

Abstract
We conducted a critical review of human papillomavirus (HPV) integration into the host genome in oral/oropharyngeal cancer, reviewed the literature for HPV-induced cancers, and obtained current data for HPV-related oral and oropharyngeal cancers. In addition, we performed studies to identify HPV integration sites and the relationship of integration to viral-host fusion transcripts and whether integration is required for HPV-associated oncogenesis. Viral integration of HPV into the host genome is not required for the viral life cycle and might not be necessary for cellular transformation, yet HPV integration is frequently reported in cervical and head and neck cancer specimens. Studies of large numbers of early cervical lesions revealed frequent viral integration into gene-poor regions of the host genome with comparatively rare integration into cellular genes, suggesting that integration is a stochastic event and that site of integration may be largely a function of chance. However, more recent studies of head and neck squamous cell carcinomas (HNSCCs) suggest that integration may represent an additional oncogenic mechanism through direct effects on cancer-related gene expression and generation of hybrid viral-host fusion transcripts. In HNSCC cell lines as well as primary tumors, integration into cancer-related genes leading to gene disruption has been reported. The studies have shown that integration-induced altered gene expression may be associated with tumor recurrence. Evidence from several studies indicates that viral integration into genic regions is accompanied by local amplification, increased expression in some cases, interruption of gene expression, and likely additional oncogenic effects. Similarly, reported examples of viral integration near microRNAs suggest that altered expression of these regulatory molecules may also contribute to oncogenesis. Future work is indicated to identify the mechanisms of these events on cancer cell behavior.
Keywords: cancer biology, genomics, infectious disease(s), neoplasms, tumor virology, oral carcinogenesis
Background
Human papillomavirus (HPV) is a double-stranded, small DNA virus transmitted through sexual contact that infects human epithelium in anogenital and oral mucosa. Although HPV is one of the most common sexually transmitted infections, persistent oral HPV infections are relatively rare in the healthy population. Data from the National Health and Nutrition Examination Survey (NHANES 2009–2010) showed that the prevalence of oral HPV infection in the United States was 6.9% (Gillison et al. 2012). There are many factors associated with increased risk of oral HPV infection, including age, sex, number of sexual partners, and current smoking habits. Oral HPV infections peak in prevalence around ages 30 to 34 y and 60 to 64 y, following a bimodal pattern. Men are more likely to be infected, and higher numbers of sexual partners and smoking are associated with increased risk of infection. Most HPV infections are naturally cleared, but in women, it has been well established that persistence of a genital HPV infection is a significant risk factor for developing cervical squamous cell carcinoma (Walboomers et al. 1999; Bosch et al. 2002). However, the factors that contribute to persistent oral infection and the natural history of oral HPV infections leading to cancer have still not been well characterized.
There are more than 200 HPV types with varying epithelial tropism and associated conditions. A subset of 13 mucosal HPV types is considered high-risk HPV (hrHPV) based on their frequent association with various cancers and transforming ability in model systems. Another subset is considered potentially high risk based on association with cancers, but evidence demonstrating carcinogenicity is lacking. Infection with 1 or more HPV strains is nearly ubiquitous in sexually active persons. These infections are usually asymptomatic and clear spontaneously. However, persistent infections may lead to a variety of HPV-mediated diseases, including genital warts, precancers, and cancers of the cervix, anus, penis, vulva, vagina, and head and neck—particularly cancers of the oropharynx (Centers for Disease Control and Prevention 2015).
hrHPV types 16 and 18 are known to cause the majority of cervical cancers and many squamous cancers of the head and neck (Syrjänen 2007; zur Hausen 2009). Among oropharynx cancers studied at the University of Michigan, roughly 90% are associated with HPV16 and 10% are associated with other high-risk HPV types (Walline et al. 2013). The HPV16 genome is a 7.9-kb circular genome organized into 6 early region genes and 2 late region genes (Fig. 1). HPV is dependent on the host cell for replication. The viral proteins manipulate cellular pathways for viral genome replication/amplification and coordinate these processes with the cellular differentiation pathway, timing viral capsid production with the later stages of epithelial differentiation. The HPV oncoproteins E6 and E7 induce host cell replication by blocking the function of key cell cycle regulators, TP53 and RB1. The viral E6 protein binds to cellular TP53 and induces ubiquitin-mediated degradation (Fig. 2A) (Scheffner et al. 1990). HPV E7 protein binds to the RB1 pocket, thereby inhibiting its interaction with the transcription factor E2F, resulting in unscheduled transcription of cell cycle entry genes (Boyer et al. 1996) (Fig. 2B). The E6 gene can be expressed as full-length or alternatively spliced forms, referred to as E6*I, E6*II, or E6*III. These alternate transcripts are thought to be drivers of oncogenic transformation (Sathish et al. 2004). The hrHPV E1 and E2 genes play an essential role in viral replication, and E2 is a transcriptional repressor of E6 and E7. Persistent HPV infection that leads to carcinoma is characterized by high expression of E6 and E7 and frequent loss of E2, leading to unregulated expression of E6 and E7, which promotes genomic instability, oncogenic transformation, and clonal expansion (Wiest et al. 2002).
Figure 1.

Human papillomavirus (HPV) genome map. The HPV genome encodes 8 major proteins. E6 and E7 are the viral oncoproteins of HPV that function to disrupt cell cycle control mechanisms in the host cell, driving cell proliferation and viral replication. The other “early” genes, E1, E2, E4, and E5, encode proteins that have roles in viral replication. E2 is also involved in controlling viral oncogene expression. One function is to serve as a transcriptional repressor of E6 and E7, coordinating the viral life cycle with keratinocyte differentiation. After viral genome amplification in the suprabasal cells, E6 and E7 expression is attenuated and viral capsid production begins. The 2 “late” genes, L1 and L2, encode for structural proteins that constitute the viral capsid necessary for packaging and transmission of the newly replicated virus (Munger et al. 2004).
Figure 2.

