

Abstract
AIM
To detect hyper-conserved regions in the hepatitis B virus (HBV) X gene (HBX) 5’ region that could be candidates for gene therapy.
METHODS
The study included 27 chronic hepatitis B treatment-naive patients in various clinical stages (from chronic infection to cirrhosis and hepatocellular carcinoma, both HBeAg-negative and HBeAg-positive), and infected with HBV genotypes A-F and H. In a serum sample from each patient with viremia > 3.5 log IU/mL, the HBX 5’ end region [nucleotide (nt) 1255-1611] was PCR-amplified and submitted to next-generation sequencing (NGS). We assessed genotype variants by phylogenetic analysis, and evaluated conservation of this region by calculating the information content of each nucleotide position in a multiple alignment of all unique sequences (haplotypes) obtained by NGS. Conservation at the HBx protein amino acid (aa) level was also analyzed.
RESULTS
NGS yielded 1333069 sequences from the 27 samples, with a median of 4578 sequences/sample (2487-9279, IQR 2817). In 14/27 patients (51.8%), phylogenetic analysis of viral nucleotide haplotypes showed a complex mixture of genotypic variants. Analysis of the information content in the haplotype multiple alignments detected 2 hyper-conserved nucleotide regions, one in the HBX upstream non-coding region (nt 1255-1286) and the other in the 5’ end coding region (nt 1519-1603). This last region coded for a conserved amino acid region (aa 63-76) that partially overlaps a Kunitz-like domain.
CONCLUSION
Two hyper-conserved regions detected in the HBX 5’ end may be of value for targeted gene therapy, regardless of the patients’ clinical stage or HBV genotype.
Keywords: Hepatitis B virus, Hepatitis B X gene, Hepatitis B X protein, Gene therapy, Next-generation sequencing, HBV conserved regions, Small interference RNA
Core tip: Hepatitis B virus (HBV) is not cured with classic treatments, and liver disease can progress by persistence and expression of covalently-closed circular DNA. Gene therapy with small interference RNA may be an effective approach to ensure inhibition of viral expression and disease progression, and hepatitis B virus X gene (HBX) transcripts could be optimal targets for this therapy. This study includes patients with different HBV genotypes and clinical stages to cover many clinical and virological situations. Using next-generation sequencing, we found two hyper-conserved HBX regions, candidates for small interference RNA therapy, which could enable pan-genotypic inhibition of HBV expression, regardless of the patients’ disease status.
INTRODUCTION
Despite the efficacy of preventive vaccines, an estimated 257 million people are living with chronic hepatitis B virus infection (CHB) and more than 880000 people die each year of hepatitis B virus (HBV)-related complications such as cirrhosis and hepatocellular carcinoma (HCC) (WHO report, July 2017).
HBV is an enveloped DNA virus with partially double-stranded circular DNA. HBV replication requires RNA intermediate and the activity of a reverse transcriptase. This implies a high probability that genetic mutations will occur, as the reverse transcriptase lacks 3’ to 5’ proofreading activity, leading to a viral mutation rate of 10-4 to 10-5 substitutions/site/year, similar to that observed for RNA viruses[1]. Inter- and intragenotype recombination events can further increase HBV variability[2]. Hence, HBV circulates as a complex mixture of genetic variants, known as a quasispecies[3], that enables the virus to escape from the host’s immune system, antiviral treatment, and vaccination, thereby promoting progression to CHB. Furthermore, the mutational profile is closely associated with HBV genotype, and the genotype is associated with differing effectiveness of the treatments used and outcomes of the infection[4,5].
The main therapeutic approach for HBV infection is based on inhibition of the viral polymerase by the action of nucleotide analogues, whose goal is to improve the patients’ quality of life and prolong survival by preventing progression of the disease[6]. However, HBV cannot be completely eradicated with these drugs because the viral intermediate known as covalently closed circular DNA (cccDNA) can persist within the nucleus of HBV-infected liver cells. cccDNA interacts with histone and non-histone proteins, including viral proteins such as the core and X protein (HBx), and forms a minichromosome that permits transcription of HBV genes[7], including pregenomic RNA, the precursor of de novo viral DNA genomes. Because cccDNA persists, it constitutes a viral reservoir that could promote reactivation of the infection after treatment interruption[8]. Within this challenging scenario, research has been aimed at deeply investigating the host-virus interactions to better understand the mechanisms that establish persistent HBV infection and to find new therapeutic targets that can cure it.
In this line, new treatment approaches are currently under development[9], with gene therapy being a promising option. Homing endonucleases, such as zinc-finger endonucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and RNA-guided clustered regulatory interspaced short palindromic repeats associated with the Cas endonuclease family (CRISPR/Cas), can cleave selected sequences in cccDNA, resulting in disruption of the gene due to nonspecific DNA repair with consequent elimination of the viral minichromosome[10,11]. However, systematic random integration of the viral genome in the host genome could represent a strong limitation to this strategy. Indeed, the activity of this “molecular scissors”, although sequence-specific, could entail a potential risk of damage for the human genes close to the viral site of integration.
Another promising gene therapy consists in silencing specific genes at the post-transcriptional level through a sequence-specific interaction between an mRNA target and small interfering RNA (siRNA)[12]. With this approach, various regions of the viral mRNA sequence can be targeted, including non-coding regions, without affecting the host DNA. Although these therapies show promise, the high variability of HBV and the association between this variability and the patients’ clinical outcome suggests that it may be important to find a highly conserved target to guarantee their efficacy.
A good candidate for targeted gene therapy could be the HBx protein, encoded by the HBV X gene (HBX). This pleiotropic and multifunctional protein trans-activates the expression of the viral genes. Together with the HBV core protein (HBc), HBx attaches to the cccDNA structure and is crucial for HBV replication[7]. In addition, this protein interacts with several cell signaling pathways and genes, thus affecting many cellular activities[13-15]. Due to its wide range of activity, HBx plays a key role in the pathogenesis of HBV infection and disease progression, and is strongly associated with HCC. Hence, it could be an optimal target for a hypothetical curative therapy for HBV infection.
The HBX gene, nucleotides (nt) 1374-1838, contains important regulatory elements[16,17]. The coded protein is comprised of 2 domains. The N-terminal domain [amino acid (aa) 1-50, encoded by the 5’ end of the gene] acts as negative regulator of the HBx transactivation function, which resides in the C-terminal domain (aa 51-154, encoded by the 3’ end). Interestingly, a significant presence of multiple variants with deletions and/or insertions (indels) has been found in the 3’ end of HBX[18-20]. Considering this variability, the 3’ coding region of the X gene would be ruled out as a possible therapeutic candidate[21]. However, the conservation at 5’ end of HBX and its potential for use as a gene therapy target remains unexplored. To silence HBX at the post-transcriptional level, the non-coding region included in HBX transcripts, upstream of the coding region, should also be considered. The HBX gene is located near the co-terminal 3’ end; hence, all HBV mRNAs produced during the infection include this sequence (Figure 1). Consequently, by targeting HBX transcripts at the coding or non-coding level, interference with expression of all the viral proteins could be achieved.
Figure 1.
