

Abstract
Reactive oxygen species (ROS) mediate redox signaling necessary for numerous cellular functions. Yet, high levels of ROS in cells and tissues can cause damage and cell death. Therefore, regulation of redox homeostasis is essential for ROS-dependent signaling that does not incur cellular damage. Cells achieve this optimal balance by coordinating ROS production and elimination. In this Minireview, we discuss the mechanisms by which proliferating cancer and T cells maintain a carefully controlled redox balance. Greater insight into such redox biology may enable precisely targeted manipulation of ROS for effective medical therapies against cancer or immunological disorders.
Keywords: reactive oxygen species (ROS), redox regulation, redox signaling, cancer, T-cell
Introduction
Reactive oxygen species (ROS)3 are oxygen (O)-containing molecules with higher chemical reactivity than molecular oxygen (O2) itself. ROS are generated upon partial reduction of O2 and include superoxide anion (O2˙̄), hydrogen peroxide (H2O2), and hydroxyl radical (OH•). Because of their reactivity toward lipid, protein, and DNA, ROS have traditionally been considered solely as toxic molecules associated with oxidative damage. However, over the past 2 decades, there has been growing appreciation for the role of H2O2 as a secondary messenger in cellular signaling. Although very high levels of ROS, especially OH•, produce oxidative damage and cell death, a pool of spatially localized and a moderate concentration of ROS are required for many biological processes, including cell survival, death, proliferation, and immune responses (1–3). Therefore, cells maintain an optimal concentration and localization of intracellular ROS that support necessary signaling pathways without causing cellular damage and death. Under physiological conditions, such redox homeostasis is achieved by the careful control of both ROS production and elimination.
In recent years, growing interest toward tumor redox biology has highlighted a unique state of redox homeostasis in cancer cells that support their pathological proliferation, proposing modulation of ROS as a promising anti-cancer therapy. Concurrently, development of cancer immunotherapies has emphasized the significance of immune cells, especially T cells, in the regulatory pathways of tumors. However, the current understanding of the redox balance in T cells is limited, challenging the prediction on how anti- or pro-oxidants would affect antitumor immunity. Therefore, understanding redox biology in both cancer and T cells is essential for the progress in treatments against cancer and furthermore immune disorders. In this Minireview, we discuss the molecular mechanisms by which cancer and T cells maintain redox homeostasis.
Production of ROS
The two main sources of intracellular ROS are mitochondria and NADPH oxidases (NOXs) (Fig. 1) (4, 5). Mammalian mitochondria have 11 known sites that produce O2˙̄. These sites include electron transport chain (ETC) complex I and complex III, which are best characterized for their role in redox signaling (4, 6). During aerobic respiration, the ETC complexes relay electrons from nicotinamide adenine dinucleotide (NADH) and dihydroflavin adenine dinucleotide (FADH2) to complex IV, where the final electron acceptor, O2, is reduced to water (H2O). However, a small percentage (<0.5%) of electrons escape from the chain, and nonenzymatically react with O2 to generate O2˙̄ (7). The O2˙̄ from complexes I and III is released into the mitochondrial matrix. Complex III also releases O2˙̄ into the mitochondrial intermembrane space where the O2˙̄ traverses through the voltage-dependent anion channels into cytoplasm (Fig. 1) (6, 8).
Figure 1.

Production, elimination, and signaling of ROS. Mitochondria and NADPH oxidases (NOXs) are the main sources of superoxide (O2˙̄), which is converted to hydrogen peroxide (H2O2) by superoxide dismutases (SODs). H2O2 can subsequently (a) oxidize thiols within redox-regulated proteins to conduct cellular signaling or (b) be reduced to water by antioxidant systems largely composed of NRF2-regulated enzymes. The peroxiredoxin (PRX)/thioredoxin (TRX) and glutathione peroxidase (GPX)/glutathione (GSH) systems are fueled by NADPH. This key reducing equivalent is generated by a complex network of metabolic pathways and enzymes involving the pentose phosphate pathway (PPP), isocitrate dehydrogenases (IDHs), malic enzymes (MEs), and one-carbon metabolism. Interestingly, NADPH is also a substrate for the ROS-generating NOXs. This suggests that the antioxidant systems and ROS producers are equally important for biological processes, and they work in concert to regulate redox environments that permit the ROS-mediated signaling without incurring oxidative damage.
Several factors regulate generation of mitochondrial ROS (mROS) to the times, levels, and locations necessary for cellular signaling. The rate at which mROS is produced depends on kinetic and thermodynamic determinants, including concentration of electron carriers within the ETC, the redox state of the electron carriers, availability of mitochondrial O2, and proximity of O2 to electron carriers (6). Mitochondrial membrane potential (ψ) is another crucial regulator of the mROS production rate. ψ is required for mROS generation (9), despite that ψ and mROS levels are negatively correlated (10). The production and release of mROS can be augmented by diverse signaling factors, including hypoxia, thermogenesis, nutrient metabolites, tumor necrosis factor-α (TNF-α), SHC-transforming protein 1, and toll-like receptors (10–15). Additionally, mitochondria are dynamic organelles that constantly move inside the cells. In response to cellular signals, mitochondria can therefore redistribute to form localized pools of mROS that influence signaling pathways (16).
In addition to mitochondria, the NOX protein family is another major producer of intracellular ROS. These transmembrane proteins catalyze the transfer of electrons from nicotinamide adenine dinucleotide phosphate (NADPH) to O2, generating O2˙̄ (Fig. 1). NOX isoforms are present in the plasma membrane, as well as intracellular membranes of the nucleus, mitochondria, and endoplasmic reticulum. Depending on its location, a NOX can release O2˙̄ into intracellular or extracellular space (5). The ROS production by NOX depends on the assembly of the functional NOX complex. This process is regulated by the availability of flavin adenine dinucleotide (FAD), an essential prosthetic group of NOX enzyme (17). Additionally, activation of small GTP-binding proteins, protein phosphorylation, and intracellular calcium concentration control incorporation of accessory subunits required for the enzymatic activity of NOX (5). NOX complex can be activated upon cellular signaling by insulin, growth factors, tumor necrosis factor (TNF), angiotensin, and toll-like receptors (5, 18–20). NOX isoform activation occurs within discrete subcellular compartments, and such spatial organization of NOX is necessary for ROS-mediated signaling (21).
Elimination of ROS
Cells utilize robust antioxidant defense systems to maintain ROS levels. Mitochondria and NOX generate O2˙̄, a free radical capable of damaging iron–sulfur cluster-containing proteins (22). The primary cellular defense against O2˙̄ accumulation is superoxide dismutase (SOD), which rapidly converts O2˙̄ to H2O2 (Fig. 1), keeping the intracellular O2˙̄ level extremely low. Different SOD isoforms reside in specific subcellular compartments: SOD1 in cytosol, SOD2 in mitochondria, and SOD3 in extracellular matrix (23).