E6 and E7 induce replication by blocking the function of cell cycle regulators. (A) The human papillomavirus (HPV) oncoprotein E6 recruits the cellular E3 ubiquitin ligase, E6-associated protein (E6AP) (Talis et al. 1998), and binds TP53, leading to TP53 polyubiquitination and subsequent degradation by the 26S proteasome. The destruction of TP53 results in failure of cell cycle arrest and apoptosis, contributing to unrestricted host cell growth and proliferation. (B) In normal cells, the cell cycle regulator RB binds the transcription factor E2F, preventing cell cycle progression. When cell growth signaling occurs, expression of cyclin D1 is initiated. Cyclin D1 activates cyclin-dependent kinase (CDK) 4/6, leading to monophosphorylation of RB. CDK2 is then activated by cyclin E and further phosphorylates RB, releasing E2F and initiating transcription of cell cycle entry genes. E2F also activates transcription of p16INK4a (CDKN2A, an inhibitor of cyclin-dependent kinase 4 and the off signal for RB phosphorylation), shutting off RB phosphorylation. Ubiquitous phosphatase activity dephosphorylates RB, which resequesters E2F and stops cell cycle entry. In the presence of HPV, E7 binds to the pocket of RB, disrupting the interaction with E2F. When E2F is liberated, it leads to continual transcription of S-phase genes, driven by other cell cycle cyclin-CDK complexes. p16INK4a is also inappropriately transcribed and expressed, making it a useful surrogate histological marker of HPV infection. The binding of E7 to RB leads to continuous cell cycle entry, progression, and cellular proliferation.
Although the incidence of HPV-induced cervical cancer has decreased due to improved screening and early intervention, the rate of HPV-associated head and neck cancers has been increasing (Chaturvedi et al. 2008; Brouwer et al. 2016). In 2009, there were more incident cases of oropharynx cancer than cervical cancer in the United States (Jemal et al. 2013). This is in contrast to HPV-negative head and neck cancers, which represent a distinct clinical entity and have been declining in incidence, primarily due to public health efforts to decrease smoking. HPV-positive cancer is more likely to develop in the oropharynx than in the oral cavity, and it is suggested that this is due to the architecture of tonsillar crypts in the oropharynx, which act as a reservoir for HPV. The palatine and lingual tonsils are the most common sites of origin for HPV-induced oropharyngeal cancer (Paz et al. 1997; Gillison et al. 2000). Patients with HPV-positive oropharyngeal squamous cell carcinoma (OPSCC) have a survival advantage over those with HPV-negative OPSCC regardless of treatment modality (Gillison et al. 2000; Licitra et al. 2006; Fakhry et al. 2008; Worden et al. 2008). Patients with HPV-positive OPSCC tend to respond better to chemoradiation therapy (Feng et al. 2010) and have enhanced radiosensitivity (Ziemann et al. 2015). HPV-negative patients generally have a higher mutational burden, particularly mutant TP53, which may contribute to their worse prognosis (Westra et al. 2008). However, patients with HPV-positive oral cavity squamous cell carcinoma (OCSCC) do not have the same survival advantage over patients with HPV-negative OCSCC (Fakhry et al. 2017); some studies suggest they have a worse prognosis (Duray et al. 2012). A meta-analysis by Ragin and Taioli (2007) showed no survival difference between HPV-positive and HPV-negative patients with cancer at nonoropharyngeal sites of the head and neck, including oral cavity. The source of the discrepancy in outcome between the oropharynx and oral cavity is not entirely clear, but differences in immune response from site to site may be an important factor (Duray et al. 2014).
Integration in Cervical Carcinomas
HPV typically persists in cells as a circular episome but can also linearize and become integrated into the host genome. It has been of great interest to understand the implications of integration and to determine whether it is involved in tumor formation. HPV is commonly found integrated into the host genome in cervical cancer (Klaes et al. 1999; Vinokurova et al. 2008). Integration of HPV is characteristic of cervical lesion progression but may not be required for tumor formation (Klaes et al. 1999; Wentzensen et al. 2004). Early studies investigating the role of integration in cervical lesions showed that integration is a stochastic process or favors a preference for common fragile sites, regions of microhomology, highly transcriptionally active regions, or near microRNAs (miRNAs) (Ziegert et al. 2003; Wentzensen et al. 2004; Hu et al. 2015). There were few reported examples of integration into genes that led to a disruption of gene expression, and in general, integration was not presumed to have any major impact on gene expression. Integration near the cMYC locus on chromosome 8q24 has been shown to lead to overexpression of cMYC; this locus may be a preferential target site for integration of HPV18 (Ferber et al. 2003). Later studies, however, showed that integration of HPV might represent an additional oncogenic mechanism through direct alteration of cancer-related gene expression. One study showed that most integration events occur in known or predicted genes or near miRNAs, which have major roles in regulation of cellular processes (Schmitz et al. 2012). A more recent study by Hu et al. (2015) showed that integration events occur in genomic hotspot regions and may function to inactivate or activate genes that favor clonal expansion. Bodelon and colleagues (2016) analyzed over 1,200 integration events in cervical cancers and reported that integration occurred most frequently at 3 loci: 3q28, 8q24.21, and 13q22.1. These regions all are gene rich and contain important tumor suppressors, including TP63, TPRG1, MYC, and KLF5 and KLF12. They also reported that integration into genes occurs more often than expected by chance and may lead to functional alteration of important genes. Integration events in cervical cancer have been better described than in head and neck cancer, but still much is not understood about the role integration plays on the progression from dysplasia to invasive carcinoma.
How Is Integration Detected?
Whether integration of HPV is required for malignant transformation in oral cancers is not clear. The wide variety of techniques used to detect integration events makes it challenging to compare results of different studies. The most commonly used methods to detect integrated HPV sequences are detection of integrated papillomavirus sequences–polymerase chain reaction (DIPS-PCR), amplification of papillomavirus oncogene transcripts (APOT), whole-genome sequencing (WGS), whole-exome sequencing, and RNA-seq. DIPS-PCR and APOT are PCR-based methods used to detect fusions at the DNA and RNA level, respectively. These 2 methods are technically simpler and cheaper options than larger-scale sequencing methods like WGS or RNA-seq but may be unable to detect all integration sites and complicated structural changes within samples. Therefore, WGS and RNA-seq may better reflect the true complexity of viral integration using tiling of paired ends across the genome. Many groups use an E2/E6 gene expression ratio to represent the physical state of HPV, based on the hypothesis that during integration, the E2 gene is disrupted, leading to increased levels of E6. In this method, a ratio is made comparing the expression levels of the E2 and E6 genes as measured by quantitative PCR (qPCR), assuming that a ratio of 1 means HPV is episomal and a ratio of less than 1 means HPV is integrated. This method is not as effective as others because it is based on the assumption that E2 is always disrupted and E6 is always increased during integration, which has been shown to not be true in all cases (Parfenov et al. 2014; Hu et al. 2015; Gao et al. 2017).
Where Does HPV Integration Occur in HNSCCs?
