

Abstract
Stimulator of interferon genes (STING) is an endoplasmic reticulum–resident membrane protein that mediates cytosolic pathogen DNA–induced innate immunity and inflammatory responses in host defenses. STING is activated by cyclic di-nucleotides and is then translocated to the Golgi apparatus, an event that triggers STING assembly with the downstream enzyme TANK-binding kinase 1 (TBK1). This assembly leads to the phosphorylation of the transcription factor interferon regulatory factor 3 (IRF3), which in turn induces expression of type-I interferon (IFN) and chemokine genes. STING also mediates inflammatory responses independently of IRF3, but these molecular pathways are largely unexplored. Here, we analyzed the RAW264.7 macrophage secretome to comprehensively identify proinflammatory factors released into the extracellular medium upon STING activation. In total, we identified 1299 proteins in macrophage culture supernatants, of which 23 were significantly increased after STING activation. These proteins included IRF3-dependent cytokines, as well as previously unknown targets of STING, such as the immune semaphorin SEMA4D/CD100, which possesses proinflammatory cytokine-like activities. Unlike for canonical cytokines, the expression of the SEMA4D gene was not up-regulated. Instead, upon STING activation, membrane-bound SEMA4D was cleaved into a soluble form, suggesting the presence of a post-translational shedding machinery. Importantly, the SEMA4D shedding was blocked by TMI-1, an inhibitor of the sheddase ADAM metallopeptidase domain 17 (ADAM17) but not by the TBK1 inhibitor BX795. These results suggest that STING activates ADAM17 and that this activation produces soluble proinflammatory SEMA4D independently of the TBK1/IRF3-mediated transcriptional pathway.
Keywords: ADAM, cell surface protein, inflammation, proteomics, post-translational modification (PTM), SEMA4D, STING
Introduction
The detection of cytosolic DNAs derived from various pathogens triggers innate immune and inflammatory responses that are essential for host defense (1). In contrast, endogenous self-DNA does not induce an immune response normally, because it is localized to the nucleus or mitochondria. However, inadequate accumulation of self-DNA in cytoplasm can also activate innate immunity (2). During animal development and tissue turnover, including definitive erythropoiesis, apoptotic cells and pyrenocytes are engulfed by macrophages (3, 4), and their DNA is degraded by the lysosomal enzyme DNase II. If this degradation machinery is impaired in macrophages, the accumulated apoptotic or pyrenocyte DNA leaks from the lysosome into the cytoplasm and triggers a strong inflammatory response, leading to severe anemia and polyarthritis (5–9). Of note, loss-of-function mutations in DNase II have been identified recently in patients with autoinflammation featuring severe neonatal anemia and deforming arthropathy (10). Understanding the immune response triggered by cytoplasmic DNA is an important issue in resolving inflammatory diseases.
STING2 is an endoplasmic reticulum (ER)–resident transmembrane protein that is essential for DNA-induced innate immune signaling (11). STING is activated by binding to cyclic di-nucleotides such as cGMP-AMP (cGAMP), which is produced as an intracellular second messenger when cGAMP synthase recognizes cytosolic DNA (12–15). Upon binding to cGAMP, STING causes its dimerization and translocation from the ER to the Golgi apparatus (16–18). After relocation, STING recruits a serine/threonine kinase, TBK1, leading to the phosphorylation of IRF3 and the up-regulation of type-I IFN and IFN-stimulated genes, including IFN-β and CXCL10 (19, 20).
IFN-β production via the STING–TBK1–IRF3 axis is crucial for the development of anemia in DNase II–deficient mice (21). However, another phenotype, polyarthritis, is dependent on STING but not on IRF3 (21). Similarly, systemic inflammation characterized by interstitial lung disease, skin lesions, and hypercytokinemia in patients with activating mutations of the STING gene has been shown to be IRF3-independent using a mouse model (22). These observations suggest that an additional signaling pathway or pathways are induced downstream of STING. Importantly, the lack of proinflammatory cytokine genes such as TNFα, IL-6, and IL-1β has been shown to inhibit arthritis in DNase II–null mice (8). Although STING activates another transcriptional factor, NF-κB, which induces proinflammatory cytokine gene expression, this activation is rather weak (11, 23, 24). Thus, it is unclear whether NF-κB–mediated gene activation is sufficient for a STING-mediated inflammatory response.
Here, we performed an unbiased comprehensive secretome analysis and determined that the activation of STING promoted the cleavage of the plasma membrane protein SEMA4D to release the inflammatory soluble form without its gene induction. We also found that TNFα protein was dramatically increased in the culture supernatant of STING-activated cells, although its mRNA expression was moderately induced in this condition. The production of soluble SEMA4D, as well as TNFα, was completely blocked by an inhibitor of the ADAM17 sheddase. On the other hand, the treatment of cycloheximide (CHX) or TBK1 inhibitor did not abrogate the shedding of SEMA4D. These results suggest that STING activates ADAM17-mediated post-translational shedding machinery. Because soluble SEMA4D is known to activate inflammatory cytokine production, the STING–ADAM17–SEMA4D axis may be required for optimal inflammatory response.
Results
Identification of SEMA4D as a novel protein released by STING agonist
To determine the inflammatory factors released from STING-activated cells, we prepared culture supernatants from Raw264.7 murine macrophage cells treated with or without DMXAA, a cell-permeable synthetic STING agonist. Like a natural ligand, DMXAA binds directly to murine STING and causes its conformational activation (15). The DMXAA-treated cells were cultured for 4 h under serum-free conditions to prevent contamination of serum-derived abundant proteins, but no significant cell death was observed, at least during the 4-h incubation period (data not shown). After collecting proteins from the conditioned medium, we performed an unbiased proteomic analysis by “shotgun” LC-MS/MS (Fig. 1A). In total, we identified 1299 proteins, of which 23 proteins were significantly increased (q < 0.05, -fold increase > 4) by DMXAA stimulation (Fig. 1B, Table 1, and Table S1). These 23 significantly increased proteins included IRF3-dependent cytokines such as IFN-β and CXCL10, and the most increased protein was TNFα (about 150-fold increase). We also identified novel downstream targets of STING including SEMA4D, LRPAP1, and Fam3C. To confirm these results, we performed targeted LC-MS/MS analysis using parallel reaction monitoring (PRM), which enables highly specific and accurate protein quantification of multiple proteins simultaneously (25). As expected, the protein levels of SEMA4D, LRPAP1, and Fam3C in culture supernatants were increased about 3–6-fold by DMXAA (Fig. 1C and Table S2). DMXAA did not induce the release of another class 4 semaphorin member, SEMA4B, or of chemokine CCL9 (Fig. 1C and Table S2), confirming that specific sets of proteins were released by stimulation with the STING agonist. Among the 23 identified proteins, SEMA4D has been well-characterized as a proinflammatory protein (26). Notably, the number of peptide spectrum matches of SEMA4D was relatively high among them (Table 1), suggesting that SEMA4D is an abundant protein in the culture supernatant of STING-activated cells. Thus, the activation of STING causes the release of the inflammatory factor SEMA4D in addition to known cytokines.
