

Abstract
Human follicle-stimulating hormone (FSH) exhibits both macro- and microheterogeneity in its carbohydrate moieties. Macroheterogeneity results in three physiologically relevant FSHβ subunit variants, two that possess a single N-linked glycan at either one of the two βL1 loop glycosylation sites or one with both glycans. Microheterogeneity is characterized by 80 to over 100 unique oligosaccharide structures attached to each of the 3 to 4 occupied N-glycosylation sites. With respect to its receptor, partially glycosylated (hypo-glycosylated) FSH variants exhibit higher association rates, greater apparent affinity, and greater occupancy than fully glycosylated FSH. Higher receptor binding-activity is reflected by greater in vitro bioactivity and, in some cases, greater in vivo bioactivity. Partially glycosylated pituitary FSH shows an age-related decline in abundance that may be associated with decreased fertility. In this review, we describe an integrated approach involving genetic models, in vitro signaling studies, FSH biochemistry, relevance of physiological changes in FSH glycoform abundance, and characterize the impact of FSH macroheterogeneity on fertility and reproductive aging. We will also address the controversy with regard to claims of a direct action of FSH in mediating bone loss especially at the peri- and postmenopausal stages.
Keywords: pituitary, N-glycosylation, follicle-stimulating hormone, bone, female Infertility
Structural Attributes of Follicle-Stimulating Hormone (FSH) and its Subunits
Follicle-stimulating hormone is one of three gonadotropins in the human glycoprotein hormone family. This hormone family is part of the cystine knot growth factor superfamily, a large group of homo- and heterodimeric signaling molecules (1). FSH plays a central role in reproduction, particularly in females. In the ovary, FSH stimulates follicle development and estrogen synthesis. In the testis, FSH maintains Sertoli cell function, which supports spermatogenesis. Although currently controversial (2, 3), FSH has been claimed to play a direct role in osteoporosis by stimulating differentiation of osteoclasts, which are responsible for removing bone (4). The idea put forth is that in the postmenopausal period when FSH levels rise, activation of osteoclasts results in bone loss. Reports of non-gonadal actions of FSH have recently been summarized (5).
Follicle-stimulating hormone is composed of two dissimilar, cystine knot motif glycoprotein subunits: a common α-subunit and hormone-specific β-subunit (Figure 1) (6). The FSHα subunit amino-acid sequence and disulfide bond organization, including a cystine knot motif, are identical to those in the other glycoprotein hormones, luteinizing hormone (LH), thyroid-stimulating hormone (TSH), and chorionic gonadotropin (CG) (7). However, the N-glycan populations at both glycosylated residues, Asn52 and Asn78, differ from those of the other glycoprotein hormone α-subunits such that these otherwise identical subunits can be distinguished from each other and from free α-subunit by their oligosaccharide populations (8–10). The hormone-specific FSHβ subunit shares 34–40% sequence homology, six conserved disulfide bonds, cystine knot motif, and seatbelt loop with the other human glycoprotein hormone β-subunits (7, 11, 12). While there are two potential N-glycosylation sites in FSHβ, partially glycosylated variants exist that are missing either one of these oligosaccharides (13). These contribute to an unknown degree of charge variation in FSH preparations and result in the classic FSH isoforms (14, 15). The classic interpretation of FSH isoforms was based solely on the notion that variant patterns of negatively charged sialic acid or, to a much lesser extent, sulfate residues terminated oligosaccharide branches, which gave rise to differentially charged isoforms. The observation of hypo-glycosylation further refines our understanding of isoforms, in that net charge may vary, due to presence or absence of entire glycans.
Figure 1.
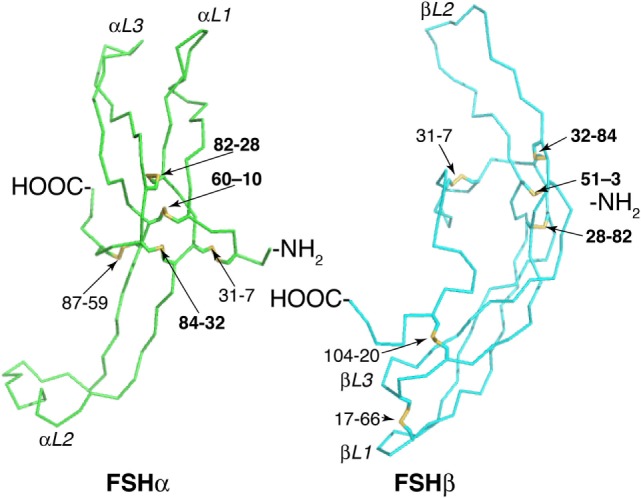
Follicle-stimulating hormone (FSH) subunit peptide moieties. Wire-frame models of FSH subunits extracted from pdb 1FL7 using MacPyMOL v1.8.2.3. FSHα backbone is green and FSHβ backbone is cyan. Disulfide bonds are indicated as yellow sticks. Cystine knot loops are designated by subunit (α or β) and number (1–3). Pairs of numbers refer to Cys residues involved in a disulfide bond. Bold numbers indicate Cys Knot disulfide bonds.
FSH Glycosylation Heterogeneity
Follicle-stimulating hormone glycosylation exhibits both macro- and microheterogeneity (Table 1). Macroheterogeneity herein refers to the presence or absence of glycosylation at any one potential glycosylation site. Examples of FSH macroheterogeneity involve the absence of either FSHβ Asn7 or Asn24 oligosaccharides in a population of fully processed and secreted FSH. Microheterogeneity herein refers to as many as 80 to over 100 unique oligosaccharide structures, which can be detected once released from each of the 3–4 glycan-occupied Asn residues in FSH.
Table 1.
Macro- and microheterogeneity of hFSH preparations.
| Macroheterogeneity (% relative abundance) |
|||||||
|---|---|---|---|---|---|---|---|
| FSH preparation | Pituitary hFSH | Urinary hFSH | Pituitary hFSH24 | Pituitary hFSH21 | Pituitary hFSH21/18 | Recombinant GH3 hFSH24 | Recombinant GH3 hFSH21 |
| FSH24 | 77 | 86 | 100 | – | – | 89 | – |
| FSH21 | 23 | 14 | – | 100 | 60 | 11 | 54 |
| FSH18 | –a | – | – | – | 40 | – | 46 |
| FSH15 | – | – | – | – | – | – | – |
|
Types of oligosaccharides (% relative abundance) |
|||||||
| Oligosaccharide type | Recombinant GH3-hFSHb | ||||||
| Biantennary | 38.2 | 37.2 | 47.1 | 51.2 | 28.6 | 55.5 | |
| Triantennary(3)c | 41.0 | 44.0 | 30.7 | 35.9 | 2.5 | 0 | |
| Triantennary(6)d | 0 | 0 | 0 | 0 | 0 | 29.7 | |
| Tetra-antennary | 15.0 | 14.8 | 10.6 | 6.0 | 0.01 | 0 | |
| Neutral | 0.3 | 2.2 | 9.9 | 4.5 | 74.2 | 12.3 | |
| Sialylated | 99.1 | 97.5 | 75.4 | 78.8 | 20.7 | 87.7 | |
| Sulfated | 6.5 | 4.2 | 39.3 | 35.0 | 9.6 | 0 | |
| Sial/sulfat | 5.9 | 3.9 | 24.0 | 18.3 | 4.5 | 0 | |
| Core fucose | 43.0 | 23.9 | 45.1 | 47.8 | 23.0 | 50.6 | |
| Antenna-fucose | 0.3 | 0 | 3.6 | 0.8 | 0.4 | 19.9 | |
| Bisect GlcNAc | 32.6 | 23.9 | 17.9 | 23.2 | 7.9 | 47.0 | |
| GalNAc | 2.8 | 1.7 | 20.3 | 13.8 | 14.1 | 10.5 | |
Relative abundance determined by Western blot and mass spectrometry, respectively. Data are limited to those preparations for which both glycoform abundance and glycan microheterogeneity exist.
a– = not detected.
b = glycoforms not separated.
cTriantennary(3) = third branch attached to Man(α1–3) branch.
dTriantennary(6) = third branch attached to Man(α1–6) branch.
Differences in electrophoretic mobility of FSH subunits, revealed by subunit-specific Western blots, provide a convenient means to distinguish four FSH variants resulting from macroheterogeneity. Fully glycosylated hFSHβ migrates as a 24-kDa band (hereinafter, 24k-FSHβ), desN24glycan-FSHβ migrates as a 21-kDa band (21k-FSHβ), and desN7glycan-FSHβ migrates as an 18-kDa band (18k-FSHβ). The FSH heterodimers that incorporate these β-subunit variants are designated, FSH24, FSH21, and FSH18, respectively (19), and are shown in Figure 2. Pituitary extracts also possess a non-glycosylated, 15-kDa FSHβ variant (20). However, the corresponding FSH15 does not appear to be physiologically relevant, because subunit association is extremely inefficient when both FSHβ glycans are missing, and little, if any, FSH heterodimer is secreted (21). FSH24 and FSH21 are detected in FSH derived from human pituitary extracts, as well as from urinary protein preparations (Table 1). When FSH is separated into fully- and hypo-glycosylated fractions, the latter often include FSH18, which can constitute as much as 40% of the hypo-glycosylated FSH preparation (13). As most hFSH21 preparations also possess hFSH18, and are not easily separated, it has become a convention to abbreviate the mixture of physiologically relevant hypo-glycosylated FSH preparations as hFSH21/18.
Figure 2.

