

Abstract
Activation of the cholecystokinin type B receptor (CCKBR) by cholecystokinin octapeptide (CCK-8) inhibits opioid analgesia. Chronic opiate treatment leads to an increase in the CCK-8 concentration and thus enhances the antagonism of CCK-8 against opioid analgesia; the underlying molecular mechanisms remain of great interest. In the present study, we validated the colocalization of the μ-opioid receptor (MOR) and CCKBR in pain signal transmission-related spinal cord dorsal horn and dorsal root ganglion neurons of rats. Co-immunoprecipitation (Co-IP) and fluorescence lifetime-imaging-microscopy-based fluorescence resonance energy transfer (FLIM-FRET) assays showed that MOR heteromerized with CCKBR directly in transfected HEK293 cells. Combined with MOR mutant construction, the third transmembrane domain of MOR (TM3MOR) was demonstrated to participate in heteromerization with CCKBR. Receptor ligand binding, ERK phosphorylation and cAMP assays showed that MOR heteromerization with CCKBR weakened the activity of MOR. A cell-penetrating interfering peptide consisting of TM3MOR and TAT (a transactivator of HIV-1) sequences from the N terminal to the C terminal disrupted the MOR–CCKBR interaction and restored the activity of MOR in transfected HEK293 cells. Furthermore, intrathecal application of the TM3MOR-TAT peptide alleviated CCK-8-injection-induced antagonism to morphine analgesia in rats. These results suggest a new molecular mechanism for CCK-8 antagonism to opioid analgesia in terms of G-protein-coupled receptor (GPCR) interaction through direct heteromerization. Our study may provide a potential strategy for pain management with opioid analgesics.
Pain medicine: Boosting the potency of opioid analgesics
A hormone known to weaken the pain-relieving effects of opioid drugs does so because of interaction between the hormone receptor and the opioid receptor. A team from Peking University in Beijing, China, led by Li Su and You Wan showed that an opioid receptor and a receptor that is activated by a peptide hormone involved in digestion are both found in rat neurons of the spinal cord and spinal ganglion. They demonstrated in human cells that the two receptors could form a single structural complex, and that this process weakened the ability of the opioid receptor to respond to drugs such as morphine. A decoy peptide that disrupted the interaction between the receptors boosted the analgesic effects of morphine in rats. Similar strategies could help with pain management.
Introduction
Opium has been used to relieve acute and chronic pain for hundreds of years. As the mainstay of pain management for severe pain, the importance of opioids has never been contested1. It is well known that nearly all opioids in clinical use mediate their analgesic effects through the μ-opioid receptor (MOR); however, long-term application of μ-opioid agonists produces tolerance, limiting their clinical application. Opioid tolerance involves a wide range of mechanisms that lead to reduction in response to opioids. In addition to receptor phosphorylation, arrestin association, endocytosis and desensitization2, anti-opioid systems contribute to the inhibition of opioid analgesia as well3.
Our previous serial investigations found the antagonism of cholecystokinin octapeptide (CCK-8) to opioid analgesia4–7. Studies using L-365,260, a specific antagonist of the cholecystokinin type B receptor (CCKBR), showed that CCK-8 inhibited opioid analgesia through CCKBR8–10. L-365,260 potentiated opioid-induced analgesia; however, its administration per se did not affect pain threshold11. In addition, the inhibitory effect of MOR on voltage-gated calcium current in dorsal root ganglion (DRG) neurons could be antagonized by CCK-8 through CCKBR located in the same neuron12. In the development of electroacupuncture analgesia tolerance, CCK-8 and its CCKBR were additionally involved13. A remarkable increase in CCK-8 immunoreactivity was observed in the perfusate of the rat spinal cord during electroacupuncture analgesia tolerance11. These reports suggest that CCKBR may mediate antagonism to opioid analgesia, specifically MOR-mediated analgesia through interaction with MOR rather than simply reducing the pain threshold.
Recent literature has reported that G-protein-coupled receptor (GPCR) heteromerization can modulate receptor functions14–16. Direct interaction between MOR and δ-opioid receptor (DOR) enhanced DOR binding through allosteric modulation of receptor function17. The activity of MOR was regulated by interaction with DOR and contributed to morphine tolerance18. The antagonistic interaction of adenosine A2A and dopamine D2 receptors depended on their heteromerization and Gq/11–PLC signaling19. Both MOR and CCKBR are GPCRs; therefore, it is possible that MOR and CCKBR may directly form heteromers and thus influence MOR functions.
In the present study, we hypothesize that CCKBR and MOR form heteromers, and the heteromerization then inhibits the function of MOR and thus contributes to the anti-opioid effects of CCK-8. We first validated the colocalization of MOR and CCKBR in neurons and then utilized co-immunoprecipitation (Co-IP) and fluorescence lifetime-imaging-microscopy-based fluorescence resonance energy transfer (FLIM-FRET) to examine whether MOR and CCKBR could form heteromers through direct protein–protein interaction and thus inhibit MOR functions. Finally, we identified the third transmembrane domain of MOR participating in binding with CCKBR.
Materials and methods
Plasmid construction
The procedures for the construction of plasmids expressing HA-MOR, FLAG-CCKBR, MOR-EGFP, CCKBR-mCherry, and MOR (TM3 to TM6, or TM4 to TM6)-EGFP are described in the Supplementary Materials. The pcDNA 3.0-HA-MOR was kindly provided by Professor Liu-Chen L-Y at the Temple University School of Medicine, USA. The pcDNA 4.0-FLAG-CCKBR was a gift from Professor Hellmich MR at the University of Texas Medical Branch, USA. The pEGFP-N1 and pmCherry-N1 were purchased from Clontech Laboratories Inc., CA, USA. All primers used in plasmid constructions are listed in the Supplementary Materials (Table S1).
Cell culture and transfection
HEK293 cells were cultured in DMEM (Gibco, Carlsbad, CA) containing 10% fetal bovine serum (HyClone, Logan, UT) in a humidified atmosphere containing 5% CO2 at 37 °C. The cells were transfected with 1 μg of plasmid per 35-mm dish or 4 μg of plasmid per 60-mm dish using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and were cultured for 48 h. For the co-expression of MOR and CCKBR, the cells were co-transfected with the relevant plasmids.
Stable expression of HA-MOR and FLAG-CCKBR in HEK293 cells
The HEK293 cells were first transfected with pcDNA 3.0-HA-MOR or pcDNA 4.0-FLAG-CCKBR using Lipofectamine 2000. Forty-eight hours after transfection, the cells were digested successively with 0.02% EDTA and 0.25% trypsin, selected in culture medium containing neomycin (Invitrogen) or zeocin (Invitrogen). To generate cells stably co-expressing two receptors, cells stably expressing FLAG-CCKBR were subjected to a second round of pcDNA 3.0-HA-MOR plasmid transfection and selected in the presence of neomycin and zeocin. Co-expression and membrane colocalization of HA-MOR and FLAG-CCKBR were confirmed by western blot and immunofluorescent staining.
Western blot
HA-MOR or FLAG-CCKBR expressed in the HEK293 cells was detected using western blot, as previously described20. Briefly, total cellular proteins extracted from relevant receptor-expressing cells (20 μg per well) were separated in SDS-PAGE electrophoresis (Bio-Rad, Hercules, CA; 10% gel) and then transferred to PVDF membranes (Millipore, Billerica, MA). The membranes were blocked with 5% non-fat milk (Applygen, Beijing, China) or bovine serum albumin (BSA; Amresco, Solon, OH) for 1 h at room temperature and incubated with relevant antibodies overnight at 4 °C. The PVDF membrane was then incubated with a relevant horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson ImmunoResearch Inc., West Grove, PA) for 1 h at room temperature. Subsequently, the target proteins on the PVDF membrane were detected using the enhanced chemiluminescence protocol (ECL kit; PerkinElmer, Waltham, MA). The protein-extracting procedures and antibody information (Table S2) are provided in Supplementary Materials.