Like in cervical cancers, there is no consensus sequence or 1 location where HPV integration is known to target in oral and oropharyngeal cancers. Integration breakpoints have been reported throughout the cellular genome. In HNSCC cell lines, Akagi et al. (2014) reported that HPV insertional breakpoints were found at regions of genomic amplification or deletion and demonstrated an association of insertional breakpoints with structural variation, including chromosomal translocations, deletions/insertions, and rearrangements. Parfenov and colleagues (2014) analyzed the genomic landscape of the 35 HPV-positive HNSCCs in The Cancer Genome Atlas (TCGA), including both OPSCCs and OCSCCs, by WGS and found over 100 integration sites in 25 of the tumors. Integration into a known gene was seen in 54% of the events, and 17% integrated within 20 kb of a gene. Walline et al. (2017) investigated 9 HNSCC cell lines by DIPS-PCR and found integration in all cell lines throughout the cellular genome, 8 of which had integration into cancer-related genes.
What Is the Mechanism of Integration?
The exact mechanism of HPV integration into the host genome is not known. In most models, both the viral and cellular genomes undergo breakage, allowing for fusion between the two. Some groups assert that fusion occurs as a result of cellular repair mechanisms, including nonhomologous end joining and homologous recombination (Senapati et al. 2016). However, others have criticized these proposed mechanisms because small numbers of breakpoints are seen even when many copies of HPV are present, which argues against random breakpoints (Akagi et al. 2014). Akagi et al. (2014) developed a looping model for focal genomic instability to explain the genomic structural variations seen in HNSCC cell lines using a chromosomal mapping technique to determine the DNA structure surrounding integration sites. In this model, both the host and viral genomes are nicked, the viral genome is inserted, and a circular piece of DNA containing both is transiently formed, resulting in rolling circle amplification. This amplification leads to concatemer formation characterized by amplified segments of a genomic sequence flanked by HPV segments. This is consistent with reports from patient tumors with focal copy number elevation at sites of HPV integration (Parfenov et al. 2014; Cancer Genome Atlas Network 2015).
How Does Integration Promote Oncogenesis in HNSCC?
Integration has been thought to promote oncogenesis through the dysregulation of the oncoproteins E6 and E7, resulting in increased cellular proliferation and genetic instability (Jeon and Lambert 1995). Dysregulation of E6 and E7 gives the cells a selective growth advantage and allows for oncogenic progression. Multiple events have been described that result in dysregulation of E6/E7, including disruption of E2 or its binding sites, disruption of E1, formation of stable viral-host transcripts, or generation of a viral super-enhancer from repeats of regulatory elements (McBride and Warburton 2017). E2 is responsible for regulation of E6/E7, so disruption of the E2 gene or its binding sites allows for unregulated E6/E7 transcription. When E1 is disrupted, lack of replicative functions can induce DNA damage and growth arrest, promoting focal instability at the site of integration (Sakakibara et al. 2011). It has been shown that integration can generate hybrid E6/E7 viral-host fusion transcripts, which are often more stable than viral E6/E7 transcripts (Jeon and Lambert 1995). Dooley et al. (2016) recently showed that a Brd4-dependent super-enhancer, capable of driving expression of viral E6/E7, can be generated from tandem integrated repeats of HPV16 DNA. However, both Parfenov et al. (2014) and Olthof et al. (2014) reported that there are tumors with HPV integration that do not have E2 disruption leading to increased E6/E7. Olthof et al. (2014) reported that there was no significant difference in E2, E6, or E7 levels between integrated versus nonintegrated tumors. This suggests that increased E6/E7 is not always the main driver of oncogenesis.
What Is the Effect of Integration on Cellular Genes?
Integration has traditionally been thought of as promoting oncogenesis through sustained expression of E6 and E7. However, integration has more recently been shown to have effects on cellular gene expression, which may represent an additional oncogenic mechanism in the development of HNSCC (Table). Parfenov et al. (2014) saw increases in somatic DNA copy number of the integrated region and reported that gene disruption occurs by integration through several key mechanisms: tumor suppressor loss of function, enhanced oncogene expression, and rearrangements that lead to altered gene expression.
Table.
Summary of Integration Events in Head and Neck Squamous Cell Carcinoma Cell Lines or Tumors.
| Author | Method | Sample | HPV Type | Tumor Site | HPV Segment(S) Involved | Human Genomic Region(s) Involved | Amplification | Deletion | Oncogene Over-expression | Trans-location | ↓Tran-scription | Alternate Transcripts | Intergenic Integration | Loss of Native Transcript | In-Frame Transcript across Junction Site | Fusion Tran-script |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parfenov et al. (2014) | WGS RNA-seq |
TCGA-BA-4077 | 16 | BOT | E1, E4, E5 | RAD51 | ✓ | ✓ | ||||||||
| TCGA-CV-6961 | 16 | TG | E1, L1 | ETS2 | ✓ | |||||||||||
| TCGA-CR-6482 | 16 | T | E1, L1 | Upstream of NR4A2 | ✓ | ✓ | ✓ | |||||||||
| TCGA-CN-4741 | 16 | AR | E6, E1 | Chrom 3, Chrom 13 | ✓ | ✓ | ✓ | |||||||||
| TCGA-CV-5443 | 16 | L | L1, LCR | PDL1 | ✓ | ✓ | ||||||||||
| Akagi et al. (2014) | WGS RNA-seq 5′/3′ RACE |
UD-SCC-2 | 16 | H | Multiple | DIAPH2 | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| UM-SCC-47 | 16 | TG | E2, E5, L1 | TP63 | ✓ | ✓ | ✓ | |||||||||
| UPCI:SCC090 | 16 | TG | Multiple | FOXE1 | ✓ | ✓ | ✓ | ✓ | ||||||||
| Multiple | PIM1 | ✓ | ✓ | ✓ | ✓ | |||||||||||
| UM-SCC-104 | 16 | OC | E2 | NOTCH1 | ✓ | ✓ | ✓ | |||||||||
| HMS001 | 16 | T | E1, L1 | Chrom 20 | ✓ | ✓ | ✓ | ✓ | ||||||||
| Tumor A | 18 | OC | Multiple | Chrom 5 | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||
| Tumor B | 16 | T | E6, E7 | Chrom 11q13, Chrom 8p11 |