Follicle-stimulating hormone (FSH) glycoform models. Models of FSH heterodimers extracted from pdb 4AY9 decorated with the most abundant glycan observed at each N-glycosylation site by nano-ESI-ion mobility-MS (Bousfield, G. R. and Harvey, D. J., unpublished). Subunits are shown as cartoons rendered by MacPyMOL with subunits and their oligosaccharides colored as in Figure 1; FSHα green and FSHβ cyan. Oligosaccharides shown as sticks were created and attached to the FSH model using GLYCAM [Woods Group. (2005–2017) GLYCAM Web. Complex Carbohydrate Research Center, University of Georgia, Athens, GA, USA. (http://glycam.org)]. (A) FSH18, which lacks Asn24 glycan. (B) FSH21, which lacks Asn7 glycan. (C) FSH24, which possesses all four N-glycans.
Follicle-stimulating hormone microheterogeneity results from a structurally heterogeneous population of oligosaccharides attached to each glycosylated Asn residue of the four glycosylation sequons in FSH. Microheterogeneity in this hormone has largely been evaluated at the whole hormone level in studies of pituitary and urinary FSH preparations (16, 22–25). Human pituitary FSH oligosaccharides are 85–98% complex-type, 88–99% are sialylated, 36–46% are biantennary, 30–49% are triantennary, 5–15% are tetra-antennary, while only 4–7% are sulfated (Table 1). The low extent of oligosaccharide sulfation appears to be a human-specific characteristic (no data exist for nonhuman primate FSH glycans), as FSH preparations from cattle, pigs, sheep, and horses possess higher levels of sulfated oligosaccharides, ranging from 13 to 58% (23, 26). Accordingly, a major factor in determining hFSH clearance rates is the extent of sialic acid termination at the non-reducing ends of oligosaccharide branches. As compared with naturally occurring hFSH preparations, recombinant hFSH preparation oligosaccharides exhibit a reduced degree of branching, consisting of largely (55%) biantennary glycans. However, the degree of sialylation in these preparations lags that of urinary hFSH to a lesser extent, because the most abundant urinary FSH triantennary and tetra-antennary glycans are one sialic acid residue short of a full complement (16, 25, 27).
As mentioned above, microheterogeneity contributes to charge variation in FSH, and this has been reported to alter FSH biological activity (14, 28, 29). Comparisons of microheterogeneity in early studies were challenged not only by the large number of oligosaccharide structures encountered, but also by the different analytical methods each group employed, as each of these exhibited bias toward or against specific families of oligosaccharides. We recently characterized microheterogeneity in three purified human pituitary FSH glycoform preparations, as well as highly purified pituitary, urinary, and recombinant hFSH preparations using nano-electrospray mass spectrometry (13, 16–18). Because over 33–109 structures were detected in each sample, comparing oligosaccharide populations derived from different FSH preparations proved challenging.
The oligosaccharide structures shown in Figure 3 represent those present in at least 1% relative abundance in at least FSH preparation. Using this criterion, a total of 54 glycans were selected for comparison. The glycans are organized by position in the N-glycan biosynthetic pathway or by the number of complex branches. Within each antennary group, 2-, 3-, or 4-branch glycans, monosaccharide composition is the basis of organization. Structures 1–7 are oligomannose glycan intermediates found in ER and cisGolgi-derived glycoprotein precursors (Figure 3A). In multi-glycosylation site glycoproteins, these can be found in glycoproteins possessing mature glycans at other sites, when glycan processing at individual sites differs (30). Structures 8 and 9 exhibit the beginnings of complex oligosaccharide synthesis (Figure 3A), structures 10–34 are biantennary glycans (Figures 3A–C), structures 35–48 are triantennary glycans (Figures 3C,D), and structures 49–54 are tetra-antennary glycans (Figure 3D). The oligosaccharide populations of fully glycosylated FSH24 and hypo-glycosylated FSH21 preparations, F and D, respectively, possessed 51 of the 54 major glycans identified in these studies, and 45 of these, representing 88% of these more abundant glycans, were detected in both preparations. Pituitary and urinary FSH preparations P and U, respectively, both possessed 38 glycans (75%) in common with glycoforms F and D, while the hypo-glycosylated hFSH21/18 preparation L, possessed 35 glycans (68%) found in glycoform preparations F and D. Recombinant hFSH preparation G, expressed by stably transfected GH3 cells, displayed the lowest qualitative similarity to FSH24 and FSH21, possessing only 28 (55%) of the glycans found in glycoforms F and D. Moreover, the triantennary recombinant hFSH oligosaccharides displayed a different branching pattern.
Figure 3.

Human follicle-stimulating hormone (FSH) oligosaccharide microheterogeneity. Summary of results of nano-ESI mass spectrometry studies showing only those oligosaccharides present at >1% relative abundance in at least one hFSH preparation. The glycan diagram indicates it was detected in the preparation. The Consortium for Functional Glycomics monosaccharide symbols are used in conjunction with Oxford Glycobiology Institute linkage indicators (1–2, —; 1–3, \; 1–4, |; 1–6,/; solid lines indicate β-linkage and dashed lines indicate α-linkage). The bar graphs at the bottom of each panel indicate the relative abundance of the structure in each preparation. The preparations are indicated by single letters as follows: G is GH3-recombinant hFSH; U is urinary hFSH; P is pituitary hFSH; F is fully glycosylated pituitary hFSH24; H is hypo-glycosylated pituitary hFSH21/18; and L is hFSH21/18 isolated from hLH preparations. The structures are distributed across four panels beginning with the high mannose precursors and ending with tetra-antennary oligosaccharides, the largest found in hFSH. (A) Structures 1–14. (B) Structures 15–28. (C) Structures 29–42. (D) Structures 43–54.
Raising the cutoff to 4% relative abundance identified four groups of highly abundant glycans. The first group revealed a unique pattern of glycosylation for hFSH21/18 preparation L, consisting of a series of high mannose oligosaccharide intermediates possessing 9, 8, 7, 6, 5, and 3 mannose residues (structures 1–7, Figure 3A). Taken in isolation, this observation suggests that these glycoforms may not have exited the biosynthetic pathway. However, complex oligosaccharides, identical to those found in all other FSH preparations examined in this study, were also present in hFSH-L, suggesting oligosaccharide processing occurred at least at one glycosylation site in the Golgi. Glycosylation site-specific glycan analysis, when sufficient samples are available, or top–down proteomics for limited samples, have the potential to demonstrate the presence of both oligomannose and complex glycans in the same hypo-glycosylated hFSH molecule to support this hypothesis. Oligosaccharide structures 2–7 were also found in two pituitary glycoform preparations, hFSH24 and hFSH21. However, in both cases, these glycans were present in very low abundance, consistent with their being N-glycan biosynthetic intermediates. Moreover, both secreted hFSH preparations, urinary hFSH and recombinant hFSH, were devoid of oligomannose structures 1–7. In the case of urinary hFSH, this could have resulted either from rapid clearance of oligomannose-containing hFSH from the circulation or bias during purification.
As only secreted recombinant hFSH was recovered from conditioned medium, the absence of oligomannose glycans indicated that mature hFSH secreted by the GH3 cell line possessed only complex N-glycans. Moreover, the antibody used to capture recombinant hFSH appeared to capture all FSH forms, reducing the likelihood of purification biasing the oligosaccharide population (13). The high abundance of biosynthetic intermediate and low abundance of complex glycans in hFSH21/18 preparation L was notable because it exhibited the highest receptor binding-activity of any hFSH preparation we have studied. This led to the concern that we were studying a physiologically irrelevant glycoform. However, subsequent demonstration of significant biological activity differences between other pituitary and recombinant FSH glycoform preparations eliminated this concern (18, 31, 32).
Three clusters of high-abundance, complex glycans were noted in the other five hFSH preparations comprising oligosaccharide structures 22–23, 31–34, and 38–42. Group 2 structure 23, a disialylated, biantennary glycan possessing one GalNAc substituted for Gal, was highly abundant in all five preparations. This was notable, because the absence of sulfated GalNAc from hFSH N-glycans has been attributed to impaired recognition of a Pro-Leu-Arg motif in the common α-subunit of hFSH by β1,4-N-acetylgalactosaminyltransferase-T3 and -T4 (βGalNAct-T3 and βGalNAc-T4, respectively), as compared with hCG and hLH. The resulting reduction in FSH oligosaccharide sulfation was proposed as a consequence of altered motif access in this hormone, probably due to conformational change (33).
Comparison of Pro-Leu-Arg motifs in both hCG crystal structures, 1hcn (12) and 1hrp (11), with those in the two hFSH structures found in 1fl7 (6) showed positions of the Pro40 and Leu41 residue side chains were very similar in all six possible alignments (Figure 4). The Arg42 side chains were closely aligned in only one comparison, u-hCGα2:r-hFSHα1 (Figure 4D), suggesting flexibility in that region of the subunit (6, 34). Indeed, molecular dynamics simulations of FSH bound and unbound to the FSH receptor (FSHR) high-affinity binding site support flexibility in residue 40–47 region as unbound FSH exhibits root mean square fluctuations >1 Å (35). Unbound FSH is the form of the heterodimer recognized by β4GalNAc transferases. When FSH is bound to FSHR, this region loses flexibility, indicating it can achieve a stable conformation when bound to another protein. Thus, pituitary βGalNAc transferases are likely to bind this motif in both hLH and hFSH, consistent with the widespread distribution of GalNAc in hFSH oligosaccharides. The frequent appearance of GalNAc in sulfate-deficient glycans suggests an alternative hypothesis to explain reduced sulfation; human sialyltransferases compete more effectively with sulfotransferase in the human pituitary, leading to preferential addition of Neu5Ac to GalNAc. As N-glycan branches terminated with Neu5Ac-GalNAc were first reported for hLH oligosaccharides, finding this type of glycan is not unprecedented (36).
Figure 4.