Immunofluorescence staining
Immunofluorescence staining of MOR and CCKBR in the frozen sections of the spinal cord and L4–L5 dorsal root ganglia (DRG) was performed as previously described21. Rats were anesthetized and sequentially transcardially perfused with saline and 4% paraformaldehyde (pH 7.4). The spinal cord and DRG were quickly removed, post-fixed with 4% paraformaldehyde, and then placed in 20% sucrose solution, followed by 30% sucrose solution. For the spinal cord, cross-sections of 15-μm thickness were cut. For the DRG, 8-μm thick sections were cut along the long axis. The sections were hydrated in phosphate-buffered saline (PBS) at room temperature for 30 min and blocked with 10% goat serum for 1 h. Then, sections were incubated with guinea pig anti-rat MOR antibody (1:50 dilution; Chemicon, Temecula, CA) overnight at 4 °C, followed by incubation with rabbit anti-human CCKBR antibody (1:50; Abcam, Cambridge, UK), FITC-conjugated goat anti-guinea pig IgG (1:2000; Jackson ImmunoResearch Inc.), and TRITC-conjugated goat anti-rabbit IgG (1:2000; Jackson ImmunoResearch Inc.). After washing with PBS, the stained slides were viewed and photographed with a CCD camera under a fluorescence microscope (DMIRB, Leica Microsystems, Wetzlar, Germany).
For immunofluorescent staining of cultured cells, the cells were seeded on poly-L-lysine (Sigma-Aldrich, St. Louis, MO)-coated glass 1 day before staining. The cells were washed with 0.01 M PBS three times, fixed with 4% cold paraformaldehyde (pH 7.4), subsequently blocked with 10% goat serum in PBS for 1 h, and incubated with anti-FLAG antibody (1:200; Sigma-Aldrich) and anti-HA antibody (1:50; Roche, Basel, Switzerland) in 1% BSA and 0.3% Triton X-100 diluted with PBS overnight at 4 °C. After washing three times with PBS, the cells were incubated with FITC-conjugated goat anti-rabbit IgG (1:2000; Jackson ImmunoResearch Inc.) and subsequently TRITC-conjugated goat anti-rat IgG (1:100; Santa Cruz, Dallas, TX) and Hoecht33342 (10 μg/ml; Dingguo, Beijing, China). After washing with PBS, the stained slides were viewed and photographed with a CCD camera under a confocal laser scanning microscope (TCS SP2, Leica Microsystems).
Co-immunoprecipitation
The suspended lysate of the cells was prepared as described for the western blot. Samples containing 500 μg of protein were diluted to 500 μL (1 μg/μL) and incubated with 3 μg of monoclonal anti-HA (Roche) or anti-FLAG (Sigma-Aldrich) antibody overnight at 4 °C. Parallel incubation with isotype IgG was used as a negative control. Protein samples were added to 20 μL A/G agarose (Santa Cruz) suspension and incubated for 4 h at 4 °C. Following centrifugation at 4 °C, the beads were washed six times with the lysis buffer. After washing, the precipitates were resuspended with SDS sample buffer, and the samples were heated twice at 95 °C for 5 min. Following centrifugation, the supernatant containing immunoprecipitates was processed for immunoblotting with anti-HA or anti-FLAG antibody, as described above.
Fluorescence lifetime imaging-microscopy-based fluorescence resonance energy transfer
Freshly split HEK293 cells were plated onto a 35-mm glass-bottom dish (043514B, Shengyou Biotechnology, Hangzhou, China) coated with cell adherent reagent (C1010, Applygen) and after 24 h were transiently transfected with the following series of plasmids: (i) pEGFP-N1 alone, (ii) pEGFP-N1 and pmCherry-N1, (iii) pEGFP-mCherry alone, (iv) pEGFP-N1-MOR alone, (v) pEGFP-N1-MOR and pmCherry-N1, (vi) pEGFP-N1-MOR and pmCherry-N1-CCKBR, (vii) pEGFP-N1-MOR (TM3 to TM6) alone, and (viii) pEGFP-N1-MOR (TM3 to TM6) and pmCherry-N1-CCKBR plasmids. The cells were fixed by 4% paraformaldehyde for 20 min at room temperature and washed three times in PBS before analysis. FLIM analysis was performed using a Leica TCS SP8 two-photon excitation scanning confocal microscope (×64 APO/NA = 1.4, Leica Microsystems) combined with PicoHarp 300 TCSPC module and Picos event timer (PicoQuant GmbH, Rudower Chaussee 29 D-12489, Germany). For lifetime imaging, the laser was tuned to 920 nm to excite EGFP with a frame size of 256 × 256 pixels, and at least 10 cells per sample were acquired. The lowest photon count limit of 500 photons per pixel was used when selecting pixels for fitting fluorescent decay curves. Regions of interest were selected from FLIM images, and time-resolved photon counts were summed up into a lifetime histogram within the SymPhoTime software (PicoQuant GmbH). FRET efficiency was calculated using the following equation:
τD represented the lifetime of the donor-alone group and was calculated and judged by the χ2-values and residuals of the fit. τDA represented the lifetime of the donor and FRET-acceptor co-expressing group and was calculated by fixing τD (no FRET) to obtain the sum of biexponential terms.
For MOR–CCKBR-binding disruption, a scrambled sequence-TAT (6 μg) or TM3MOR-TAT (6 μg) was added into the medium 4 h before evaluation.
Radio-ligand-binding assay
HEK293 cells stably expressing HA-MOR or co-expressing HA-MOR and FLAG-CCKBR were harvested and lysed using the binding buffer containing 5 mM Tris-HCl (pH 7.4), 5 mM EDTA, and 5 mM EGTA at 4 °C for 30 min. The harvested cells were centrifuged at 40,000×g for 20 min at 4 °C, and the pellet was homogenized by filtering through a needle. The homogenate was then centrifuged at 40,000×g for 20 min at 4 °C. The pellet was resuspended in the binding buffer and used as the membrane preparation for the radio-ligand-binding assay. Protein concentration in the prepared membrane was measured by the Coomassie Brilliant Blue method. For the saturation-binding analysis, cell membranes (50–80 μg of protein per assay tube) were incubated with gradient concentrations of [3H]-[D-Ala2,N-Me-Phe4,Gly-ol5]-enkephalin (DAMGO; PerkinElmer) for 1 h at 37 °C. Nonspecific binding was determined by adding 100 nM DAMGO (Sigma-Aldrich) to the reaction mixture. The binding reaction was terminated by the rapid filtration through glass microfiber filters. Filters were washed with ice-cold binding buffer three times, and the bound radioactivity was measured using a liquid scintillation counter. The Prism program (GraphPad Software, La Jolla, CA) was used to analyze the data derived from the saturation-binding assays and obtain Bmax and KD values.
ERK1/2 phosphorylation assay
Cells were cultured overnight with serum-free DMEM and then exposed to DAMGO at 37 °C. After being washed twice with PBS, cells were lysed in the SDS sample buffer containing 31 mM Tris-HCl (pH 6.8), 1% SDS, 5% glycerol, and 2.5% β-mercaptoethanol. Approximately 40 μg protein per well was separated in 10% SDS-PAGE gel and then transferred to PVDF membrane. After being blocked with block buffer (5% BSA dissolved in TBST) for 1 h at room temperature, the membrane was incubated with anti-phospho-ERK1/2 mitogen-activated protein kinase (MAPK) polyclonal antibody (1:2000 dilution; CST, Danvers, MA) overnight at 4 °C. After being washed, the membrane was incubated with HRP-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch Inc.). Subsequently, immunoreactive proteins on the membrane were visualized using the ECL protocol (ECL kit, CST). To confirm the equal amount of MAPK loaded, the blots were stripped and re-blotted with anti-total ERK1/2 polyclonal antibody (1:2000 dilution; CST). The phospho-ERK1/2 MAPK gel bands were normalized with total ERK1/2 MAPK and expressed as percentage of the basal value of untreated cells.