✓ | ✓ | ||||||||||
| Walline et al. (2016) | DIPS-PCR RT-PCR |
Tumor 1733 | 16 | OP | E2 | Chrom 2p16 | ✓ | |||||||||
| Tumor 1769 | 16 | OP | E2 | Chrom 9q21 | ✓ | |||||||||||
| L1 | Chrom 16q11.2 | ✓ | ||||||||||||||
| L1 | Chrom 4q27 | ✓ | ||||||||||||||
| Tumor 1804 | 16 | OP | E1 | Chrom 6q16 | ✓ | |||||||||||
| L2 | Chrom 10p11.1 | ✓ | ||||||||||||||
| E2, E5 | Chrom 16q11.2 | ✓ | ||||||||||||||
| Tumor 1971 | 16 | OP | E1 | Chrom 7p22.3 | ✓ | |||||||||||
| L2 | Chrom 4p16.3 | ✓ | ||||||||||||||
| L1 | TP63 | X | ✓ | X | ||||||||||||
| Tumor 2148 | 16 | OP | L2 | Chrom 7p22 | ✓ | |||||||||||
| Tumor 0732 | 16 | OP | E2 | Chrom 10p11.1 | ✓ | |||||||||||
| L2 | TNFRSF13B | X | ✓ | X | ||||||||||||
| Tumor 0843 | 16 | OP | L2 | SCN2A | ✓ | ✓ | X | ✓ | ||||||||
| Tumor 1040 | 16 | OP | L2 | SH2B1 | X | ✓ | X | |||||||||
| L1 | Chrom 10p11.1 | ✓ | ||||||||||||||
| Tumor 2049 | 16 | OP | E1 | UBE2V2 | ✓ | X | ✓ | ✓ | ||||||||
| E1 | SMOC1 | ✓ | ✓ | X | ✓ | |||||||||||
| Tumor 2238 | 16 | OP | E2 | SEMA6A | X | ✓ | X | X | ||||||||
| E2, L1/L2 | NFIA | ✓ | ✓ | X | X | |||||||||||
| Walline et al. (2017) | DIPS-PCR RT-PCR |
UM-SCC-105 | 18 | L | L1 | Chrom 8q12.3, Chrom 4p15.33 | ✓ | ✓ | ||||||||
| L1 | Chrom 17q12 | ✓ | ||||||||||||||
| UM-SCC-104 | 16 | OC | E1 | DCC | ✓ | X | X | |||||||||
| UM-SCC-47 | 16 | TG | E2 | TP63 | X | ✓ | ✓ | |||||||||
| UD-SCC-2 | 16 | H | E1 | JAK1 | ✓ | X | X | |||||||||
| UPCI:SCC152 | 16 | OP | E1 | ETV6 | X | ✓ | X | |||||||||
| LCR | ATR | ✓ | X | X | ||||||||||||
| UPCI:SCC090 | 16 | OP | E1 | ETV6 | X | X | X | |||||||||
| UPCI:SCC154 | 16 | TG | E1 | PGR | ✓ | X | X | |||||||||
| E2 | PTPRN2 | X | ✓ | X | ||||||||||||
| E2 | TMEM237 | X | ✓ | X | ||||||||||||
| VU-SCC-147 | 16 | FOM/AR | E2 | TERT | X | ✓ | X |
For each sample reported, sample information is listed (ID, HPV type, tumor site). HPV segment(s) and human genomic region(s) involved in each integration events are listed. Oncogene overexpression refers to cellular oncogene expression. For each integration event, the effect of the event is checked in the far-right columns. ✓ = present, X = not present. If the box is empty, the information was not available.
AR, alveolar ridge; BOT, base of tongue; DIPS-PCR, detection of integrated papillomavirus sequences–polymerase chain reaction; FOM, floor of mouth; H, hypopharynx; HPV, human papillomavirus; L, larynx; OC, oral cavity; OP, oropharynx; RACE, Rapid Amplification of cDNA Ends; RT-PCR, reverse transcriptase polymerase chain reaction; T, tonsil; TG, tongue; WGS, whole-genome sequencing.
Loss of function of a tumor suppressor occurs when HPV integration into a gene results in deletion of gene regions and generates truncated transcripts, as well as host-viral fusion transcripts. Parfenov et al. (2014) reported integration into RAD51, resulting in a 28-fold amplification extrachromosomally, leading to alternate transcripts being generated and likely nonfunctional RAD51 protein. They also reported integration into ETS2, which led to deletion of exons 7 and 8. The overall expression of the gene was unaffected, but transcription of exons 7 and 8 was decreased, likely leading to a truncated protein.
HPV integration upstream of an oncogene can lead to oncogene overexpression via amplification of the nearby downstream region, leading to elevated transcripts. Parfenov et al. (2014) reported viral integration upstream of NR4A2, leading to a 250-fold amplification of the downstream region and subsequent overexpression of NR4A2. NR4A2 is a transcription factor that is overexpressed in a wide variety of human cancers (Safe et al. 2014). Parfenov et al. also reported interchromosomal translocation of chromosomes 3 and 13, which caused overexpression of key oncogenes KLF5, TP63, and TPRG1.
Walline et al. (2017) characterized integration sites of 8 HPV-positive HNSCC cell lines (7 HPV16 and 1 HPV18) by DIPS-PCR. Integration into cancer-related genes was detected in all of the HPV16 cell lines. The HPV18 cell line, UM-SCC-105, had 2 integration events, but both were intergenic. In UM-SCC-104, viral integration of HPV16 E1 into the tumor suppressor DCC was detected. When the transcripts of the DCC gene were interrogated, no transcripts were generated. This demonstrates an example of viral integration leading to disruption of a tumor suppressor, potentially providing a growth advantage for those cells. In UM-SCC-47, integration into TP63 resulted in the generation of a hybrid viral-host fusion transcript between HPV16 E2 and exon 14 of TP63, which resulted in a truncated ΔNTP63 protein as shown by Western blot. The other cell lines did not exhibit viral-host fusion transcripts, potentially due to integration in frame into introns that were subsequently spliced out.
Akagi et al. (2014) investigated whether the rearrangements resulting from integration generated cell-virus fusion transcripts and altered cellular gene expression. In all 10 HNSCC cell lines analyzed and in 1 primary tumor, they found virus-host fusion transcript expression, which frequently confirmed the rearrangements described by WGS. They also reported multiple examples of gene disruption at sites of integration. In UD-SCC-2, HPV integration led to deletions and rearrangements of the segment of DIAPH2, which resulted in viral-fusion transcripts but no native transcripts or functional protein. In UM-SCC-47, they reported aberrant TP63 expression due to HPV integration-mediated amplification, leading to viral-host transcripts and a truncated TP63 protein. They saw additional examples of gene disruption, including amplification of the oncogenes FOXE1 and PIM1 in UPCI:SCC090 cells.
However, Olthof et al. (2014) examined patient tumors and saw no significant effect of integration on gene expression, nor were messenger RNA (mRNA) levels of disrupted genes significantly different. Even when HPV was integrated directly into a gene, the mRNA expression levels were not significantly different from a nondisrupted gene elsewhere in the genome. Either there are other expressed gene copies present that allow overall expression levels to be unchanged, or viral integration did not deregulate genes as assessed by their method.