Comparison of Pro-Leu-Arg motif in hCG and follicle-stimulating hormone (FSH) crystal structures. Cystine knot loop αL2 in the common α-subunits from each hormone structure were aligned using MacPyMOL. The backbone traces are shown and the side chains for Pro40, Leu41, and Arg42 shown as sticks. The residues are labeled because the flattening effect of printing appears to invert the order of Leu41 and Arg42. Chemically deglycosylated recombinant selenomethionine hCGα is r-hCGα1 (1hcn), chemically deglycosylated urinary hCGα is u-hCGα2 (1hrp), recombinant insect cell hFSH (1fl7) resulted in two models identified as r-hFSHα1 and r-hFSHα2, respectively. (A–F) α-subunit models aligned as indicated.
In fact, hLH possesses the greatest abundance of sialic acid of all characterized mammalian LH preparations (23, 36, 37). Moreover, structure 23 is part of a series of 15 GalNAc-containing, biantennary glycans observed in at least one of the six hFSH preparations (structures 10–25, Figures 3A,B). While two other structures are possible for the m/z 1130.9 ion associated with structure 23 (17), they do not permit addition of the two sialic acid residues associated with this oligosaccharide because the 5th hexosamine in the alternative structures is a bisecting GlcNAc residue and the single antenna possessing a Gal residue provides attachment for only one Neu5Ac residue. Group 3 glycan structures 31–34, are conventional, disialylated, biantennary oligosaccharides in which Neu5Ac residues are attached to Gal residues (Figure 3C). Structures 31 and 32 were the most abundant oligosaccharides derived from recombinant, urinary, and pituitary hFSH (Figure 3C). As 85–100% core-fucosylated glycans are found on the other human pituitary hormone LHβ and TSHβ subunits, structure 31 most likely reflects FSHα subunit glycosylation, while structure 32 reflects FSHβ subunit glycosylation (36, 38). The 4th high abundance glycan cluster, comprising structures 38–42, includes triantennary oligosaccharides possessing only two sialic acid residues. For this group of oligosaccharides, recombinant hFSH differed in the location of the two branch-mannose residues. In pituitary hFSH, GlcNAc transferase IV initiated a third glycan branch on Man (α1–3), while in recombinant hFSH GlcNAc transferase V initiated a third branch on Man (α1–6) (Figure 3C, compare row G with the other five rows). This suggested a difference in the relative activities of GlcNAc transferases IV and V between pituitary gonadotropes and somatotrope-derived GH3 cells, despite the expression of both transferase genes in GH3 cells (18). Another feature of recombinant hFSH glycans was antenna-linked fucose residues, such as observed in structure 43, one of the >1% abundance class of oligosaccharides (18).
Impact of FSH Glycosylation Heterogeneity on Cognate Receptor Binding
The FSHR is a G-protein-coupled receptor (GPCR) with a leucine-rich repeat extracellular domain comprising 358 amino-acid residues. This ligand binding domain is connected to a 337-residue, hepta-helical transmembrane domain (39, 40). Crystal structures of the high-affinity FSH binding domain in complex with FSH revealed that the interface of the complex involves contacts exclusively via protein–protein interactions (41, 42). FSH oligosaccharides added by modeling do not appear to interact with the extracellular domain engaged with FSH, as they are located on a face of the hormone, which is oriented away from the hormone receptor interface (Figure 5). Since it is well established that FSH carbohydrate is necessary for full FSHR activation (43–46), it seems reasonable to assume that the carbohydrate affects hormone conformation, which in turn modulates activity. The structure of the entire FSHR (extracellular domain and transmembrane domains) in complex with FSH has yet to be determined, and until then, carbohydrate interaction with the transmembrane domain cannot be ruled out. Alternatively, carbohydrate modulation of FSH conformation may affect the final disposition of FSHR extracellular domain (FSHRECD) hinge region putative interactions with extracellular loops of the transmembrane domains (34, 47). Consistent with the absence of FSH carbohydrate interaction with FSHRECD, isolated hybrid-type oligosaccharides related to structure 12 in Figure 3 have no effect on FSHR binding (48). Nevertheless, these oligosaccharides significantly inhibit both basal granulosa cell steroidogenesis, as well as FSH-stimulated steroidogenesis (48). The low affinity of carbohydrate–protein interactions requires sufficiently high oligosaccharide concentrations in inhibition studies that hormone contamination can inhibit binding assays. In our hands, a minimum of two purification steps is necessary to eliminate residual hormone assay interference (48). Accordingly, we attributed hormone contamination in the oligosaccharide preparation as the reason for a report that hCG-derived oligosaccharides inhibited both receptor binding and cellular activation (49).
Figure 5.

Follicle-stimulating hormone (FSH) glycoform models bound to monomeric FSH receptor (FSHR) extracelluar domain model (FSHRECD). FSH glycoform models are oriented as in Figure 2. The FSHRECD model was extracted from pdb 4AY9 and rendered as cartoon using MacPyMOL. The FSH glycoform models were aligned to the FSH model extracted from the pdb file along with the FSHRECD to illustrate the positions of oligosaccharides relative to the high-affinity binding site in the FSHR. (A) Glycosylated model of FSH18 and FSHR extracellular domain. (B) Glycosylated model of FSH21 and FSHR extracellular domain. (C) Glycosylated model of FSH24 and FSHR extracellular domain.
Loss of a single FSHβ oligosaccharide has three effects on FSH binding to its receptor. First, hypo-glycosylated hFSH immediately engages FSHR preparations, whereas fully glycosylated hFSH24 exhibits about a 30-min lag before FSHR binding begins in earnest (13). Second, hypo-glycosylated hFSH21/18 exhibits a 2.8- to over 14-fold higher apparent affinity for the FSHR as compared with hFSH24 (Table 2). Third, hypo-glycosylated hFSH21/18 occupies 2- to threefold more FSHRs than FSH24 (13, 18). A glance at the structures of FSH glycoforms bound to the FSHRECD immediately raises the question of how loss of either FSHβ N-glycan facilitates FSH association with the receptor, as neither glycan is close to the binding site (Figure 5). This leaves yet to be defined hindrance by the FSHR transmembrane domain or FSHR oligomerization as potential mechanisms.
Table 2.
Follicle-stimulating hormone (FSH) receptor-binding activities of pituitary and recombinant hFSH glycoform preparations.
| FSH preparation | Pituitary hFSH | Urinary hFSH | Pituitary hFSH24 | Pituitary hFSH21/18 | Recombinant GH3 hFSH24 | Recombinant GH3 hFSH21 |
|---|---|---|---|---|---|---|
| FSH RLA potency (IU/mg) | 8,560 | 10,000 | 18,737 | 269,445 | 20,844 | 57,942 |
| FSH21/FSH24 ratio | 14.4 | 2.8 | ||||
The crystal structure of the high-affinity binding site of the FSHRECD comprised two FSHR domains associated back to back, sandwiched by FSH ligands (41). There was no indication of FSH oligosaccharide interaction with the receptor. The crystal structure of the entire FSHRECD with FSH bound revealed a strikingly different FSHRECD conformation as trimeric FSHR–FSH complexes (42). To obtain diffractable crystals in both studies, endoglycosidase-F digestion reduced FSH and FSHRECD N-glycans to single GlcNAc residues, which eliminated oligosaccharide influence on hormone-receptor binding. The trimeric FSHR crystal structure suggested FSH αAsn52 oligosaccharide, when present, would restrict ligand binding to one glycosylated FSH ligand per FSHR trimer as a biantennary glycan attached to this Asn residue would occupy the center of the trimeric complex (47). While no subsequent studies supporting the dimeric FSHR model have been reported, several lines of evidence appear to support the trimeric FSHRECD model. Biochemical data in support of the trimeric FSHR model were provided when recombinant-mutant des-αN52-hFSH exhibited threefold greater binding to CHO cells expressing hFSHRs as compared with recombinant wt-hFSH (47). Small molecule allosteric FSHR modulators were reported to increase FSH binding ~threefold, suggesting trimeric FSHR complexes dissociating to form FSHR monomers (50–52). Incorporating a transmembrane domain model to the FSHRECD trimer model predicted that only a single β-arrestin could bind to the trimeric FSHR. Addition of an allosteric modulator to β-arrestin binding assays produced a threefold increase in β-arrestin binding, supporting a model that allosteric small molecule FSHR modulators dissociate FSHR trimers into monomers, thereby increasing FSH access (47). However, a superresolution microscopic technique, dual-color photoactivatable dyes, and localization microscopy (PD-PALM) revealed the closely related LHR existed as a variety of oligomeric forms as well as monomers in the cell membrane (53). Docking of complete LHR models in this study provided a variety of conformations of LHR oligomers, including trimeric LHRs. Similar studies with FSHRs would help clarify the relationship of FSHRs.
As greater FSHR occupancy is directly proportional to FSH-stimulated cAMP production by target cells, increased hypo-glycosylated hFSH binding to FSHR is expected to provide a correspondingly greater cellular activation than fully glycosylated hFSH (54). However, since the model of an FSHR trimer can only accommodate one G protein, it is unlikely that the increase in cAMP is due to occupancy alone. Another possibility is that occupancy by hypo-glycosylated FSH fails to engage the GRK/arrestin pathway which would otherwise attenuate the reengagement of G protein subsequent to activation of adenyl cyclase. Another possibility is that hypo-glycosylated FSH creates a more stable complex with FSHR such that during intracellular trafficking, cAMP- and arrestin-mediated persistent signaling (55) is enhanced. Finally, one may also suggest that since the FSH/FSHR complex appears to recycle to the cell surface (56, 57), the high-affinity binding of hypo-glycosylated FSH may have a proclivity for FSHR, thus failing to dissociate upon relocation to the plasma membrane and perhaps reformation of the putative trimeric structures. This could affect the dynamic stoichiometry of the cell surface unoccupied receptor cohort whose ontogeny resets not only with new FSHR synthesis but also by occupancy/recycling engaged by other members of the orchestra1 of glycoforms.