For MOR–CCKBR-binding disruption, a scrambled sequence-TAT (6 μg/mL) or TM3MOR-TAT (6 μg/mL) was added into the medium 4 h before tests.
Intracellular cAMP assay
HEK293 cells stably expressing HA-MOR alone or co-expressed with FLAG-CCKBR were used to investigate DAMGO inhibition of forskolin-induced cAMP increase. Cells were seeded onto 10-cm dishes. On the experiment day, cells were pretreated with serum-free DMEM media containing 500 μM IBMX (Sigma-Aldrich) for 10 min, followed by incubation with 100 μM forskolin (Sigma-Aldrich) in the presence or absence of different concentrations of DAMGO (from 10−11 to 10−6 M) for 30 min. Reactions were stopped with ice-cold PBS. Cell pellets were collected and resuspended with lysis buffer (provided in the ELISA kit). Subsequently, cells were frozen/thawed twice, and the supernatant was obtained by centrifugation (4 °C, 600×g for 10 min) and immediately subjected to a cAMP assay. The cAMP levels were determined using a sensitive ELISA kit according to the manufacturer’s instructions (KGE002B, R&D Systems Inc., Minneapolis, MN). A four-parameter logistic (4-PL) curve was generated to create the standard curve and fitted to an optical density-cAMP concentration functional equation. The cAMP concentrations of the samples were calculated according to the equation. The Prism program (GraphPad Software) was used to analyze the differences in forskolin responses22 between MOR alone and MOR–CCKBR-co-expressing cells treated with each concentration of DAMGO.
For MOR–CCKBR-binding disruption, a scrambled sequence-TAT (6 μg/mL) or TM3MOR-TAT (6 μg/mL) was added into the medium of MOR and CCKBR-co-expressing cells 4 h before treatment with DAMGO and forskolin.
Animals and intrathecal catheter implantation surgery
All animal experiments were performed according to the guidelines of the International Association for the Study of Pain and were approved by the Animal Care and Use Committee of our University.
Adult male Sprague–Dawley rats (8–10 weeks, 160–180 g) were provided by the Department of Laboratory Animal Sciences, Peking University Health Science Center. The rats were allowed to adapt to the environment for 1 week and were handled at least 5 min per day for 3 days before any experiments were performed. The rats were housed in a separate cage on a 12-h light/dark cycle with access to food and water ad libitum.
For the intrathecal catheter implantation surgery, the rats were anesthetized with chloral hydrate (0.3 g/kg, intraperitoneal), and the surgical procedures were performed as previously described in our laboratory23. A guide cannula was used to puncture the dura at the cauda equine level, and a PE-10 catheter was implanted to the lumbar enlargement level (3.5–4.0 cm rostral than the cannula) at the subarachnoid space through the cannula. The outer part of the catheter was plugged and fixed onto the skin between the rat ears. A 5-day recovery was given before further experimental procedures.
Drug delivery and behavior tests
Cholecystokinin octapeptide (C-2175, Sigma-Aldrich) and morphine (Qinghai Pharmaceutical, Qinghai, China) were dissolved in 0.9% physiological saline to a final concentration of 1 ng/μL and 10 mg/mL, respectively. The third transmembrane sequence of MOR-based TM3MOR-TAT (KIVISIDYYNMFTSIFTLCTMSV-RKKRRQRRR) and scrambled sequence-TAT (VYISLTVSITDFIIKSFCMYNMT-RKKRRQRRR) were synthesized and purified by GL Biochem, Shanghai, China. A 5-FAM fluorophore labeled at the N terminal of the TM3MOR-TAT peptide was synthesized to show the distribution of the interfering peptide in cells. Interfering peptides of 1 mg were first dissolved in 25 μL DMSO and diluted to 1 mL with PBS, sonicating for 5 min to aid in dissolving.
The thermal pain threshold test was performed as previously described24. The rat was allowed to acclimate for 30 min within an acrylic enclosure on a clear glass plate. A radiant heat source (Series 8 Model 390, IITC Life Science, Woodland Hills, CA) was focused onto the plantar surface of the left hind paw. The time from the application of the heat source to the rat paw withdrawal was recorded as the paw withdrawal latency (PWL) to reflect the pain threshold. The test was repeated three times with a minimum 5-min interval, and the pain threshold for each rat was determined as the mean value of three PWL tests.
%MPE = (test PWL − baseline PWL)/(cutoff time – baseline PWL) × 100
The power of the heat source was set to 50 W, and the cutoff time was set to 30 s. The rats were randomly divided into two groups. On the first day, baselines of thermal pain threshold were tested; CCK-8 (4 ng) or 0.9% physiological saline (equal volume) was injected (intrathecally (i.t.)) through the implanted catheter into the animals in two groups, respectively. Morphine (2 mg/kg) was injected (subcutaneously (s.c.)) 30 min before the pain threshold test. Leaving 1 day for the drugs to metabolize completely, on the third day, the baselines of the pain threshold were tested again, and TM3MOR-TAT (20 μg) or a scrambled sequence-TAT (20 μg) was injected (i.t.) through the implanted catheter into animals in two groups 4 h before CCK-8 (4 ng) injection (i.t.), respectively. Next, morphine (2 mg/kg) was injected (s.c.), and pain thresholds were tested at 30 min. The behavior test experimenter was blinded to drug delivery. A total of 20 rats were used. Two of them failed in the catheter-implanting surgery, and one of them was killed for excessive weight loss; these three rats were censored in the data analysis.
Statistics
Data are presented as the mean ± SEM. Statistical analysis was performed using Prism 5 for Windows (GraphPad Software). A two-tailed, paired or unpaired t-test was used for comparing the two groups. Differences were considered significant at p < 0.05.
Results
MOR and CCKBR heteromerized in transfected HEK293 cells
Colocalization of two receptors is the precondition of their heteromerization. To assess whether MOR and CCKBR could colocalize on the membrane of cells, we performed double-labeling immunofluorescence staining. Colocalization of MOR and CCKBR was detected in two pain-modulation-related regions including the spinal dorsal horn and the DRG in rats (Fig. 1a).
Fig. 1. Expression and colocalization of MOR and CCKBR on the membrane of cells.

(a) Colocalization of MOR (green) and CCKBR (red) examined with double immunofluorescent staining in the spinal dorsal horn (top panel) and in the DRG (bottom panel). Scale bar = 10 μm. b Co-expression of HA-MOR and FLAG-CCKBR in co-transfected HEK293 cells revealed by western blots using anti-HA or anti-FLAG antibody. Compared with wild-type HEK293 cells, MOR and CCKBR protein expression could be detected. c Distribution of HA-MOR and FLAG-CCKBR in co-transfected HEK293 cells examined with immunofluorescent labeling with anti-HA or anti-FLAG antibody. Scale bar = 25 μm. d Expressions of MOR-EGFP and CCKBR-mCherry in co-transfected HEK293 cells examined with western blot. e Colocalization of MOR-EGFP and CCKBR-mCherry in co-transfected HEK293 cells shown by confocal microscopy imaging. Scale bar = 25 μm
We further examined the co-expression of MOR and CCKBR in HEK293 cells stably expressing HA-MOR and FLAG-CCKBR with immunocytochemical techniques and immunoblotting. Approximately 70% of the cells expressed HA-MOR or co-expressed HA-MOR and FLAG-CCKBR in the stable transfected cells (Figure S1a, b and c). HA-MOR and FLAG-CCKBR were colocalized on the cell membrane (Fig. 1b, c). Colocalization is the basis of the functional regulation of two receptors through interaction. Fluorescent-protein-fused receptors, MOR-EGFP and CCKBR-mCherry, were transiently transfected in HEK293 cells. Both receptors were expressed and colocalized on the membranes (Fig. 1d, e). These results indicate that MOR and CCKBR colocalize on the membrane of the same neuron or a transfected HEK293 cell, which makes it possible to form heteromers of these two receptors.