The removal of O2˙̄ results in the formation of H2O2. Accumulation of H2O2 can also be deleterious as it can disrupt cellular signaling. Furthermore, when in excess, H2O2 can react with ferrous and cuprous ions, which yields OH•, a strong oxidant that can irreversibly damage lipids, protein, and DNA (24). Therefore, cells have multiple antioxidant systems to regulate intracellular H2O2 levels, including peroxiredoxin (PRX)/thioredoxin (TRX) systems and glutathione peroxidase (GPX)/glutathione (GSH) systems. PRX is a H2O2 scavenger, which works in concert with TRX to relay a series of redox reactions to reduce H2O2. In the first reaction, cysteine residues of PRX undergo oxidation by H2O2, reducing H2O2 to H2O. The process removes H2O2 but inactivates the PRX. In the second reaction, the cysteine residues of TRX are oxidized as the inactivated PRX is reduced and reactivated. Finally, the oxidized and inactivated TRX is reduced by thioredoxin reductase, which is fueled by the oxidation of a reducing equivalent, NADPH (25). GPX is another H2O2 scavenger. Similar to the PRX and TRX, GPX and GSH cooperate to detoxify H2O2 to H2O. This process yields an oxidized GSH (GSSG), which is subsequently reduced by glutathione reductase (GR) and NADPH (26). NADPH is generated by a closely regulated metabolic network of the pentose phosphate pathway (PPP), one-carbon metabolism, isocitrate dehydrogenases (IDHs), and malic enzymes (Fig. 1) (7). Importantly, protein families of PRX and GPX are widely distributed throughout the cells. In mammalian cells, six isoforms of PRXs and eight isoforms of GPXs have distinct cellular localization, including cytosol, mitochondria, endoplasmic reticulum, peroxisomes, and extracellular space (25, 27). As a result, cells have robust antioxidant systems throughout the cell, by which SODs convert O2˙̄ to H2O2, and PRXs and GPXs reduce H2O2 to H2O (Fig. 1).
To effectively manage the intracellular ROS in a temporal, quantitative, and spatial manner, antioxidants are regulated at both the mRNA expression and protein enzymatic activity level. One of the major regulators of the antioxidants is nuclear factor erythroid 2–related factor 2 (NRF2) (Fig. 1). Under homeostatic conditions, this transcription factor is constitutively ubiquitinated for degradation by Kelch-like ECH-associated protein (KEAP)-1 and Cullin (CUL)-3 E3 ligase complex. However, upon accumulation of ROS, KEAP1 is oxidized by H2O2, thereby inhibiting its ability to ubiquitinate NRF2. As a result, NRF2 is stabilized and translocated into the nucleus, where it binds antioxidant-responsive elements within regulatory regions of genes whose protein products are involved in many different antioxidant systems (28). NRF2 is responsible for the synthesis, regeneration, and utilization of GSH, as it regulates the expression of GR, glutamate cysteine ligase (catalyzes the first step of de novo GSH synthesis), solute carrier family 7 member 11 (SLC7A11, imports cysteine, a building block of GSH), and GPX2 (a GPX isoform expressed in the gastrointestinal system) (29–31). NRF2 also supports the PRX/TRX system by promoting transcription of isoforms PRX1 and TRX1 (32, 33). Importantly, NRF2 induces the expression of multiple metabolic enzymes involved in production of NADPH (34). Thus, the extensive array of antioxidant pathways controlled by NRF2 makes NRF2 arguably the master regulator of antioxidants.
Signaling of ROS
H2O2 is the most stable form of ROS with an ability to freely diffuse through membranes. Such a characteristic makes H2O2 an ideal intracellular signaling molecule. Indeed, H2O2 mediates signal transduction by selectively oxidizing cysteine residues within proteins, leading to alteration of their structure and, importantly, activity (35). One of the determinants of the specificity of oxidation necessary for this process is logarithmic acid dissociation constant (pKa) of the cysteine residues. Under physiological pH, the thiol group (SH) of cysteine residues with low pKa exists as a thiolate (S−), which is highly susceptible to H2O2-mediated oxidation (36). The oxidation process can generate reversible modifications of S−, including sulfenic acid (SOH), disulfide bonds (S–S) (Fig. 1), and sulfenamide (S–N). The oxidized forms of S− can be reduced back by TRX and glutaredoxin (GRX) (35). The best-characterized targets of this redox-regulation include phosphatases, kinases, and antioxidants (37, 38).
The molecular mechanisms by which the ROS producers and antioxidant systems coordinate to conduct redox signaling are important research areas. There are two proposed mechanisms: redox relay and floodgate. In the redox relay model, H2O2 scavengers PRX or GPX act as primary H2O2 receptors that specifically transfer the oxidation to the redox-regulated target protein (35). For example, a cytoplasmic PRX isoform PRX2 gets oxidized by H2O2, and subsequently it transfers this oxidizing equivalent to signal transducer and activator of transcription (STAT)-3, inhibiting its transcriptional activity (39). The floodgate model hypothesizes that “flooding” the proximity of a redox-regulated protein with H2O2 oxidizes and inactivates ROS scavengers in the area, thereby allowing H2O2 oxidation of the target (35). This model is evidenced by a redox-signaling pathway activated in the adrenal cortex mitochondria during steroidogenesis. Upon generation of corticosterone, cytochrome P450 produces a localized pool of H2O2, which oxidizes and inactivates PRX3 leading to further accumulation of H2O2, p38 activation, and suppression of steroidogenesis (40). These models and examples suggest that the specificity and efficiency of redox signaling are dependent on precise organization of ROS producers and scavengers, further highlighting the significance of their regulatory mechanisms.
Regulation of redox balance in cancer cells
In various types of cancer cells, ROS support their survival, proliferation, and metastasis through activating pro-tumorigenic cellular signaling. The classic examples are phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT), mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK), and hypoxia-inducible factor (HIF)-1α signaling (Fig. 2), wherein ROS oxidizes and inactivates their negative regulators phosphatase and tensin homolog (PTEN), MAPK phosphatase, and prolyl hydroxylase (PHD)-2, respectively (37, 41, 42). ROS also promote cancer cell proliferation through activation of nuclear factor κ-light chain enhancer of activated B cells (NF-κB) (Fig. 2). A recent study demonstrated that mROS activate protein kinase D (PKD)-1 and NF-κB to up-regulate epidermal growth factor receptor signaling, inducing formation of pancreatic pre-neoplastic lesions. The in vivo development of abnormal pancreatic structures was abrogated by a mitochondria-targeted antioxidant, mitoQ (43). In addition, ROS participate in a pro-metastatic signaling of protein-tyrosine kinase SRC/focal adhesion kinase PYK2 pathway. mROS is required to up-regulate the SRC/PYK2 signaling, leading to migration and invasion of different types of cancer cells. Importantly, administration of mROS scavenger, mitoTEMPO, prevents metastatic tumor dissemination of breast cancer xenograft (44). Therefore, ROS has causative impact on tumorigenesis and progression.
Figure 2.
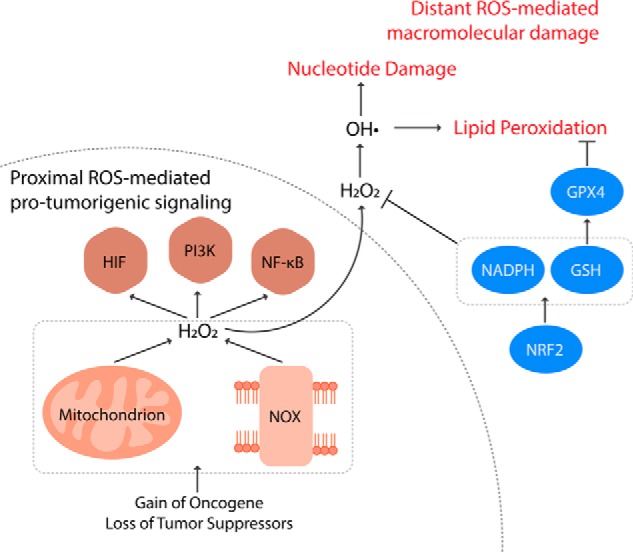
Regulation of redox balance in cancer cells. Compared with nontransformed cells, cancer cells have elevated levels of ROS instigated by acquisition of oncogenes and loss of tumor suppressors. ROS from mitochondria and NOXs oxidize co-localized redox-regulated target proteins to activate pro-tumorigenic signaling pathways, including HIF-1α, PI3K, and NF-κB. Distant from the sites of production, however, ROS nonspecifically react with nucleotides and lipids inducing oxidative damage and even cell death. These distant damaging ROS can be controlled by antioxidant systems such as NRF2, NADPH generation, GSH synthesis/regeneration, and GPX4. Therefore, cancer cells producing elevated levels of ROS concomitantly increase such antioxidant capacities. This shift in redox balance enables cancer cells to hyper-activate the proximal ROS-mediated pro-survival and proliferation signaling without experiencing ROS toxicity.
