII-induced Ca2+ signals. To guarantee Ca2+ was measured in cells expressing wild-type or mutant AT1R, full-length AT1Rs with wild-type or mutant sequences were fused at their N termini with the fluorescence protein mKate2. N-terminal fusion was chosen to avoid potential interference of mKate2 with important binding partners at the submembrane domain level. HEK293 cells expressing plasmids encoding mKate2 alone or fused with wild-type or mutant AT1R were loaded with fura-2/AM for Ca2+ imaging. mKate2 fluorescence was first identified and marked in the RFP channel (Fig. 8A), followed by switching filter cube for imaging of fura-2 fluorescence in the same microscopic field (Fig. 8B). This guaranteed that Ca2+ signals were examined in cells expressing the intended AT1R version. Merged images (Fig. 8C) show high transfection efficiency. To allow correlation with EC50(Ca2+) values determined using purified CaM and BSAT1RX, measured fura-2 signals were converted to free intracellular Ca2+ concentrations (Materials and Methods). To ensure signals were compared among cells expressing equal amounts of wild-type and mutant AT1R, cells were selected with mKate2 fluorescence intensities in the same range prior to switching to measurement of Ca2+ signals (see also Methods). Figure 8D shows average mKate2 intensity in HEK293 cells expressing mKate2 or mKate2-AT1R with wild-type or mutant sequences selected for measurement of AngII-induced Ca2+ signals. In cells expressing only the mKate2 moiety, 100 nM AngII induced a small Ca2+ signal, due most likely to the presence of functional wild-type AT1R in these cells (our AT1R cDNA was reverse-transcribed from mRNA isolated from the host HEK293 cells). AngII treatment of cells expressing mKate2 fused with wild-type AT1R produced a large transient Ca2+ signal. The AngII-induced Ca2+ signal was reduced in cells expressing mKate2-AT1R plasmids containing CaM binding-reducing mutations in SMD3 or SMD4. Surprisingly, this signal became a very slow-rising signal with low amplitude, in cells expressing mKate2-AT1R with mutant sequence on SMD2 (Fig. 8D). To quantitate these signals, integrated areas under curve were calculated for the entire time courses (Fig. 8E). AngII-induced Ca2+ signals were significantly reduced in cells expressing mKate2 in fusion with AT1R containing CaM binding-reducing substitutions in SMD2, 3 or 4 (Fig. 8E). Notably, in the case of AT1R with mutant SMD2 sequence, the AngII-induced Ca2+ signal was very slow-rising and low amplitude, even compared to cells expressing only the mKate2 moiety (Fig. 8D–E).
Fig. 8.

Effects of reduced CaM binding at individual SMDs on AngII-induced Ca2+ signals. HEK293 cells expressing plasmids encoding mKate2 alone or in fusion with the N terminus of full-length AT1R with wild-type or mutant sequence at SMD2, or SMD4JM were loaded with fura-2/AM as described under Materials and Methods. (A) Epifluorescence of mkate2 moiety. (B) Corresponding epifluorescence of fura-2 in the same microscopic field. (C) Merged mKate2 and fura-2 fluorescence images. (D) Average mKate2 fluorescence intensities of all individual cells selected for Ca2+ measurements. (E) Average time courses of Ca2+ signals stimulated by AngII as indicated. (F) Corresponding average total integrated areas under curve (AUC). N = 60 cells from 6 independent experiments for each paradigm; * and ϕ, p < 0.05 vs AUC values of cells expressing mKate2 only and mKate2-wildtype AT1R fusion, respectively.
4. Discussion
In this study, we have identified several new aspects of the interactions between CaM and AT1R: 1) a new CaM binding site is found spanning a.a. 125–141 on SMD2; 2) full interaction between CaM and SMD3 requires the entire domain, a.a. 215–242; 3) CaM interactions with all three domains require Ca2+ and can occur at different Ca2+ concentrations in the physiological range, with interaction at a.a. 309–327 in SMD4JM having a biphasic sensitivity to Ca2+; and 4) CaM interaction with each binding site is important for AT1R-mediated Ca2+ signaling and ERK1/2 phosphorylation.