To determine the MOR–CCKBR heteromers, we first tested whether MOR formed a protein complex with CCKBR using Co-IP. An anti-FLAG antibody co-immunoprecipitated with HA-MOR in proteins extracted from HA-MOR and FLAG-CCKBR stably co-expressed HEK293 cells (Fig. 2a), suggesting the existence of a MOR–CCKBR complex. Conversely, anti-HA antibody co-immunoprecipitated with FLAG-CCKBR (Fig. 2b), confirming the existence of a MOR–CCKBR complex in HEK293 cells as well.
Fig. 2. Heteromerization of MOR and CCKBR in co-transfected HEK293 cells.

(a) Co-IP experiments with the anti-FLAG antibody. Compared to the IgG group, HA-MOR could be co-immunoprecipitated with FLAG-CCKBR from the lysate of HEK293 cells, stably overexpressing HA-MOR and FLAG-CCKBR. Supernatants after Co-IP reaction indicated that not all MORs form heteromers with CCKBRs. b Co-IP experiments with the anti-HA antibody. FLAG-CCKBR was also co-immunoprecipitated with HA-MOR. c FLIM image of EGFP in different co-expressing cells. Compared with the group expressing MOR-EGFP alone or the MOR-EGFP- and mCherry-co-expressing group, the lifetime of MOR-EGFP in the MOR-EGFP- and CCKBR-mCherry-co-expressing group showed blueshifting, indicating the shortened EGFP fluorescence lifetime. d Statistics of EGFP fluorescence lifetime in (c). e FRET assay results. FRET efficiency of the MOR-EGFP- and CCKBR-mCherry-co-expressing group was significantly higher than the EGFP- and mCherry-co-expressing group (negative control) or the MOR-EGFP- and mCherry-co-expressing group. The EGFP-mCherry-fusion-protein-expressing group was a positive control. ***p < 0.0001, t-test. Data are represented as the mean ± SEM
To further validate the interaction of MOR and CCKBR in intact cells, we performed two-photon FLIM-FRET in MOR-EGFP and CCKBR-mCherry-co-expressing HEK293 cells. FRET is usually limited to a distance less than ~10 nm; thus, the FLIM-FRET technique increases the spatial resolution to 10 nm and provides the means to detect direct protein–protein interactions25,26. The fluorescent images showed that ~40% of the cells expressed the transfected MOR-EGFP and CCKBR-mCherry (Figure S1d). Using FLIM-FRET, we confirmed that MOR and CCKBR were within close proximity, as measured by the shortened lifetime of the donor fluorophore EGFP that was fused with MOR (Fig. 2c). The statistical fluorescence lifetime of EGFP showed a significant decrease in EGFP-mCherry-expressing cells compared with cells expressing EGFP alone, as a positive control. Similarly, the fluorescence lifetime of EGFP significantly decreased in MOR-EGFP- and CCKBR-mCherry-co-expressing cells compared with MOR-EGFP- and mCherry-co-expressing cells (Fig. 2d). Significantly increased FRET efficiency (p < 0.0001) in the MOR-EGFP- and CCKBR-mCherry-co-expressing group compared with the MOR-EGFP- and mCherry-co-expressing group indicates a direct MOR–CCKBR interaction in the co-transfected HEK293 cells (Fig. 2e).
TM3 of MOR mediated the MOR–CCKBR heteromerization
To further validate the heteromerization of MOR–CCKBR, we aimed to identify the domain of MOR that participates in the interaction with CCKBR.
We first used a threading method to simulate a model of the MOR–CCKBR complex27. Subsequently, a computational prediction was performed to analyze the interface of the MOR–CCKBR interaction. The prediction results showed that the intracellular loop 2 (IL2), extracellular loop 2 (EL2), transmembrane domain 3 (TM3), and TM4 of MOR were most likely the domains constituting the binding interface with CCKBR (Fig. 3a). According to the predicted structure of the MOR–CCKBR complex, the TM6 of MOR was located the farthest from the binding interface and was the least likely to bind with CCKBR. We constructed MOR with IL2 or EL2 amino-acid fragment deletion, TM3 replaced by TM6 mutation of MOR and TM4 replaced by TM6 mutation of MOR, all of which were fused with EGFP at the C terminal of the MOR sequence (Fig. 3b and Figure S3). FLIM assays using the mutant MORs demonstrated that only TM3 replaced with TM6-MOR-EGFP showed significantly less shortened lifetime of the donor fluorophore co-expressed with CCKBR-mCherry compared to the wild-type MOR group (Fig. 3c, d and Figure S4). FRET efficiency significantly decreased (p = 0.0029) in the group in which TM3 was replaced with TM6 mutation compared to that in the wild-type MOR group (Fig. 3e). These results suggest that the TM3 domain of MOR acts as a key factor in the heteromerization of MOR and CCKBR.
Fig. 3. Transmembrane domain 3 (TM3) of MOR participated in the heteromerization of MOR and CCKBR.

a The computational predicted model of the MOR–CCKBR complex structure. The blue- and red-colored structures represent MOR and CCKBR, respectively. The green domains represent receptor–receptor-binding sites. b Expression and colocalization of MOR (TM3 replaced with TM6)-EGFP and CCKBR-mCherry in HEK293 cells. c FLIM images of EGFP alone or linked with mutant MOR or wild-type MOR in different overexpressing HEK293 cells. The lifetime of MOR-EGFP co-expressed with CCKBR-mCherry was shorter than that of MOR-EGFP expression alone, as indicated by a blueshift in the color. However, mutant MOR co-expressed with CCKBR-mCherry did not show significant blueshifting compared with the group expressing mutant MOR alone. d Statistics of EGFP fluorescence lifetime in (c). e FRET efficiency showed a significant decrease in energy transfer between mutant MOR-EGFP and CCKBR-mCherry. **p < 0.01, t-test. Data are represented as the mean ± SEM
Hence, we developed a cell-penetrating interfering peptide (TM3MOR-TAT) according to the sequence of the MOR TM3 by adding the TAT sequence (RKKRRQRRR) to the C terminal of the entire TM3. A 5-FAM fluorophore labeled on the N terminal of the peptide provided its location image in the HEK293 cells under laser scanning confocal microscopy (Fig. 4a). As the fluorescent images showed, the TM3MOR-TAT peptide penetrated the cell membrane, and the membrane-located peptide made it possible to maintain its function in disrupting the MOR–CCKBR interaction.
Fig. 4. TM3MOR-TAT peptide disrupted MOR–CCKBR interaction.