Figure 1.

Secretome analysis of DMXAA-treated cells. A, schematic diagram of the label-free quantitative proteomics workflow. Raw264.7 cells were cultured in serum-free DMEM containing DMSO or 100 μg/ml DMXAA for 4 h, and the conditioned medium was collected. The precipitated proteins were digested directly with trypsin/Lys-C. Three biological replicates for each sample were prepared individually and analyzed by LC-MS/MS. Label-free quantification was performed using Proteome Discoverer 2.2 software. B, volcano plot showing differential protein profiles in DMSO- and DMXAA-treated cell culture medium. The x axis indicates log2 -fold change upon DMXAA stimulation. The y axis indicates negative log10 of the t test q value. Proteins increased by more than 4-fold with high significance (q < 0.05) are shown by blue circles. C, an accurate amount of protein in the samples used for proteomics was measured by target LC-MS/MS using the PRM method. The relative abundance was calculated as compared with DMSO control. Scatter plots show the individual data, and bar graphs indicate mean ± S.D. (n = 3).
Table 1.
Proteins increased by DMXAA stimulation on secretome analysis
Proteins increased by more than 4-fold with high significance (q < 0.05) are listed.
| Gene symbol | Description | No. of PSMsa | No. of peptides | % Coverage | -Fold increase DMXAA/DMSO |
|---|---|---|---|---|---|
| Ccl4 | C-C motif chemokine 4 | 914 | 6 | 55 | 80.1 |
| Tnf | Tumor necrosis factor | 424 | 13 | 58 | 141.9 |
| Glg1 | Golgi apparatus protein 1 | 264 | 42 | 45 | 9.4 |
| Sema4d | Semaphorin 4D | 218 | 20 | 31 | 4.3 |
| Sdc4 | Syndecan-4 | 168 | 5 | 22 | 22.3 |
| Mthfd1 | C-1-tetrahydrofolate synthase, cytoplasmic | 140 | 28 | 41 | 5.0 |
| H2-T23 | H-2 class I histocompatibility antigen, d-37 α-chain | 55 | 3 | 19 | 5.6 |
| Cxcl10 | C-X-C motif chemokine 10 | 45 | 5 | 34 | 94.2 |
| Ccl2 | C-C motif chemokine 2 | 45 | 4 | 21 | 8.4 |
| Fam3c | Protein FAM3C | 36 | 5 | 26 | 6.6 |
| Hist1h1d | Histone H1.3 | 25 | 1 | 26 | 5.0 |
| Tor3a | Torsin 3A | 23 | 5 | 18 | 6.0 |
| Ifnb1 | Interferon β | 18 | 4 | 18 | 28.0 |
| Lrpap1 | α-2-Macroglobulin receptor-associated protein | 14 | 4 | 12 | 5.9 |
| Cxcl2 | C-X-C motif chemokine 2 | 13 | 2 | 36 | 18.9 |
| Ccl5 | C-C motif chemokine 5 | 7 | 2 | 21 | 41.9 |
| Icam2 | Intercellular adhesion molecule 2 | 7 | 2 | 6 | 4.8 |
| Golim4 | Golgi integral membrane protein 4 | 6 | 4 | 10 | 4.2 |
| Acvr1 | Activin receptor type-1 | 6 | 1 | 2 | 6.4 |
| Lif | Leukemia inhibitory factor | 5 | 1 | 5 | 8.3 |
| Ube2d3 | Ubiquitin-conjugating enzyme E2 D3 | 4 | 2 | 29 | 19.7 |
| Qsox2 | Sulfhydryl oxidase 2 | 4 | 3 | 6 | 4.4 |
| Lrig2 | Leucine-rich repeats and immunoglobulin-like domains protein 2 | 1 | 1 | 2 | 4.7 |
a PSM, peptide spectrum match.
STING induces SEMA4D shedding independent of gene expression
To further confirm the STING-mediated release of SEMA4D, we performed Western blot analysis of culture supernatant using an antibody that recognizes the extracellular domain of SEMA4D. As shown in Fig. 2A, we detected SEMA4D release at 2 and 4 h after DMXAA stimulation. To determine whether STING is essential for DMXAA-induced release of SEMA4D, Raw264.7 cells lacking STING were generated using the CRISPR/Cas9 system. We confirmed again that SEMA4D was released along with TBK1 activation in DMXAA-stimulated wildtype (WT) cells (Fig. 2B). In contrast, neither SEMA4D release nor TBK1 activation was observed in STING-deficient cells; this phenotype was rescued by ectopic expression of STING (Fig. 2B). Because STING activates the transcription of type-I IFN and inflammatory cytokine genes, we checked the induction of the SEMA4D gene after DMXAA stimulation. Real-time PCR analysis showed that DMXAA did not induce the expression of SEMA4D mRNA (Fig. 3A). As a control, we examined the expression levels of the canonical cytokine genes, and the IFN-β and CXCL10 mRNA were dramatically increased, about 500–1500-fold, after DMXAA stimulation (Fig. 3A). The expression of TNFα mRNA was also up-regulated but only by an 8-fold increase, although the protein level in the cell medium was increased about 150-fold, which is comparable to IFN-β and CXCL10 (Figs. 1C and 3A). These observations prompted us to speculate that STING activates not only transcription but also post-translational machinery to release SEMA4D and TNFα. To confirm this possibility, the cells were treated with CHX to inhibit de novo protein synthesis and then stimulated with DMXAA. As expected, the production of CXCL10 was blocked by treatment with CHX in a dose-dependent manner, whereas SEMA4D was still released even in the presence of high-dose CHX (Fig. 3B). Because SEMA4D is a plasma membrane protein and its soluble ectodomain is generated by proteolytic cleavage (26), we tested for whether the STING agonist promotes shedding of SEMA4D. The total cell lysates and culture supernatants were prepared at the same time, and the cleavage was analyzed by Western blotting. As shown in Fig. 3C, the full-length membrane form of SEMA4D was reduced by DMXAA treatment in the cell lysates, concomitant with an increase of the cleaved form of SEMA4D in the culture supernatants. Although an intracellular cleaved form was observed in the cell lysates, its amount did not change after DMXAA stimulation. These results indicated that STING activates the post-translational processing of SEMA4D on the cell surface to release the soluble ectodomain.