The identification of a.a. 125–141 on SMD2 as a new binding site for CaM on AT1R is interesting in that the amino acid sequence of SMD2 does not conform to known CaM-binding motifs. This is similar to our identification of SMD1 on GPER as a CaM-binding domain of only 12 residues that can interact with CaM at basal Ca2+ levels [27]. The case of AT1R requiring the entire SMD3 (a.a. 215–242) for full interaction with CaM at this domain is also reminiscent of our observation that fragment a.a. 150–170 of GPER’s SMD2 binds CaM, but with a substantially lower affinity than the full-length SMD2, a.a. 150–175, indicating an important role of the basic patch a.a. 170–175 in this process [27]. In the case of AT1R’s SMD3, a.a. 233–242 contains a high percentage of hydrophobic and charged residues, which apparently contribute significantly to CaM binding, adding a 3-fold increase in binding affinity and doubling Ca2+ sensitivity for the interaction with the full SMD3, a.a. 215–242, vs the previously reported segment, a.a. 215–232. These findings further support the value of using FRET biosensors to screen for CaM-binding sites in GPCRs.
Simultaneous measurements of BSAT1RX-CaM interaction and corresponding free Ca2+ concentrations indicate that the interactions between CaM and the three identified domains on AT1R can all occur during a typical cytoplasmic Ca2+ signal in VSMCs. With disparate affinities and Ca2+ sensitivities, CaM binding may occur at different submembrane domains at different levels of free Ca2+. SMD3-CaM interaction has an EC50(Ca2+) value of 150 nM (Fig. 5 and Table-4) and at 50 nM free Ca2+, there is significant binding at this domain in vitro. These values are well within the resting range of cytoplasmic Ca2+ in VSMCs, suggesting that AT1R-CaM interaction can occur in basal conditions in cells. Considering a competitive environment for CaM among CaM-dependent proteins due to limiting CaM [14, 23–25], CaM interactions with EC50(Ca2+) values in the range of basal Ca2+ levels are more likely to occur since fewer target proteins bind CaM at this level of Ca2+ compared to during a large Ca2+ signals. For the remaining domains, EC50(Ca2+) values of 0.31, 3.18 and 4.1 μM indicate that CaM-SMD4JMSp1, CaM-SMD4JMSp2 and CaM-SMD2 complexes, respectively, are likely to take place at different points during the time course of a typical agonist-stimulated response. Based on Ca2+ titration curve, full association between CaM and a.a. 125–141 on SMD2 requires ~ 10 μM free Ca2+. This is attainable in cells, as sub-PM Ca2+ in VSMCs reaches 45 μM and remains above 5 μM throughout the time course of the vasopressin-induced Ca2+ signal [16].
What are the in-situ effects of multi-site CaM associations with AT1R? Kai et al. showed that synthetic peptides corresponding to residues 125–137 (rat sequence) in SMD2, 217–227 in the N-terminal side of SMD3, and 304–316 in SMD4JM inhibit to various degrees AngII-induced GTPase activity of isolated vascular smooth muscle membrane [9]. This indicates that these segments constitute, at least in part, G protein interaction sites for AT1R. Our data now show that the full SMD2 and SMD3, and a.a. 309–327 on SMD4JM can all interact with CaM at physiological Ca2+ concentrations. This suggests that CaM binding at these locations may interfere with G protein coupling. Consistently, Zhang et al. have shown in vitro that CaM can compete with peptides corresponding to a.a. 215–232 on SMD3 and a.a. 305–317 on SMD4JM for interaction with Gβγ subunit [32]. It has been postulated that CaM interaction with SMDs in GPCRs could interfere with G protein coupling via two mechanisms prevention of G protein-receptor preassociation or promotion of dissociation [25]. Given multi-site interactions between CaM and AT1R with disparate Ca2+ sensitivities, both mechanisms might be in play. For example, with the ability to interact with a.a. 215–242 (SMD3) at resting Ca2+ levels, CaM interaction here might prevent Gβγ preassociation at rest. On the other hand, CaM binding at a.a. 125–241 (SMD2) and a.a. 309–327 on SMD4JM may promote Gβγ dissociation upon AT1R stimulation.