To drive the pro-tumorigenic redox signaling, cancer cells have elevated levels of intracellular ROS. Increased ROS production is instigated by acquisition of oncogenes such as the constitutively active isoforms of RAS (Fig. 2). Upon overexpression of H-RASV12, human fibroblasts produce large amounts of O2˙̄ by NOX (45). Furthermore, in mouse embryonic fibroblasts lacking functional p53, expression of H-RASV12 or K-RASV12 increases production of mROS (46). The oncogenic K-RAS–induced mROS has been found to be necessary for the formation of lung adenocarcinoma and pancreatic pre-neoplastic lesions (43, 46). The increase in ROS production can also be driven by tumor hypoxia (11). Additionally, cancer cells can further potentiate ROS production by down-regulating antioxidant systems at the sites of ROS generation. Mitochondrial sirtuin (SIRT)-3 deacetylates and activates SOD2, regulating mitochondrial O2˙̄ levels (47). Thus, loss of SIRT3, as observed in many breast cancers, induces mROS accumulation, and HIF-1α stabilization (48).
Elevated intracellular ROS in cancer cells is also contingent on suppressing global antioxidant systems. Many tumor suppressors serve antioxidant functions in nontransformed cells. A redox-regulated DNA damage–sensing protein, ataxia-telangiectasia mutated (ATM) kinase, can inhibit ROS production (49–51). An ATM-regulated tumor suppressor, breast cancer type 1 susceptibility protein (BRCA1), interferes with KEAP1-mediated ubiquitination of NRF2, stabilizing and activating the master regulator of antioxidants (52). Tumor suppressor p53 is another potential activator of NRF2 and increases the expression of antioxidant enzymes, including SOD2 and GPX1, as well as production of NADPH, the reducing equivalent necessary to reactivate antioxidant systems (53–55). It is important to note that p53 can also reduce the expression of SLC7A11, which uptakes cysteine for GSH synthesis, thereby playing a pro-oxidant role under certain physiological contexts (56). However, the antioxidant function of p53 has been found to be necessary for its ability to prevent cancer (57). Therefore, loss of tumor suppressors promotes tumorigenesis by elevating intracellular ROS levels (Fig. 2).
Paradoxically, cancer cells concomitantly elevate their antioxidant capacity to buffer ROS from rising to levels that are toxic. Excessive amount of ROS can be detrimental to cancer cell viability and proliferation. Such vulnerability can posit a great challenge during metastasis as cancer cells detached from extracellular matrix experience elevated ROS levels (58). Moreover, blood and viscera are oxidizing environments hostile to the survival and proliferation of circulating cancer cells (59). As a result, excess ROS limits metastasis as well as tumorigenesis. Consequently, up-regulation of NRF2 has been observed in a broad spectrum of tumor types, including skin, lung, pancreas, breast, ovarian, and prostate (Fig. 2). Such NRF2 dysregulation can be mediated by loss-of-function mutations in KEAP1 or gain-of-function mutations in NRF2 that allow constitutive stabilization of NRF2 (60). Acquisition of the K-RAS, BRAF, and MYC oncogenes can also activate NRF2 antioxidant program. Importantly, NRF2 is required for the pancreatic and lung tumorigenesis in vivo (61). This has led to investigations on targeting NRF2-dependent cancers. Indeed, a recent study used chemical proteomics to identify a NRF2-regulated protein, nuclear receptor subfamily 0 group B member 1 (NROB1), as a druggable target in KEAP1-null nonsmall cell lung cancers (62).
GSH is another important antioxidant molecule for cancer cells. Elevation of GSH has been observed in tissues of breast, ovarian, head and neck, and lung cancer (Fig. 2) (63). The abundant GSH in tumor tissues is supported by the increased cellular availability of its biosynthetic precursors: glutamate, glycine, and cysteine. The cystine/glutamate antiporter SLC7A11, the main route of cysteine acquisition, is highly expressed in human tumors (56). Furthermore, the glutamate cysteine ligase modifier subunit, which is necessary for the efficient synthesis of GSH, is up-regulated across multiple types of human cancer (64). Aside from the de novo biosynthesis, another process that affects the cellular GSH level is its regeneration. Oxidized GSSG is reduced back to GSH by GR and NADPH. To facilitate this process, cancer cells up-regulate the production of NADPH, which will be discussed below. The elevation and maintenance of cellular GSH levels are essential for tumor initiation and proliferation (64, 65). Additionally, a GPX isoenzyme, GPX4, has been found to be required for the survival of therapy-resistant cancer cells, further highlighting the importance of GSH-mediated antioxidant pathways in cancer progression (Fig. 2) (66).
Cancer cells up-regulate the metabolic pathways necessary to produce the reducing potential, NADPH (Fig. 2). Oxidative PPP, one of the major sources of NADPH, branches from glycolysis. Certain regulatory enzymes of glycolysis, including glyceraldehyde-3-phosphate dehydrogenase (GAPDH), pyruvate kinase isoform M2 (PKM2), and TP53-induced glycolysis regulatory phosphatase (TIGAR), can redirect glycolytic intermediates into oxidative PPP to generate the reducing potential (67–70). Interestingly, such up-regulation of PPP by GAPDH and PKM2 is dependent on oxidation of specific cysteine residues (68, 69). The activators of PPP are often overexpressed in many types of cancer cells. Furthermore, the NADPH-generating/antioxidant capacity of TIGAR and PKM2 are essential for the development of intestinal and lung cancer, respectively (69, 70). On an important note, PKM2 can channel glycolytic precursors into another NADPH-generating pathway branching from glycolysis: one-carbon metabolism (71). One-carbon metabolism produces NADPH as phosphoglycerate dehydrogenase (PHGDH) catalyzes serine biosynthesis, and serine hydroxymethyltransferase (SHMT) subsequently incorporates a carbon unit from the serine into folate cycle. In the folate cycle, 5,10-methenyl-THF is oxidized by methylene-THF dehydrogenase, generating NADPH (72). When MYC-transformed cells are exposed to hypoxia, HIF-1α and MYC cooperatively increase expression of the mitochondrial isoform of SHMT, SHMT2. This facilitates production of mitochondrial NADPH, which contributes to taming the elevated mROS during hypoxia. Targeting SHMT2 reduced survival of the hypoxic transformed cells, which were rescued by the antioxidant N-acetylcysteine (73). This supports that hypoxic cancer cells up-regulate one-carbon metabolism and the mitochondrial NADPH level to evade ROS-induced cell death. The generation of mitochondrial NADPH is also important for metastasis. In anchorage-independent tumor spheroids, cytosolic IDH (IDH1), mitochondrial IDH (IDH2), and mitochondrial citrate transporter allow reductive carboxylation to generate NADPH in mitochondria. This process enables cells to maintain mitochondrial redox balance and evade the oxidative stress induced by detachment from extracellular matrix (74). Furthermore, folate cycle inhibition limits distant metastasis of melanoma cells in vivo (59), and targeting PHGDH abrogates the metastatic capacity of breast cancer cells (75).