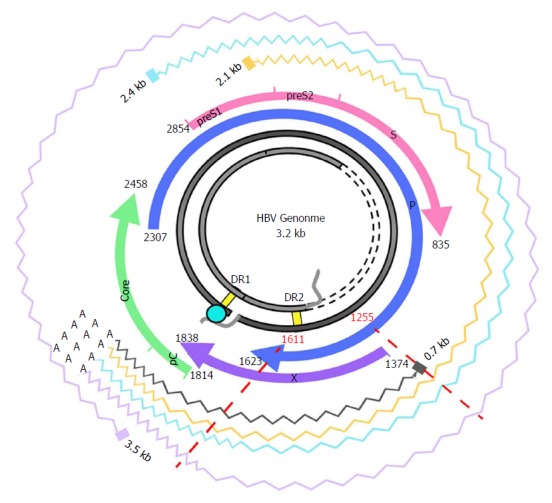
Hepatitis B virus genome and transcripts. The figure shows the HBV genome (in grey), with the DR1 and DR2 (direct repeat) regions, necessary for viral DNA synthesis. The viral ORFs are highlighted with colored arrows, and nucleotide positions are reported. Wavy lines show the various HBV transcripts: the 3.5-kb transcript, corresponding to the pregenomic RNA, which is translated to the core and polymerase and later subjected to reverse-transcription in the viral capsid, or to the precore/core transcript, which is translated to the precore protein; the 2.4-kb and 2.1-kb transcripts, which are translated, respectively, to large and medium/small HBsAg); and the 0.7-kB transcript, which is translated to the HBx protein. The region analyzed in this study and its corresponding nt positions are indicated by red dashed lines. Note that the region of interest is included in all the viral transcripts. HBV: Hepatitis B virus; ORF: Open reading frame.
The aim of this study was to determine the conservation of a region of the HBV genome encompassing the HBX 5’ coding region and upstream non-coding region (included in all HBV transcripts) in samples from HBV-infected patients in various clinical stages and with different viral genotypes. The ultimate objective was to find hyper-conserved regions that might be feasible targets for gene therapy, which could be used whatever the patient’s clinical status or HBV genotype.
MATERIALS AND METHODS
Patients and samples
From a cohort of 46 well-characterized CHB patients attending the outpatient clinic of Vall d’Hebron University Hospital (Barcelona, Spain), we selected a group of 27 patients in various clinical stages and with different viral genotypes. The samples included were 17 from HBeAg-negative patients (3 with chronic infection and14 chronic hepatitis, 2 of them with cirrhosis and 1 with HCC), and 10 from HBeAg positive (2 with chronic infection and 8 with chronic hepatitis, 3 of them with cirrhosis and 2 with HCC, characterized according to the latest EASL guidelines[6]), infected with several HBV genotypes: 5 A, 1 B, 7 C, 8 D, 2 E, 3 F, 1 H (Table 1).
Table 1.
Main clinical and virological characteristics of the hepatitis B virus infected patients enrolled
| Patient | Age | Sex | Origin | Clinical stage | HBeAg | HBV DNA (log IU/mL) | ALT (IU/L) | Genotype1 |
| 1 | 27 | M | Sub-Saharan | Chronic hepatitis | Negative | 7.8 | 170 | E |
| 2 | 31 | M | Asian | Chronic hepatitis | Negative | 6.6 | 103 | C |
| 3 | 51 | M | Caucasian | Chronic hepatitis | Negative | 6.6 | 262 | D |
| 4 | 28 | F | Caucasian | Chronic hepatitis | Negative | 7.9 | 126 | D |
| 5 | 47 | F | Caucasian | Chronic hepatitis | Negative | 6.3 | 170 | D |
| 6 | 37 | F | Caucasian | Chronic hepatitis | Negative | 4.5 | 53 | D |
| 7 | 37 | M | Caucasian | Chronic hepatitis | Negative | 4.5 | 33 | F |
| 8 | 38 | M | Caucasian | Chronic hepatitis | Negative | 5.0 | 29 | D |
| 9 | 46 | M | Caucasian | Chronic hepatitis | Negative | 5.8 | 88 | F |
| 10 | 46 | F | Caucasian | Chronic hepatitis | Negative | 5.3 | 23 | H |
| 11 | 71 | F | Caucasian | Chronic hepatitis | Negative | 6.2 | 87 | F |
| 12 | 51 | M | Asian | Chronic hepatitis | Negative | 5.7 | 435 | C |
| 13 | 52 | M | Caucasian | Chronic infection | Negative | 4.4 | 18 | A |
| 14 | 40 | F | Caucasian | Chronic infection | Negative | 4.2 | 29 | D |
| 15 | 33 | M | Asian | Chronic infection | Negative | 4.3 | 25 | D |
| 16 | 63 | M | Hispanic | Cirrhosis | Negative | 4.0 | 16 | A |
| 17 | 53 | M | Caucasian | Cirrhosis/HCC | Negative | 3.7 | 36 | A |
| 18 | 35 | M | Sub-Saharan | Chronic hepatitis | Positive | 5.7 | 36 | E |
| 19 | 37 | M | Caucasian | Chronic hepatitis | Positive | 8.4 | 32 | C |
| 20 | 45 | M | Caucasian | Chronic hepatitis | Positive | 5.6 | 35 | A |
| 21 | 29 | F | Asian | Chronic hepatitis | Positive | 6.9 | 355 | B |
| 22 | 28 | M | Asian | Chronic hepatitis | Positive | > 8.0 | 341 | C |
| 23 | 28 | M | Asian | Chronic infection | Positive | 8.7 | 24 | C |
| 24 | 28 | F | Asian | Chronic infection | Positive | 8.8 | 22 | C |
| 25 | 55 | F | Caucasian | Cirrhosis | Positive | 5.4 | 73.9 | A |
| 26 | 82 | F | Caucasian | Cirrhosis/HCC | Positive | 4.8 | 24 | C |
| 27 | 64 | M | Caucasian | Cirrhosis/HCC | Positive | 6.3 | 45 | D |
Genotype determined by Sanger sequencing of the X region (same region as was analyzed by next-generation sequencing). ALT: Alanine aminotransferase; HBV: Hepatitis B virus; HBeAg: Hepatitis B e antigen; M: Male; F: Female; HBV: Hepatitis B virus.
All 27 patients were treatment-naïve, tested negative for hepatitis D virus (HDV), hepatitis C virus (HCV), and human immunodeficiency virus (HIV), and had a serum sample with viremia levels > 3.5 logIU/mL, the sensitivity limit of the PCR to amplify the studied region (described below). The study was approved by the Ethics Committee of Vall d'Hebron Research Institute, and all patients signed a consent form to participate.
Serological and virological determinations
HBV serological markers (HBsAg, HBeAg, and anti-HBe) and anti-HCV antibodies were tested using commercial chemiluminescent assays on a COBAS 8000 analyzer (Roche Diagnostics, Rotkreuz, Switzerland). Antibodies against HDV were tested using the HDV Ab kit (Dia.Pro Diagnostic Bioprobes, Sesto San Giovanni, Italy), and anti-HIV antibodies were tested by the Liaison XL murex HIV Ab/Ag kit (DiaSorin, Saluggia, Italy). HBV-DNA was quantified by real-time PCR with a detection limit of 10 IU/mL (COBAS 6800, Roche Diagnostics). HBV genotypes in the region of interest were determined by Sanger sequencing and by phylogenetic analysis with the same regions extracted from 102 full-length HBV genome sequences representative of HBV genotypes A to H, obtained from GenBank (Supplementary Table 1 and Supplementary Figure 1).
Amplification of the region of interest
In this study we analyzed a portion of the HBX gene encompassing HBX gene encompassed nt 1255 to nt 1611, a region included in the 5’ end of all the viral transcripts. It covered a non-coding upstream region (nt 1255-1373) and the 5’end of the HBX coding region (nts 1374-1611), encoding aa 1 to 79 of HBx.