Fshr-Mediated Signaling in vitro and in vivo
Biased signaling has underpinned GPCR drug development for years but only recently has the mechanism of this phenomenon been revealed in the GPCR field, including the FSHR (51, 58–60). The realization that one GPCR can activate several effector proteins to activate different pathways has prompted the challenging of previously accepted dogma and may help to explain previously unexplained observations. An example of such dogma is that both FSHR and LH/CGR primarily signal via Gαs leading to the activation of the cAMP/protein kinase A (PKA) pathway and subsequently leading to steroidogenesis (51, 61–64). Alternative pathways, such as phospholipase C/inositol trisphosphate metabolism were first recognized over 25 years ago (65, 66); however, most studies examining the actions of gonadotropin glycosylation variants remain fixed on the primary pathway. The concept of biased signaling predicts that the specificity of signal transduction depends on, at least in part, the structure of the ligand [reviewed in Ref. (58, 59)]. In support of this idea, a partially deglycosylated eLH variant (67) (eLHdg) was found to exhibit biased signaling through the FSHR (68). While incapable of activating the cAMP/PKA pathway and eliciting steroidogenesis in granulosa cells, binding of eLHdg to FSHR recruited β-arrestins and activated ERK MAPK signaling via a cAMP-independent pathway (68).
Another recent study showed that the oligosaccharide complexity of recombinant hFSH preparations differentially affected gene expression and steroidogenesis in human granulosa cells (69). Our own studies with hFSH glycoforms have found evidence for biased signaling, albeit in different cell types. The hFSH21/18 glycoforms were more active than hFSH24 in activating the cAMP/PKA pathway and phosphorylation of PKA substrates via Gαs in human KGN granulosa cells (31). The actions of FSH21/18 were 10-fold greater than FSH24 on induction of CYP19A1 and estrogen (31). The obvious next step is to determine if this biased signaling by hFSH24 occurs in gonadal cells, which is an active area of pursuit using both in vitro and in vivo genetic approaches.
Genetic Models to Study the Physiology of FSH Glycoforms
Fshb Knockout Mice
As mentioned above, hypo-glycosylated FSH21/18 has been shown to be more avid compared with fully glycosylated FSH24 in several receptor binding assays (13, 18), and more potent when tested using primary granulosa cell- or immortalized granulosa cell-based in vitro assays (31). Translation of these in vitro observations from biochemistry to physiology required the development of new models as well as implementation of existing mouse models. Accordingly, in vivo effects of FSH glycoforms FSH21/18 and FSH24 were evaluated using the experimental design of an in vivo pharmacological rescue approach. In this experimental paradigm, first, immature Fshb null female mice (at 21 days of age) were injected i.p. with different doses of FSH glycoforms separately and at different times postinjection, ovaries were collected for subsequent selected gene expression analysis by quantitative real-time PCR. In these studies, hypo-glycosylated FSH21/18 elicited in vivo bioactivity comparable to that of FSH24; however, these analyses also indicated that differences exist between FSH21/18 and FSH24 glycoforms in inducing a unique subset of FSH-responsive genes (32). Second, to assess the upstream signaling pathways which control FSH-induced gene expression, immunofluorescence analysis was performed on ovarian sections obtained from Fshb null female mice injected with FSH21/18 and FSH24 glycoforms using p-CREB and p-PKA substrate antibodies. At three different time points tested (0.5, 1, and 2 h), both glycoforms were equally effective and significantly upregulated p-PKA and p-PKA substrates (nuclear accumulation in granulosa cells) over PBS-injected controls, with maximal induction observed at the 1-h time point (32).
In a third set of experiments, ovarian protein extracts were obtained from Fshb null female mice at different time points after injecting with FSH glycoforms separately. These extracts were subjected to Western blot analysis followed by densitometry quantification. When induction of p-CREB, p-PKA substrate and p-p38, p-p44/42, and p-AKT was compared, FSH21/18 hypo-glycosylated FSH, similar to the above assays, was as active as that of FSH24, the fully glycosylated FSH (32). Finally, in ovarian weight gain response assays, FSH21/18 was equally potent as that of the FSH24, although FSH21/18 elicited better estradiol induction compared with that by FSH24 (32). Thus, the in vivo pharmacological rescue experiments suggest biased agonism exhibited by different FSH glycoforms and, as would be expected, these are nuanced. In addition to determining if this phenomenon occurs in vivo as a function of age (particularly in regard to bone density given the correlation of age with changing FSH glycoform abundance), it will also be critical to determine if these nuances correlate with fertility or embryo quality, having great potential impact on therapeutic use.
In vivo pharmacological rescue of Fshb null male mice was also performed using recombinant human FSH glycoforms and measurement of testicular weight gain between postnatal day 5 and 10 in Fshb null male mice (32). When injected separately into Fshb null male mice at postnatal day 5, both FSH glycoforms significantly induced testicular weight gain by day 10 compared with that in PBS-injected controls (32). Testis weight correlated well with testis tubule size, as well as number of germ cells per tubule. Hypo-glycosylated FSH21/18 was more active than FSH24 (32). Similarly, a subset of FSH-responsive genes in mouse Sertoli cells responded much better to hypo-glycosylated FSH21/18 than fully glycosylated FSH24. Furthermore, the number of BrdU+ Sox9+ proliferating Sertoli cells was also found significantly higher in testes of mice injected with FSH21/18 compared with FSH24 (32). It is likely that different human FSH glycoforms act via different FSHR-mediated downstream signaling pathways in mouse Sertoli cells, similar to granulosa cells, and elicit distinct gene/protein expression changes. These observations suggest there may be a therapeutic potential advantage of using glycoform-specific hFSH preparations for treatment of male factor fertility, such as marginal sperm counts.
Evaluation of FSH15 in Fshb Null Mice
In vitro expression, purification and characterization of recombinant human FSH glycoforms in somatotrope-derived GH3 cells often results in FSH21/18 and FSH24 as the most abundant FSH glycoforms identified by mass spectrometry (18). However, according to the all or none FSHβ glycosylation concept, FSH dimers containing non-glycosylated FSHβ (expected to be 15 kDa in denaturing gels) could also exist in pituitaries (20). To test the biological significance of non-glycosylated FSHβ, separate lines of transgenic mice were first generated that expressed, either a human FSHB-mutant transgene (HFSHB7Δ24Δ) encoding a glycosylation defective 15k-FSHβ subunit or a human FSHB WT transgene (HFSHBWT)-encoding wild-type (WT) FSHβ subunit, specifically in gonadotropes. The transgenes were subsequently introduced onto an Fshb null genetic background by intercrossing using a genetic rescue strategy (70).
Real-time qPCR assays, immuno co-localization, and Western blot analyses under denaturating conditions confirmed that the transgene encoded mRNA and the corresponding subunits were abundantly expressed in pituitaries (21). While WT human FSHβ subunit-containing, inter-species hybrid FSH was readily detectable by Western blot analysis under non-denaturing conditions of HFSHB WT mouse pituitaries, FSH dimer containing double N-glycosylation-mutant human FSHβ subunit was barely detectable in pituitaries of HFSHBWT mice on an Fshb null genetic background (21). Consistent with these expression data, mutant FSHβ subunit-containing FSH dimer was not detectable in either short-term pituitary organ culture media or serum samples by specific RIAs (21). Furthermore, gonad histology, gonad gene expression, and fertility assays all indicated that the double N-glycosylation-mutant HFSHB transgene failed to rescue Fshb null mice (21). Taken together, these genetic experiments confirmed that the double N-glycosylation-mutant human FSHβ subunit-containing FSH dimer is unstable in vivo. Such a dimer is also secretion incompetent and even when secreted in low amounts, it fails to rescue mice lacking FSH. Thus, at least one N-glycosylation site on human FSHβ subunit is essential for efficient FSH dimer assembly, secretion, and biological activity in vivo.
Summary of Integrated Results
Implementation of Glycoforms in ART/IVF
Fundamental and heretofore unrecognized differences in human FSH relating to the number and location of FSH glycans resulting in FSH glycoforms, FSH24, FSH21, and FSH18 (16, 19) have been summarized. Moreover, the seminal observation from analysis of individual human pituitaries was that the abundance of FSH21 declines with age in women [Table 3 and (16)] raises the question whether this had implications for therapeutic intervention. FSH21 is elevated in young women of reproductive age, but declines thereafter leading to a condition of FSH24 dominance. Thus, the active reproductive period is characterized by the presence of FSH21, while the period of declining fertility and reproductive senescence is characterized by significantly diminished FSH21 along with FSH24 dominance.
Table 3.
Relative abundance of FSH21 in individual human pituitaries.
| No. of pituitaries | 2 | 4 | 4 |
| Age range (years) | 21–24 | 39–43 | 58–71 |
| FSH21 | 62 ± 10.5 | 41 ± 8.2 | 17 ± 3.7 |
Based on band density in Western blots using anti-FSHβ monoclonal antibody RFSH20 (16).
In this regard, it is noteworthy that current hFSH products available commercially for clinical use, whether they are of menopausal or recombinant origin, consist overwhelmingly of FSH24 (18). Thus, despite the general success of IVF, there has not been a systematic clinical trial which considers that a form of the hormone associated physiologically with a period of decreased reproductive function rather than the form of the hormone present during the reproductive period may be compromising both yield and quality of embryos. It is believed that the clinical utilization of hypo-glycosylated FSH21/18 preparations for IVF would represent a paradigm shift in the treatment of infertility. The use of something truly different, an apparently more active and more physiologically relevant FSH, might provide the basis for improved ovarian stimulation and overall pregnancy outcome. Thus, an emerging question is whether the shift from FSH21 to FSH24 dominance occurs as a result of normal aging or a premature change and represents an underlying cause of subfertility/infertility. To place this in context, a brief overview of controlled ovarian stimulation (COS) is warranted.