Figure 2.

STING-dependent release of SEMA4D. A, Raw264.7 cells were cultured in 0.1% FCS/DMEM containing 100 μg/ml DMXAA for 0, 15, 30, 60, 120, and 240 min. Soluble SEMA4D in the culture supernatants was detected by Western blotting with anti-SEMA4D antibody. B, WT, STING−/− (SKO), and STING−/− Raw264.7 cells ectopically expressing STING-FLAG (SKO/STING-Flag) were cultured in 0.1% FCS/DMEM containing DMSO (−) or 100 μg/ml DMXAA for 4 h. The culture supernatants and the cell lysates were analyzed by Western blotting with anti-SEMA4D, anti-phospho-TBK1, anti-STING, or anti-α-tubulin.
Figure 3.

Activation of STING induces shedding of SEMA4D in a transcription-independent manner. A and C, Raw264.7 cells were cultured in 0.1% FCS/DMEM containing DMSO (−) or 100 μg/ml DMXAA for 4 h. In A, the mRNA levels of the indicated genes were determined by real-time PCR. The data are normalized by β-actin mRNA and shown as -fold change relative to DMSO control. Scatter plots show the individual data, and bar graphs indicate mean ± S.D. (n = 3). B, Raw264.7 cells were cultured in 0.1% FCS/DMEM, with or without the indicated concentrations of CHX for 30 min, and then further incubated with DMSO (−) or 100 μg/ml DMXAA for 4 h. SEMA4D and CXCL10 levels in the culture supernatants and the α-tubulin level in cell lysates were determined by Western blotting with anti-SEMA4D, anti-CXCL10, and anti-α-tubulin. In C, the protein levels of soluble SEMA4D in culture supernatants and membrane SEMA4D in cell lysates from three biological replicates were analyzed by Western blotting with anti-SEMA4D. The arrowhead indicates an intracellular cleaved form of SEMA4D.
Disease-associated mutant of STING induces SEMA4D shedding
Several autosomal dominant mutations of STING have been reported in patients with autoinflammatory diseases (27, 28). These mutations encode an active form of the STING protein that triggers IRF3-dependent gene induction at steady state. However, a recent study revealed that IRF3 is dispensable for the development of systemic inflammation caused by the disease-associated mutant of STING in mice (22). Therefore, it is possible that the shedding of SEMA4D might be involved in inflammation caused by this STING mutant. To test whether the STING mutant could induce SEMA4D shedding, we established stable cell lines that lack endogenous STING but inducibly express WT or the corresponding disease-associated mutant of mouse STING (V146L). After treatment with doxycycline, the STING V146L mutant but not STING WT accumulated in the perinuclear region, where most of it was co-localized with a cis-Golgi matrix protein, GM130 (Fig. 4A). We also observed phosphorylation of TBK1 in cells expressing STING V146L but not STING WT (Fig. 4B), confirming the constitutive activation of this STING mutant. Then, we collected the culture supernatants and found that release of soluble SEMA4D was provoked by the expression of the STING mutant. Thus, SEMA4D shedding is an additional phenotype induced by the disease-associated mutant of STING.
Figure 4.
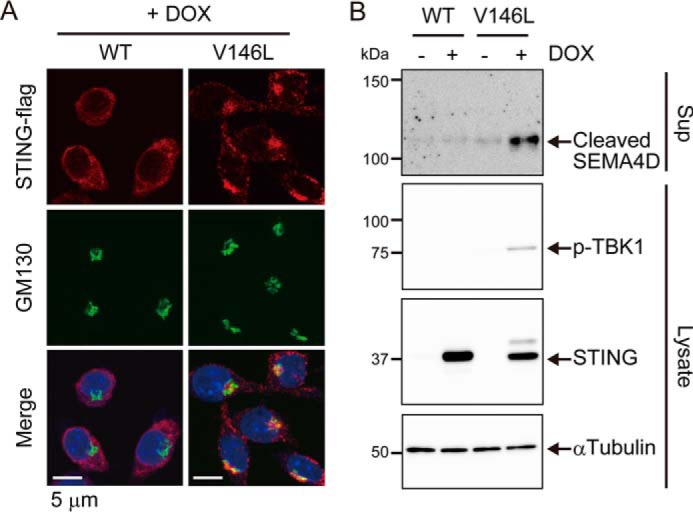
Disease-associated mutant of STING activates SEMA4D shedding. A and B, STING−/− Raw264.7 cells expressing the Tet-on–STING-FLAG WT or Tet-on–STING-FLAG V146L mutant (V146L) were cultured in 0.1% FCS/DMEM with or without 1 μg/ml doxycycline (DOX) for 4 h. In A, the cells were fixed and co-stained with anti-FLAG and anti-GM130 antibodies followed by examination under confocal microscopy. Scale bars, 5 μm. In B, the culture supernatants and the cell lysates were analyzed by Western blotting with anti-SEMA4D, anti-phospho-TBK1, anti-STING, or anti-α-tubulin.