HBV DNA was extracted from 500 μL of serum with the QIAamp UltraSens Virus Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. Molecular amplification was performed by nested PCR. The first PCR round used primers carrying the universal adaptor M13 (underlined sequence) in their 5’ end (forward 5’-GTTGTAAAACGACGGCCAGTATGCGTGGAACCTTTGTGGCT-3’ and reverse 5’-CACAGGAAACAGCTATGACCATGGGCGTTCACGGTGGTCT-3’) using the following protocol: 95 °C for 2 min, followed by 30 cycles of 95 °C for 15 s, 60 °C for 20 s, and 72 °C for 15 s, and finally, 72 °C for 3 min. The second PCR round was performed using the primers: forward 5’-CGTATCGCCTCCCTCGCGCCATCAG-MID-GTTGTAAAACGACGGCCAGT-3’ and reverse 5’-CTATGCGCCTTGCCAGCCCGCTCAG-MID-CACAGGAAACAGCTATGACC-3’. These primers included the 2 adaptors for the ultra-deep pyrosequencing system at their 5’ ends, followed by a unique identifier multiplex identifier sequence (MID), which enabled grouping the sequences for each sample/patient, and the same M13 universal adaptor sequences as those used in the first PCR in the 3’ ends. This second amplification protocol comprised one denaturation step of 95 °C for 2 min, followed by 20 cycles of 95 °C for 15 s, 60 °C for 20 s, and 72 °C for 15 s, and finally, 72 °C for 3 min. All PCR steps were performed using high-fidelity Pfu Ultra II DNA polymerase (Stratagene, Agilent Technologies, Santa Clara, United States). The final PCR products (amplicons) were purified with Agencourt AMPure XP magnetic beads (Beckman Coulter, Beverly, United States). The quality of the purified products was verified with the Agilent 2200 TapeStation System using the D1000 ScreenTape kit (Agilent Technologies, Waldbronn, Germany).
Next-generation sequencing and sequence quality control
Purified DNA from each sample was quantified using the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific - Life Technologies, Austin, United States), and a pool was formed in which each amplicon was adequately represented in the analysis. The pool was sequenced by next-generation sequencing (NGS) based on ultra-deep pyrosequencing (UDPS) on the GS-Junior or GS FLX platforms (454 Life sciences-Roche, Branford, United States), following the manufacturer’s protocol. The two platforms are reported to be interchangeable[22].
The sequences (reads) obtained after UDPS underwent an in-house bioinformatics filtering procedure, based on scripts developed in R language[23], as previously described by our group[22]. Briefly, the sequences were assigned to each patient (demultiplexed) according to their specific MID, and primers were trimmed. After a general quality filter step, reads with the same nt sequence were collapsed into haplotypes (unique sequences covering the full amplicon observed on the clean set of sequences). Only haplotypes common to the forward and reverse strands and present in abundances of at least 0.1% were accepted; their final frequencies were calculated as the sum of reads observed in each strand. Finally, haplotypes with abundances below 0.25% were excluded.
To analyze the aa sequence of HBx, all individual nt haplotypes from each patient were translated into aa sequences in the HBX gene open reading frame (ORF), which was translated from frame 2. In the fragment analyzed (nt 1255-1611) this ORF expanded from nt 1374 to 1611, encoding aa 1 to 79 of the HBx protein. The upstream sequence was not translated, as it corresponded to a non-coding region whose sequence is included in the HBX transcripts. Once translated, identical aa sequences were recollapsed into aa haplotypes and their frequencies were updated.
Genotyping of the region haplotypes
The genotype of the nt haplotypes obtained by UDPS was determined by discriminant analysis with the same regions extracted from the 102 full-length patterns used for Sanger sequencing (Supplementary Table 1 and Supplementary Figure 1). We determined the maximum genetic distances between sequences from the same HBV genotype in this region and the minimum genetic distances between sequences from different genotypes, in order to set a sequence identity threshold: sequences with an identity above this threshold were clustered together. Genotyping of each cluster centroid was done by distance-based discriminant analysis (DB rule)[24,25], which takes into account the inter- and intra-class variability of all genotypes. Genetic distances were computed according to the Kimura-80 model[26].
Conservation analysis
Sequence conservation was determined by calculating the information content (IC) of each position in a multiple alignment of all the different sequences found in the patients. This analysis, based on Shannon’s uncertainty, was done for a multiple alignment of nt and aa sequences, and is defined as[27]:
Math 1
Math 1.
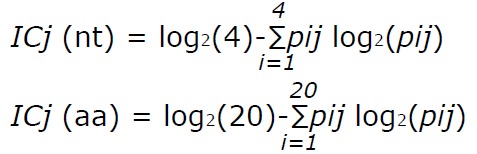
Math(A1).
where j stands for the j-th position in the alignment, i runs over the 4 nucleotides (or over the 20 aa), and pij is the frequency of the i-th nucleotide (or aa) in the j-th alignment position. IC ranges from 0, indicating maximum uncertainty or variability, to log2 4 (i.e., 2 bits) for nt or log2 20 (i.e., 4.32 bits) for aa, indicating maximum information or conservation.
When considering variability in human genetics, a mutation is commonly considered fixed if it is found in at least 1% of the population[28]. However, in viral quasispecies, variants can be present at any abundance in a patient, and the limit for defining a fixed mutation has not yet been established. Taking that into account, we considered two scenarios providing limiting values in our analysis. In the first scenario, we only included the most abundant nucleotide at each position in each patient (consensus approach). The IC values computed in this way would be the upper limit of conservation. In the second scenario, we included all variants in the haplotypes from each patient that were present at abundance greater than 0.25% (quasispecies approach). The IC values computed in this way would be the lower limit of conservation.
Sliding window analysis was then carried out to locate the fragment of at least 25 nt or 10 aa (which corresponds to the length of a possible target for siRNA therapy) with the highest IC within the multiple alignments. This analysis uses windows of 25 nt (or 10 aa) starting from the first position in the multiple alignments and moves forward in steps of 1 (nt or aa). For each window, the analysis computes the mean IC of each position within the window. In addition, the results are represented as sequence logos created using the R language package motifStack[27].
The bioinformatics methods used in this study were reviewed by Dr. Josep Gregori from the Liver Disease-Viral Hepatitis Laboratory of Vall d’Hebron Hospital (Barcelona, Spain), CIBERehd research group, and Roche Diagnostics SL.
RESULTS
Analysis of the NGS sequences obtained and genotyping results
After applying the quality filters, 1333069 sequences were obtained from the 27 serum samples, yielding a median (IQR) of 4578 (2478-9279) sequences per patient.
In the region from nt 1255 to 1611 extracted from the 102 full-length HBV genome sequences from GenBank, analysis of the maximum genetic distance within the same genotype (data not shown) resulted in a sequence identity threshold of 96%. Therefore, for each patient, haplotypes with a sequence identity > 96% were clustered together and were considered to belong to the same HBV genotype. Results of the phylogenetic analysis of master sequences from each cluster in each patient and the 102 GenBank patterns are shown in Table 2. Genotype D nt haplotypes were the most frequent in our patients, followed by genotypes C, A, E, F, B, and H. None of the patients included showed genotype G haplotypes. Moreover, in 14/27 cases (51.8%), some haplotypes were found corresponding to different genotypes than those previously identified by Sanger sequencing, thus yielding a complex mixture of genotypic variants.
Table 2.