The use of COS began in the 1980s as a means to enhance/improve the chances of generating a pregnancy via the combination of procedures involved in in vitro fertilization (IVF). Prior to this, “natural cycle” IVF was utilized, which generated on average, a single utilizable oocyte (71). Not surprisingly, success via this method was severely limited. COS was developed as a means to generate multiple oocytes, which would increase the chances for successful fertilization, enhance embryo development, and coupled with multiple embryo transfer to the uterus, increase pregnancy rates. Indeed, COS proved invaluable as the preferred mechanism underlying IVF (72, 73). In parallel to COS, increased focus on IVF Laboratory practice coupled with IVF Laboratory Certification greatly moved IVF from “experimental procedure” status to that of standard of care (74). At the core of COS is the utilization of hFSH, the fundamental endocrine driver of ovarian follicle development (72).
The history of COS has witnessed a number of modifications aimed at increasing IVF success. Among these are: the utilization of GnRH agonist or antagonists to block endogenous gonadotropin production; utilization of urinary-derived human menopausal gonadotropin or well-controlled recombinant cDNA-driven expression of hFSH produced primarily using cells of Chinese hamster ovary origin; the use of FSH alone or the combined use of FSH coupled with LH; variable gonadotropin dosage and administration regimens; and utilization of supplemental progesterone to offset or oppose estradiol levels (72, 73, 75). Often, modifications have been undertaken to treat women with special conditions that impact success including women with PCOS, older women, and women with cancer (76). Indeed, a women’s age is one of the most predictive factors underlying success with IVF due in large part to the diminishing pool of primordial follicles. The common and overriding feature of the above modifications is the utilization of FSH.
The mechanistic functions and potential differences among FSH glycoforms remain largely unknown. As noted above, differences in receptor binding and the subsequent impact upon certain intracellular signaling systems and cell function can and have been demonstrated (77, 78). The fundamental mechanisms underlying female fertility in terms of producing a viable oocyte still remain largely unknown. However, there are clearly defined stages, which offer targets for differential regulation. These stages include primordial follicle activation, preantral follicle growth, antral follicle growth, and dominant follicle selection. An intriguing hypothesis is that hFSH glycoforms function during different stages of follicle development. This might explain in part, the reported differences in glycoform stimulation of ovarian gene expression and cellular signaling pathways observed in the immature Fshb null mice (32).
Follicle development up to the antral stage is not dependent upon FSH in the mouse (79, 80). Nevertheless, preantral follicles are responsive to FSH (81, 82). Owing to the recently reported in vivo activities of the glycoforms, could FSH21/18 preparations function to drive preantral follicle development to provide follicles appropriately responsive to FSH24? Might supplementation with FSH21/18 for one or two cycles prior to COS overcome what appears to be a natural decline in fertility with age concomitant with a decline in the levels of hFSH21/18? One proposes supplementation in the event that FSH21/18 drives preantral follicle development, so that replacement of FSH24 by FSH21/18 under standard COS strategies may not provide for improved results if FSH21/18 is needed during the earlier stages of follicle development and ineffective in later stages. Furthermore, such treatment paradigms might serve to ameliorate the decreased responsiveness of older women to COS with commercially available FSH, which is essentially FSH24. There is, for example, some evidence that microheterogeneity differences affect estradiol production (78).
FSH21/18 supplementation over an extended period to promote preantral follicle development, which would serve to provide appropriately developed follicles for continued development, perhaps with either glycoform. Owing to potential differences in uptake and circulating half-life, and whether the glycoforms are under episodic as opposed to a more tonic secretion, differences in hFSH glycoform dose and administration regimen may be needed to provide for a more physiological representation. Clearly, the discovery of FSH21/18 and the initial characterization of its activity provide the basis for new ideas concerning COS and IVF. These data indicate that FSH21/18 and FSH24 exist, and they exhibit differences in both in vitro and in vivo activities, and their relative abundance changes with age. These data provide a compelling basis for continued investigation. Central to the improvement of IVF outcomes will be the understanding of how and when these two glycoforms function to promote the proper developmental program of the follicle.
Implementation of FSH Glycoforms to Preserve Bone
Follicle-stimulating hormone has been reported to have direct effects on bone, attributed to FSH-driven (83–85) osteoclast development and activity (86–89). During the premenopausal period, when ovarian reserve is waning and FSH levels are rising because of the lack of negative feedback by ovarian estrogen (90), the abundance of fully glycosylated hFSH24 in the pituitary also rises. It is well established that declining levels of estradiol during the menopausal transition affects bone mineral density, and other metabolic parameters (91). Since the 1940s it has been assumed that reduced bone mineral density was due to a simple sex steroid deficiency (92). Previous reports, largely from one laboratory, have challenged this view by providing evidence that elevated FSH during menopause or ovarian deficiency might explain the bone loss (86, 93). A number of observations highlight the potential importance of FSH in mediating, at least in part, bone loss in humans (94) not associated with changes in steroid hormones (84). A recent study found that FSH, but not estrogen, was strongly associated with bone loss in postmenopausal women treated for breast cancer (95). Furthermore, polymorphisms in the FSHR are associated with accelerated bone loss in women (96). As such, the levels of estrogen and FSH may contribute in multiple ways to bone mineral density during aging.
It should be appreciated that the extra-gonadal actions of FSH have only been recently identified and the actions of FSH on bone have been controversial [reviewed in Ref. (97)]. Allan et al. (98) reported that FSH produced anabolic effects on bone that correlated with inhibin and testosterone levels. Ritter et al. (99) found that treatment of mice with FSH had no effect on bone loss or gain and did not increase osteoclast formation. Two other groups found little correlation of FSH levels and bone mineral density (2, 100). In contrast, other studies provide evidence that FSH can promote the development of human osteoclast precursor cells (89) and induce the production of bone-resorbing cytokines (87, 88, 93). These are relevant observations since the immune system plays a role in a variety of disease states linking inflammatory responses and bone loss (101). Furthermore, several lines of evidence support the initial observations that loss of either Fshb or Fshr confers protection from bone loss in mice (86).
Geng et al. (102) showed that exogenous FSH enhanced osteoclast differentiation and treatment with neutralizing antibodies to FSH or a GST–FSHβ fusion protein prevented bone loss in ovariectomized rats. Likewise, Zhu et al. (103) reported that treatment of ovariectomized mice with an FSH antibody prevented bone loss. Our data show that treatment of murine and human osteoclast precursor cells with FSH24, but not FSH21, increases the formation of multi-nucleated, TRAP (tartrate-resistant acid phosphatase-5b, a bone resorption marker) positive osteoclasts (Davis et al., unpublished). FSH also works together with receptor activator of nuclear factor-κB (NFκB) ligand (RANKL) to induce expression of MMP9 and cathepsin-k (CTSK) in osteoclasts. These data are in agreement with our own and indicate that FSH24 increases TNFα and IRAK mRNA in human CD14+ osteoclast precursors. TNFα is important for osteoclast formation (93, 104, 105). These findings indicate that the age-related increase in hFSH24 may regulate bone, a nontraditional FSH target. Evidence points to the ability of FSH to activate Gαi in bone cells, resulting in a reduction in cAMP levels (86), which contrasts to the activation of Gαs and increase in cAMP in granulosa cells. In bone, FSH stimulates MAPK and NFκB osteoclastogenic intracellular signaling pathways (86). Our data indicate that FSH24 is responsible for activating these signaling pathways and formation of osteoclasts. Hence, there is a critical need to settle the controversy regarding a role for FSH in targeting osteoclasts in women.
Author Contributions
GB, JM, JSD, JD, and TK authored individual sections of the review. GB, JM, JD, JD, and TK reviewed the entire manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding. This work was supported by the National Institute on Aging (grant number AG029531 to GB, JM, JD, and TK) and The Edgar L. & Patricia M. Makowski Endowment (to TK). This work was supported in part by a Senior Career Scientist Award (to JSD) from the United States (US) Department of Veterans Affairs Biomedical Laboratory Research and Development Service.
1Each of the FSH molecules is analogous to a member of the orchestra. Their seat is the receptor and their glycoforms are the instruments which they bring to play. The role of each molecule in the orchestra performance will be dependent on the glycoform instrument they bring with them. Like an orchestral performance, one must envision FSH signaling as a complex symphony which may be deconstructed but with loss to the nuance and impact of the full symphony.