Deregulation of miRNAs in HPV-positive HNSCCs could result from HPV integration near miRNA sites as has been shown in cervical cancer (Schmitz et al. 2012; Hu et al. 2015) and HNSCC cell lines (Wald et al. 2011). HPV-positive and HPV-negative HNSCCs have distinct miRNA expression patterns, and miRNA subsets were significantly associated with overall survival, disease-free survival, and distant metastasis in HPV-positive HNSCCs (Lajer et al. 2012; Hui et al. 2013). Hui et al. (2013) reported 128 miRNAs that were differentially expressed between tumor and normal tissue in OPSCCs and speculated that integration of HPV into the genome near these miRNAs contribute to their deregulation. Wald et al. (2011) reported a subset of miRNAs that had altered expression in HPV16-positive HNSCC cell lines compared to both HPV-negative HNSCC cell lines and immortalized normal keratinocytes. The HPV16-positive cell lines used in this study all have been reported to contain integrated HPV, suggesting a possible role of integration on the deregulation of miRNAs.
How Does Integration Affect Viral Gene Expression?
Many studies investigating HPV integration report breakpoints throughout the viral genome, with an increased incidence in E1 (Parfenov et al. 2014). The effects of integration on viral gene expression are still not entirely known. Akagi et al. (2014) reported that loss of viral segments upon integration or rearrangement contributes to nonuniform coverage of the viral genome when analyzed by RNA-seq. Despite this, viral fragments containing E6 and E7 were retained, and all samples had strong E6/E7 expression. Walline et al. (2017) also reported enhanced E6/E7 expression upon integration, particularly the splice isoform E6*I, and reduced E1/E2 expression in integration-positive cell lines. E6* transcripts are thought to be drivers of tumor development, so the expression of this isoform at the expense of full-length E6 is significant. Despite many reports of enhanced oncoprotein expression, Parfenov et al. (2014) reported that this does not occur in all integration-positive tumors. Although integration-negative tumors tended to have higher E2/E5 expression levels and lower E6/E7 than integration-positive tumors, this was not always the case. They reported no correlation between the presence of integration within specific HPV genes and their expression level. These results further support the view that HPV plays a larger role in oncogenesis beyond viral oncoprotein expression and subsequent disruption of the p53 and Rb axes.
Can Integration Site Be Clinically Predictive?
After observing integration of HPV16 into cancer-related genes in 7 HNSCC cell lines, 6 established from patients who had progressed, our group investigated integration events in HPV16-positive oropharynx tumors (Walline et al. 2016). We hypothesized that responsive tumors are driven primarily by viral oncoprotein expression, but recurrent tumors harbor additional carcinogenic events as a result of HPV integration into cancer-related genes. We expected to see integration into cancer-related genes leading to an alteration in gene expression and potential generation of fusion transcripts in tumors that later recurred but no integration or integration only into cellular intergenic regions in responsive tumors. The integration events in HNSCC tumors from 10 patients were characterized; 5 were responsive after therapy, and 5 recurred after treatment. Our results supported our hypothesis; tumors from responsive patients had integration events into mainly intergenic loci, and tumors from recurrent patients had integration events into cancer-related genes. Only 1 of the responsive tumors had an integration event into a gene; HPV L1 was found integrated into intron 4 of TP63 on chromosome 3q28. However, when transcript analysis of the region was performed, no fusion transcript was produced, and transcripts across exons 4 and 5, spanning the integration site in intron 4, were produced and were in frame. This suggests that TP63 may not be disrupted by this integration or that at least 1 intact copy of TP63 remains unaltered. All other responsive tumors had only intergenic integration events.
In contrast, all 5 of the tumors from recurrent patients had at least 1 integration event into an intron of a cancer-related gene. There were 7 total gene integration events detected in the 5 tumors, and upon transcript analysis, 4 of the events led to gene disruption (Fig. 3). The other 3 events did not produce fusion transcripts and retained intact, in-frame cellular gene exon-exon transcripts spanning each respective intronic integration site as well as exon-exon transcripts downstream of the integration site. In tumor 2049 from a recurrent patient, viral integration into SMOC1 led to generation of a SMOC1-HPV E1 fusion transcript. The result of this fusion transcript was inactivation of the gene, demonstrated by the absence of intact cellular exon-exon transcripts surrounding the integration site (Fig. 3A). Tumor 0843 had integration into SCN2A; transcript analysis revealed a complex rearrangement that produced a fusion transcript containing SCN2A, HPV L1, and fragments of chromosomes 2q34 and 1q32. This integration event failed to yield intact SCN2A exon-exon transcripts downstream of the integration site, suggesting gene disruption (Fig. 3B). A third tumor, 2238, had 2 integration events that each resulted in gene disruption. In this tumor, HPV L1 was integrated into NF1A and E2 integrated into SEMA6D. Neither of these integration events produced fusion transcripts, but disruption of both genes was evident from the lack of cellular exon-exon transcripts spanning the integration sites. This demonstrates that generation of viral/cellular fusion transcripts is not required for cellular gene disruption to occur (Fig. 3C, D). All of the tumors, including those from responsive patients, displayed strong E6/E7 gene expression; E6*I was the highest expressed viral gene in 8 of the 10 tumors (Walline et al. 2016). Taken together, these results suggest that there are multiple mechanisms leading to integration-mediated cellular gene disruption and that viral integration events can alter gene expression in the host cell. Furthermore, the consequence of these alterations in cellular gene expression may mediate additional carcinogenic mechanisms, leading to a more aggressive tumor phenotype (Fig. 4). This study, although small, highlights the potential for site of HPV integration to explain clinical outcomes and warrants further investigation.
Figure 3.