The downstream functional impact of CaM binding to AT1R in living cells has not been studied. Inhibition of CaM using W-7 in primary vascular smooth muscle cells virtually abolishes AngII-induced ERK1/2 phosphorylation. Consistently, mutagenesis data indicate that reduction in CaM binding affinity and Ca2+ sensitivity for interaction at each identified domain is associated with significantly reduced AT1R-mediated ERK1/2 phosphorylation. In addition, AngII-induced Ca2+ signals are also significantly reduced in cells expressing mKate2-AT1R with mutant sequence at each domain. In our Ca2+ measurements, cells heterologously expressing a mutant AT1R were recognized by mKate2 fluorescence and selected prior to switching to fura-2 channel, so that the measured signals reflect effect of the intended expressed receptor. Furthermore, the presence of some non-transfected cells in the same microscopic field (absence of mKate2 fluorescence) allowed for comparing Ca2+ signals from cells expressing or not expressing exogenous AT1R in the same experiments. Interestingly, reduced CaM binding at a.a. 125–141 (SMD2) drastically alters the dynamics of AngII-induced Ca2+ signal, from a typical transient into a slow-rising, low amplitude signal. This finding first confirms that signals from the expressed receptors dominated over any endogenous AT1R in this system. However, the finding is surprising considering the lower affinity, dynamic range and Ca2+ sensitivity of CaM interaction with SMD2 than with the other two locations. We do not know the explanation for this. Speculatively, given proximity of the submembrane domains in cells, CaM binding at SMD2 might affect associations at the other domains and thus have impact in cells beyond what biochemical properties of its interaction with SMD2 in isolation would predict. While this is an attractive hypothesis, it is technically challenging at present to test in cells, given that insertion of reporters in an SMD is likely to alter the relative association of binding partners to the remaining SMDs substantially.
In conclusion, AT1R possesses up to three CaM-binding domains located at a.a. 125–141 (SMD2), 215–242 (SMD3), and 309–327 (SMD4JM). These domains interact with CaM with disparate affinities and Ca2+ sensitivities in the physiological range of Ca2+ signals in cells. CaM interaction with SMD3 can occur at resting Ca2+ concentration. Functionally, interaction at each domain is important for AngII-stimulated Ca2+ signaling and ERK1/2 phosphorylation.
Acknowledgments
This study was supported by National Institutes of Health Grant HL112184 and Iowa Osteopathic and Educational Research Funds to QK-T. We thank Vahe Matnishian and Briana Gebert-Oberle for assistance in some experimental paradigms.
Footnotes
Conflict of Interest
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ali MS, Sayeski PP, Dirksen LB, Hayzer DJ, Marrero MB, Bernstein KE. Dependence on the motif YIPP for the physical association of Jak2 kinase with the intracellular carboxyl tail of the angiotensin II AT1 receptor. J Biol Chem. 1997;272:23382–23388. doi: 10.1074/jbc.272.37.23382. [DOI] [PubMed] [Google Scholar]
- 2.Aricescu AR, Lu W, Jones EY. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta crystallographica. Section D, Biological crystallography. 2006;62:1243–1250. doi: 10.1107/S0907444906029799. [DOI] [PubMed] [Google Scholar]