References
- 1.Hearn MTW, Gomme PT. Molecular architecture and biorecognition processes of the cystine knot protein superfamily: part I. The glycoprotein hormones. J Mol Recognit (2000) 13:223–78. [DOI] [PubMed] [Google Scholar]
- 2.Gao J, Tiwari-Pandey R, Samadfam R, Yang Y, Miao D, Karaplis AC, et al. Altered ovarian function affects skeletal homeostasis independent of the action of follicle-stimulating hormone. Endocrinology (2007) 148(6):2613–21. 10.1210/en.2006-1404 [DOI] [PubMed] [Google Scholar]
- 3.Williams GR. Hypogonadal bone loss: sex steroids or gonadotropins? Endocrinology (2007) 148(6):2610–2. 10.1210/en.2007-0337 [DOI] [PubMed] [Google Scholar]
- 4.Agrawal M, Zhu G, Sun L, Zaidi M, Iqbal J. The role of FSH and TSH in bone loss and its clinical relevance. Curr Osteoporos Rep (2010) 8(4):205–11. 10.1007/s11914-010-0028-x [DOI] [PubMed] [Google Scholar]
- 5.Kumar TR. Extragonadal actions of FSH: a critical need for novel genetic models. Endocrinology (2018) 159(1):2–8. 10.1210/en.2017-03118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fox KM, Dias JA, Van Roey P. Three-dimensional structure of human follicle-stimulating hormone. Mol Endocrinol (2001) 15:378–89. 10.1210/mend.15.3.0603 [DOI] [PubMed] [Google Scholar]
- 7.Bousfield GR, Jia L, Ward DN. Gonadotropins: chemistry and biosynthesis. 3rd ed In: Neill JD, editor. Knobil and Neill: Physiology of Reproduction. San Diego: Elsevier; (2006). p. 1581–634. [Google Scholar]
- 8.Maghuin-Rogister G, Closset J, Hennen G. Differences in the carbohydrate portion of the α subunit of porcine lutropin (LH), follitropin (FSH) and thyrotropin (TSH). FEBS Lett (1975) 60(2):263–6. 10.1016/0014-5793(75)80727-7 [DOI] [PubMed] [Google Scholar]
- 9.Nilsson B, Rosen SW, Weintraub BD, Zopf DA. Differences in the carbohydrate moieties of the common α-subunits of human chorionic gonadotropin, luteinizing hormone, follicle-stimulating hormone, and thyrotropin: preliminary structural inferences from direct methylation analysis. Endocrinology (1986) 119:2737–43. 10.1210/endo-119-6-2737 [DOI] [PubMed] [Google Scholar]
- 10.Gotschall RR, Bousfield GR. Oligosaccharide mapping reveals hormone-specific glycosylation patterns on equine gonadotropin α-subunit Asn56. Endocrinology (1996) 137(6):2543–57. 10.1210/endo.137.6.8641208 [DOI] [PubMed] [Google Scholar]
- 11.Lapthorn AJ, Harris DC, Littlejohn A, Lustbader JW, Canfield RE, Machin KJ, et al. Crystal structure of human chorionic gonadotropin. Nature (1994) 369:455–61. 10.1038/369455a0 [DOI] [PubMed] [Google Scholar]
- 12.Wu H, Lustbader JW, Liu Y, Canfield RE, Hendrickson WA. Structure of human chorionic gonadotropin at 2.6 Å resolution from MAD analysis of the selenomethionyl protein. Structure (1994) 2:545–58. 10.1016/S0969-2126(00)00054-X [DOI] [PubMed] [Google Scholar]
- 13.Bousfield GR, Butnev VY, Butnev VY, Hiromasa Y, Harvey DJ, May JV. Hypo-glycosylated human follicle-stimulating hormone (hFSH21/18) is much more active in vitro than fully-glycosylated hFSH (hFSH24). Mol Cell Endocrinol (2014) 382:989–97. 10.1016/j.mce.2013.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulloa-Aguirre A, Midgley AR, Jr, Beitins IZ, Padmanbhan V. Follicle-stimulating isohormones: characterization and physiological relevance. Endocr Rev (1995) 16(6):765–87. 10.1210/edrv-16-6-765 [DOI] [PubMed] [Google Scholar]
- 15.Bousfield GR, Butnev VY, Bidart JM, Dalpathado D, Irungu J, Desaire H. Chromatofocusing fails to separate hFSH isoforms on the basis of glycan structure. Biochemistry (2008) 47(6):1708–20. 10.1021/bi701764w [DOI] [PubMed] [Google Scholar]
- 16.Bousfield GR, Butnev VY, Rueda-Santos MA, Brown A, Smalter Hall A, Harvey DJ. Macro and micro heterogeneity in pituitary and urinary follicle-stimulating hormone glycosylation. J Glycomics Lipidomics (2014) 4:125. 10.4172/2153-0637.1000125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bousfield GR, Butnev VY, White WK, Smalter Hall A, Harvey DJ. Comparison of follicle-stimulating hormone glycosylation microheterogeneity by quantitative negative mode nano-electrospray mass spectrometry of peptide-N-glycanase-released oligosaccharides. J Glycomics Lipidomics (2015) 5(1):129. 10.4172/2153-0637.1000129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butnev VY, Butnev VY, May JV, Shuai B, Tran P, White WK, et al. Production, purification, and characterization of recombinant hFSH glycoforms for functional studies. Mol Cell Endocrinol (2015) 405:41–52. 10.1016/j.mce.2015.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis JS, Kumar TR, May JV, Bousfield GR. Naturally occurring follicle-stimulating hormone glycosylation variants. J Glycomics Lipidomics (2014) 4:e117. 10.4172/2153-0637.1000e117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walton WJ, Nguyen VT, Butnev VY, Singh V, Moore WT, Bousfield GR. Characterization of human follicle-stimulating hormone isoforms reveals a non-glycosylated β-subunit in addition to the conventional glycosylated β-subunit. J Clin Endocrinol Metab (2001) 86:3675–85. 10.1210/jcem.86.8.7712 [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Butnev VY, Bousfield GR, Kumar TR. A human FSHB transgene encoding the double N-glycosylation mutant (Asn7Δ Asn25Δ) FSHβ fails to rescue Fshb null mice. Mol Cell Endocrinol (2016) 426:113–24. 10.1016/j.mce.2016.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renwick AGC, Mizuochi T, Kochibe N, Kobata A. The asparagine-linked sugar chains of human follicle-stimulating hormone. J Biochem (1987) 101:1209–21. 10.1093/oxfordjournals.jbchem.a121985 [DOI] [PubMed] [Google Scholar]
- 23.Green ED, Baenziger JU. Asparagine-linked oligosaccharides on lutropin, follitropin, and thyrotropin II. Distributions of sulfated and sialylated oligosaccharides on bovine, ovine, and human pituitary glycoprotein hormones. J Biol Chem (1988) 263(1):36–44. [PubMed] [Google Scholar]
- 24.Wolfenson C, Groisman J, Couto AS, Hedenfalk M, Cortvrindt RG, Smitz JE, et al. Batch-to-batch consistency of human-derived gonadotrophin preparations compared with recombinant preparations. Reprod Biomed Online (2005) 10(4):442–54. 10.1016/S1472-6483(10)60819-X [DOI] [PubMed] [Google Scholar]
- 25.Lombardi A, Andreozzi C, Pavone V, Triglione V, Angiolini L, Caccia P. Evaluation of the oligosaccharide composition of commercial follicle stimulating hormone preparations. Electrophoresis (2013) 34(16):23–94–2406. 10.1002/elps.201300045 [DOI] [PubMed] [Google Scholar]
- 26.Dalpathado DS, Irungu JA, Go EP, Butnev VY, Norton K, Bousfield GR, et al. Comparative glycomics of the glycoprotein hormone follicle-stimulating hormone (FSH): glycopeptide analysis of isolates from two mammalian species. Biochemistry (2006) 45(28):8665–73. 10.1021/bi060435k [DOI] [PubMed] [Google Scholar]
- 27.Gervais A, Hammel Y-A, Pelloux S, Lepage P, Baer G, Carte N, et al. Glycosylation of human recombinant gonadotrophins: characterization and batch-to-batch consistency. Glycobiology (2003) 13(3):179–89. 10.1093/glycob/cwg020 [DOI] [PubMed] [Google Scholar]
- 28.Ulloa-Aguirre A, Timossi C, Damian-Matsumura P, Dias JA. Role of glycosylation in function of follicle-stimulating hormone. Endocrine (1999) 11(3):205–15. 10.1385/ENDO:11:3:205 [DOI] [PubMed] [Google Scholar]
- 29.Ulloa-Aguirre A, Dias JA, Bousfield GR. Gonadotropins. In: Simoni M, Huhtaniemi I, editors. Endocrinology of the Testis and Male Reproduction. Cham: Springer; (2017). p. 1–52. [Google Scholar]
- 30.Bousfield GR, Butnev VY, Butnev VY, Nguyen VT, Gray CM, Dias JA, et al. Differential effects of a asparagine56 oligosaccharide structure on equine lutropin and follitropin hybrid conformation and receptor-binding activity. Biochemistry (2004) 43:10817–33. 10.1021/bi049857p [DOI] [PubMed] [Google Scholar]
- 31.Jiang C, Hou X, Wang C, May JF, Butnev VY, Bousfield GR, et al. Hypo-glycosylated hFSH has greater bioactivity than fully-glycosylated recombinant hFSH in human granulosa cells. J Clin Endocrinol Metab (2015) 100(6):E852–60. 10.1210/jc.2015-1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, May J, Shuai B, May JV, Bousfield GR, Kumar TR. Evaluation of in vivo bioactivities of recombiant hypo-(FSH21/18) and fully-(FSH24) glycosylated human FSH glycoforms in Fshb null mice. Mol Cell Endocrinol (2016) 437:224–36. 10.1016/j.mce.2016.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mengeling BJ, Manzella SM, Baenziger JU. A cluster of basic amino acids within an α-helix is essential for α-subunit recognition by the glycoprotein hormone N-acetylgalactosaminyltransferase. Proc Natl Acad Sci U S A (1995) 92:502–6. 10.1073/pnas.92.2.502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang X, Dias JA, He X. Structural biology of glycoprotein hormones and their receptors: insights to signaling. Mol Cell Endocrinol (2014) 384(1):424–51. 10.1016/j.mce.2013.08.021 [DOI] [PubMed] [Google Scholar]