Collectively, cancer cells expand both their pro-oxidant and antioxidant capacities. An example that illustrates the importance of this redox balance comes from the observation that adenomatous polyposis coli (APC)-deficient intestinal cells use aberrant WNT signaling to up-regulate both mitochondria and NOX-mediated ROS production and TIGAR-mediated antioxidant defense (76, 77). Elimination of either pro-oxidant or antioxidant driver attenuates the proliferation of the APC-deficient intestinal crypts in vivo. Yet intriguingly, a simultaneous removal of both synergizes to induce more severe proliferative defects (77). This suggest that NOX and TIGAR likely regulate independent ROS pools with opposing functions of pro-proliferation and anti-proliferation, respectively: NOX generates ROS that drive proliferation, whereas TIGAR limits the damaging ROS. The data are consistent with a model in which the ROS productions localized to the redox-regulated target proteins activate pro-tumorigenic signaling pathways, whereas the antioxidant pathways prevent accumulation of distant ROS from nonspecifically oxidizing macromolecules and inducing cell death (Fig. 2) (78). Further dissection of the molecular mechanisms regulating the redox balance in cancer cells with high spatial and temporal resolutions will provide a more comprehensive blueprint of the cancer redox biology.
Regulation of redox balance in T cells
T cells are critical for the establishment of host resistance against infectious agents or tumors. To initiate the T cell-mediated adaptive immunity, T cell receptors (TCRs) of naive CD4+ and CD8+ T cells engage with the peptide–major histocompatibility complex ligands displayed by antigen-presenting cells. The TCR stimulation activates multiple signaling pathways and transcription factors, enabling T cells to proliferate and acquire effector or regulatory functions. The T cell activation is accompanied by rapid generation of ROS (79), implying a crucial role of ROS in TCR signaling. This was first evidenced by an observation that primary T cells treated with antioxidants following TCR stimulation exhibit reduced interleukin (IL)-2 receptor expression and proliferation (80). Antioxidants also suppress the T cell expansion post-viral infection in vivo (81). Interestingly, such an immunosuppressive effect of antioxidants may be attributable to the necessity of redox signaling in MYC-dependent metabolic reprogramming. MYC is an essential transcription factor in activated T cells that up-regulates glycolysis and mitochondrial metabolism to generate the biosynthetic intermediates and ATP needed for cell growth and proliferation (82). A negative regulator of MYC, AMP-activated protein kinase (AMPK), has been found to be inhibited by ROS post-TCR engagement (83). Therefore, ROS potentially amplify the MYC-signaling pathway, inducing the metabolic shift, and thus the antigen-stimulated T cell expansion.
A major source of ROS production upon TCR stimulation is mitochondria (Fig. 3) (84). Indeed, the ROS generated by mitochondria are necessary for T cell activation and subsequent proliferation. TCR engagement induces a rapid influx of calcium to increase the release of mROS. T cells lacking complex III, one of the major sources of mROS, have limited activation of nuclear factor of activated T cells (NFAT), resulting in reduced IL-2 expression and proliferation (Fig. 3). Importantly, such a phenotype is rescued by exogenous H2O2, whereas wild-type T cells treated with a mitochondria-targeted antioxidant mimic the complex III-deficient T cells. These data strongly support that the complex III-generated mROS is required for the T cell response (85). In addition, mROS from complex I and glycerol-3-phosphate dehydrogenase contribute to IL-2 and IL-4 production through activation of NF-κB and activator protein (AP)-1 (86, 87). Furthermore, upon TCR stimulation, mitochondria translocate to immune synapses where increasing their generation of H2O2 is sufficient to potentiate the c-Jun N-terminal kinase (JNK) and MAPK signaling (88, 89). Taken together, mROS have a prominent signaling function during T cell activation.
Figure 3.
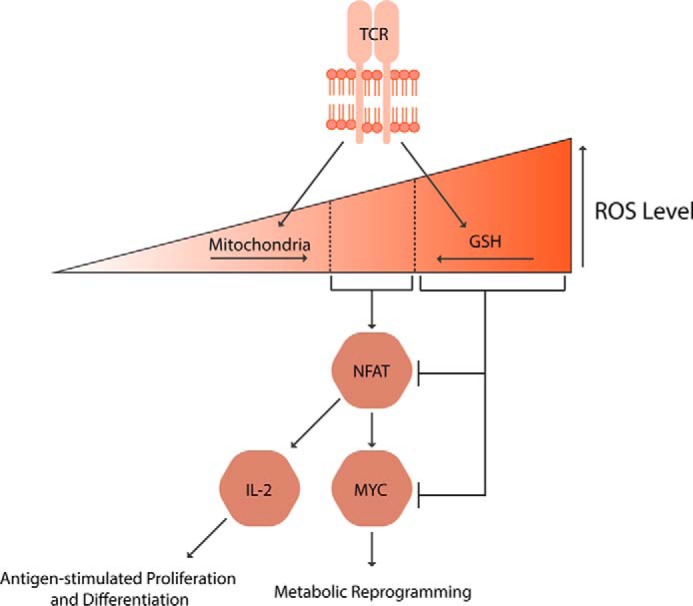
Regulation of redox balance in T cells. Both ROS generation by mitochondria and ROS scavenging by GSH are essential for the T cell activation. The ROS at the defined window activates nuclear factor of activated T cells (NFAT), which in turn induces IL-2 and MYC expressions, leading to T cell metabolic reprogramming, expansion, and differentiation into effector or regulator cells.
NOXs also contribute to T cell activation. The elevation of ROS generation induced by TCR stimulation is partly dependent on a phagocyte-type NOX isoform, NOX2. NOX2 can be activated by the TCR-triggered mROS (84), thereby maintaining the ROS levels to sustain T cell activation. Additionally, DUOX1, a nonphagocytic isoform of NOX, is an integral part of the redox signaling post-TCR stimulation. TCR engagement activates DUOX1, which is required for the phosphorylation of ζ-chain-associated protein kinase (ZAP)-70 and ERK, leading to secretion of interferon (IFN)-γ, TNF-α, IL-4, and IL-10 (90).
Although ROS is required for the T cell activation and subsequent expansion, excessive levels of ROS can jeopardize their viability. In the context of viral infection, elevated ROS productions by NOXs in granulocytes and macrophages impede the survival of infiltrating T cells, and thus the rate of viral clearance in vivo (91). Therefore, T cell functionality paradoxically depends on not only ROS but also the capacities to limit ROS. Activated T cells restrict ROS flow across the mitochondrial permeability transition pores to prevent excess mROS from entering the cytosol. Deregulation of the pore permeability leads to increased cell death upon TCR stimulation (92), supporting the necessity of ROS compartmentalization during T cell activation.