Results of genotyping of nucleotide haplotypes obtained in each patient, extracted by next-generation sequencing based on ultra-deep pyrosequencing analysis %
| Patient | A | B | C | D | E | F | H |
| 1 | 0 | 0 | 0 | 0 | 100 | 0 | 0 |
| 2 | 0 | 0 | 100 | 0 | 0 | 0 | 0 |
| 3 | 7.1 | 0 | 0 | 92.9 | 0 | 0 | 0 |
| 4 | 0 | 0 | 0 | 100 | 0 | 0 | 0 |
| 5 | 0 | 0 | 0 | 100 | 0 | 0 | 0 |
| 6 | 1.7 | 0 | 0 | 98.3 | 0 | 0 | 0 |
| 7 | 0 | 0 | 0.3 | 51.3 | 0 | 48.4 | 0 |
| 8 | 7.1 | 0 | 0 | 92.9 | 0 | 0 | 0 |
| 9 | 0 | 0 | 0.3 | 7 | 0 | 92.7 | 0 |
| 10 | 0.9 | 0 | 0 | 50.9 | 0 | 0 | 48.2 |
| 11 | 0 | 0 | 0.3 | 7 | 0 | 92.7 | 0 |
| 12 | 95.1 | 0 | 4.4 | 0.5 | 0 | 0 | 0 |
| 13 | 0 | 0 | 100 | 0 | 0 | 0 | 0 |
| 14 | 46.6 | 0 | 8.2 | 33.2 | 0 | 12 | 0 |
| 15 | 89.7 | 0 | 4.4 | 8 | 0 | 1.5 | 0 |
| 16 | 0 | 0 | 0 | 100 | 0 | 0 | 0 |
| 17 | 0 | 0 | 0 | 100 | 0 | 0 | 0 |
| 18 | 0 | 0 | 0 | 0 | 100 | 0 | 0 |
| 19 | 0 | 0 | 95.3 | 3,6 | 0.9 | 0 | 0 |
| 20 | 100 | 0 | 0 | 0 | 0 | 0 | 0 |
| 21 | 0 | 99.6 | 0.4 | 0 | 0 | 0 | 0 |
| 22 | 0 | 0 | 100 | 0 | 0 | 0 | 0 |
| 23 | 0 | 0 | 87.8 | 12.2 | 0 | 0 | 0 |
| 24 | 0 | 0 | 100 | 0 | 0 | 0 | 0 |
| 25 | 97.9 | 0 | 0 | 2.1 | 0 | 0 | 0 |
| 26 | 0 | 0 | 100 | 0 | 0 | 0 | 0 |
| 27 | 0 | 0 | 0 | 100 | 0 | 0 | 0 |
%A to %H indicates the percentage of nucleotide haplotypes from each patient, classified as HBV genotype A, B, C, D, E, F or H. HBV: Hepatitis B virus.
Conservation of the HBX nucleotide sequence in the region of interest
The region of interest was studied in multiple nt alignments of the entire quasispecies in order to highlight the most highly conserved regions. Sliding windows analysis was implemented in two scenarios: using the consensus approach (n = 27 sequences) and using the quasispecies approach (n = 720 sequences). Of note, the relative frequency of each haplotype was not considered in the multiple alignments, so that the conservation results would not be influenced by haplotype fitness. As no differences were seen when the analyses by the 2 approaches were superimposed (2 highly conserved regions with a mean IC near 2 bits were observed in both; Figure 2), the results reported below all refer to the analysis in the quasispecies scenario.
Figure 2.
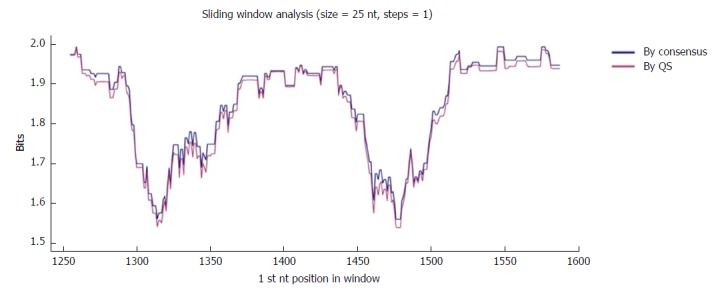
Sliding window analysis of the nucleotide region of interest in the hepatitis B virus X gene (nt 1255-1611). Each point on the graph is the result of the mean information content (IC, bits) of the windows 25-nt in size, with displacement between them in 1-nt steps. The purple line represents the mean IC from the multiple alignments of all haplotypes (in abundance > 0.25%) in the quasispecies (QS) of all patients (n = 720), whereas the blue line shows the mean IC obtained from the multiple alignments of the consensus obtained for each patient (n = 27).
The first hyper-conserved region identified was between nt 1255 and 1286 (23 nt in length) (Figure 3A). Most of the nucleotide positions showed high conservation, yielding IC values near 2 bits (100% maximum conservation), with the exception of position 1272 which showed an IC between 1.6 and 1.8 bits (80%-90% maximum conservation) and positions 1258 and 1284, with an IC between 1.4 and 1.6 bits (70%-80% maximum conservation).
Figure 3.
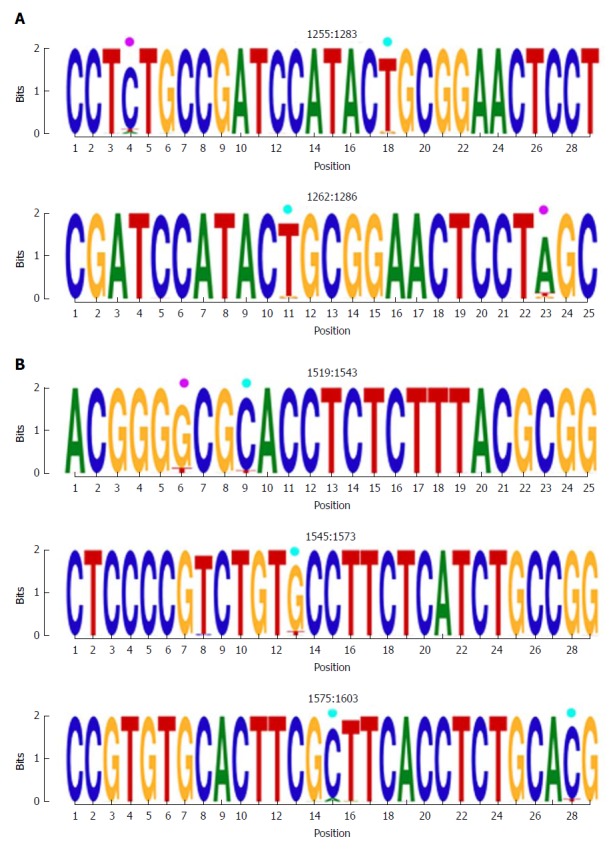
Representation by sequence logos of the information content of the most conserved regions detected in the multiple alignment of all nucleotide haplotypes obtained (quasispecies), nts 1255-1286 and nts 1519-1603. The relative sizes of the letters in each stack of nt sequence logos indicate their relative frequencies at each position within the multiple alignments of nt haplotypes. The total height of each stack of letters depicts the IC of each nt position, measured in bits (Y-axis): 0 bits being the minimum and 2, the maximum conservation. Nucleotide positions with an IC between 1.6 and 1.8 (80%-90% of maximum conservation) are indicated by light blue circles and those with an IC between 1.4 and 1.6 bits (70%-80% of maximum conservation) by pink circles. IC: Information content.