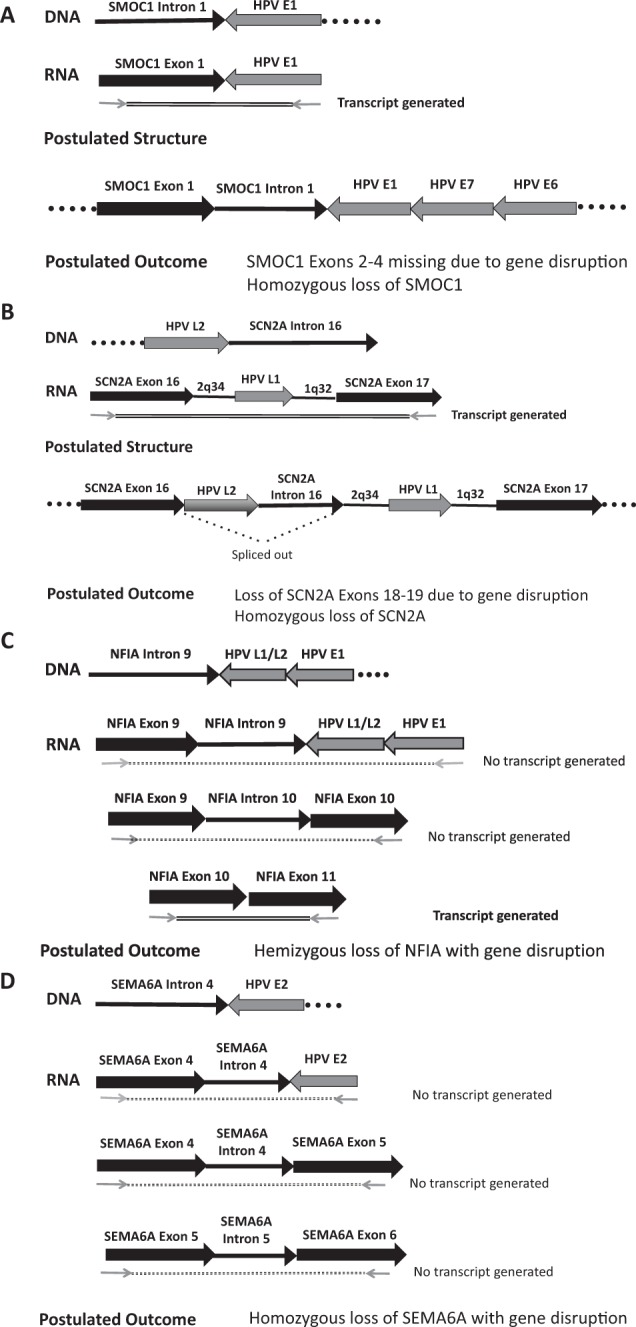
Transcript analysis of recurrent tumors that revealed gene disruption following viral integration. (A) Tumor 2049. Detection of integrated papillomavirus sequences–polymerase chain reaction (DIPS-PCR) identified a viral integration event linking human papillomavirus (HPV) E1 to SMOC1 intron 1 in the DNA. Reverse transcriptase PCR converted the RNA to complementary DNA (cDNA), and primers spanning the integration site produced a viral-host fusion transcript containing E1 and SMOC1. However, primers from SMOC1 exon 1 to exon 2 and from exon 3 to exon 4 produced no native transcripts of exon junctions spanning the site or of exons downstream of the site. We postulate that the structure includes SMOC1 exon 1 and intron 1 fused to HPV E1-E7-E6, including the viral promoter, with a loss or disruption of SMOC1 downstream exons. Since no normal transcripts were produced, we conclude that both copies of SMOC1 were lost in this tumor. (B) Tumor 0843. A fusion of HPV L2 into SCN2A intron 16 was found at the DNA level. Upon PCR testing of the cDNA using primers spanning SCN2A exon 16_exon 17, a fusion transcript containing SCN2A, HPV L1, and fragments of chromosome 2q32 and 1q32 was produced. No intact SCN2A exon-exon transcripts downstream of the integration site were produced. We postulate that HPV L2 and SCN2A intron 16 are spliced out of the structure shown above, leading to gene disruption that was accompanied by loss of the other copy of SCN2A. (C) Tumor 2238, integration event 1, NFIA intron 9 fused to HPVL1/L2 and HPVE1. This event failed to yield viral-host fusion transcripts when a primer from exon 9 was paired with an HPV E1 primer. Similarly, PCR testing using primers spanning exons 9 and exon 10 failed to produce a transcript. However, downstream exons 10 to 11 showed in-frame transcripts. It is unclear whether these transcripts come from the affected chromosome or the other copy but suggests that at least 1 copy of the gene is intact, leading us to suspect hemizygous loss of NFIA. (D) Tumor 2238, integration event 2, SEMA6A intron 4 fused to HPVE2. This integration event failed to yield viral-host fusion transcripts by PCR (SEMA6A exon 4 to E2), as well as cellular exon-exon transcripts spanning the integration site (exon 4 to exon 5). In addition, the SEMA6D-HPV E2 integration event also led to lack of cellular exon-exon transcripts downstream of the integration site (exon 5 to exon 6). Because of this, we postulate that gene disruption occurred by homozygous loss of SEMA6D.
Figure 4.

Model of role of human papillomavirus (HPV) in carcinogenesis. Our model predicts that tumors containing episomal HPV or HPV integrated in intergenic regions are primarily driven by E6/E7 oncogene expression, which are responsive to current therapies. However, tumors with HPV integration into cancer-related genes (or tumors that have integration events that alter expression of microRNAs [Wald et al. 2011]) will be also driven by altered cellular gene expression. These changes in gene expression can have further oncogenic effects, and therefore the tumor will be more aggressive and nonresponsive to therapy. These tumors are still driven by E6/E7 proteins, due to E1/E2 disruption, unregulated expression of E6/E7, and expression of E6* alternate transcripts linked to increased E7 protein expression. In addition, these tumors often have loss of disrupted genes or overexpressed fusion transcripts that contribute to oncogenesis by mechanisms that are still poorly understood.
Conclusion
There is evidence that HPV integration is implicated in oral/oropharyngeal cancer oncogenesis, but its exact role remains largely unknown. A variety of mechanisms of integration and their effects on both the viral and cellular genome, as well as likely outcomes, are summarized in Figure 5. Integration of HPV into the host genome may lead to increased expression of viral oncoproteins, and recent data suggest that viral integration contributes to alterations in host cell gene expression and generation of viral-host fusion transcripts. It is unclear whether integration is required for oncogenesis or if it is consistently associated with a more aggressive, treatment-resistant phenotype. Our work has shown that tumors from patients with recurrent disease are more likely to exhibit integration into cancer-related cellular genes than those from patients who respond to treatment, which contain integration events primarily at intergenic sites. Further investigation of integration-mediated alteration of cellular gene expression, production of viral-host fusion transcripts, and the subsequent effects that contribute to oncogenesis and tumor progression is needed, as are correlative studies on the outcome and survival of patients based on HPV integration status and site. This will establish the feasibility of developing viral integration evaluation as a clinically relevant predictive or prognostic indicator.
Figure 5.