a An example of confocal microscopy image of 5-FAM-labeled TM3MOR-TAT-treated HEK293 cells showing successful penetration of the TM3MOR-TAT peptides into the cells (left). The magnified area indicates the membrane location of the peptide (right). b An example of the FLIM image of TAT-fused peptide-treated cells co-expressing MOR-EGFP and CCKBR-mCherry. Compared with the untreated or scrambled-TAT-treated group, EGFP in the TM3MOR-TAT-treated cells showed longer lifetime of fluorescence. c Statistics of EGFP fluorescence lifetime in (b). d FRET efficiency of MOR-EGFP and CCKBR-mCherry in interfering peptide-treated cells. Compared with the non-peptide-treated or scrambled-TAT-treated group, the TM3MOR-TAT-treated group exhibited a significant decrease in FRET efficiency. *p < 0.05, **p < 0.01, t-test. Data are represented as the mean ± SEM
FLIM images showed that MOR-fused EGFP in the TM3MOR-TAT-treated group exerted a longer fluorescence lifetime than in the group pretreated with an amino-acid scrambled control peptide (scrambled-TAT; Fig. 4b, c). FRET efficiency calculation showed that the interaction of MOR and CCKBR was effectively interfered with by TM3MOR-TAT pretreatment (Fig. 4d). These data further confirm that TM3 is the interface of MOR interacting with CCKBR and indicate that the TM3MOR-TAT treatment could be a convenient approach to interfere with the heteromerization of MOR and CCKBR.
Heteromerization of MOR and CCKBR inhibited MOR activity in transfected HEK293 cells
To determine whether heteromerization with CCKBR alters pharmacological properties of MOR, a radio-ligand-binding assay was performed using a MOR-selective agonist [3H]-DAMGO. The density of binding sites (Bmax) and the affinity to radioligands were determined through saturation analysis with membranes of HEK293 cells expressing HA-MOR alone or co-expressing HA-MOR and FLAG-CCKBR (Table 1). Although expression of total cellular HA-MOR was upregulated in the HA-MOR- and FLAG-CCKBR-co-expressed cells (Figure S2a and b), the Bmax values were not significantly influenced (Figure S2c), indicating that the heteromerization of MOR and CCKBR did not increase the density of MOR ligand-binding sites on the cell membrane. However, the KD values for [3H]-DAMGO decreased significantly. The KD values of MOR were significantly higher in the cells stably co-transfected with CCKBR and MOR (2.27 ± 0.28 nM) than those in the cells expressing MOR alone (1.28 ± 0.11 nM). These results indicate that heteromerization of MOR and CCKBR leads to a decrease of MOR affinity, while it has no significant effects on the maximum binding sites of MOR.
Table 1.
Results of radio-ligand-binding assay of MOR
| Receptor | [3H]-DAMGO | |
|---|---|---|
| Bmax (fmol/mg protein) | KD (nM) | |
| MOR | 1851 ± 110 | 1.28 ± 0.11 |
| MOR + CCKBR | 2550 ± 720 | 2.27 ± 0.28* |
Saturation-binding assays were performed on membrane samples prepared from stably transfected cells. The dissociation constant (KD) and the number of [3H]-DAMGO-binding sites (Bmax) were calculated. Co-expressing cells were compared with cells expressing MOR alone. *p < 0.05, t-test, n = 4
To confirm the radio-ligand-binding results, we further assayed the agonist-mediated phosphorylation of ERK1/2 and measured the effect of CCKBR-co-expression on MOR-dependent phosphorylation of ERK1/2 (Fig. 5a). In HEK293 cells stably transfected with only MOR or co-transfected with MOR and CCKBR, DAMGO enhanced ERK1/2 phosphorylation in a concentration-dependent manner. Significantly lower ERK1/2 phosphorylation responses were observed in the MOR- and CCKBR-co-expressing cells compared to those in the cells expressing MOR alone under stimulation of DAMGO at 10−10, 10−9, and 10−8 M, respectively (Fig. 5b).
Fig. 5. Heteromerization of MOR and CCKBR inhibited MOR activities in co-transfected HEK293 cells.

a Immunoblot assays showing the ERK1/2 phosphorylation level in cells expressing HA-MOR alone (top) or co-expressing HA-MOR and FLAG-CCKBR (bottom). Cells were exposed to different concentrations of DAMGO for 5 min, and pERK1/2 and total ERK1/2 were determined. Concentrations of DAMGO were indicated above the bands. b Densitometric analysis was quantified by normalizing with total ERK1/2 and expressed as folds of phosphorylated ERK1/2 over basal in vehicle-treated cells. ERK1/2 phosphorylation in cells expressing HA-MOR alone was significantly higher than that in HA-MOR- and FLAG-CCKBR-co-expressing cells at concentrations of 10−10, 10−9, and 10−8 M. *p < 0.05, t-test, n = 4. c Immunoblot assays showing ERK1/2 phosphorylation in HA-MOR-expressing cells (top) or HA-MOR- and FLAG-CCKBR-co-expressing cells (bottom). Time points of DAMGO stimulation are labeled above the bands. d Statistical results. ERK1/2 phosphorylation in cells expressing HA-MOR alone significantly increased compared with that in HA-MOR- and FLAG-CCKBR-co-expressing cells at 5 min after DAMGO exposure. **p < 0.01, t-test, n = 3. Data are represented as the mean ± SEM
We further examined whether MOR co-expression with CCKBR could result in differences in the time-response curve of MOR to DAMGO, an MOR agonist. The cells were incubated with 10−10 M DAMGO for different time intervals from 1 to 60 min and then were lysed to determine pERK1/2 levels (Fig. 5c). After incubation, DAMGO induced ERK1/2 phosphorylation with a similar response pattern in these two types of cells, i.e., ERK1/2 phosphorylation appeared to increase at 1 min, reached a peak at 5 min, then gradually declined and almost returned to the basal level at 15 min. Although the two cell lines shared similar response curves to DAMGO, the ERK1/2 phosphorylation response decreased significantly in cells co-transfected with MOR and CCKBR with a 5-min-DAMGO incubation (Fig. 5d), suggesting that co-expression with CCKBR could diminish DAMGO-induced ERK1/2 phosphorylation by decreased affinity for DAMGO.
To accurately quantify receptor activation, we further performed cAMP assays with an ELISA experiment. It is known that DAMGO activation of MOR decreased forskolin-induced intracellular cAMP accumulation. Percent inhibition of forskolin-induced cAMP accumulation by DAMGO-stimulated MOR was calculated to evaluate the activity of MOR. In accordance with the results of the ERK1/2 phosphorylation assays, 10−11 M DAMGO-induced inhibition of cAMP accumulation decreased in the MOR- and CCKBR-co-expressing cells compared with that in the cells expressing MOR alone (Fig. 6). The original cAMP concentrations were provided in the Figure S5a. When there was no DAMGO treatment, the basal cAMP concentration in the cells expressing MOR alone was slightly higher than in the MOR- and CCKBR-co-expressing cells. However, the decrease in the cAMP concentration was greater in the cells expressing MOR alone than in the MOR- and CCKBR-co-expressing cells, after DAMGO treatment.
Fig. 6.
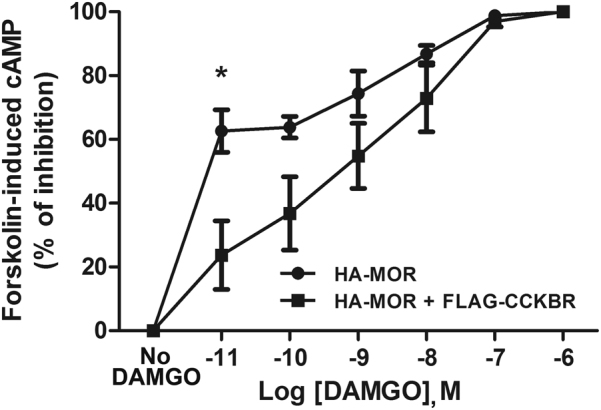
Heteromerization of MOR and CCKBR reduced inhibition of cAMP accumulation by DAMGO-activated MOR
These results suggest that the MOR co-expression with CCKBR decreases the signaling transduction coupled with MOR.