Intracellular GSH is another antioxidant defense pivotal for T cell functionality, particularly for its antigen-stimulated proliferative response (Fig. 3). The GSH was first implicated in T cell proliferation when l-buthionine-(S,R)-sulfoximine, an inhibitor of de novo GSH biosynthesis, was found to induce T cell cytosis (93). Subsequently, it was uncovered that antigen-presenting dendritic cells in contact with T cells secrete cysteine, a precursor of GSH, to be taken up by the T cells. When the cysteine release is inhibited, T cells are unable to proliferate post-TCR stimulation (94), implying the importance of GSH synthesis in the T cell response. Indeed, the de novo GSH biosynthesis is required to induce metabolic rewiring needed for the activated T cells to undergo rapid proliferation. T cells lacking the glutamate cysteine ligase catalytic subunit, which is necessary for GSH synthesis, exhibit limited activation of NFAT and the mechanistic target of rapamycin, resulting in a dramatic decrease of MYC expression. Consequently, the GSH deficiency impairs MYC-dependent metabolic reprogramming (Fig. 3). With the un-fulfilled biosynthetic and bioenergetic needs, the GSH-deficient T cells fail to proliferate post-activation and clear viral infection in vivo (95). Moreover, the necessity of GPX4, in T cell survival, maintenance, and antigen-stimulated expansion further highlights the crucial role of GSH-mediated antioxidant pathways in T cell functionality (96). Collectively, antioxidants, as well as localized production of mROS, are essential for the T cell-mediated adaptive immunity.
Looking forward
A central theme in redox biology is its translational potential. Can we manipulate the redox balance in medicine? Although ROS are necessary signaling molecules, they can turn cytotoxic at the wrong time, the wrong place, and the wrong amount. This duality of ROS leads healthy and diseased cells to rely on precisely coordinated regulations of both ROS generation and elimination. Therefore, to achieve therapeutic benefits with minimum adverse effects, it is crucial to target or foster ROS at the time and/or cellular location that selectively benefits diseased cells. For example, successful cancer redox therapy may target the localized ROS pool that potentiates pro-tumorigenic redox signaling while fostering distant ROS that induce oxidative damage to cancer cells (78). This therapeutic approach is made plausible by the recent advances in targeted antioxidants that scavenge ROS at specific sites such as mitochondrial complex I or complex III (97, 98). Furthermore, identification of cancer cell dependences on specific antioxidants such as GPX4 encourages the development of molecules that disable such antioxidant proteins to potentiate the distant damaging ROS (66, 99). Importantly, the dosage of the anti- and/or pro-oxidants must be regulated to preserve the redox signaling necessary for the healthy cells, including anti-tumor T cells. Similarly, immunomodulation therapy with anti- and/or pro-oxidants must be administered at specific timing that has become dysregulated and at a dose that does not impede normal immune responses. Therefore, significant medical advances could arise from an improved understanding of redox regulation with high temporal, spatial, and quantitative resolution.
Acknowledgment
We apologize for any references not cited due to space limitations.
This work was supported in part by National Institute of Health Grants 5R35CA197532-02, 5P01AG049665-03, and 5P01HL071643-13 (to N. S. C.). This is the second article in the Thematic Minireview Series “Redox metabolism and signaling.” The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ROS
- reactive oxygen species
- NOX
- NADPH oxidase
- SOD
- superoxide dismutase
- IDH
- isocitrate dehydrogenase
- PPP
- pentose phosphate pathway
- PRX
- peroxiredoxin
- TRX
- thioredoxin
- GPX
- glutathione peroxidase
- SHMT
- serine hydroxymethyltransferase
- PHGDH
- phosphoglycerate dehydrogenase
- APC
- adenomatous polyposis coli
- TCR
- T cell receptor
- ETC
- electron transport chain
- mROS
- mitochondrial ROS
- ATM
- ataxia-telangiectasia mutated
- NFAT
- nuclear factor of activated T cell
- GR
- glutathione reductase
- APC
- adenomatous polyposis coli
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- TIGAR
- TP53-induced glycolysis regulatory phosphatase.
References
- 1. Holmström K. M., and Finkel T. (2014) Cellular mechanisms and physiological consequences of redox-dependent signaling. Nat. Rev. Mol. Cell Biol. 15, 411–421 10.1038/nrm3801 [DOI] [PubMed] [Google Scholar]
- 2. Raimundo N., Song L., Shutt T. E., McKay S. E., Cotney J., Guan M. X., Gilliland T. C., Hohuan D., Santos-Sacchi J., and Shadel G. S. (2012) Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 148, 716–726 10.1016/j.cell.2011.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., Logan A., Nadtochiy S. M., Ord E. N. J., Smith A. C., Eyassu F., Shirley R., Hu C. H., Dare A. J., James A. M., et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brand M. D. (2016) Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 100, 14–31 10.1016/j.freeradbiomed.2016.04.001 [DOI] [PubMed] [Google Scholar]
- 5. Bedard K., and Krause K. H. (2007) The NOX family of ROS-generating NADPH oxidase: physiology and pathophysiology. Physiol. Rev. 87, 245–313 10.1152/physrev.00044.2005 [DOI] [PubMed] [Google Scholar]
- 6. Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chandel N. S. (2015) Navigating Metabolism, 1st Ed., Cold Spring Harbor Laboratory Press, pp. 37–84, Cold Spring Harbor, NY [Google Scholar]
- 8. Han D., Antunes F., Canali R., Rettori D., and Cadenas E. (2003) Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. chem. 278, 5557–5563 10.1074/jbc.M210269200 [DOI] [PubMed] [Google Scholar]
- 9. Martínez-Reyes I., Diebold L. P., Kong H., Schieber M., Huang H., Hensley C. T., Mehta M. M., Wang T., Santos J. H., Woychik R., Dufour E., Spelbrink J. N., Weinberg S. E., Zhao Y., Deberardinis R. J., et al. (2016) TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell 61, 199–209 10.1016/j.molcel.2015.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gottlieb E., Vander Heiden M. G., and Thompson C. B. (2000) Bcl-x(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor α-induced apoptosis. Mol. Cell. Biol. 20, 5680–5689 10.1128/MCB.20.15.5680-5689.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandel N. S., Maltepe E., Goldwasser E., Mathieu C. E., Simon M. C., and Schumacker P. T. (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. U.S.A. 95, 11715–11720 10.1073/pnas.95.20.11715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chouchani E. T., Kazak L., Jedrychowski M. P., Lu G. Z., Erickson B. K., Szpyt J., Pierce K. A., Laznik-Bogoslavski D., Vetrivelan R., Clish C. B., Robinson A. J., Gygi S. P., and Spiegelman B. M. (2016) Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 532, 112–116 10.1038/nature17399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Görlach A., Dimova E. Y., Petry A., Martínez-Ruiz A., Hernansanz-Agustín P., Rolo A. P., Palmeira C. M., and Kietzmann T. (2015) Reactive oxygen species, nutrition, hypoxia and diseases: problems solved? Redox Biol. 6, 372–385 10.1016/j.redox.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giorgio M., Migliaccio E., Orsini F., Paolucci D., Moroni M., Contursi C., Pelliccia G., Luzi L., Minucci S., Marcaccio M., Pinton P., Rizzuto R., Bernardi P., Paolucci F., and Pelicci P. G. (2005) Electron transfer between cytochrome c and p66Shc generated reactive oxygen species that trigger mitochondrial apoptosis. Cell 122, 221–233 10.1016/j.cell.2005.05.011 [DOI] [PubMed] [Google Scholar]