The second hyper-conserved region consisted of 3 conserved nt fragments (1519-1543, 1545-1573, and 1575-1603: 25, 29, and 29 nt in length, respectively) spanning a region between nt 1519 and 1603 (85 nt). Five of these 85 nt positions (5.9%) showed an IC below 1.8 bits: positions 1527, 1557, 1589, and 1602 between 1.6 and 1.8 bits, and position 1524 between 1.4 and 1.6 bits (Figure 2B).
Conservation of the HBx amino acid sequence
To further confirm the nt conservation found, we also analyzed aa conservation in the same 2 scenarios considered for nt variants (n = 27 sequences for the consensus and n = 330 for the quasispecies approach). As was seen with the nt sequences, there were no difference when the 2 analyses (quasispecies vs consensus) were superimposed (Figure 4), which highlighted a single highly conserved region. Again, the results reported refer to the analysis using the quasispecies approach.
Figure 4.
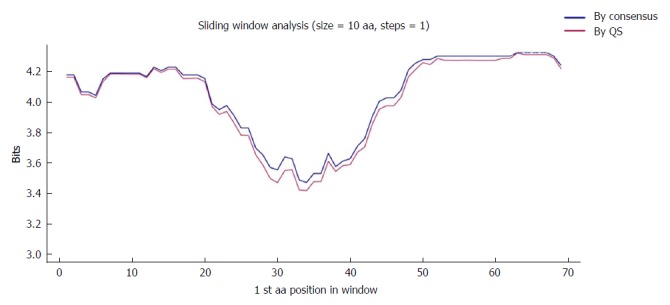
Sliding window of the coded the core and X protein amino acid sequence (aa 1-79). Each point on the graph is the result of the mean information content (IC, bits) between windows of 10-aa size with displacement between them in 1-aa steps. The purple line represents the mean IC from the multiple alignments of all haplotypes (in abundance > 0.25%) in the quasispecies (QS) of all patients (n = 330), whereas the blue line shows the mean IC obtained from the multiple alignments of the consensus obtained for each patient (n = 27).
One highly conserved region was identified between aa 63 and 76 (13 aa), which included a portion of a Kunitz-like domain (Figure 5). All aa showed conservation near 4 bits (100% maximum conservation). This region in the HBx protein corresponded to the hyper-conserved nt sequence between positions 1563 and 1602. The first hyper-conserved nt region observed (nt 1255-1286) was not taken into account in this analysis, as it corresponded to a non-coding region and therefore, was not translated into aa.
Figure 5.
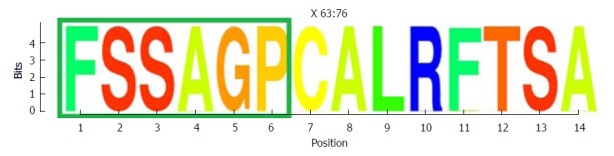
Representation by sequence logos of the information content of the conserved region in the the core and X protein amino acid sequence (aa 63-76). The relative sizes of the letters in each stack in the sequence logos indicate their relative frequencies at each position within the multiple alignments of aa haplotypes obtained. The total height of each stack of letters depicts the IC of each aa position, measured in bits (Y-axis): 0 bits being the minimum and 4.32 bits the maximum conservation. Amino acids belonging to the Kunitz-like domain portion are framed in green. IC: Information content.
DISCUSSION
Although classic nucleotide analogue-based therapies can effectively control HBV infection, eradication of the virus is not achieved because of persistence of the viral minichromosome, cccDNA. Furthermore, even though HBV replication can be inhibited by drug treatment, production of viral antigens may be maintained, and this could lead to progression of the disease[29]. To overcome this challenge, new therapeutic approaches are needed, and gene therapy has emerged as an interesting option.
Ramanan et al[30] proposed a gene therapy based on CRISPR/Cas9 to specifically target a conserved region in HBV cccDNA. These authors reported an anti-HBV effect both in vitro and in vivo, together with inhibition of de novo infection in HepG2-hNTCP cells. However, in HBV infection, the viral genome may be inserted in the host genome. Hence, it is possible that a molecular scissors strategy, such as the CRISPR/Cas9 approach, might imply a risk of affecting the host genome in the regions of viral genome insertion.
With the siRNA approach, viral replication could be hampered and disease progression limited by direct interference with the viral messengers. As has been seen in both cell and mouse models[12,31-33], this interfering RNA regulates the expression of specific viral genes by promoting cleavage of targeted mRNAs, thus inhibiting HBV replication. Specifically, siRNA promotes target mRNA cleavage in a sequence-specific manner through the RNA-induced silencing complex (RISC)[34].
Definition of an extremely conserved region in an optimal HBV genomic region, such as the HBX gene, could be very useful for siRNA-based gene therapy strategies, and some authors have investigated this concept. In a recent study using predictive software, Thongthae et al[33] estimated potential siRNA target sites in the HBX gene (positions: 1317-1337, 1357-1377, and 1644-1664) from an HBV genotype A sequence. These were later tested in vitro, and a reduction in HBV expression was observed. In another effort, the Arbutus Biopharma Corporation recently published a phase-two study in this line. An siRNA was used as treatment for patients with chronic HBV infection, and the preliminary data indicated that the therapy was well tolerated and led to a significant reduction in HBsAg levels[35,36].
HBX is located near the co-terminal 3’ end of all the HBV mRNAs, which implies that interference at this level could abrogate the production of all the viral antigens. In addition, the HBX gene encodes a protein, HBx, which plays a key role in the HBV viral cycle. However, previous data reported by our group[37] and supported in other studies[17,38-40] have described considerable variability in the HBx transactivating C-terminal domain (encoded by the 3’ end of the gene), with multiple insertions and deletions. Because of this variability, this region would not be considered an appropriate gene therapy target.
In light of the importance of the HBx protein for viral replication, it would be reasonable to posit that the gene encoding this protein would have a conserved region. On that basis and after excluding the 3’ end region, we focused our study on the 5’ end region of HBX and its upstream non-coding region (nt 1255-1611). For a gene therapy to be effective in a broad range of conditions, the target sequence should remain conserved in a wide spectrum of clinical and virological situations. Hence, we analyzed samples from a heterogeneous group of 27 HBV-infected patients (in different clinical stages of HBV infection and with different viral genotypes) to seek a conserved target sequence over this spectrum. Two hyper-conserved regions were found. The first was located between nucleotides 1255 and 1286 in the non-coding region. Of note, HBX transcripts initiate at several different sites (between nt 1250-1350)[41], which means that this conserved region might be not present in all of them, but would likely be present in the other viral transcripts. The second hyper-conserved region was located between nucleotides 1519 and 1603, within the coding region.
Conserved regions in this portion of the HBV gene have been reported previously. Karinova et al[42] observed two conserved regions in the S and X ORF of the HBV genotype A genome. These authors found that a CRISPR/Cas9 molecular scissor directed to this conserved region in HBX was able to modify both episomal cccDNA and chromosomally-integrated HBV DNA in reporter cell lines, thereby interfering with HBV replication and with de novo infection of hepatoma cell lines. In addition, with the use of predictive software, Thongthae et al[33] estimated some potential siRNA targets in the HBX gene (including the non-coding region identified here) in a single viral sequence, and reported the efficacy of this approach in an in vitro study. The value of the present study is that conservation of the regions examined was directly substantiated by sequencing analysis of patient samples, taking into account different HBV genotypes and different clinical stages of the infection. Furthermore, the nucleotide conservation documented here was supported by detection of a conserved region in the HBx protein sequence between aa 63 and 76, which is encoded by nt 1563-1602 (within the second hyper-conserved region). Of note, this fragment includes some aa from one of the HBx Kunitz-like domains (aa 58-70)[43], which are able to inhibit the function of cellular degrading enzymes, such as proteases[44]. This suggests that this portion of the HBx protein may be conserved to preserve the integrity of the protein, protecting it from undesired degradation.