- 3.Becamel C, Alonso G, Galeotti N, Demey E, Jouin P, Ullmer C, Dumuis A, Bockaert J, Marin P. Synaptic multiprotein complexes associated with 5-HT(2C) receptors: a proteomic approach. EMBO J. 2002;21:2332–2342. doi: 10.1093/emboj/21.10.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Black DJ, LaMartina D, Persechini A. The IQ domains in neuromodulin and PEP19 represent two major functional classes. Biochemistry. 2009;48:11766–11772. doi: 10.1021/bi9014874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Black DJ, Tran QK, Persechini A. Monitoring the total available calmodulin concentration in intact cells over the physiological range in free Ca2+ Cell Calcium. 2004;35:415–425. doi: 10.1016/j.ceca.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Bockaert J, Fagni L, Dumuis A, Marin P. GPCR interacting proteins (GIP) Pharmacol Ther. 2004;103:203–221. doi: 10.1016/j.pharmthera.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Bofill-Cardona E, Kudlacek O, Yang Q, Ahorn H, Freissmuth M, Nanoff C. Binding of calmodulin to the D2-dopamine receptor reduces receptor signaling by arresting the G protein activation switch. J Biol Chem. 2000;275:32672–32680. doi: 10.1074/jbc.M002780200. [DOI] [PubMed] [Google Scholar]
- 8.Bolzon BJ, Cheung DW. Isolation and characterization of single vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension. 1989;14:137–144. doi: 10.1161/01.hyp.14.2.137. [DOI] [PubMed] [Google Scholar]
- 9.Kai H, Alexander RW, Ushio-Fukai M, Lyons PR, Akers M, Griendling KK. G-Protein binding domains of the angiotensin II AT1A receptors mapped with synthetic peptides selected from the receptor sequence. Biochem J. 1998;332(Pt 3):781–787. doi: 10.1042/bj3320781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kakiuchi S, Yasuda S, Yamazaki R, Teshima Y, Kanda K, Kakiuchi R, Sobue K. Quantitative determinations of calmodulin in the supernatant and particulate fractions of mammalian tissues. J Biochem (Tokyo) 1982;92:1041–1048. doi: 10.1093/oxfordjournals.jbchem.a134019. [DOI] [PubMed] [Google Scholar]
- 11.Kendall RT, Strungs EG, Rachidi SM, Lee MH, El-Shewy HM, Luttrell DK, Janech MG, Luttrell LM. The beta-arrestin pathway-selective type 1A angiotensin receptor (AT1A) agonist [Sar1,Ile4,Ile8]angiotensin II regulates a robust G protein-independent signaling network. J Biol Chem. 2011;286:19880–19891. doi: 10.1074/jbc.M111.233080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SA, Heinze KG, Waxham MN, Schwille P. Intracellular calmodulin availability accessed with two-photon cross-correlation. Proc Natl Acad Sci U S A. 2004;101:105–110. doi: 10.1073/pnas.2436461100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leik CE, Willey A, Graham MF, Walsh SW. Isolation and culture of arterial smooth muscle cells from human placenta. Hypertension. 2004;43:837–840. doi: 10.1161/01.HYP.0000119191.33112.9c. [DOI] [PubMed] [Google Scholar]
- 14.Luby-Phelps K, Hori M, Phelps JM, Won D. Ca(2+)-regulated dynamic compartmentalization of calmodulin in living smooth muscle cells. J Biol Chem. 1995;270:21532–21538. doi: 10.1074/jbc.270.37.21532. [DOI] [PubMed] [Google Scholar]
- 15.Mahon MJ, Shimada M. Calmodulin interacts with the cytoplasmic tails of the parathyroid hormone 1 receptor and a sub-set of class b G-protein coupled receptors. FEBS Lett. 2005;579:803–807. doi: 10.1016/j.febslet.2004.12.056. [DOI] [PubMed] [Google Scholar]
- 16.Marsault R, Murgia M, Pozzan T, Rizzuto R. Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. Embo J. 1997;16:1575–1581. doi: 10.1093/emboj/16.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 18.Minakami R, Jinnai N, Sugiyama H. Phosphorylation and calmodulin binding of the metabotropic glutamate receptor subtype 5 (mGluR5) are antagonistic in vitro. J Biol Chem. 1997;272:20291–20298. doi: 10.1074/jbc.272.32.20291. [DOI] [PubMed] [Google Scholar]