- 35.Meher BR, Dixit A, Bousfield GR, Lushington GH. Glycosylation effects on FSH-FSHR interaction dynamics: a case study of different FSH glycoforms by molecular dynamics simulations. PLoS One (2015) 10(9):e0137897. 10.1371/journal.pone.0137897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weisshaar G, Hiyama J, Renwick AGC, Nimtz M. NMR investigations of the N-linked oligosaccharides at individual glycosylation sites of human lutropin. Eur J Biochem (1991) 195:257–68. 10.1111/j.1432-1033.1991.tb15702.x [DOI] [PubMed] [Google Scholar]
- 37.Weisshaar G, Hiyama J, Renwick AGC. Site-specific N-glycosylation of ovine lutropin: structural analysis by one- and two-dimensional 1H-NMR spectroscopy. Eur J Biochem (1990) 192:741–51. 10.1111/j.1432-1033.1990.tb19285.x [DOI] [PubMed] [Google Scholar]
- 38.Hiyama J, Weisshaar G, Renwick AGC. The asparagine-linked oligosaccharides at individual glycosylation sites in human thyrotropin. Glycobiology (1992) 2(5):401–9. 10.1093/glycob/2.5.401 [DOI] [PubMed] [Google Scholar]
- 39.Sprengel R, Braun T, Nikolics K, Segaloff DL, Seeburg PH. The testicular receptor for follicle stimulating hormone: structure and functional expression of cloned cDNA. Mol Endocrinol (1990) 4:525–30. 10.1210/mend-4-4-525 [DOI] [PubMed] [Google Scholar]
- 40.Minegishi T, Nakamura K, Takakura Y, Ibuki Y, Igarashi M. Cloning and sequencing of human FSH receptor cDNA. Biochem Biophys Res Commun (1991) 175(3):1125–30. 10.1016/0006-291X(91)91682-3 [DOI] [PubMed] [Google Scholar]
- 41.Fan QR, Hendrickson WA. Structure of human follicle-stimulating hormone in complex with its receptor. Nature (2005) 433:269–77. 10.1038/nature03206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang X, Liu H, Chen X, Chen PH, Fischer D, Sriraman V, et al. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc Natl Acad Sci U S A (2012) 109(31):12491–6. 10.1073/pnas.1206643109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manjunath P, Sairam MR, Sairam J. Studies on pituitary follitropin. X. Biochemical, receptor binding and immunological properties of deglycosylated ovine hormone. Mol Cell Endocrinol (1982) 28:125–38. 10.1016/0303-7207(82)90026-0 [DOI] [PubMed] [Google Scholar]
- 44.Calvo FO, Keutmann HT, Bergert ER, Ryan RJ. Deglycosylated human follitropin: characterization and effects on adenosine cyclic 3’,5’-phosphate production in porcine granulosa cells. Biochemistry (1986) 25:3938–43. 10.1021/bi00361a030 [DOI] [PubMed] [Google Scholar]
- 45.Bishop LA, Robertson DM, Cahir N, Schofield PR. Specific roles for the asparagine-linked carbohydrate residues of recombinant human follicle stimulating hormone in receptor binding and signal transduction. J Mol Endocrinol (1994) 8(6):722–31. 10.1210/me.8.6.722 [DOI] [PubMed] [Google Scholar]
- 46.Flack MR, Froehlich J, Bennet AP, Anasti J, Nisula BC. Site-directed mutagenesis defines the individual roles of the glycosylation sites on follicle-stimulating hormone. J Biol Chem (1994) 269(19):14015–20. [PubMed] [Google Scholar]
- 47.Jiang X, Fischer D, Chen X, McKenna SD, Liu H, Sriraman V, et al. Evidence for follicle-stimulating hormone receptor as a functional trimer. J Biol Chem (2014) 289(20):14273–82. 10.1074/jbc.M114.549592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen VT, Singh V, Butnev VY, Gray CM, Westfall S, Davis JS, et al. Inositol phosphate stimulation by LH requires the entire α Asn56 oligosaccharide. Mol Cell Endocrinol (2003) 199(1–2):73–86. 10.1016/S0303-7207(02)00297-6 [DOI] [PubMed] [Google Scholar]
- 49.Thotakura NR, Weintraub BD, Bahl OP. The role of carbohydrate in human choriogonadotropin (hCG) action. Effects of N-linked carbohydrate chains from hCG and other glycoproteins on hormonal activity. Mol Cell Endocrinol (1990) 70(3):263–72. 10.1016/0303-7207(90)90217-V [DOI] [PubMed] [Google Scholar]
- 50.Janovick JA, Maya-Nunez G, Ulloa-Aguirre A, Huhtaniemi IT, Dias JA, Verbost PM, et al. Increased plasma membrane expression of human follicle-stimulating hormone receptor by a small molecule thienopyr(im)idine. Mol Cell Endocrinol (2009) 298(1–2):84–8. 10.1016/j.mce.2008.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dias JA, Bonnet B, Weaver BA, Watts J, Kuuetzman K, Thomas RM, et al. A negative allosteric modulator demonstrates biased antagonism of the follicle stimulating hormone receptor. Mol Cell Endocrinol (2011) 333(2):143–50. 10.1016/j.mce.2010.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Koppen CJ, Verbost PM, van de Lagemaat R, Karsents WJ, Loozen HJ, vn Acterberg TA, et al. Signaling of an allosteric, nanomolar potent, low molecular weight agonist for the follicle-stimulating hormone receptor. Biochem Pharmacol (2013) 85(8):1162–70. 10.1016/j.bcp.2013.02.001 [DOI] [PubMed] [Google Scholar]
- 53.Jonas KC, Fanelli F, Huhtaniemi IT, Hanyaloglu AC. Single molecule analysis of functionally asymmetric G protein-coupled receptor (GPCR) oligomers reveals diverse spatial and structural assemblies. J Biol Chem (2015) 290:3875–92. 10.1074/jbc.M114.622498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bhaskaran RS, Ascoli M. The post-endocytotic fate of the gonadotropin receptors is an important determinant of the desensitization of gonadotropin responses. J Mol Endocrinol (2005) 34(2):447–57. 10.1677/jme.1.01745 [DOI] [PubMed] [Google Scholar]
- 55.Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol (2009) 7(8):e1000172. 10.1371/journal.pbio.1000172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krishnamurthy H, Kishi H, Shi M, Galet C, Bhaskaran RS, Hirakawa T, et al. Postendocytotic trafficking of the follicle-stimulating hormone (FSH)-FSH receptor complex. Mol Endocrinol (2003) 17(11):2162–76. 10.1210/me.2003-0118 [DOI] [PubMed] [Google Scholar]
- 57.Kluetzman KS, Thomas RM, Nechamen CA, Dias JA. Decreased degradation of internalized follicle-stimulating hormone caused by mutation of aspartic acid 6.30(550) in a protein kinase-CK2 consensus sequence in the third intracellular loop of human follicle-stimulating hormone receptor. Biol Reprod (2011) 84:1154–63. 10.1095/biolreprod.110.087965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Landomiel F, Gallay N, Jégot G, Tranchant T, Durand G, Bourquard T, et al. Biased signalling in follicle stimulating hormone action. Mol Cell Endocrinol (2014) 382(1):452–9. 10.1016/j.mce.2013.09.035 [DOI] [PubMed] [Google Scholar]
- 59.Luttrell LM. Minireview: more than just a hammer: ligand ‘bias’ and pharmaceutical discovery. Mol Endocrinol (2014) 28(3):281–94. 10.1210/me.2013-1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shukla AK. Biasing GPCR signaling from inside. Sci Signal (2014) 7:e3. 10.1126/scisignal.2005021 [DOI] [PubMed] [Google Scholar]
- 61.Means AR, MacDonald E, Soderling TR, Corbin JD. Testicular adenosine 3’:5’-monophosphate-dependent protein kinase. Regulation by follicle-stimulating hormone. J Biol Chem (1974) 249(4):1131–8. [PubMed] [Google Scholar]
- 62.Marsh JM. The role of cyclic AMP in gonadal steroidogenesis. Biol Reprod (1976) 14(1):30–53. 10.1095/biolreprod14.1.30 [DOI] [PubMed] [Google Scholar]
- 63.Dattatreyamurty B, Figgs LW, Reichert LE, Jr. Physical and functional association of follitropin receptors with cholera toxin-sensitive guanine nucleotide-binding protein. J Biol Chem (1987) 262(24):11737–45. [PubMed] [Google Scholar]
- 64.Hunzicker-Dunn M, Maizels ET. FSH signaling pathways in immature granulosa cells that regulate target gene expression: branching out from protein kinase A. Cell Signal (2006) 18(9):1351–9. 10.1016/j.cellsig.2006.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davis JS, Weakland LL, West LA, Farese RV. Luteinizing hormone stimulates the formation of inositol trisphosphate and cyclic AMP in rat granulosa cells. Biochem J (1986) 238:597–604. 10.1042/bj2380597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davis JS, Weakland LL, Farese RV, West LA. Luteinizing hormone increases inositol trisphosphate and cytosolic Ca2+ in isolated bovine luteal cells. J Biol Chem (1987) 262(18):8515–21. [PubMed] [Google Scholar]
- 67.Butnev VY, Singh V, Nguyen VT, Bousfield GR. Truncated eLHβ and asparagine56-deglycosylated eLHα combine to produce a potent follicle-stimulating hormone antagonist. J Endocrinol (2002) 172(3):545–55. 10.1677/joe.0.1720545 [DOI] [PubMed] [Google Scholar]
- 68.Wehbi V, Tranchant T, Durand G, Musnier A, Decourtye G, Piketty V, et al. Partially deglycosylated equine LH preferentially activates beta-arrestin-dependent signaling at the follicle-stimulating hormon receptor. Mol Endocrinol (2010) 24(3):561–73. 10.1210/me.2009-0347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loreti N, Fresno C, Barrera D, Andreone L, Albarran SL, Fernandez EA, et al. The glycan structure in recombinant human FSH affects endocrine activity and global gene expression in human granulosa cells. Mol Cell Endocrinol (2013) 366(1):68–80. 10.1016/j.mce.2012.11.021 [DOI] [PubMed] [Google Scholar]