As a limitation of the study, we should mention the relatively small sample size. From the initial group of 46 well-characterized treatment-naïve CHB patients available, only those with viremia levels high enough to amplify the HBV genome region of interest by our PCR technique could be included. Furthermore, we wished to have a representation of various clinical stages of HBV infection and most HBV genotypes (A to F and H), which yielded a sample of 27 patients. Larger samples should be analyzed in future studies to confirm conservation of the regions investigated. We also have to point out that the NGS technology used in this study (GS-Junior platform, 454/Roche) has been discontinued by the supplier; nonetheless, the protocol described here can be adapted to currently available platforms, such as the Illumina MiSeq (San Diego, United States). Finally, in vitro functional studies should be performed to test the potential usefulness of the 2 hyper-conserved domains described here as targets for siRNA-based antiviral gene therapy.
In summary, this study, performed in serum samples from HBV patients infected by different viral genotypes and in different clinical stages, identified regions in the HBX gene with high levels of conservation in all these circumstances. We found 2 hyper-conserved regions, the first in the non-coding region of HBX transcripts, and the second in the HBX coding region, which was conserved at both the nt and aa level. These hyper-conserved regions could be candidates for targeted gene therapies such as the siRNA approach. Of particular interest, because of the co-terminal localization of the HBX gene, a siRNA system designed to target these regions could interfere with expression of all the HBV viral transcripts.
ARTICLE HIGHLIGHTS
Research background
Hepatitis B virus (HBV) infection can be controlled with current treatments, but cure is not achieved due to persistence of covalently closed circular DNA (cccDNA) in the nuclei of infected hepatocytes. This minichromosome forms a viral reservoir that is a source of residual viral replication and expression of viral proteins; thus, it has a key role in liver disease progression. To surmount this circumstance, new anti-HBV therapeutic approaches are under development, with gene therapy being a promising option. Among these approaches, small interference RNA (siRNA) can be used to silence specific genes at the post-transcriptional level through a sequence-specific interaction with target mRNAs, resulting in inhibition of viral protein expression. Among all the HBV proteins, Hepatitis B X protein (HBx), coded by the HBV X gene (HBX), is a determining factor in the infection. It regulates cccDNA expression and interacts with several cellular pathways, facilitating liver disease progression. Of particular note, because of its location near the co-terminal 3’ end, all HBV transcripts include the HBX sequence. Hence, it could be a valuable target for a hypothetical curative treatment based on gene therapy. In this sense, identification of hyper-conserved regions within HBX is needed to define a new gene therapy system that would be effective whatever the patient’s clinical stage or HBV genotype.
Research motivation
Although antiviral therapy can suppress viral replication, the risk of liver disease progression and development of hepatocellular carcinoma (HCC) remains due to cccDNA-related expression of viral antigens. Interference with expression of the viral proteins could be helpful to limit progression of the disease, and siRNAs would be valid tools in this sense. To design an effective siRNA, an appropriate target must be found. The HBX sequence is included in all the viral transcripts due to its co-terminal localization in the viral genome. siRNAs targeting hyper-conserved regions of this gene would interfere with expression of all the viral proteins. Furthermore, as these regions are conserved in the spectrum of clinical disease phases and viral genotypes, it would be a valid therapeutic approach for a wide range of situations. This could profoundly limit the risk of HCC, particularly in patients with low viremia due to antiretroviral efficacy.
Research objectives
Considering the essential role of HBx in viral infection and its potential utility as target for gene therapy, the aim of this study was to identify hyper-conserved regions within the HBV genome encompassing the HBX 5’ coding region and the upstream non-coding region (included in all HBV transcripts) in samples from HBV-infected patients in various clinical stages and with different viral genotypes. The regions identified might be feasible targets for a gene therapy able to inhibit viral protein expression in a wide spectrum of clinical and virological circumstances, thus limiting liver disease progression and the risk of HCC.
Research methods
The study included 27 treatment-naïve chronic hepatitis B monoinfected patients in different clinical stages and with several HBV genotypes (from A-F and H). A serum sample from each patient with viremia > 3.5 log IU/mL was analyzed. The HBX 5’ end region [nucleotide (nt) 1255-1611] was PCR-amplified and later analyzed using next-generation sequencing (NGS). The sequences (reads) obtained after sequencing underwent an in-house bioinformatics filtering procedure, and haplotypes with a relative frequency ≥ 0.25% were maintained in the analysis. Haplotypes were genotyped by discriminant analysis with the same regions extracted from the 102 full-length patterns. Conservation of the quasispecies sequences was determined by calculating the information content (IC), based on Shannon’s uncertainty, of each position in a multiple alignment of all different sequences found in the patients. Sliding window analysis was then carried out to locate the fragment of at least 25 nt or 10 aa (which corresponds to the length of a possible target for siRNA therapy) with the highest IC within the multiple alignments, moving forward in steps of 1 (nt or aa). This method enables detection of conserved regions within the 5’ HBX gene by directly analyzing the viral quasispecies obtained with NGS.
Research results
After applying the quality filter, 1333069 haplotype sequences were obtained. Genotyping analysis highlighted a complex mixture of HBV genotypes. By studying the nt conservation, we identified two hyper-conserved nucleotide regions in HBX. The first one, between nt 1255 and 1286, corresponded to a non-coding region, whereas the second one, consisting of 3 conserved fragments (spanning an overall portion between 1519 and 1603), coincided with a coding region. Of note, the fragment between nt 1563 and 1602 was also conserved at the amino acid level, identifying a region between residues 63 and 76, which included a portion of a Kunitz-like domain. These results highlight new potential targets for gene therapy, mainly based on siRNA. Of note, in vitro and in vivo functional studies of the specific siRNAs should be performed to test their potential usefulness for therapy.
Research conclusions
Gene therapy represents a highly promising therapeutic tool to achieve a cure against HBV infection. Several sequence-specific treatment systems are currently in development, and identification of conserved sequences would provide useful therapeutic targets. Detection of a target present in all the clinical disease stages and HBV genotypes could lead to development of a therapy that would be effective in a wide range of situations. Considering the key role of HBx in viral infection and disease progression, we focused the study on analyzing conservation of the HBX gene. Of note, considering the high variability previously observed in the 3’end of HBX, we speculated that the 5’end could be a better subject for study. Moreover, thanks to the co-terminality of this viral gene, a siRNA targeting this gene could interfere with all the viral transcripts. Here, we investigated conservation of a portion of the HBV genome encompassing the HBX 5’ coding region and upstream non-coding region, both of which are included in all HBV transcripts. By NGS analysis, we identified two hyper-conserved regions in our region of interest in serum samples from HBV patients with different clinical and virological characteristics. This new therapeutic tool could have relevant applicability in clinical practice. Together with inhibition of the expression of one of the main viral proteins involved in HBV replication and disease progression, it could block the expression of the other viral antigens, thus profoundly interfering with disease evolution and the appearance of HCC. Furthermore, the NGS method developed here could be used to find other hyper-conserved regions within the HBV genome that could be potential targets for gene therapy based on siRNA.
Research perspectives
This study describes a method that can be used to find other conserved sequences in the HBV genome, making it a starting point in the search for other possible targets for gene therapy. Here, the hyper-conserved regions were found by directly analyzing the viral quasispecies sequences obtained using NGS. These regions can then be used to produce siRNA molecules for in vitro and in vivo testing of antiviral activity.