TM3MOR-TAT peptide rescued the MOR activity by disrupting MOR–CCKBR interaction and remitted CCK-8-induced antagonism to morphine analgesia
If the MOR–CCKBR interaction plays a role in the functional inhibition of MOR, we predicted that disruption of the interaction should abolish the inhibition of MOR in heteromers with CCKBR. More importantly, if the MOR–CCKBR heteromerization indeed plays a role in CCK-8-mediated antagonism to opioid analgesia, disrupting that the heteromerization should improve opioid analgesic effects.
To determine whether the inhibited signaling of MOR could be restored when MOR heteromerization with CCKBR was interfered with by TM3MOR-TAT, we performed the ERK1/2 phosphorylation assay in the MOR- and CCKBR-co-expressing cells. DAMGO-induced ERK1/2 phosphorylation through MOR was significantly enhanced in the TM3MOR-TAT-pretreated group compared to that in the scrambled-TAT-pretreated group (p = 0.0477; Fig. 7a, b), indicating an enhanced signaling of MOR in TM3MOR-TAT-pretreated group.
Fig. 7. The TM3MOR-TAT peptide rescued MOR activity by disrupting the MOR–CCKBR interaction and remitted CCK-8-induced antagonism to morphine analgesia.

a A representative result of ERK1/2 phosphorylation immunoblot assay. The top panel showed phosphorylated ERK1/2 bands, and the bottom panel showed total ERK1/2 bands. b Statistical results. The gray density values of the bands were obtained by Quantity One software and analyzed with GraphPad Prism 5.0 after gray density values normalized. Compared with the scrambled-TAT treatment, the TM3MOR-TAT treatment significantly increased the phosphorylation of ERK1/2. *p < 0.05, t-test, n = 3. Data are expressed as the mean ± SEM. c TM3MOR-TAT pretreatment enhanced DAMGO-induced inhibition of cAMP accumulation. d TM3MOR-TAT reversed CCK-8 antagonism to morphine analgesia. Percent of maximum possible effect (%MPE) 30 min after morphine (2 mg/kg, s.c.) application. Compared with saline group, CCK-8 (i.t.) pretreatment decreased the analgesic effects of morphine. However, pretreatment with TM3MOR-TAT 4 h before CCK-8 and morphine injection showed significantly stronger analgesic effects compared to that with scrambled-TAT pretreatment. *p < 0.05, t-test. Data are represented as the mean ± SEM
Further cAMP assays were performed using MOR and CCKBR-co-expressing cells with scrambled-TAT or TM3MOR-TAT pretreatment. DAMGO-induced inhibition of cAMP accumulation was enhanced in TM3MOR-TAT-treated groups with DAMGO concentrations at 10−11, 10−9, 10−8, and 10−7 M (Fig. 7c). The original cAMP concentrations were provided in the Figure S5b. The TM3MOR-TAT treatment enhanced the cAMP inhibition of MOR compared with the scrambled-TAT treatment.
These results suggest that the inhibition of MOR activity could be reversed when the MOR–CCKBR heteromerization is interfered.
On the basis of the above experimental results, we further hypothesized that CCK-8-induced antagonism to morphine analgesia would be diminished when the MOR–CCKBR interaction was disrupted. 5-FAM-labeled TM3MOR-TAT peptides were applied intrathecally (i.t.) to the rats and were observed to localize in the spinal cord dorsal horn 5 h after injection (Figure S6). Morphine-induced antinociception was attenuated by i.t. CCK-8 pre-injection, but the inhibition was diminished when the TM3MOR-TAT peptides were injected (i.t.) 4 h before the CCK-8 injection. However, there was no change in morphine antinociception when the scrambled-TAT peptides were injected (Fig. 7d). These data suggest that interfering with the MOR–CCKBR interaction in the spinal cord using TM3MOR-TAT can prevent CCK-8-induced antagonism against morphine analgesia in rats.
Discussion
Since several studies indicated that CCKBR antagonized MOR-mediated analgesia through interaction with MOR rather than simply reducing pain thresholds11,12, we first investigated whether a direct interaction existed between MOR and CCKBR. Because highly specific and efficient antibodies for MOR and CCKBR are not available yet, we tagged the receptors with HA and FLAG, respectively, and co-transfected their genes into HEK293 cells to facilitate the investigation of MOR–CCKBR interaction. In western blot experiments, MOR expression in the MOR- and CCKBR-co-expressed cells is higher than that in the cells expressing MOR alone (Figure S2a and b). This may result from the difference in transfection efficiency. Although we aimed to ensure similar conditions for transfection, we could still not exclude the variation occurring after transfection. It is notable that the existence of CCKBR may affect the membrane expression of MOR. In the radio-ligand-binding assays, the Bmax of MOR, which reflects the receptor density on the membrane, showed no significant difference in the MOR- and CCKBR-co-expressing cells or in the cells expressing MOR alone (Table 1). The ERK1/2 phosphorylation assays and cAMP assays showed no significant difference between the MOR- and CCKBR-co-expressing cells and cells expressing MOR alone at a higher dose of DAMGO stimulation; this indicated that the membrane density of MOR in the two cell types was the same (Figs. 5 and 6). Because total cellular proteins were used in the western blot analysis, while cell membranes were used in the radio-ligand-binding assay, this indicated that most MORs were located intracellularly in the MOR- and CCKBR-co-expressing cells. Co-IP results demonstrated the existence of the MOR and CCKBR protein interaction in co-transfected cells (Fig. 2). It is well known that co-IP of two proteins does not indicate a direct protein–protein interaction; there are possibly one or more other proteins acting as a bridge to link the two co-precipitated proteins. To clearly illuminate the modality of the MOR–CCKBR interaction, we used FLIM-FRET for the direct protein–protein interaction study. FLIM-FRET provides a non-invasive way to study the interaction of proteins in their natural environment and makes it possible to observe nanoscale resolution of interactions in real time28,29. Importantly, we observed strong FRET signals in the MOR-EGFP- and CCKBR-mCherry-co-expressing cells using FLIM-FRET (Fig. 2). On the basis of the improved resolution of this method, it is reasonable to consider that MOR and CCKBR form heteromers through direct interaction.
According to the predicted MOR–CCKBR interaction from the computational model, amino acids in IL2 (172–176 a.a.) and EL2 (228–236 a.a.) of the MOR participated in the heteromerization of MOR with CCKBR (Fig. 3); however, the relevant amino-acid-deleted mutants appear to have a stronger interaction with CCKBR. Moreover, the TM3, TM4, and TM5 of MOR lie in the interface of the MOR–CCKBR interaction. It was additionally proved that the TM1 domain of MOR mediated its interaction with the DOR30. We additionally compared the amino-acid sequences of different types of opioid receptors and selected TM3, which is more highly conserved across GPCRs, and TM4, which has a sequence more unique to MOR for further investigation. As predicted, TM6 of MOR did not lie in the protein–protein interacting interface. We constructed two types of MOR mutants, i.e., using TM6 to replace either TM3 or TM4. The FLIM-FRET assay demonstrated that the MOR–CCKBR interaction was significantly disrupted when using TM6 to replace TM3 of MOR (Fig. 3). On the other hand, the TM4-replaced mutation of MOR showed no changes in interaction with CCKBR (Figure S4).
In a general view of protein–protein interactions, the GST-pulldown technique should be adopted to determine the domains that participate in interaction binding31. However, considering the hydrophobic property of the transmembrane domains of GPCRs, false-positive results are unavoidable if using GST-pulldown in analyzing membrane protein interactions. Therefore, our present study used FLIM-FRET and direct receptor mutations instead to investigate the membrane receptor–receptor interaction. At the same time, the FLIM-FRET assay is based on detection of the fluorescence lifetime imaging of EGFP, which is only influenced by molecule–molecule direct interaction. Therefore, using this more reliable FLIM-FRET technique combined with fragment mutations, our study provides strong evidence that MOR and CCKBR form heteromers through direct protein–protein interactions.