- 70.Kumar TR, Low MJ, Matzuk MM. Genetic rescue of follicle-stimulating hormone beta-deficient mice. Endocrinology (1998) 139(7):3289–95. 10.1210/endo.139.7.6111 [DOI] [PubMed] [Google Scholar]
- 71.Zech NH, Zech M, Baldauf S, Comploj G, Murtinger M, Spitzer D, et al. Ovarian stimulation in ART––Unwinding pressing issues. Minerva Ginecol (2015) 67(2):127–47. [PubMed] [Google Scholar]
- 72.Grainger DA, Tjaden BL, Ttpati LL. Assisted reproductive technologies. 2nd ed In: Goldman MB, Troisi R, Rexfode KM, editors. Women & Health. Waltham, MA: Academic Press; (2013). p. 307–20. [Google Scholar]
- 73.Jungheim ES, Meyer M, Broughton DE. Best practices for controlled ovarian stimulation in IVF. Semin Reprod Med (2015) 33:77–82. 10.1055/s-0035-1546424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.May JV. Ovarian hyperstimulation: effects on oocyte quality and communication between physician and embryologist to optimize oocyte quality. In: May JV, editor. Infertility and Reproductive Medicine Clinics of North America: Assisted Reproduction, Laboratory Considerations. Philadelphia, PA: W.B Saunders Co; (1998):163–79. [Google Scholar]
- 75.Vuong TNL, Phung HT, Ho MT. Recombinant follicle-stimulating hormone and recombinant luteinizing hormone versus recombinant follicle-stimulating hormone alone during GnRH antagonist ovarian stimulation in patients aged >= 35 years: a randomized controlled trial. Hum Reprod (2015) 30:1188–95. 10.1093/humrep/dev038 [DOI] [PubMed] [Google Scholar]
- 76.Cakmak H, Rosen MP. Ovarian stimulation in cancer patients. Fert Seril (2013) 99:1476–84. 10.1016/j.fertnstert.2013.03.029 [DOI] [PubMed] [Google Scholar]
- 77.Williams RS, Vensel T, Sistrom CL, Kipersztok S, Rhoton-Vlasak A, Drury K. Pregnancy rates in varying age groups after in vitro fertilization: a comparison of follitropin alfa (Gonal F) and follitropin beta (Follistim). Am J Obstet Gynecol (2003) 189(2):342–6; discussion 346–7. 10.1067/S0002-9378(03)00728-2 [DOI] [PubMed] [Google Scholar]
- 78.Orvieto R, Nahum R, Rabinson J, Ashkenazi J, Anteby EY, Meltcer S. Follitropin-alpha (Gonal-F) versus follitropin-beta (Puregon) in controlled ovarian hyperstimulation for in vitro fertilization: is there any difference? Fertil Steril (2009) 91(4 Suppl):1522–5. 10.1016/j.fertnstert.2008.08.112 [DOI] [PubMed] [Google Scholar]
- 79.Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet (1997) 15(2):201–4. 10.1038/ng0297-201 [DOI] [PubMed] [Google Scholar]
- 80.Kumar TR. Mouse models for gonadotropins: a 15-year saga. Mol Cell Endocrinol (2007) 260-262:249–54. 10.1016/j.mce.2006.09.002 [DOI] [PubMed] [Google Scholar]
- 81.McGee EA, Perlas E, LaPolt PS, Tsafriri A, Hsueh AJ. Follicle-stimulating hormone enhances the development of preantral follicles in juvenile rats. Biol Reprod (1997) 57(5):990–8. 10.1095/biolreprod57.5.990 [DOI] [PubMed] [Google Scholar]
- 82.Abel MH, Wootton AN, Wilkins V, Huhtaniemi I, Kinight PG, Charlton HM. The effect of a null mutation in the follicle-stimulating hormone receptor gene on mouse reproduction. Endocrinology (2000) 141(5):1795–803. 10.1210/endo.141.5.7456 [DOI] [PubMed] [Google Scholar]
- 83.Ebeling PR, Atley LM, Guthrie JR, Burger HG, Dennerstein L, Hopper JL, et al. Bone turnover markers and bone density across the menopausal transition. J Clin Endocrinol Metab (1996) 81(9):3366–71. 10.1210/jc.81.9.3366 [DOI] [PubMed] [Google Scholar]
- 84.Sowers MR, Jannausch M, McConnell D, Little R, Greendale GA, Finkelstein JS, et al. Hormone predictors of bone mineral density changes during the menopausal transition. J Clin Endocrinol Metab (2006) 91(4):1261–7. 10.1210/jc.2005-1836 [DOI] [PubMed] [Google Scholar]
- 85.Lo JC, Burnett-Bowie SAM, Finkelstein JS. Bone and the perimenopause. Obstet Gynecol Clin North Am (2011) 38(3):503–17. 10.1016/j.ogc.2011.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ, et al. FSH directly regulates bone mass. Cell (2006) 125(2):247–60. 10.1016/j.cell.2006.01.051 [DOI] [PubMed] [Google Scholar]
- 87.Cannon JG, Cortez-Cooper M, Meaders E, Stallings J, Haddow S, Kraj B, et al. Follicle-stimulating hormone, interleukin-1, and bone density in adult women. Am J Physiol Regul Integr Comp Physiol (2010) 298(3):R790–8. 10.1152/ajpregu.00728.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sun L, Zhang Z, Zhu LL, Peng Y, Liu X, Li J, et al. Further evidence for direct pro-resorptive actions of FSH. Biochem Biophys Res Commun (2010) 394(1):6–11. 10.1016/j.bbrc.2010.02.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cannon JG, Kraj B, Sloan G. Follicle-stimulating hormone promotes RANK expression on human monocytes. Cytokine (2011) 53(2):141–4. 10.1016/j.cyto.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Crandall CJ, Tseng CH, Karlamangla AS, Finkelstein JS, Randolph JF, Jr, Thurston RC, et al. Serum sex steroid levels and longitudinal changes in bone density in relation to the final menstrual period. J Clin Endocrinol Metab (2013) 98(4):E654–63. 10.1210/jc.2012-3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dennerstein L, Lehert P, Guthrie JR, Burger HG. Modeling women’s health during the menopausal transition: a longitudinal analysis. Menopause (2007) 14(1):53–62. 10.1097/01.gme.0000229574.67376.ba [DOI] [PubMed] [Google Scholar]
- 92.Riggs BL, Khosla S, Melton LJ, III. Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev (2002) 23(3):279–302. 10.1210/edrv.23.3.0465 [DOI] [PubMed] [Google Scholar]
- 93.Iqbal J, Sun L, Kumar TR, Blair HC, Zaidi M. Follicle-stimulating hormone stimulates TNF production from immune cells to enhance osteoblast and osteoclast formation. Proc Natl Acad Sci U S A (2006) 103(40):14925–30. 10.1073/pnas.0606805103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Devleta B, Adem B, Senada S. Hypergonadotropic amenorrhea and bone density: new approach to an old problem. J Bone Miner Metab (2004) 22(4):360–4. 10.1007/s00774-004-0495-1 [DOI] [PubMed] [Google Scholar]
- 95.Tabatabai LS, Bloom J, Stewart S, Sellmeyer DE. FSH levels predict bone loss in premenopausal women treated for breast cancer more than one year after treatment. J Clin Endocrinol Metab (2016) 101(3):1257–62. 10.1210/jc.2015-3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rendina D, Gianfrancesco F, De Filippo G, Merlotti D, Esposito T, Mingione A, et al. FSHR gene polymorphisms influence bone mineral density and bone turnover in postmenopausal women. Eur J Endocrinol (2010) 163(1):165–72. 10.1530/eje-10-0043 [DOI] [PubMed] [Google Scholar]
- 97.Colaianni G, Cuscito C, Colucci S. FSH and TSH in the regulation of bone mass: the pituitary/immune/bone axis. Clin Dev Immunol (2013) 2013:382698. 10.1155/2013/382698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Allan CM, Kalak R, Dunstan CR, McTavish KJ, Zhou H, Handelsman DJ, et al. Follicle-stimulating hormone increases bone mass in female mice. Proc Natl Acad Sci U S A (2010) 107(52):22629–34. 10.1073/pnas.1012141108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ritter V, Thuering B, Saint Mezard P, Luong-Nguyen NH, Seltenmeyer Y, Junker U, et al. Follicle-stimulating hormone does not impact male bone mass in vivo or human male osteoclasts in vitro. Calcif Tissue Int (2008) 82(5):383–91. 10.1007/s00223-008-9134-5 [DOI] [PubMed] [Google Scholar]
- 100.Gourlay ML, Specker BL, Li C, Hammett-Stabler CA, Renner JB, Rubin JE. Follicle-stimulating hormone is independently associated with lean mass but not BMD in younger postmenopausal women. Bone (2012) 50(1):311–6. 10.1016/j.bone.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Faienza MF, Ventura A, Marzano F, Cavallo L. Postmenopausal osteoporosis: the role of immune system cells. Clin Dev Immunol (2013) 2013:575936. 10.1155/2013/575936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Geng W, Yan X, Du H, Cui J, Li L, Chen F. Immunization with FSHbeta fusion protein antigen prevents bone loss in a rat ovariectomy-induced osteoporosis model. Biochem Biophys Res Commun (2013) 434(2):280–6. 10.1016/j.bbrc.2013.02.116 [DOI] [PubMed] [Google Scholar]
- 103.Zhu LL, Blair H, Cao J, Yuen T, Latif R, Guo L, et al. Blocking antibody to the beta-subunit of FSH prevents bone loss by inhibiting bone resorption and stimulating bone synthesis. Proc Natl Acad Sci U S A (2012) 109(36):14574–9. 10.1073/pnas.1212806109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ralston SH, Russell RG, Gowen M. Estrogen inhibits release of tumor necrosis factor from peripheral blood mononuclear cells in postmenopausal women. J Bone Miner Res (1990) 5(9):983–8. 10.1002/jbmr.5650050912 [DOI] [PubMed] [Google Scholar]
- 105.Roggia C, Gao Y, Cenci S, Weitzmann MN, Toraldo G, Isaia G, et al. Up-regulation of TNF-producing T cells in the bone marrow: a key mechanism by which estrogen deficiency induces bone loss in vivo. Proc Natl Acad Sci U S A (2001) 98(24):13960–5. 10.1073/pnas.251534698 [DOI] [PMC free article] [PubMed] [Google Scholar]