ACKNOWLEDGMENTS
The statistical and bioinformatics methods used in this study were reviewed by Dr. Josep Gregori from the liver disease-viral hepatitis laboratory (Vall d’Hebron Institut Recerca-Hospital Universitari Vall d’Hebron), CIBERehd and Roche Diagnostics SL. The authors thank Celine Cavallo for English language support and helpful editing suggestions.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Spain
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Supported by the Instituto de Salud Carlos III, No. PI15/00856; and the European Regional Development Fund (ERDF), No. PI15/00856.
Institutional review board statement: The study was reviewed and approved by the Clinical Research Ethics Committee (CEIC) of Hospital Universitari Vall d’Hebron.
Conflict-of-interest statement: Josep Gregori is an employee of Roche Diagnostics, SL.
Data sharing statement: No additional data are available.
Peer-review started: February 17, 2018
First decision: March 9, 2018
Article in press: May 6, 2018
P- Reviewer: Enomoto H S- Editor: Wang XJ L- Editor: A E- Editor: Huang Y
Contributor Information
Carolina González, Liver Pathology Unit, Departments of Biochemistry and Microbiology, Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain.
David Tabernero, Liver Pathology Unit, Departments of Biochemistry and Microbiology, Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain; Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Instituto de Salud Carlos III, Madrid 28029, Spain.
Maria Francesca Cortese, Liver Pathology Unit, Departments of Biochemistry and Microbiology, Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain; Marçall Yll, Josep Quer, Liver Unit, Liver Disease Laboratory-Viral Hepatitis, Vall d’Hebron Institut Recerca-Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain. maria.cortese@vhir.org.
Josep Gregori, Marçall Yll, Josep Quer, Liver Unit, Liver Disease Laboratory-Viral Hepatitis, Vall d’Hebron Institut Recerca-Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain; Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Instituto de Salud Carlos III, Madrid 28029, Spain; Roche Diagnostics SL, Sant Cugat del Vallès 08174, Spain.
Rosario Casillas, Liver Pathology Unit, Departments of Biochemistry and Microbiology, Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain; Marçall Yll, Josep Quer, Liver Unit, Liver Disease Laboratory-Viral Hepatitis, Vall d’Hebron Institut Recerca-Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain.
Mar Riveiro-Barciela, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Instituto de Salud Carlos III, Madrid 28029, Spain; Rafael Esteban and Maria Buti, Liver Unit, Department of Internal Medicine, Hospital Universitari Vall d'Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain.
Cristina Godoy, Liver Pathology Unit, Departments of Biochemistry and Microbiology, Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain; Marçall Yll, Josep Quer, Liver Unit, Liver Disease Laboratory-Viral Hepatitis, Vall d’Hebron Institut Recerca-Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain.
Sara Sopena, Liver Pathology Unit, Departments of Biochemistry and Microbiology, Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain; Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Instituto de Salud Carlos III, Madrid 28029, Spain.
Ariadna Rando, Liver Pathology Unit, Departments of Biochemistry and Microbiology, Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain.
Marçal Yll, Liver Pathology Unit, Departments of Biochemistry and Microbiology, Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain.
Rosa Lopez-Martinez, Liver Pathology Unit, Departments of Biochemistry and Microbiology, Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain.
Josep Quer, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Instituto de Salud Carlos III, Madrid 28029, Spain.
Rafael Esteban, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Instituto de Salud Carlos III, Madrid 28029, Spain.
Maria Buti, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Instituto de Salud Carlos III, Madrid 28029, Spain.
Francisco Rodríguez-Frías, Liver Pathology Unit, Departments of Biochemistry and Microbiology, Hospital Universitari Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona 08035, Spain; Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Instituto de Salud Carlos III, Madrid 28029, Spain.
References
- 1.Lin YY, Liu C, Chien WH, Wu LL, Tao Y, Wu D, Lu X, Hsieh CH, Chen PJ, Wang HY, et al. New insights into the evolutionary rate of hepatitis B virus at different biological scales. J Virol. 2015;89:3512–3522. doi: 10.1128/JVI.03131-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kay A, Zoulim F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007;127:164–176. doi: 10.1016/j.virusres.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 3.Locarnini S, Zoulim F. Molecular genetics of HBV infection. Antivir Ther. 2010;15 Suppl 3:3–14. doi: 10.3851/IMP1619. [DOI] [PubMed] [Google Scholar]
- 4.Sunbul M. Hepatitis B virus genotypes: global distribution and clinical importance. World J Gastroenterol. 2014;20:5427–5434. doi: 10.3748/wjg.v20.i18.5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pujol FH, Navas MC, Hainaut P, Chemin I. Worldwide genetic diversity of HBV genotypes and risk of hepatocellular carcinoma. Cancer Lett. 2009;286:80–88. doi: 10.1016/j.canlet.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 6.European Association for the Study of the Liver. European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67:370–398. doi: 10.1016/j.jhep.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 7.Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, Raimondo G, Levrero M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci USA. 2009;106:19975–19979. doi: 10.1073/pnas.0908365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang HC, Kao JH. Persistence of hepatitis B virus covalently closed circular DNA in hepatocytes: molecular mechanisms and clinical significance. Emerg Microbes Infect. 2014;3:e64. doi: 10.1038/emi.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bloom K, Ely A, Arbuthnot P. Recent advances in use of gene therapy to treat hepatitis B virus infection. Adv Exp Med Biol. 2015;848:31–49. doi: 10.1007/978-1-4939-2432-5_2. [DOI] [PubMed] [Google Scholar]
- 10.Dong C, Qu L, Wang H, Wei L, Dong Y, Xiong S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res. 2015;118:110–117. doi: 10.1016/j.antiviral.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 11.Weber ND, Stone D, Sedlak RH, De Silva Feelixge HS, Roychoudhury P, Schiffer JT, Aubert M, Jerome KR. AAV-mediated delivery of zinc finger nucleases targeting hepatitis B virus inhibits active replication. PLoS One. 2014;9:e97579. doi: 10.1371/journal.pone.0097579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gish RG, Yuen MF, Chan HL, Given BD, Lai CL, Locarnini SA, Lau JY, Wooddell CI, Schluep T, Lewis DL. Synthetic RNAi triggers and their use in chronic hepatitis B therapies with curative intent. Antiviral Res. 2015;121:97–108. doi: 10.1016/j.antiviral.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 13.Poch O, Sauvaget I, Delarue M, Tordo N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989;8:3867–3874. doi: 10.1002/j.1460-2075.1989.tb08565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tu H, Bonura C, Giannini C, Mouly H, Soussan P, Kew M, Paterlini-Bréchot P, Bréchot C, Kremsdorf D. Biological impact of natural COOH-terminal deletions of hepatitis B virus X protein in hepatocellular carcinoma tissues. Cancer Res. 2001;61:7803–7810. [PubMed] [Google Scholar]