Several studies focused on functional regulation of receptors by interacting with each other. For instance, stimulation of adenosine A2 receptors potently reduced the affinity of dopamine D2 receptor with its agonist through receptor–receptor interaction in striatal neuronal membranes32. In addition, ERK1/2 phosphorylation induced by adenosine A2 receptor activation was synergistically potentiated by simultaneous activation of subtype 5 metabotropic glutamate receptor through the same mechanism33. The ligand-binding features and agonist-induced ERK1/2 phosphorylation of receptors in our present study in the MOR- and CCKBR-co-expressing cells (Table 1 and Fig. 5) provide insights into MOR activity regulation by heteromerizing with other receptors. Coupled with the results of the intracellular cAMP assays, it can be deduced that the potency of DAMGO, an MOR agonist, is strengthened when MOR is not heteromered with CCKBR (i.e., in the cells expressing MOR alone or in TM3MOR-TAT pretreated MOR- and CCKBR-co-expressing cells). Heteromerization with CCKBR may modulate activation of MOR through allosteric regulation. These results shed new light on potential mechanisms of CCK-8-induced anti-opioid effects. The diminished activity of MOR in the MOR–CCKBR complex can lead to a decline in morphine analgesia, which may be a morphine tolerance mechanism.
Intrathecal application of TM3MOR-TAT reversed CCK-8-induced inhibition of morphine analgesia (Fig. 7), suggesting the contribution of CCKBR-mediated antagonisms to morphine analgesia. This result was in accordance with a previous study that focused on CCK-8-induced antagonism to electroacupuncture analgesia in the caudate nucleus through CCKBR34. Another study reported that opioid-induced placebo responses were completely abolished by administration of a CCKBR agonist35. In contrast, administration of a CCKAR antagonist potentiated electroacupuncture analgesia36. Interestingly, several researchers reported the important role of CCK-8 and CCKBR in opioid tolerance8,37–41; therefore, disruption of the MOR–CCKBR interaction, as in the present study, might provide a new strategy to prevent opioid tolerance in clinical practice.
In summary, our present study proposes a molecular mechanism underlying MOR regulation (Fig. 8). The heteromerization of MOR and CCKBR decreases the MOR activity, such as decreasing the ligand-binding affinity and agonist-induced ERK1/2 phosphorylation. Disrupting the MOR–CCKBR heteromerization with MOR TM3-based interfering peptide could restore the activity of MOR and relieve the anti-opioid effects of CCK-8. Drugs targeting MOR–CCKBR heteromerization may be developed to improve the effect of opioid analgesics or to reduce tolerance progress.
Fig. 8. Schematic diagram: MOR–CCKBR heteromerization antagonizes MOR activity and morphine analgesia.

a When MOR is not heteromerized with CCKBR, MOR has high binding affinity with its agonist DAMGO (shown here as an example) and highly activates signal transmissions, such as strong DAMGO-induced ERK1/2 phosphorylation and inhibition of cAMP accumulation. b When MOR directly forms heteromers with CCKBR, the ligand-binding affinity of MOR is decreased, and agonist-induced ERK1/2 phosphorylation and cAMP inhibition are diminished, exhibiting CCK-8 antagonism to morphine analgesia. c Disruption of the heteromerization of MOR–CCKBR with TM3MOR-TAT treatment enhances the activities of the MOR and thus improves the analgesic effects of μ-opioid agonists such as DAMGO or morphine
Electronic supplementary material
Acknowledgements
This work was supported by grants from the National Basic Research Program of the Ministry of Science and Technology of China (2014CB548200 and 2015CB554503), National Natural Science Foundation of China (91732107, 81521063, 81571067, and 30700206), and “111” Project of the Ministry of Education of China (B07001). We thank Professor Lee-Yuan Liu-Chen (Temple University School of Medicine, Philadelphia, USA) for kindly providing the MOR plasmid and Professor Mark R. Hellmich (Department of Surgery, University of Texas Medical Branch, Galveston, TX, USA) for providing the CCKBR plasmid as gifts. We would additionally like to thank Professor Ze-Hui Gong (Beijing Institute of Pharmacology and Toxicology, Beijing, China) for help in radio-ligand-binding assays, Professor Tai-Jiao Jiang (Institute of Biophysics, Chinese Academy of Sciences, Beijing, China), and for assistance in prediction of the MOR–CCKBR interaction interface.
Authors' contributions
Y.Y. performed the plasmid construction, co-IP, FLIM-FRET, drug delivery, and behavior tests, participated in the ERK1/2 phosphorylation assay and the design of the study, and drafted the manuscript. Q.L. performed stable receptor-expressing cell construction, immunofluorescent staining, and radio-ligand-binding assay and participated in the ERK1/2 phosphorylation assay and the design of the study. Q-.H.H. participated in the FLIM-FRET assay. J-.S.H. participated in the design of the study. L.S. participated in the design of the study, performed experiments, and revised the manuscript. Y.W. conceived the study, participated in the design, and revised the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
These authors contributed equally: Yin Yang, Qian Li.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s12276-018-0090-5.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Li Su, Phone: +86 10 82805034, Email: lisu76@bjmu.edu.cn.
You Wan, Phone: +86 10 82805185, Email: ywan@hsc.pku.edu.cn.
References
- 1.Pasternak GW, Pan YX. Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev. 2013;65:1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dang VC, Christie MJ. Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br. J. Pharmacol. 2012;165:1704–1716. doi: 10.1111/j.1476-5381.2011.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faris PL, Komisaruk BR, Watkins LR, Mayer DJ. Evidence for the neuropeptide cholecystokinin as an antagonist of opiate analgesia. Science. 1983;219:310–312. doi: 10.1126/science.6294831. [DOI] [PubMed] [Google Scholar]
- 4.Wang XJ, Wang XH, Han JS. Cholecystokinin octapeptide antagonized opioid analgesia mediated by mu- and kappa- but not delta-receptors in the spinal cord of the rat. Brain Res. 1990;523:5–10. doi: 10.1016/0006-8993(90)91629-U. [DOI] [PubMed] [Google Scholar]
- 5.Li Y, Han JS. Cholecystokinin-octapeptide antagonizes morphine analgesia in periaqueductal gray of the rat. Brain Res. 1989;480:105–110. doi: 10.1016/0006-8993(89)91572-2. [DOI] [PubMed] [Google Scholar]
- 6.Han JS, Ding XZ, Fan SG. Cholecystokinin octapeptide (CCK-8): antagonism to electroacupuncture analgesia and a possible role in electroacupuncture tolerance. Pain. 1986;27:101–115. doi: 10.1016/0304-3959(86)90227-7. [DOI] [PubMed] [Google Scholar]
- 7.Han JS, Ding XZ, Fan SG. Is cholecystokinin octapeptide (CCK-8) a candidate for endogenous anti-opioid substrates? Neuropeptides. 1985;5:399–402. doi: 10.1016/0143-4179(85)90038-1. [DOI] [PubMed] [Google Scholar]
- 8.Huang C, et al. CCK(B) receptor antagonist L365,260 potentiates the efficacy to and reverses chronic tolerance to electroacupuncture-induced analgesia in mice. Brain Res. Bull. 2007;71:447–451. doi: 10.1016/j.brainresbull.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Pu SF, Zhuang HX, Han JS. Cholecystokinin octapeptide (CCK-8) antagonizes morphine analgesia in nucleus accumbens of the rat via the CCK-B receptor. Brain Res. 1994;657:159–164. doi: 10.1016/0006-8993(94)90963-6. [DOI] [PubMed] [Google Scholar]