Human papillomavirus (HPV) integration into the host genome: effects and potential outcomes. Integration events have been reported to occur as a result of a stochastic process, inserting into readily available regions of the genome or via directed integration into fragile sites, highly transcriptionally active regions with open chromatin, regions of microhomology between HPV and human sequence, or near microRNAs (miRNAs). Integration has been reported to cause a variety of effects on both the HPV and cellular genomes. This includes E1/E2 disruption, expression of alternate E6 transcripts, or creation of viral super-enhancers that drive oncoprotein transcription. Integration can lead to chromosome instability, gene disruption, or regional amplifications within the cellular genome. These effects serve as possible alternate mechanisms increasing HPV-mediated carcinogenesis and largely depend on site of integration, either within an intergenic region or near/within genes. The integration event could have no effect on cellular gene expression if it occurs in an intergenic, nonregulatory region of the genome, although there are reports of integration affecting miRNA expression, which can then alter expression of other genes. Other integration events can lead to E6/E7 viral oncogene overexpression, expression of viral-host fusion transcripts, or gene disruption, including tumor suppressor loss of function and cellular oncogene overexpression.
Author Contributions
L.M. Pinatti, T.E. Carey, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; H.M. Walline, contributed to conception, design, data acquisition, analysis, and interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
Studies carried out by the authors were supported by R01 CA 194538 and P50 CA 097248. L. Pinatti was supported by the Cancer Biology Training Grant NIH-NCI Training Grant T32-CA009676.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, Rocco JW, Teknos TN, Kumar B, Wangsa D, et al. 2014. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 24(2):185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodelon C, Untereiner ME, Machiela MJ, Vinokurova S, Wentzensen N. 2016. Genomic characterization of viral integration sites in HPV-related cancers. Int J Cancer. 139(9):2001–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch FX, Lorincz A, Munoz N, Meijer CJ, Shah KV. 2002. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol. 55(4):244–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer SN, Wazer DE, Band V. 1996. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 56(20):4620–4624. [PubMed] [Google Scholar]
- Brouwer AF, Eisenberg MC, Meza R. 2016. Age effects and temporal trends in HPV-related and HPV-unrelated oral cancer in the United States: a multistage carcinogenesis modeling analysis. PLoS One. 11(3):e0151098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. 2015. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 517(7536):576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. 2015. Sexually transmitted diseases treatment guidelines: HPV-associated cancers and precancers [accessed 2017 Mar 11]. https://www.cdc.gov/std/tg2015/hpv-cancer.htm.
- Chaturvedi AK, Engels EA, Anderson WF, Gillison ML. 2008. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the united states. J Clin Oncol. 26(4):612–619. [DOI] [PubMed] [Google Scholar]
- Dooley KE, Warburton A, McBride AA. 2016. Tandemly integrated HPV16 can form a brd4-dependent super-enhancer-like element that drives transcription of viral oncogenes. MBio. 7(5). pii: e01446–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duray A, Descamps G, Decaestecker C, Remmelink M, Sirtaine N, Lechien J, Ernoux-Neufcoeur P, Bletard N, Somja J, Depuydt CE, et al. 2012. Human papillomavirus DNA strongly correlates with a poorer prognosis in oral cavity carcinoma. Laryngoscope. 122(7):1558–1565. [DOI] [PubMed] [Google Scholar]
- Duray A, Lacremans D, Demoulin S, Delvenne P, Saussez S. 2014. Prognosis of HPV-positive head and neck cancers: implication of smoking and immunosuppression. Adv Cell Mol Otolaryng. 2(1) 25717 http://www.tandfonline.com/doi/abs/10.3402/acmo.v2.25717 [Google Scholar]
- Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, Forastiere A, Gillison ML. 2008. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 100(4):261–269. [DOI] [PubMed] [Google Scholar]
- Fakhry C, Westra WH, Wang SJ, van Zante A, Zhang YH, Rettig E, Yin LDX, Ryan WR, Ha PK, Wentz A, et al. 2017. The prognostic role of sex, race, and human papillomavirus in oropharyngeal and nonoropharyngeal head and neck squamous cell cancer. Cancer. 123(9):1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng FY, Kim HM, Lyden TH, Haxer MJ, Worden FP, Feng M, Moyer JS, Prince ME, Carey TE, Wolf GT, et al. 2010. Intensity-modulated chemoradiotherapy aiming to reduce dysphagia in patients with oropharyngeal cancer: clinical and functional results. J Clin Oncol. 28(16):2732–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferber MJ, Thorland EC, Brink AA, Rapp AK, Phillips LA, McGovern R, Gostout BS, Cheung TH, Chung TK, Fu WY, et al. 2003. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene. 22(46):7233–7242. [DOI] [PubMed] [Google Scholar]
- Gao G, Johnson SH, Vasmatzis G, Pauley CE, Tombers NM, Kasperbauer JL, Smith DI. 2017. Common fragile sites (cfs) and extremely large cfs genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes Chromosomes Cancer. 56(1):59–74. [DOI] [PubMed] [Google Scholar]
- Gillison ML, Broutian T, Pickard RK, Tong ZY, Xiao W, Kahle L, Graubard BI, Chaturvedi AK. 2012. Prevalence of oral HPV infection in the United States, 2009-2010. JAMA. 307(7):693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, et al. 2000. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 92(9):709–720. [DOI] [PubMed] [Google Scholar]
- Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, Ding W, Yu L, Wang X, Wang L, et al. 2015. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet. 47(2):158–163. [DOI] [PubMed] [Google Scholar]
- Hui AB, Lin A, Xu W, Waldron L, Perez-Ordonez B, Weinreb I, Shi W, Bruce J, Huang SH, O’Sullivan B, et al. 2013. Potentially prognostic miRNAs in HPV-associated oropharyngeal carcinoma. Clin Cancer Res. 19(8):2154–2162. [DOI] [PubMed] [Google Scholar]
- Jemal A, Simard EP, Dorell C, Noone AM, Markowitz LE, Kohler B, Eheman C, Saraiya M, Bandi P, Saslow D, et al. 2013. Annual report to the nation on the status of cancer, 1975–2009, featuring the burden and trends in human papillomavirus (HPV)–associated cancers and HPV vaccination coverage levels. J Natl Cancer Inst. 105(3):175–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S, Lambert PF. 1995. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of e6 and e7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci USA. 92(5):1654–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaes R, Woerner SM, Ridder R, Wentzensen N, Duerst M, Schneider A, Lotz B, Melsheimer P, von Knebel Doeberitz M. 1999. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res. 59(24):6132–6136. [PubMed] [Google Scholar]