ADAM17 but not TBK1 is required for SEMA4D shedding
Because the active mutant STING that was constitutively localized to the Golgi apparatus was able to induce SEMA4D shedding, we propose that STING trafficking may regulate its shedding machinery. As shown in Fig. 5A, treatment with a pharmacological inhibitor, brefeldin A (BFA), which inhibits anterograde transport from the ER to the Golgi apparatus, completely blocked DMXAA-induced release of CXCL10, TNFα, and SEMA4D, suggesting that the Golgi apparatus acts as a platform for the induction of all downstream signaling events of STING. It should be noted that BFA also seemed to block SEMA4D trafficking to the plasma membrane, as the smear bands of SEMA4D migrated faster in the BFA-treated cells, which may imply incomplete glycosylation or degradation in the ER. After translocation to the Golgi apparatus, STING associates with TBK1, triggering CXCL10 and TNFα gene induction by phosphorylation of IRF3 and NF-κB, respectively (29). Surprisingly, DMXAA-induced SEMA4D shedding was not blocked by BX795, a specific TBK1 inhibitor, although it inhibited the induction of CXCL10 (Fig. 5A), indicating that SEMA4D shedding is TBK1-independent. Blocking of NF-κB–dependent transcription by BX795 partially but not completely inhibited the release of TNFα, further supporting the idea that STING promotes its production through a transcription-independent pathway. In addition to SEMA4D, our proteomic analysis revealed that TNFα, SDC4, TGFBR1, and SORL1 were also released into extracellular space by STING activation (Table 1 and Table S1). All of them are plasma membrane proteins that commonly undergo proteolytic cleavage by ADAM17 (30–32). Therefore, we tested the possibility that ADAM17 is required for STING-mediated SEMA4D release. The inhibition of ADAM17 sheddase activity by a pharmacological inhibitor, TMI-1, abolished DMXAA-induced release of TNFα and SEMA4D but not CXCL10 (Fig. 5A). We also confirmed that TMI-1 inhibited the SEMA4D shedding induced by the active mutant of STING without affecting TBK1 phosphorylation (Fig. 5B). These results suggest that STING activates two independent downstream signaling pathways after trafficking to the Golgi: one is TBK1-mediated gene induction, and another is ADAM17-mediated cleavage of plasma membrane proteins (Fig. 5C).
Figure 5.

ADAM17 but not TBK1 activity is required for STING-dependent SEMA4D shedding. A, Raw264.7 cells were cultured in 0.1% FCS/DMEM with or without the indicated inhibitors for 30 min and then further incubated with DMSO (−) or 100 μg/ml DMXAA for 4 h. The indicated protein levels in culture supernatants (Sup) and cell lysates were determined by Western blotting with anti-SEMA4D, anti-TNFα, anti-CXCL10 and anti-α-tubulin. B, STING−/− Raw264.7 cells expressing the Tet-on–STING-FLAG V146L mutant (V146L) were cultured in 0.1% FCS/DMEM with or without TMI-1 (0.5 μm) for 30 min and then further incubated with or without 1 μg/ml doxycycline (DOX) for 4 h. The culture supernatants and the cell lysates were analyzed by Western blotting with anti-SEMA4D, anti-phospho-TBK1, or anti-α-Tubulin. C, proposed model for STING-mediated inflammatory signaling pathways. Once STING is activated by agonist or genetic mutation, it relocates from the ER to the Golgi apparatus, which induces the ADAM17-mediated post-translational shedding pathway independent of the TBK1-mediated transcriptional pathway. This ectodomain shedding is required for the production of biologically active inflammatory proteins such as SEMA4D and TNFα. Arrowheads indicate an intracellular cleaved form of SEMA4D.
Discussion
In this study, we identified novel STING-dependent signaling in which a cell surface protein, SEMA4D, is cleaved to produce its soluble ectodomain. The increased concentration of soluble SEMA4D in serum or plasma is associated with numerous inflammatory conditions in humans and mice (26), and the soluble SEMA4D has been described as a stimulator of both innate and acquired immunity (26). STING-dependent polyarthritis in DNase II–null mice is triggered by innate immune response–producing proinflammatory cytokines, including TNFα, IL-6, and IL-1β, but not by acquired immunity (8). In this case, soluble SEMA4D may have contributed to such cytokine production rather than to acquired immunity. Indeed, when CD14+ monocytes from rheumatoid arthritis patients were treated with recombinant soluble SEMA4D protein, the cells produced high levels of TNFα and IL-6 through a poorly understood mechanism, although its receptor, CD72, has been implicated (33). Furthermore, the administration of anti-SEMA4D antibodies to mice with collagen-induced arthritis significantly reduces the arthritis score and serum level of TNFα and IL-6 (33). Thus, the soluble SEMA4D released from STING-activated cells may induce the production of inflammatory cytokines in adjacent cells or SEMA4D-releasing cells in a paracrine or autocrine manner.
It has been reported that membrane SEMA4D is cleaved by several proteinases including ADAM17, MT1-MMP, and ATAMTS4 (26). Because the potent ADAM17 inhibitor TMI-1 used in this study also inhibits matrix metalloproteinases (MMP) (34), we could not exclude the possibility that certain MMPs are involved in the STING-mediated SEMA4D shedding. However, our secretome analysis showed that other ADAM17 substrates such as TNFα, SDC4, TGFBR1, and SORL1 were also released into culture supernatant by STING activation, suggesting that STING activates ADAM17 to promote shedding of these plasma membrane proteins in a cell-intrinsic manner. Although STING is known to activate NF-κB to induce TNFα gene expression, its activation is very weak compared with IRF3-mediated IFN-β or CXCL10 gene activation (11, 23, 24). This is supported by our gene expression experiment shown in Fig. 2A. However, the protein level of TNFα in the cell medium was dramatically increased by DMXAA stimulation compared with the mRNA level (Fig. 1C). From these observations, we propose the model that STING activates both transcriptional and post-translational machineries for efficient release of TNFα. This two-step mechanism comprising NF-κB–mediated TNFα mRNA induction followed by ADAM17-mediated ectodomain shedding is analogous to the case of another inflammatory cytokine, IL-1β, in which release is mediated by NF-κB–dependent transcription and caspase-1-dependent proteolytic processing.