- 10.Dourish CT, et al. The selective CCK-B receptor antagonist L-365,260 enhances morphine analgesia and prevents morphine tolerance in the rat. Eur. J. Pharmacol. 1990;176:35–44. doi: 10.1016/0014-2999(90)90129-T. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Y, Sun YH, Shen JM, Han JS. Increased release of immunoreactive CCK-8 by electroacupuncture and enhancement of electroacupuncture analgesia by CCK-B antagonist in rat spinal cord. Neuropeptides. 1993;24:139–144. doi: 10.1016/0143-4179(93)90077-N. [DOI] [PubMed] [Google Scholar]
- 12.Liu NJ, et al. Cholecystokinin octapeptide reverses mu-opioid-receptor-mediated inhibition of calcium current in rat dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 1995;275:1293–1299. [PubMed] [Google Scholar]
- 13.Han JS. Acupuncture analgesia: areas of consensus and controversy. Pain. 2011;152:S41–S48. doi: 10.1016/j.pain.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalrymple MB, Pfleger KD, Eidne KA. G protein-coupled receptor dimers: functional consequences, disease states and drug targets. Pharmacol. Ther. 2008;118:359–371. doi: 10.1016/j.pharmthera.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Navarro G, et al. Quaternary structure of a G-protein-coupled receptor heterotetramer in complex with Gi and Gs. BMC Biol. 2016;14:26. doi: 10.1186/s12915-016-0247-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomes I, Ijzerman AP, Ye K, Maillet EL, Devi LA. G protein-coupled receptor heteromerization: a role in allosteric modulation of ligand binding. Mol. Pharmacol. 2011;79:1044–1052. doi: 10.1124/mol.110.070847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Bao L. Interaction and regulatory functions of mu- and delta-opioid receptors in nociceptive afferent neurons. Neurosci. Bull. 2012;28:121–130. doi: 10.1007/s12264-012-1206-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferre S, et al. An update on adenosine A2A-dopamine D2 receptor interactions: implications for the function of G protein-coupled receptors. Curr. Pharm. Des. 2008;14:1468–1474. doi: 10.2174/138161208784480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu L, et al. The role of TRPV1 in different subtypes of dorsal root ganglion neurons in rat chronic inflammatory nociception induced by complete Freund’s adjuvant. Mol. Pain. 2008;4:61. doi: 10.1186/1744-8069-4-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu FY, et al. Activation of satellite glial cells in lumbar dorsal root ganglia contributes to neuropathic pain after spinal nerve ligation. Brain Res. 2012;1427:65–77. doi: 10.1016/j.brainres.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 22.Li N, et al. BN-9, a chimeric peptide with mixed opioid and neuropeptide FF receptor agonistic properties, produces nontolerance-forming antinociception in mice. Br. J. Pharmacol. 2016;173:1864–1880. doi: 10.1111/bph.13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao X, et al. Shp-1 dephosphorylates TRPV1 in dorsal root ganglion neurons and alleviates CFA-induced inflammatory pain in rats. Pain. 2015;156:597–608. doi: 10.1097/01.j.pain.0000460351.30707.c4. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, et al. Lysine-specific demethylase 1 in breast cancer cells contributes to the production of endogenous formaldehyde in the metastatic bone cancer pain model of rats. PLoS ONE. 2013;8:e58957. doi: 10.1371/journal.pone.0058957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herman B, Krishnan RV, Centonze VE. Microscopic analysis of fluorescence resonance energy transfer (FRET) Methods Mol. Biol. 2004;261:351–370. doi: 10.1385/1-59259-762-9:351. [DOI] [PubMed] [Google Scholar]
- 26.Sun Y, Periasamy A. Localizing protein-protein interactions in living cells using fluorescence lifetime imaging microscopy. Methods Mol. Biol. 2015;1251:83–107. doi: 10.1007/978-1-4939-2080-8_6. [DOI] [PubMed] [Google Scholar]
- 27.Hu Y, et al. Incorporation of local structural preference potential improves fold recognition. PLoS ONE. 2011;6:e17215. doi: 10.1371/journal.pone.0017215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Omer T, Zhao L, Intes X, Hahn J. Reduced temporal sampling effect on accuracy of time-domain fluorescence lifetime Forster resonance energy transfer. J. Biomed. Opt. 2014;19:86023. doi: 10.1117/1.JBO.19.8.086023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schoberer J, Botchway SW. Investigating protein-protein interactions in the plant endomembrane system using multiphoton-induced FRET-FLIM. Methods Mol. Biol. 2014;1209:81–95. doi: 10.1007/978-1-4939-1420-3_6. [DOI] [PubMed] [Google Scholar]
- 30.He SQ, et al. Facilitation of mu-opioid receptor activity by preventing delta-opioid receptor-mediated codegradation. Neuron. 2011;69:120–131. doi: 10.1016/j.neuron.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Liu F, et al. Direct protein-protein coupling enables cross-talk between dopamine D5 and gamma-aminobutyric acid A receptors. Nature. 2000;403:274–280. doi: 10.1038/35002014. [DOI] [PubMed] [Google Scholar]
- 32.Ferre S, von Euler G, Johansson B, Fredholm BB, Fuxe K. Stimulation of high-affinity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proc. Natl Acad. Sci. USA. 1991;88:7238–7241. doi: 10.1073/pnas.88.16.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferre S, et al. Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: implications for striatal neuronal function. Proc. Natl. Acad. Sci. USA. 2002;99:11940–11945. doi: 10.1073/pnas.172393799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang CX, et al. Cholecystokinin-8 antagonizes electroacupuncture analgesia through its B receptor in the caudate nucleus. Neuromodulation. 2010;13:93–98. doi: 10.1111/j.1525-1403.2009.00247.x. [DOI] [PubMed] [Google Scholar]
- 35.Benedetti F, Amanzio M, Thoen W. Disruption of opioid-induced placebo responses by activation of cholecystokinin type-2 receptors. Psychopharmacology. 2011;213:791–797. doi: 10.1007/s00213-010-2037-y. [DOI] [PubMed] [Google Scholar]
- 36.Shi TF, et al. L-364,718 potentiates electroacupuncture analgesia through cck-a receptor of pain-related neurons in the nucleus parafascicularis. Neurochem. Res. 2011;36:129–138. doi: 10.1007/s11064-010-0281-5. [DOI] [PubMed] [Google Scholar]
- 37.Ding XZ, Fan SG, Zhou JP, Han JS. Reversal of tolerance to morphine but no potentiation of morphine-induced analgesia by antiserum against cholecystokinin octapeptide. Neuropharmacology. 1986;25:1155–1160. doi: 10.1016/0028-3908(86)90164-4. [DOI] [PubMed] [Google Scholar]
- 38.Dourish CT, Hawley D, Iversen SD. Enhancement of morphine analgesia and prevention of morphine tolerance in the rat by the cholecystokinin antagonist L-364,718. Eur. J. Pharmacol. 1988;147:469–472. doi: 10.1016/0014-2999(88)90183-5. [DOI] [PubMed] [Google Scholar]
- 39.Idanpaan-Heikkila JJ, Guilbaud G, Kayser V. Prevention of tolerance to the antinociceptive effects of systemic morphine by a selective cholecystokinin-B receptor antagonist in a rat model of peripheral neuropathy. J. Pharmacol. Exp. Ther. 1997;282:1366–1372. [PubMed] [Google Scholar]
- 40.Xiong W, Yu LC. Involvement of endogenous cholecystokinin in tolerance to morphine antinociception in the nucleus accumbens of rats. Behav. Brain Res. 2006;173:116–121. doi: 10.1016/j.bbr.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 41.DeSantana JM, Da SL, Sluka KA. Cholecystokinin receptors mediate tolerance to the analgesic effect of TENS in arthritic rats. Pain. 2010;148:84–93. doi: 10.1016/j.pain.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.