- Lajer CB, Garnaes E, Friis-Hansen L, Norrild B, Therkildsen MH, Glud M, Rossing M, Lajer H, Svane D, Skotte L, et al. 2012. The role of miRNAs in human papilloma virus (HPV)–associated cancers: bridging between HPV-related head and neck cancer and cervical cancer. Br J Cancer. 106(9):1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licitra L, Perrone F, Bossi P, Suardi S, Mariani L, Artusi R, Oggionni M, Rossini C, Cantu G, Squadrelli M, et al. 2006. High-risk human papillomavirus affects prognosis in patients with surgically treated oropharyngeal squamous cell carcinoma. J Clin Oncol. 24(36):5630–5636. [DOI] [PubMed] [Google Scholar]
- McBride AA, Warburton A. 2017. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 13(4):e1006211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. 2004. Mechanisms of human papillomavirus–induced oncogenesis. J Virol. 78(21):11451–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olthof NC, Speel EJ, Kolligs J, Haesevoets A, Henfling M, Ramaekers FC, Preuss SF, Drebber U, Wieland U, Silling S, et al. 2014. Comprehensive analysis of HPV16 integration in OSCC reveals no significant impact of physical status on viral oncogene and virally disrupted human gene expression. PLoS One. 9(2):e88718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, Lee S, Hadjipanayis AG, Ivanova EV, Wilkerson MD, et al. 2014. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci USA. 111(43):15544–15549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz IB, Cook N, Odom-Maryon T, Xie Y, Wilczynski SP. 1997. Human papillomavirus (HPV) in head and neck cancer: an association of HPV 16 with squamous cell carcinoma of Waldeyer’s tonsillar ring. Cancer. 79(3):595–604. [DOI] [PubMed] [Google Scholar]
- Ragin CC, Taioli E. 2007. Survival of squamous cell carcinoma of the head and neck in relation to human papillomavirus infection: review and meta-analysis. Int J Cancer. 121(8):1813–1820. [DOI] [PubMed] [Google Scholar]
- Safe S, Jin UH, Hedrick E, Reeder A, Lee SO. 2014. Minireview: role of orphan nuclear receptors in cancer and potential as drug targets. Mol Endocrinol. 28(2):157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara N, Mitra R, McBride AA. 2011. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol. 85(17):8981–8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathish N, Abraham P, Peedicayil A, Sridharan G, John S, Chandy G. 2004. Human papillomavirus 16 E6/E7 transcript and E2 gene status in patients with cervical neoplasia. Mol Diagn. 8(1):57–64. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus type-16 and type-18 promotes the degradation of p53. Cell. 63(6):1129–1136. [DOI] [PubMed] [Google Scholar]
- Schmitz M, Driesch C, Jansen L, Runnebaum IB, Durst M. 2012. Non-random integration of the HPV genome in cervical cancer. PLoS One. 7(6):e39632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senapati R, Senapati NN, Dwibedi B. 2016. Molecular mechanisms of HPV mediated neoplastic progression. Infect Agent Cancer. 11:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrjänen S. 2007. Human papillomaviruses in head and neck carcinomas. N Engl J Med. 356(19):1993–1995. [DOI] [PubMed] [Google Scholar]
- Talis AL, Huibregtse JM, Howley PM. 1998. The role of E6AP in the regulation of p53 protein levels in human papillomavirus (HPV)–positive and HPV-negative cells. J Biol Chem. 273(11):6439–6445. [DOI] [PubMed] [Google Scholar]
- Vinokurova S, Wentzensen N, Kraus I, Klaes R, Driesch C, Melsheimer P, Kisseljov F, Durst M, Schneider A, von Knebel Doeberitz M. 2008. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. 68(1):307–313. [DOI] [PubMed] [Google Scholar]
- Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Muñoz N. 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 189(1):12–19. [DOI] [PubMed] [Google Scholar]
- Wald AI, Hoskins EE, Wells SI, Ferris RL, Khan SA. 2011. Alteration of microRNA profiles in squamous cell carcinoma of the head and neck cell lines by human papillomavirus. Head Neck. 33(4):504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walline HM, Goudsmit CM, McHugh JB, Tang AL, Owen JH, Teh BT, McKean E, Glover TW, Graham MP, Prince ME, et al. 2017. Integration of high-risk human papillomavirus into cellular cancer-related genes in head and neck cancer cell lines. Head Neck. 39(5):840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walline HM, Komarck CM, McHugh JB, Bellile EL, Brenner JC, Prince ME, McKean EL, Chepeha DB, Wolf GT, Worden FP, et al. 2016. Genomic integration of high-risk HPV alters gene expression in oropharyngeal squamous cell carcinoma. Mol Cancer Res. 14(10):941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walline HM, Komarck CM, McHugh JB, Byrd SA, Spector ME, Hauff SJ, Graham MP, Belille E, Moyer JS, Prince ME, et al. 2013. High-risk human papillomavirus detection in oropharyngeal, nasopharyngeal, and, oral cavity cancers: comparison of multiple methods. JAMA Otolaryngol Head Neck Surg. 139(12):1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzensen N, Vinokurova S, von Knebel Doeberitz M. 2004. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 64(11):3878–3884. [DOI] [PubMed] [Google Scholar]
- Westra WH, Taube JM, Poeta ML, Begum S, Sidransky D, Koch WM. 2008. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin Cancer Res. 14(2):366–369. [DOI] [PubMed] [Google Scholar]
- Wiest T, Schwarz E, Enders C, Flechtenmacher C, Bosch FX. 2002. Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene. 21(10):1510–1517. [DOI] [PubMed] [Google Scholar]
- Worden FP, Kumar B, Lee JS, Wolf GT, Cordell KG, Taylor JM, Urba SG, Eisbruch A, Teknos TN, Chepeha DB, et al. 2008. Chemoselection as a strategy for organ preservation in advanced oropharynx cancer: response and survival positively associated with HPV16 copy number. J Clin Oncol. 26(19):3138–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegert C, Wentzensen N, Vinokurova S, Kisseljov F, Einenkel J, Hoeckel M, von Knebel Doeberitz M. 2003. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene. 22(25):3977–3984. [DOI] [PubMed] [Google Scholar]
- Ziemann F, Arenz A, Preising S, Wittekindt C, Klussmann JP, Engenhart-Cabillic R, Wittig A. 2015. Increased sensitivity of HPV-positive head and neck cancer cell lines to x-irradiation ± cisplatin due to decreased expression of E6 and E7 oncoproteins and enhanced apoptosis. Am J Cancer Res. 5(3):1017–1031. [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. 2009. Papillomaviruses in the causation of human cancers—a brief historical account. Virology. 384(2):260–265. [DOI] [PubMed] [Google Scholar]