How does STING activate ADAM17-dependent shedding machinery? Given that the STING mutant that constitutively localized to the Golgi induced ADAM17-mediated shedding of SEMA4D (Figs. 4 and 5B), STING trafficking from the ER to the Golgi may activate ADAM17. STING activates TBK1 at the Golgi (20); however, inhibition of TBK1 activity by BX795 did not suppress the release of SEMA4D, suggesting the involvement of another downstream signaling pathway. Although BX795 partially blocked the release of TNFα, this discrepancy may be explained by the suppression of its transcriptional activation. The enzymatic activity of ADAM17 is tightly regulated by iRhom2, which mediates its maturation, localization, and conformation (35–39). Upon binding to iRhom2, pro-ADAM17 is exported from the ER to the Golgi, where pro-protein convertases such as furin cleave the autoinhibitory pro-domain of ADAM17 to the mature form. The interaction with iRhom2 is also required for post-Golgi trafficking of mature ADAM17 to the plasma membrane and regulates its conformation and proteolytic activity at the cell surface. Interestingly, a recent report shows that iRhom2 interacts directly with STING and mediates its trafficking from the ER to the Golgi (40). In other words, the activation of STING induces enrichment of iRhom2 in the Golgi apparatus, which may promote ER–Golgi or post-Golgi trafficking of ADAM17. Alternatively, MAPK-mediated regulation may be involved. The activity of ADAM17 has been proposed to be regulated by the phosphorylation of ADAM17 and iRhom2 by either ERK or p38 (38, 39, 41–43). Importantly, STING agonists induce MAPK activation independent of TBK1, whose biological significance remains unclear (29).
In addition to membrane proteins cleaved by ADAM17, our comprehensive proteomic approach identified Fam3C and LRPAP1 as intracellular proteins secreted from STING-activated cells. Fam3C was originally identified as the interleukin-like epithelial-to-mesenchymal transition (EMT) inducer that is essential for EMT and tumor metastasis (44). Inflammatory injury triggers EMT to promote tissue remodeling, and sustained EMT causes pathologic fibrosis (45). It would be interesting to know whether Fam3C is involved in tissue repair or fibrosis during STING-dependent inflammation. LRPAP1, also known as RAP, interacts with members of the LDL receptor family such as LRP1 within the ER to assist the folding of such receptor proteins (46). Because of its high-affinity binding, recombinant LRPAP1 protein is used for LRP1 ligand and induces proinflammatory cytokine expression (47). However, whether ER chaperone protein LRPAP1 is secreted under physiological conditions was unclear. This is the first evidence that LRPAP1 is secreted in response to inflammatory stimuli.
So far, most studies have focused on the IRF3-dependent transcriptional program in the STING pathway, and thus no one has found release of SEMA4D, Fam3C, and LRPAP1, for which gene expression levels are not altered during STING activation (Fig. 3A). This study provides new insight into the STING-associated inflammatory response and diseases.
Experimental procedures
Reagents
Anti-SEMA4D mAb (clone no. 547406) and biotinylated anti-CXCL10 pAb were purchased from R&D Systems (Minneapolis, MN). Anti-α-tubulin mAb (DM1A) and anti-FLAG pAb were from Sigma-Aldrich. Anti-phospho-TBK1 (Ser-172) mAb (D52C2) and anti-TNFα mAb (D2D4) were from Cell Signaling (Danvers, MA). Anti-STING pAb was from Proteintech (Chicago, IL). Anti-GM130 mAb (35/GM130) was from BD Biosciences (Franklin Lakes, NJ). DMXAA was from Ark Pharm, Inc. (Arlington Heights, IL). Doxycycline was from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). BFA was from Cayman Chemical (Ann Arbor, MI). BX795, and TMI-1 and CHX were from Sigma-Aldrich.
Cell lines
Raw264.7 cells were cultured in DMEM containing 10% FCS. CRISPR/Cas9-mediated gene editing was used to establish Raw264.7 cells lacking STING. Px330 vector expressing Cas9 and sgRNA for mouse STING (16) was transfected into Raw264.7 cells by electroporation using the NEPA21 system (135 V, 10 ms; Nepagene, Chiba, Japan). After a 24-h incubation at 37 °C, the cells were subjected to limiting dilution, and the mutated clones were identified by Western blotting. For the rescue experiment, the C-terminal FLAG-tagged mouse STING (STING-FLAG) was stably expressed in STING-deficient cells using the PiggyBac transposon system. To generate the inducible gene expression cells, Tet-on expression vector carrying STING-FLAG WT or V146L mutant was introduced into the STING-deficient cells. The V146L mutant STING was constructed with recombinant PCR using these primers: 5′-CACCGGCCAGTGTGGTGGAAGATGCCATACTCCAACCT-3′ (BstXI-Start) and 5′-CAGCGGAAGTCTCTGCACTCTGTGAAGAAAAGAAGTT-3′ (V146L); and 5′-AACTTCTTTTCTTCACAGAGTGCAGAGACTTCCGCTG-3′(V146L) and 5′-GCTAACCACTGTGCTGGCTACTTATCGTCGTCATCCTTGTAATCGATGAGGTCAGTGCGGAGTG-3′ (FLAG-Stop-BstXI).
Sample preparation for secretome analysis
Raw264.7 cells were seeded in 6-cm dishes (2.5 × 106 cells/well) and cultured overnight. After washing with PBS twice, the cells were cultured with FCS-free DMEM containing DMSO or 100 μg/ml DMXAA for 4 h. The conditioned medium was collected and centrifuged at 400 × g for 10 min at 4 °C. The supernatants were concentrated to 100 μl using an Amicon Ultra 3K filter (Millipore, Burlington, MA), and proteins were purified by methanol/chloroform precipitation. The protein pellets were solubilized in 20 μl of 8 m urea followed by reduction in 5 mm DTT at 37 °C for 30 min and alkylation in 27.5 mm iodoacetamide at room temperature for 30 min in the dark. After reducing the urea concentration to 1 m with 50 mm Tris-HCl (pH 8.0), the proteins were digested with 50 ng of trypsin/Lys-C mix, Mass Spec Grade (Promega, Madison, WI) at 37 °C overnight. The peptides were desalted using GL-Tip SDB (GL Sciences, Tokyo, Japan) according to the manufacturer's protocol, and the eluates were evaporated in a SpeedVac concentrator (Thermo Fisher Scientific). The residues were dissolved in 0.1% TFA.