- 15. West A. P., Brodsky I. E., Rahner C., Woo D. K., Erdjument-Bromage H., Tempst P., Walsh M. C., Choi Y., Shadel G. S., and Ghosh S. (2011) TLR signaling augments macrophage bacterial activity through mitochondrial ROS. Nature 472, 476–480 10.1038/nature09973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Al-Mehdi A. B., Pastukh V. M., Swiger B. M., Reed D. J., Patel M. R., Bardwell G. C., Pastukh V. V., Alexeyev M. F., and Gillespie M. N. (2012) Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 5, ra47 10.1126/scisignal.2002712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yazdanpanah B., Wiegmann K., Tchikov V., Krut O., Pongratz C., Schramm M., Kleinridders A., Wunderlich T., Kashkar H., Utermöhlen O., Brüning J. C., Schütze S., and Krönke M. (2009) Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 460, 1159–1163 10.1038/nature08206 [DOI] [PubMed] [Google Scholar]
- 18. Bae Y. S., Oh H., Rhee S. G., and Yoo Y. D. (2011) Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 32, 491–509 10.1007/s10059-011-0276-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J. M., Fan L. M., Christie M. R., and Shah A. M. (2005) Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: role of p47phox phosphorylation and binding to TRAF4. Mol. Cell. Biol. 25, 2320–2330 10.1128/MCB.25.6.2320-2330.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Imai Y., Kuba K., Neely G. G., Yaghubian-Malhami R., Perkmann T., van Loo G., Ermolaeva M., Veldhuizen R., Leung Y. H., Wang H., Liu H., Sun Y., Pasparakis M., Kopf M., Mech C., et al. (2008) Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133, 235–249 10.1016/j.cell.2008.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen K., Kirber M. T., Xiao H., Yang Y., and Keaney J. F. Jr., (2008) Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 181, 1129–1139 10.1083/jcb.200709049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fridovich I. (1997) Superoxide anion radical (O2˙̄), superoxide dismutases, and related matters. J. Biol. Chem. 272, 18515–18517 10.1074/jbc.272.30.18515 [DOI] [PubMed] [Google Scholar]
- 23. Zelko I. N., Mariani T. J., and Folz R. J. (2002) Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 33, 337–349 [DOI] [PubMed] [Google Scholar]
- 24. Winterbourn C. C. (2013) The biological chemistry of hydrogen peroxide. Methods Enzymol. 528, 3–25 10.1016/B978-0-12-405881-1.00001-X [DOI] [PubMed] [Google Scholar]
- 25. Rhee S. G., Woo H. A., Kil I. S., and Bae S. H. (2012) Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 287, 4403–4410 10.1074/jbc.R111.283432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lubos E., Loscalzo J., and Handy D. E. (2011) Glutathione peroxidase-1 health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 15, 1957–1997 10.1089/ars.2010.3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brigelius-Flohé R., and Maiorino M. (2013) Glutathione peroxidases. Biochim. Biophys. Acta 1830, 3289–3303 10.1016/j.bbagen.2012.11.020 [DOI] [PubMed] [Google Scholar]
- 28. Taguchi K., Motohashi H., and Yamamoto M. (2011) Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 16, 123–140 10.1111/j.1365-2443.2010.01473.x [DOI] [PubMed] [Google Scholar]
- 29. Yates M. S., Tran Q. T., Dolan P. M., Osburn W. O., Shin S., McCulloch C. C., Silkworth J. B., Taguchi K., Yamamoto M., Williams C. R., Liby K. T., Sporn M. B., Sutter T. R., and Kensler T. W. (2009) Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 30, 1024–1031 10.1093/carcin/bgp100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sasaki H., Sato H., Kuriyama-Matsumura K., Sato K., Maebara K., Wang H., Tamba M., Itoh K., Yamamoto M., and Bannai S. (2002) Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 277, 44765–44771 10.1074/jbc.M208704200 [DOI] [PubMed] [Google Scholar]
- 31. McGrath-Morrow S., Lauer T., Yee M., Neptune E., Podowski M., Thimmulappa R. K., O'Reilly M., and Biswal S. (2009) Nrf2 increases survival and attenuates alveolar growth inhibition in neonatal mice exposed to hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, L565–L573 10.1152/ajplung.90487.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim Y. J., Ahn J. Y., Liang P., Ip C., Zhang Y., and Park Y. M. (2007) Human Prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 67, 546–554 10.1158/0008-5472.CAN-06-2401 [DOI] [PubMed] [Google Scholar]
- 33. Chorley B. N., Campbell M. R., Wang X., Karaca M., Sambandan D., Bangura F., Xue P., Pi J., Kleeberger S. R., and Bell D. A. (2012) Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor α. Nucleic Acids Res. 40, 7416–7429 10.1093/nar/gks409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., and Motohashi H. (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79 10.1016/j.ccr.2012.05.016 [DOI] [PubMed] [Google Scholar]
- 35. Reczek C. R., and Chandel N. S. (2015) ROS-dependent signal transduction. Curr. Opin. Cell Biol. 33, 8–13 10.1016/j.ceb.2014.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Finkel T. (2012) From sulfenylation to sulfhydration: what a thiolate needs to tolerate. Sci. Signal. 5, pe10 10.1126/scisignal.2002943 [DOI] [PubMed] [Google Scholar]
- 37. Lee S. R., Yang K. S., Kwon J., Lee C., Jeong W., and Rhee S. G. (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277, 20336–20342 10.1074/jbc.M111899200 [DOI] [PubMed] [Google Scholar]
- 38. Paulsen C. E., Truong T. H., Garcia F. J., Homann A., Gupta V., Leonard S. E., and Carroll K. S. (2011) Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 8, 57–64 10.1038/nchembio.736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sobotta M. C., Liou W., Stöcker S., Talwar D., Oehler M., Ruppert T., Scharf A. N., and Dick T. P. (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70 10.1038/nchembio.1695 [DOI] [PubMed] [Google Scholar]
- 40. Kil I. S., Lee S. K., Ryu K. W., Woo H. A., Hu M. C., Bae S. H., and Rhee S. G. (2012) Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell 46, 584–594 10.1016/j.molcel.2012.05.030 [DOI] [PubMed] [Google Scholar]
- 41. Seth D., and Rudolph J. (2006) Redox regulation of MAP kinase phosphatase 3. Biochemistry 45, 8476–8487 10.1021/bi060157p [DOI] [PubMed] [Google Scholar]
- 42. Guzy R. D., and Schumacker P. T. (2006) Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 91, 807–819 10.1113/expphysiol.2006.033506 [DOI] [PubMed] [Google Scholar]
- 43. Liou G. Y., Döppler H., DelGiorno K. E., Zhang L., Leitges M., Crawford H. C., Murphy M. P., and Storz P. (2016) Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 14, 2325–2336 10.1016/j.celrep.2016.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Porporato P. E., Payen V. L., Pérez-Escuredo J., De Saedeleer C. J., Danhier P., Copetti T., Dhup S., Tardy M., Vazeille T., Bouzin C., Feron O., Michiels C., Gallez B., and Sonveaux P. (2014) A mitochondrial switch promotes tumor metastasis. Cell Rep. 8, 754–766 10.1016/j.celrep.2014.06.043 [DOI] [PubMed] [Google Scholar]