- 15.Wang Q, Zhang WY, Ye LH, Zhang XD. A mutant of HBx (HBxDelta127) promotes hepatoma cell growth via sterol regulatory element binding protein 1c involving 5-lipoxygenase. Acta Pharmacol Sin. 2010;31:367–374. doi: 10.1038/aps.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panjaworayan N, Roessner SK, Firth AE, Brown CM. HBVRegDB: annotation, comparison, detection and visualization of regulatory elements in hepatitis B virus sequences. Virol J. 2007;4:136. doi: 10.1186/1743-422X-4-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim H, Lee SA, Kim BJ. X region mutations of hepatitis B virus related to clinical severity. World J Gastroenterol. 2016;22:5467–5478. doi: 10.3748/wjg.v22.i24.5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali A, Abdel-Hafiz H, Suhail M, Al-Mars A, Zakaria MK, Fatima K, Ahmad S, Azhar E, Chaudhary A, Qadri I. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. World J Gastroenterol. 2014;20:10238–10248. doi: 10.3748/wjg.v20.i30.10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SA, Mun HS, Kim H, Lee HK, Kim BJ, Hwang ES, Kook YH, Kim BJ. Naturally occurring hepatitis B virus X deletions and insertions among Korean chronic patients. J Med Virol. 2011;83:65–70. doi: 10.1002/jmv.21938. [DOI] [PubMed] [Google Scholar]
- 20.Peng Y, Liu B, Hou J, Sun J, Hao R, Xiang K, Yan L, Zhang J, Zhuang H, Li T. Naturally occurring deletions/insertions in HBV core promoter tend to decrease in hepatitis B e antigen-positive chronic hepatitis B patients during antiviral therapy. Antivir Ther. 2015;20:623–632. doi: 10.3851/IMP2955. [DOI] [PubMed] [Google Scholar]
- 21.Homs M, Caballero A, Gregori J, Tabernero D, Quer J, Nieto L, Esteban R, Buti M, Rodriguez-Frias F. Clinical application of estimating hepatitis B virus quasispecies complexity by massive sequencing: correlation between natural evolution and on-treatment evolution. PLoS One. 2014;9:e112306. doi: 10.1371/journal.pone.0112306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramírez C, Gregori J, Buti M, Tabernero D, Camós S, Casillas R, Quer J, Esteban R, Homs M, Rodriguez-Frías F. A comparative study of ultra-deep pyrosequencing and cloning to quantitatively analyze the viral quasispecies using hepatitis B virus infection as a model. Antiviral Res. 2013;98:273–283. doi: 10.1016/j.antiviral.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 23.Team RC. 2016. A language and environment for statistical computing. R Found. Stat. Comput. Vienna, Austria. Available from: https://www.r-project.org/ [Google Scholar]
- 24.Cuadras C. A distance approach to discriminant analysis and its properties. In: Mathematics preprint series. Barcelona: 1991 [Google Scholar]
- 25.Cuadras C. Distance analysis in discrimination and classification using both continuous and categorical variables. In: Statistical AData analysis and Interference. Amsterdam; 1989. pp. 459–473. [Google Scholar]
- 26.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 27.Schneider TD. Information content of individual genetic sequences. J Theor Biol. 1997;189:427–441. doi: 10.1006/jtbi.1997.0540. [DOI] [PubMed] [Google Scholar]
- 28.Griffiths AJF, Miller JH, Suzuki DT et al. An Introduction to Genetic Analysis. 7th edition. New York; WH. Freeman: 2000. [Google Scholar]
- 29.Kwon H, Lok AS. Hepatitis B therapy. Nat Rev Gastroenterol Hepatol. 2011;8:275–284. doi: 10.1038/nrgastro.2011.33. [DOI] [PubMed] [Google Scholar]
- 30.Ramanan V, Shlomai A, Cox DB, Schwartz RE, Michailidis E, Bhatta A, Scott DA, Zhang F, Rice CM, Bhatia SN. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci Rep. 2015;5:10833. doi: 10.1038/srep10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein C, Bock CT, Wedemeyer H, Wüstefeld T, Locarnini S, Dienes HP, Kubicka S, Manns MP, Trautwein C. Inhibition of hepatitis B virus replication in vivo by nucleoside analogues and siRNA. Gastroenterology. 2003;125:9–18. doi: 10.1016/s0016-5085(03)00720-0. [DOI] [PubMed] [Google Scholar]
- 32.Shlomai A, Shaul Y. Inhibition of hepatitis B virus expression and replication by RNA interference. Hepatology. 2003;37:764–770. doi: 10.1053/jhep.2003.50146. [DOI] [PubMed] [Google Scholar]
- 33.Thongthae N, Payungporn S, Poovorawan Y, T-Thienprasert NP. A rational study for identification of highly effective siRNAs against hepatitis B virus. Exp Mol Pathol. 2014;97:120–127. doi: 10.1016/j.yexmp.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Tan FL, Yin JQ. RNAi, a new therapeutic strategy against viral infection. Cell Res. 2004;14:460–466. doi: 10.1038/sj.cr.7290248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agarwal K, Gane E, Cheng W, Sievert W, Roberts S, Ahn SH, Kim YJ, Streinu-cercel A, Denning J, Symonds W, et al. HBcrAg, HBV-RNA Declines in A Phase 2a Study Evaluating the Multi-Dose Activity of ARB-1467 in HBeAg-Positive and Negative Virally Suppressed Patients With Hepatitis B. Hepatology. 2017;66:22A–23A. [Google Scholar]
- 36.Eley T, Russ R, Streinu-cercel A, Gane EJ, Roberts SK, Ahn SH, Kim YJ, Symonds W, Mendez P. Pharmacokinetics and exploratory exposure-response of siRNAs administered monthly as ARB-001467 ( ARB-1467 ) in a Phase 2a study in HBeAg positive and negative virally suppressed subjects with chronic hepatitis B. Hepatology. 2017;66:23A–24A. [Google Scholar]
- 37.Caballero A, Gregori J, Buti M, Tabernero D, Quer J, Blasi M, Rodriguez-Algarra F, Casillas R, González C, Belmonte I, et al. Insertions and/or deletions in the main regulatory region of hepatitis B virus suggest multicoding of the X protein. J Hepatol. 2015;62:S523. [Google Scholar]
- 38.Zhang ZH, Wu CC, Chen XW, Li X, Li J, Lu MJ. Genetic variation of hepatitis B virus and its significance for pathogenesis. World J Gastroenterol. 2016;22:126–144. doi: 10.3748/wjg.v22.i1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lazarevic I. Clinical implications of hepatitis B virus mutations: recent advances. World J Gastroenterol. 2014;20:7653–7664. doi: 10.3748/wjg.v20.i24.7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salarnia F, Besharat S, Zhand S, Javid N, Khodabakhshi B, Moradi A. Mutations in Hepatitis-B X-Gene Region: Chronic Hepatitis-B versus Cirrhosis. J Clin Diagn Res. 2017;11:OC31–OC34. doi: 10.7860/JCDR/2017/22570.9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Treinin M, Laub O. Identification of a promoter element located upstream from the hepatitis B virus X gene. Mol Cell Biol. 1987;7:545–548. doi: 10.1128/mcb.7.1.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karimova M, Beschorner N, Dammermann W, Chemnitz J, Indenbirken D, Bockmann JH, Grundhoff A, Lüth S, Buchholz F, Schulze zur Wiesch J, et al. CRISPR/Cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci Rep. 2015;5:13734. doi: 10.1038/srep13734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Datta S, Banerjee A, Chandra PK, Biswas A, Panigrahi R, Mahapatra PK, Panda CK, Chakrabarti S, Bhattacharya SK, Chakravarty R. Analysis of hepatitis B virus X gene phylogeny, genetic variability and its impact on pathogenesis: implications in Eastern Indian HBV carriers. Virology. 2008;382:190–198. doi: 10.1016/j.virol.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 44.Lee J, Chung JH, Kim HM, Kim DW, Kim H. Designed nucleases for targeted genome editing. Plant Biotechnol J. 2016;14:448–462. doi: 10.1111/pbi.12465. [DOI] [PMC free article] [PubMed] [Google Scholar]