Data-dependent LC-MS/MS analysis
LC-MS/MS analysis of the resultant peptides (200 ng each) was performed on an EASY-nLC 1200 UHPLC connected to a Q Exactive Plus mass spectrometer through a nanoelectrospray ion source (Thermo Fisher Scientific). The peptides were separated on a 75-μm inner diameter × 120-mm C18 reversed-phase column (Nikkyo Technos, Tokyo, Japan) with a linear gradient from 4 to 28% acetonitrile for 0–150 min followed by an increase to 80% acetonitrile during min 150–170. The mass spectrometer was operated in a data-dependent acquisition mode with a top 10 MS/MS method. Raw data were analyzed directly against the SwissProt database restricted to Mus musculus using Proteome Discoverer, version 2.2 (Thermo Fisher Scientific) for identification and label-free precursor ion quantification. Normalization was performed such that the total sum of abundance values for each sample over all peptides was the same. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the jPOST partner repository with the dataset identifier PXD009169. The data were analyzed by a two-tailed Student's t test, and the resulting p values were adjusted using the Benjamini-Hochberg method for controlling the discovery rate (FDR). A volcano plot was used for showing the -fold changes and q values (FDR-corrected p values) of each protein. To select significant proteins, we set a -fold change threshold of 4 and a q value threshold of 0.05.
PRM analysis
To quantify SEMA4D, LRPAP1, Fam3C, SEMA4B, CCL9, IFN-β, CXCL10, and TNFα, at least three peptides/protein were measured by PRM, an MS/MS-based targeted quantification method using high-resolution MS. Time alignment and relative quantification of the transitions were performed with PinPoint, version 1.4 (Thermo Fisher Scientific). The MS proteomics data for PRM analysis have been deposited to the ProteomeXchange Consortium via the jPOST partner repository with the dataset identifier PXD009170.
SDS-PAGE and Western blotting
The culture medium was replaced with 0.1% FCS/DMEM when the cells were treated with DMXAA in the presence or absence of inhibitors, and the conditioned media were concentrated using an Amicon Ultra 3K filter or Vivaspin 10K filter (Sartorius, Goettingen, Germany). Under the same conditions, the cells were lysed on ice for 10 min in lysis buffer (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1% NP-40, 10% glycerol, a protease inhibitor mixture (Nacalai Tesque, Kyoto, Japan), and a phosphatase inhibitor mixture (Nacalai Tesque)) and centrifuged at 20,000 × g for 15 min at 4 °C. The concentrated media or the cell lysates were mixed with 5× SDS sample buffer (200 mm Tris-HCl buffer (pH 6.8), 10% SDS, 25% glycerol, and 0.05% bromphenol blue) with 5% β-mercaptoethanol and heated at 95 °C for 5 min. The samples were separated by electrophoresis on a 6% (for SEMA4D), 10% (for α-tubulin, phospho-TBK1, and STING) or 16% (for CXCL10 and TNFα) polyacrylamide gel and transferred onto a polyvinylidene difluoride membrane (Millipore). After blocking, the membrane was incubated with primary antibodies in 5% skim milk or in Signal Enhancer HIKARI (Nacalai Tesque) overnight at 4 °C followed by incubation with horseradish peroxidase-conjugated secondary antibodies. Protein bands in the membrane was detected using ImageQuant LAS 4000 mini (GE Healthcare) after incubating the membrane with Clarity Western ECL substrate (Bio-Rad) or Immunostar LD (Wako Pure Chemical Industries, Ltd.).
Real-time PCR
Total RNA was isolated using RNAiso Plus (Takara, Shiga, Japan) according to the manufacturer's protocol. The RNA was reverse-transcribed using ReverTra Ace qPCR RT Master Mix (Toyobo, Osaka, Japan), and real-time PCR was performed with Light-Cycler 96 (Roche Diagnostics) using FastStart Essential DNA Green Master mix (Roche Diagnostics). The primers used for real-time PCR were as follows: IFN-β, 5′-CCACCACAGCCCTCTCCATCAACTAT-3′ and 5′-CAAGTGGAGAGCAGTTGAGGACATC-3′; CXCL10, 5′-CCATCAGCACCATGAACCCAAGT-3′ and 5′-CACTCCAGTTAAGGAGCCCTTTTAGACC-3′; SEMA4D, 5′-TTGGGCAGTGAACCCATCATC-3′ and 5′-GGATCACGTCAGCAAAGACGA-3′; TNFα, 5′-CACAGAAAGCATGATCCGCGACGT-3′ and 5′-CGGCAGAGAGGAGGTTGACTTTCT-3′; and β-actin, 5′-TGTGATGGTGGGAATGGGTCAG-3′ and 5′-TTTGATGTCACGCACGATTTCC-3′.
Immunostaining
The STING-null Raw264.7 cells expressing Tet-on–STING-FLAG WT or Tet-on–STING-FLAG V146L were seeded on coverslips in a 12-well plate. After treatment with doxycycline, the cells were fixed with 3.7% formaldehyde/PBS for 15 min at 37 °C and permeabilized with 0.2% Triton X-100/PBS for 10 min at room temperature. The coverslips were incubated with primary antibodies in 2% goat serum/PBS for 1 h at 37 °C. After washing with PBS three times, the coverslips were incubated with Alexa Fluor-488 goat anti-mouse IgG and Alexa Fluor-555 goat anti-rabbit IgG for 1 h at room temperature in the dark. After washing with PBS three times and subsequent rinsing with distilled water, the coverslips were mounted on glass slides with ProLong Gold antifade mountant with 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific). The immunofluorescence images were obtained using a confocal laser-scanning microscope (FV1200, Olympus, Tokyo, Japan).
Author contributions
K. M. conceptualization; K. M. and H. K. data curation; K. M. formal analysis; K. M. and H. K. supervision; K. M. and H. K. funding acquisition; K. M. validation; K. M. and H. K. investigation; K. M. visualization; K. M. and H. K. methodology; K. M. writing-original draft; K. M. project administration; K. M. and H. K. writing-review and editing; H. K. resources.
Supplementary Material
Acknowledgments
We thank M. Kajimoto and M. Kawano for technical assistance. We also thank M. Iwata for secretarial assistance.
This work was supported by KAKENHI Grants-in-aid for Scientific Research 15K19024 and 17K08661 from the Japan Society for the Promotion of Science (JSPS) and by the Takeda Science Foundation (to K. M.). This work also was supported by KAKENHI Grant-in-aid for Scientific Research 17K08635 from the JSPS and by the Japan Foundation for Applied Enzymology (to H. K.). The authors declare that they have no conflicts of interest with the contents of this article.
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the jPOST partner repository with the data set identifier PXD009169 and PXD009170.
This article contains Tables S1 and S2.