- 45. Ogrunc M., Di Micco R., Liontos M., Bombardelli L., Mione M., Fumagalli M., Gorgoulis V. G., and d'Adda di Fagagna F. (2014) Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 21, 998–1012 10.1038/cdd.2014.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weinberg F., Hamanaka R., Wheaton W. W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G. M., Budinger G. R., and Chandel N. S. (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 8788–8793 10.1073/pnas.1003428107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tao R., Coleman M. C., Pennington J. D., Ozden O., Park S. H., Jiang H., Kim H. S., Flynn C. R., and Hill S., Hayes McDonald W., Olivier A. K., Spitz D. R., and Gius D. (2010) Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 40, 893–904 10.1016/j.molcel.2010.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Finley L. W., Carracedo A., Lee J., Souza A., Egia A., Zhang J., Teruya-Feldstein J., Moreira P. I., Cardoso S. M., Clish C. B., Pandolfi P. P., and Haigis M. C. (2011) SIRT3 oppose reprogramming of cancer cell metabolism through HIF-1α destabilization. Cancer Cell 19, 416–428 10.1016/j.ccr.2011.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. D'Souza A. D., Parish I. A., Krause D. S., Kaech S. M., and Shadel G. S. (2013) Reducing mitochondrial ROS improves disease-related pathology in a mouse model of ataxia telangiectasia. Mol. Ther. 21, 42–48 10.1038/mt.2012.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guo Z., Kozlov S., Lavin M. F., Person M. D., and Paull T. T. (2010) ATM activation by oxidative stress. Science 330, 517–521 10.1126/science.1192912 [DOI] [PubMed] [Google Scholar]
- 51. Alexander A., Cai S. L., Kim J., Nanez A., Sahin M., MacLean K. H., Inoki K., Guan K. L., Shen J., Person M. D., Kusewitt D., Mills G. B., Kastan M. B., and Walker C. L. (2010) ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. U.S.A. 107, 4153–4158 10.1073/pnas.0913860107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gorrini C., Baniasadi P. S., Harris I. S., Silvester J., Inoue S., Snow B., Joshi P. A., Wakeham A., Molyneux S. D., Martin B., Bouwman P., Cescon D. W., Elia A. J., Winterton-Perks Z., Cruickshank J., et al. (2013) BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 210, 1529–1544 10.1084/jem.20121337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen W., Sun Z., Wang X. J., Jiang T., Huang Z., Fang D., and Zhang D. D. (2009) Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 34, 663–673 10.1016/j.molcel.2009.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hussain S. P., Amstad P., He P., Robles A., Lupold S., Kaneko I., Ichimiya M., Sengupta S., Mechanic L., Okamura S., Hofseth L. J., Moake M., Nagashima M., Forrester K. S., and Harris C. C. (2004) p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 64, 2350–2356 10.1158/0008-5472.CAN-2287-2 [DOI] [PubMed] [Google Scholar]
- 55. Bensaad K., Tsuruta A., Selak M. A., Vidal M. N., Nakano K., Bartrons R., Gottlieb E., and Vousden K. H. (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120 10.1016/j.cell.2006.05.036 [DOI] [PubMed] [Google Scholar]
- 56. Jiang L., Kon N., Li T., Wang S. J., Su T., Hibshoosh H., Baer R., and Gu W. (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 10.1038/nature14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sablina A. A., Budanov A. V., Ilyinskaya G. V., Agapova L. S., Kravchenko J. E., and Chumakov P. M. (2005) The antioxidant function of the p53 tumor suppressor. Nat. Med. 11, 1306–1313 10.1038/nm1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schafer Z. T., Grassian A. R., Song L., Jiang Z., Gerhart-Hines Z., Irie H. Y., Gao S., Puigserver P., and Brugge J. S. (2009) Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 10.1038/nature08268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Piskounova E., Agathocleous M., Murphy M. M., Hu Z., Huddlestun S. E., Zhao Z., Leitch A. M., Johnson T. M., DeBerardinis R. J., and Morrison S. J. (2015) Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gorrini C., Harris I. S., and Mak T. W. (2013) Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 10.1038/nrd4002 [DOI] [PubMed] [Google Scholar]
- 61. DeNicola G. M., Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K. H., Yeo C. J., Calhoun E. S., Scrimieri F., Winter J. M., Hruban R. H., Iacobuzio-Donahue C., Kern S. E., et al. (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 10.1038/nature10189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bar-Peled L., Kemper E. K., Suciu R. M., Vinogradova E. V., Backus K. M., Horning B. D., Paul T. A., Ichu T. A., Svensson R. U., Olucha J., Chang M. W., Kok B. P., Zhu Z., Ihle N. T., Dix M. M., et al. (2017) Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer. Cell 171, 696–709 10.1016/j.cell.2017.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gamcsik M. P., Kasibhatla M. S., Teeter S. D., and Colvin O. M. (2012) Glutathione levels in human tumors. Biomarkers 17, 671–691 10.3109/1354750X.2012.715672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Harris I. S., Treloar A. E., Inoue S., Sasaki M., Gorrini C., Lee K. C., Yung K. Y., Brenner D., Knobbe-Thomsen C. B., Cox M. A., Elia A., Berger T., Cescon D. W., Adeoye A., et al. (2015) Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 10.1016/j.ccell.2014.11.019 [DOI] [PubMed] [Google Scholar]
- 65. Cramer S. L., Saha A., Liu J., Tadi S., Tiziani S., Yan W., Triplett K., Lamb C., Alters S. E., Rowlinson S., Zhang Y. J., Keating M. J., Huang P., DiGiovanni J., Georgiou G., et al. (2017) Systemic depletion of l-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 23, 120–127 10.1038/nm.4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Viswanathan V. S., Ryan M. J., Dhruv H. D., Gill S., Eichhoff O. M., Seashore-Ludlow B., Kaffenberger S. D., Eaton J. K., Shimada K., Aguirre A. J., Viswanathan S. R., Chattopadhyay S., Tamayo P., Yang W. S., Rees M. G., et al. (2017) Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 10.1038/nature23007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ralser M., Wamelink M. M., Kowald A., Gerisch B., Heeren G., Struys E. A., Klipp E., Jakobs C., Breitenbach M., Lehrach H., and Krobitsch S. (2007) Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 6, 10 10.1186/jbiol61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Peralta D., Bronowska A. K., Morgan B., Dóka É., Van Laer K., Nagy P., Gräter F., and Dick T. P. (2015) A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol. 11, 156–163 10.1038/nchembio.1720 [DOI] [PubMed] [Google Scholar]
- 69. Anastasiou D., Poulogiannis G., Asara J. M., Boxer M. B., Jiang J. K., Shen M., Bellinger G., Sasaki A. T., Locasale J. W., Auld D. S., Thomas C. J., Vander Heiden M. G., and Cantley L. C. (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283 10.1126/science.1211485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cheung E. C., Athineos D., Lee P., Ridgway R. A., Lambie W., Nixon C., Strathdee D., Blyth K., Sansom O. J., and Vousden K. H. (2013) TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev. Cell 25, 463–477 10.1016/j.devcel.2013.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ye J., Mancuso A., Tong X., Ward P. S., Fan J., Rabinowitz J. D., and Thompson C. B. (2012) Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 109, 6904–6909 10.1073/pnas.1204176109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Boroughs L. K., and DeBerardinis R. J. (2015) Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 17, 351–359 10.1038/ncb3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ye J., Fan J., Venneti S., Wan Y. W., Pawel B. R., Zhang J., Finley L. W., Lu C., Lindsten T., Cross J. R., Qing G., Liu Z., Simon M. C., Rabinowitz J. D., and Thompson C. B. (2014) Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 4, 1406–1417 10.1158/2159-8290.CD-14-0250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jiang L., Shestov A. A., Swain P., Yang C., Parker S. J., Wang Q. A., Terada L. S., Adams N. D., McCabe M. T., Pietrak B., Schmidt S., Metallo C. M., Dranka B. P., Schwartz B., and DeBerardinis R. J. (2016) Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258 10.1038/nature17393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Samanta D., Park Y., Andrabi S. A., Shelton L. M., Gilkes D. M., and Semenza G. L. (2016) PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res. 76, 4430–4442 10.1158/0008-5472.CAN-16-0530 [DOI] [PubMed] [Google Scholar]