- 19.Paradis P, Dali-Youcef N, Paradis FW, Thibault G, Nemer M. Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc Natl Acad Sci U S A. 2000;97:931–936. doi: 10.1073/pnas.97.2.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Persechini A. Monitoring the intracellular free Ca(2+)-calmodulin concentration with genetically-encoded fluorescent indicator proteins. Methods Mol Biol. 2002;173:365–382. doi: 10.1385/1-59259-184-1:365. [DOI] [PubMed] [Google Scholar]
- 21.Shen X, Valencia CA, Szostak JW, Dong B, Liu R. Scanning the human proteome for calmodulin-binding proteins. Proc Natl Acad Sci U S A. 2005;102:5969–5974. doi: 10.1073/pnas.0407928102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas WG, Pipolo L, Qian H. Identification of a Ca2+/calmodulin-binding domain within the carboxyl-terminus of the angiotensin II (AT1A) receptor. FEBS Lett. 1999;455:367–371. doi: 10.1016/s0014-5793(99)00904-7. [DOI] [PubMed] [Google Scholar]
- 23.Tran QK, Black DJ, Persechini A. Intracellular coupling via limiting calmodulin. J Biol Chem. 2003;278:24247–24250. doi: 10.1074/jbc.C300165200. [DOI] [PubMed] [Google Scholar]
- 24.Tran QK, Black DJ, Persechini A. Dominant affectors in the calmodulin network shape the time courses of target responses in the cell. Cell Calcium. 2005;37:541–553. doi: 10.1016/j.ceca.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Tran QK, Firkins R, Giles J, Francis S, Matnishian V, Tran P, VerMeer M, Jasurda J, Burgard MA, Gebert-Oberle B. Estrogen Enhances Linkage in the Vascular Endothelial Calmodulin Network via a Feedforward Mechanism at the G Protein-Coupled Estrogen Receptor 1. J Biol Chem. 2016;291:10805–10823. doi: 10.1074/jbc.M115.697334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran QK, VerMeer M, Burgard MA, Hassan AB, Giles J. Hetero-oligomeric Complex between the G Protein-coupled Estrogen Receptor 1 and the Plasma Membrane Ca2+-ATPase 4b. J Biol Chem. 2015;290:13293–13307. doi: 10.1074/jbc.M114.628743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tran QK, VerMeer M. Biosensor-based approach identifies four distinct calmodulin-binding domains in the G Protein-Coupled Estrogen Receptor 1. PloS one. 2014;9:e89669. doi: 10.1371/journal.pone.0089669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner JH, Gelasco AK, Raymond JR. Calmodulin interacts with the third intracellular loop of the serotonin 5-hydroxytryptamine1A receptor at two distinct sites: putative role in receptor phosphorylation by protein kinase C. J Biol Chem. 2004;279:17027–17037. doi: 10.1074/jbc.M313919200. [DOI] [PubMed] [Google Scholar]
- 29.Ulrich-Merzenich G, Metzner C, Bhonde RR, Malsch G, Schiermeyer B, Vetter H. Simultaneous isolation of endothelial and smooth muscle cells from human umbilical artery or vein and their growth response to low-density lipoproteins. In Vitro Cell Dev Biol Anim. 2002;38:265–272. doi: 10.1290/1071-2690(2002)038<0265:SIOEAS>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 30.Wang D, Sadee W, Quillan JM. Calmodulin binding to G protein-coupling domain of opioid receptors. J Biol Chem. 1999;274:22081–22088. doi: 10.1074/jbc.274.31.22081. [DOI] [PubMed] [Google Scholar]
- 31.Wu X, Bers DM. Free and bound intracellular calmodulin measurements in cardiac myocytes. Cell Calcium. 2007;41:353–364. doi: 10.1016/j.ceca.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang R, Liu Z, Qu Y, Xu Y, Yang Q. Two Distinct Calmodulin Binding Sites in the Third Intracellular Loop and Carboxyl Tail of Angiotensin II (AT1A) Receptor. PloS one. 2013;8:e65266. doi: 10.1371/journal.pone.0065266. [DOI] [PMC free article] [PubMed] [Google Scholar]