- STING
- stimulator of interferon genes
- ER
- endoplasmic reticulum
- TBK1
- TANK-binding kinase 1
- SEMA4D
- semaphoring 4D
- ADAM17
- a disintegrin and metalloproteinase 17
- cGAMP
- cyclic GMP-AMP
- IFN
- interferon
- IRF
- interferon regulatory factor
- CHX
- cycloheximide
- DMXAA
- 5,6-dimethylxanthenone-4-acetic acid
- PRM
- parallel reaction monitoring
- BFA
- brefeldin A
- MMP
- matrix metalloproteinase
- MAPK
- mitogen-activated protein kinase
- EMT
- epithelial-to-mesenchymal transition
- LRPAP1
- LDL receptor-related protein–associated protein-1
- DMEM
- Dulbecco's modified Eagle's medium
- FCS
- fetal calf serum
- pAb
- polyclonal antibody
- FDR
- false discovery rate.
References
- 1. Wu J., and Chen Z. J. (2014) Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 32, 461–488 10.1146/annurev-immunol-032713-120156 [DOI] [PubMed] [Google Scholar]
- 2. Ablasser A., Hertrich C., Wassermann R., and Hornung V. (2013) Nucleic acid driven sterile inflammation. Clin. Immunol. 147, 207–215 10.1016/j.clim.2013.01.003 [DOI] [PubMed] [Google Scholar]
- 3. Nagata S., Hanayama R., and Kawane K. (2010) Autoimmunity and the clearance of dead cells. Cell 140, 619–630 10.1016/j.cell.2010.02.014 [DOI] [PubMed] [Google Scholar]
- 4. Chasis J. A., and Mohandas N. (2008) Erythroblastic islands: Niches for erythropoiesis. Blood 112, 470–478 10.1182/blood-2008-03-077883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kawane K., Fukuyama H., Kondoh G., Takeda J., Ohsawa Y., Uchiyama Y., and Nagata S. (2001) Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 292, 1546–1549 10.1126/science.292.5521.1546 [DOI] [PubMed] [Google Scholar]
- 6. Kawane K., Ohtani M., Miwa K., Kizawa T., Kanbara Y., Yoshioka Y., Yoshikawa H., and Nagata S. (2006) Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443, 998–1002 10.1038/nature05245 [DOI] [PubMed] [Google Scholar]
- 7. Yoshida H., Okabe Y., Kawane K., Fukuyama H., and Nagata S. (2005) Lethal anemia caused by interferon-β produced in mouse embryos carrying undigested DNA. Nat. Immunol. 6, 49–56 10.1038/ni1146 [DOI] [PubMed] [Google Scholar]
- 8. Kawane K., Tanaka H., Kitahara Y., Shimaoka S., and Nagata S. (2010) Cytokine-dependent but acquired immunity-independent arthritis caused by DNA escaped from degradation. Proc. Natl. Acad. Sci. U.S.A. 107, 19432–19437 10.1073/pnas.1010603107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawane K., Motani K., and Nagata S. (2014) DNA degradation and its defects. Cold Spring Harb. Perspect. Biol. 6, a016394 10.1101/cshperspect.a016394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rodero M. P., Tesser A., Bartok E., Rice G. I., Della Mina E., Depp M., Beitz B., Bondet V., Cagnard N., Duffy D., Dussiot M., Fremond M. L., Gattorno M., Guillem F., Kitabayashi N., et al. (2017) Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat. Commun. 8, 2176 10.1038/s41467-017-01932-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ishikawa H., and Barber G. N. (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun L., Wu J., Du F., Chen X., and Chen Z. J. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu J., Sun L., Chen X., Du F., Shi H., Chen C., and Chen Z. J. (2013) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 10.1126/science.1229963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao P., Ascano M., Wu Y., Barchet W., Gaffney B. L., Zillinger T., Serganov A. A., Liu Y., Jones R. A., Hartmann G., Tuschl T., and Patel D. J. (2013) Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107 10.1016/j.cell.2013.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gao P., Ascano M., Zillinger T., Wang W., Dai P., Serganov A. A., Gaffney B. L., Shuman S., Jones R. A., Deng L., Hartmann G., Barchet W., Tuschl T., and Patel D. J. (2013) Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 154, 748–762 10.1016/j.cell.2013.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Motani K., Ito S., and Nagata S. (2015) DNA-mediated cyclic GMP-AMP synthase-dependent and -independent regulation of innate immune responses. J. Immunol. 194, 4914–4923 10.4049/jimmunol.1402705 [DOI] [PubMed] [Google Scholar]
- 17. Saitoh T., Fujita N., Hayashi T., Takahara K., Satoh T., Lee H., Matsunaga K., Kageyama S., Omori H., Noda T., Yamamoto N., Kawai T., Ishii K., Takeuchi O., Yoshimori T., and Akira S. (2009) Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. U.S.A. 106, 20842–20846 10.1073/pnas.0911267106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ishikawa H., Ma Z., and Barber G. N. (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 10.1038/nature08476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tanaka Y., and Chen Z. J. (2012) STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5, ra20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mukai K., Konno H., Akiba T., Uemura T., Waguri S., Kobayashi T., Barber G. N., Arai H., and Taguchi T. (2016) Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 7, 11932 10.1038/ncomms11932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Okabe Y., Kawane K., and Nagata S. (2008) IFN regulatory factor (IRF) 3/7-dependent and -independent gene induction by mammalian DNA that escapes degradation. Eur. J. Immunol. 38, 3150–3158 10.1002/eji.200838559 [DOI] [PubMed] [Google Scholar]
- 22. Warner J. D., Irizarry-Caro R. A., Bennion B. G., Ai T. L., Smith A. M., Miner C. A., Sakai T., Gonugunta V. K., Wu J., Platt D. J., Yan N., and Miner J. J. (2017) STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med. 214, 3279–3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roberts Z. J., Goutagny N., Perera P. Y., Kato H., Kumar H., Kawai T., Akira S., Savan R., van Echo D., Fitzgerald K. A., Young H. A., Ching L. M., and Vogel S. N. (2007) The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J. Exp. Med. 204, 1559–1569 10.1084/jem.20061845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Prantner D., Perkins D. J., Lai W., Williams M. S., Sharma S., Fitzgerald K. A., and Vogel S. N. (2012) 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J. Biol. Chem. 287, 39776–39788 10.1074/jbc.M112.382986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rauniyar N. (2015) Parallel reaction monitoring: A targeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int. J. Mol. Sci. 16, 28566–28581 10.3390/ijms161226120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maleki K. T., Cornillet M., and Björkström N. K. (2016) Soluble SEMA4D/CD100: A novel immunoregulator in infectious and inflammatory diseases. Clin. Immunol. 163, 52–59 10.1016/j.clim.2015.12.012 [DOI] [PubMed] [Google Scholar]