- 76. Woo D. K., Green P. D., Santos J. H., D'Souza A. D., Walther Z., Martin W. D., Christian B. E., Chandel N. S., and Shadel G. S. (2012) Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am. J. Pathol. 180, 24–31 10.1016/j.ajpath.2011.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cheung E. C., Lee P., Ceteci F., Nixon C., Blyth K., Sansom O. J., and Vousden K. H. (2016) Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Genes Dev. 30, 52–63 10.1101/gad.271130.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chandel N. S., and Tuveson D. A. (2014) The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 371, 177–178 10.1056/NEJMcibr1405701 [DOI] [PubMed] [Google Scholar]
- 79. Devadas S., Zaritskaya L., Rhee S. G., Oberley L., and Williams M. S. (2002) Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J. Exp. Med. 195, 59–70 10.1084/jem.20010659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chaudhri G., Clark I. A., Hunt N. H., Cowden W. B., and Ceredig R. (1986) Effect of antioxidants on primary alloantigen-induced T cell activation and proliferation. J. Immunol. 137, 2646–2652 [PubMed] [Google Scholar]
- 81. Laniewski N. G., and Grayson J. M. (2004) Antioxidant treatment reduces expansion and contraction of antigen-specific CD8+ T cells during primary but not secondary viral infection. J. Virol. 78, 11246–11257 10.1128/JVI.78.20.11246-11257.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang R., Dillon C. P., Shi L. Z., Milasta S., Carter R., Finkelstein D., McCormick L. L., Fitzgerald P., Chi H., Munger J., and Green D. R. (2011) The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 10.1016/j.immuni.2011.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Previte D. M., O'Connor E. C., Novak E. A., Martins C. P., Mollen K. P., and Piganelli J. D. (2017) Reactive oxygen species are required for driving efficient and sustained aerobic glycolysis during CD4+ T cell activation. PLoS ONE 12, e0175549 10.1371/journal.pone.0175549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kaminski M., Kiessling M., Süss D., Krammer P. H., and Gülow K. (2007) Novel role for mitochondria: protein kinase Cθ-dependent oxidative signaling organelles in activation-induced T-cell death. Mol. Cell. Biol. 27, 3625–3639 10.1128/MCB.02295-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sena L. A., Li S., Jairaman A., Prakriya M., Ezponda T., Hildeman D. A., Wang C. R., Schumacker P. T., Licht J. D., Perlman H., Bryce P. J., and Chandel N. S. (2013) Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236 10.1016/j.immuni.2012.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kaminski M. M., Sauer S. W., Klemke C. D., Süss D., Okun J. G., Krammer P. H., and Gülow K. (2010) Mitochondrial reactive oxygen species control T cells activation by regulating IL-2 and IL-4 expression: mechanism of ciprofloxacin-mediated immunosuppression. J. Immunol. 184, 4827–4841 10.4049/jimmunol.0901662 [DOI] [PubMed] [Google Scholar]
- 87. Kamiński M. M., Sauer S. W., Kamiński M., Opp S., Ruppert T., Grigaravičius P., Grudnik P., Gröne H. J., Krammer P. H., and Gülow K. (2012) T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2, 1300–1315 10.1016/j.celrep.2012.10.009 [DOI] [PubMed] [Google Scholar]
- 88. Baixauli F., Martín-Cófreces N. B., Morlino G., Carrasco Y. R., Calabia-Linares C., Veiga E., Serrador J. M., and Sánchez-Madrid F. (2011) The mitochondrial fission factor dynamin-related protein 1 modulates T-cell receptor signaling at the immune synapse. EMBO J. 30, 1238–1250 10.1038/emboj.2011.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gill T., and Levine A. D. (2013) Mitochondria-derived hydrogen peroxide selectively enhances T cell receptor initiated signal transduction. J. Biol. Chem. 288, 26246–26255 10.1074/jbc.M113.476895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kwon J., Shatynski K. E., Chen H., Morand S., de Deken X., Miot F., Leto T. L., and Williams M. S. (2010) The nonphagocytic NADPH oxidase Duox1 mediates a positive feedback loop during T cell receptor signaling. Sci. Signal. 3, ra59 10.1126/scisignal.2000976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lang P. A., Xu H. C., Grusdat M., McIlwain D. R., Pandyra A. A., Harris I. S., Shaabani N., Honke N., Maney S. K., Lang E., Pozdeev V. I., Recher M., Odermatt B., Brenner D., Häussinger D., et al. (2013) Reactive oxygen species delay control of lymphocytic choriomeningitis virus. Cell Death Differ. 20, 649–658 10.1038/cdd.2012.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang B., Liu S. Q., Li C., Lykken E., Jiang S., Wong E., Gong Z., Tao Z., Zhu B., Wan Y., and Li Q. J. (2016) MicroRNA-23a curbs necrosis during early T cell activation by enforcing intracellular reactive oxygen species equilibrium. Immunity 44, 568–581 10.1016/j.immuni.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hamilos D. L., Zelarney P., and Mascali J. J. (1989) Lymphocyte proliferation in glutathione-depleted lymphocytes: direct relationship between glutathione availability and the proliferative response. Immunopharmacology 18, 223–235 10.1016/0162-3109(89)90020-9 [DOI] [PubMed] [Google Scholar]
- 94. Angelini G., Gardella S., Ardy M., Ciriolo M. R., Filomeni G., Di Trapani G., Clarke F., Sitia R., and Rubartelli A. (2002) Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc. Natl. Acad. Sci. U.S.A. 99, 1491–1496 10.1073/pnas.022630299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mak T. W., Grusdat M., Duncan G. S., Dostert C., Nonnenmacher Y., Cox M., Binsfeld C., Hao Z., Brüstle A., Itsumi M., Jäger C., Chen Y., Pinkenburg O., Camara B., Ollert M., et al. (2017) Glutathione primes T cell metabolism for inflammation. Immunity 46, 675–689 10.1016/j.immuni.2017.03.019 [DOI] [PubMed] [Google Scholar]
- 96. Matsushita M., Freigang S., Schneider C., Conrad M., Bornkamm G. W., and Kopf M. (2015) T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 212, 555–568 10.1084/jem.20140857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Orr A. L., Vargas L., Turk C. N., Baaten J. E., Matzen J. T., Dardov V. J., Attle S. J., Li J., Quackenbush D. C., Goncalves R. L., Perevoshchikova I. V., Petrassi H. M., Meeusen S. L., Ainscow E. K., and Brand M. D. (2015) Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 11, 834–836 10.1038/nchembio.1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Brand M. D., Goncalves R. L., Orr A. L., Vargas L., Gerencser A. A., Borch Jensen M., Wang Y. T., Melov S., Turk C. N., Matzen J. T., Dardov V. J., Petrassi H. M., Meeusen S. L., Perevoshchikova I. V., Jasper H., et al. (2016) Suppressors of superoxide-H2O2 production at site IQ of mitochondrial complex I protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab. 24, 582–592 10.1016/j.cmet.2016.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hangauer M. J., Viswanathan V. S., Ryan M. J., Bole D., Eaton J. K., Matov A., Galeas J., Dhruv H. D., Berens M. E., Schreiber S. L., McCormick F., and McManus M. T. (2017) Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 10.1038/nature24297 [DOI] [PMC free article] [PubMed] [Google Scholar]