- 27. Liu Y., Jesus A. A., Marrero B., Yang D., Ramsey S. E., Sanchez G. A. M., Tenbrock K., Wittkowski H., Jones O. Y., Kuehn H. S., Lee C. R., DiMattia M. A., Cowen E. W., Gonzalez B., Palmer I., DiGiovanna J. J., et al. (2014) Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 371, 507–518 10.1056/NEJMoa1312625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jeremiah N., Neven B., Gentili M., Callebaut I., Maschalidi S., Stolzenberg M. C., Goudin N., Frémond M. L., Nitschke P., Molina T. J., Blanche S., Picard C., Rice G. I., Crow Y. J., Manel N., et al. (2014) Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Invest. 124, 5516–5520 10.1172/JCI79100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abe T., and Barber G. N. (2014) Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 88, 5328–5341 10.1128/JVI.00037-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dreymueller D., Pruessmeyer J., Groth E., and Ludwig A. (2012) The role of ADAM-mediated shedding in vascular biology. Eur. J. Cell Biol. 91, 472–485 10.1016/j.ejcb.2011.09.003 [DOI] [PubMed] [Google Scholar]
- 31. Liu C., Xu P., Lamouille S., Xu J., and Derynck R. (2009) TACE-mediated ectodomain shedding of the type I TGF-β receptor downregulates TGF-β signaling. Mol. Cell 35, 26–36 10.1016/j.molcel.2009.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsukamoto S., Takeuchi M., Kawaguchi T., Togasaki E., Yamazaki A., Sugita Y., Muto T., Sakai S., Takeda Y., Ohwada C., Sakaida E., Shimizu N., Nishii K., Jiang M., Yokote K., Bujo H., and Nakaseko C. (2014) Tetraspanin CD9 modulates ADAM17-mediated shedding of LR11 in leukocytes. Exp. Mol. Med. 46, e89 10.1038/emm.2013.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshida Y., Ogata A., Kang S., Ebina K., Shi K., Nojima S., Kimura T., Ito D., Morimoto K., Nishide M., Hosokawa T., Hirano T., Shima Y., Narazaki M., Tsuboi H., et al. (2015) Semaphorin 4D contributes to rheumatoid arthritis by inducing inflammatory cytokine production: Pathogenic and therapeutic implications. Arthritis Rheum. 67, 1481–1490 10.1002/acr.22608,10.1002/art.39086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moss M. L., and Rasmussen F. H. (2007) Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal. Biochem. 366, 144–148 10.1016/j.ab.2007.04.043 [DOI] [PubMed] [Google Scholar]
- 35. Adrain C., Zettl M., Christova Y., Taylor N., and Freeman M. (2012) Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228 10.1126/science.1214400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McIlwain D. R., Lang P. A., Maretzky T., Hamada K., Ohishi K., Maney S. K., Berger T., Murthy A., Duncan G., Xu H. C., Lang K. S., Häussinger D., Wakeham A., Itie-Youten A., Khokha R., Ohashi P. S., Blobel C. P., and Mak T. W. (2012) iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232 10.1126/science.1214448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brooke M. A., Etheridge S. L., Kaplan N., Simpson C., O'Toole E. A., Ishida-Yamamoto A., Marches O., Getsios S., and Kelsell D. P. (2014) iRHOM2-dependent regulation of ADAM17 in cutaneous disease and epidermal barrier function. Hum. Mol. Genet. 23, 4064–4076 10.1093/hmg/ddu120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grieve A. G., Xu H., Künzel U., Bambrough P., Sieber B., and Freeman M. (2017) Phosphorylation of iRhom2 at the plasma membrane controls mammalian TACE-dependent inflammatory and growth factor signalling. Elife 6, e23968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cavadas M., Oikonomidi I., Gaspar C. J., Burbridge E., Badenes M., Félix I., Bolado A., Hu T., Bileck A., Gerner C., Domingos P. M., von Kriegsheim A., and Adrain C. (2017) Phosphorylation of iRhom2 controls stimulated proteolytic shedding by the metalloprotease ADAM17/TACE. Cell Rep. 21, 745–757 10.1016/j.celrep.2017.09.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luo W. W., Li S., Li C., Lian H., Yang Q., Zhong B., and Shu H. B. (2016) iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 17, 1057–1066 10.1038/ni.3510 [DOI] [PubMed] [Google Scholar]
- 41. Soond S. M., Everson B., Riches D. W., and Murphy G. (2005) ERK-mediated phosphorylation of Thr735 in TNFα-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 118, 2371–2380 10.1242/jcs.02357 [DOI] [PubMed] [Google Scholar]
- 42. Xu P., and Derynck R. (2010) Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 37, 551–566 10.1016/j.molcel.2010.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu P., Liu J., Sakaki-Yumoto M., and Derynck R. (2012) TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 5, ra34 10.1126/scisignal.2002689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Waerner T., Alacakaptan M., Tamir I., Oberauer R., Gal A., Brabletz T., Schreiber M., Jechlinger M., and Beug H. (2006) ILEI: A cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell 10, 227–239 10.1016/j.ccr.2006.07.020 [DOI] [PubMed] [Google Scholar]
- 45. Stone R. C., Pastar I., Ojeh N., Chen V., Liu S., Garzon K. I., and Tomic-Canic M. (2016) Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 365, 495–506 10.1007/s00441-016-2464-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bu G., and Rennke S. (1996) Receptor-associated protein is a folding chaperone for low density lipoprotein receptor-related protein. J. Biol. Chem. 271, 22218–22224 10.1074/jbc.271.36.22218 [DOI] [PubMed] [Google Scholar]
- 47. Brifault C., Gilder A. S., Laudati E., Banki M., and Gonias S. L. (2017) Shedding of membrane-associated LDL receptor-related protein-1 from microglia amplifies and sustains neuroinflammation. J. Biol. Chem. 292, 18699–18712 10.1074/jbc.M117.798413 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.