

Abstract
Codon usage bias affects the genomes of organisms from all kingdoms of life and results from both background substitution biases and natural selection. Natural selection on codon usage to increase translation accuracy and efficiency has long been known to affect gene sequences. Such selection is stronger on highly, compared with lowly expressed genes, resulting in higher levels of codon bias within genes with higher expression levels. Additionally, selection on translation accuracy affects more strongly codons encoding conserved amino acids, since these will more often affect protein folding and/or function. By applying tests of selection on the gene sequences of the bacterium Escherichia coli, we demonstrate that both highly and lowly expressed genes display signals of selection on codon usage. Such signals are found for both conserved and less conserved amino acid positions, even within the 10% of E. coli genes expressed at the lowest levels. We further demonstrate experimentally that single synonymous codon replacements within a lowly expressed, essential gene can carry substantial effects on bacterial fitness. Combined, our results demonstrate that even within genes expressed at relatively low levels there is substantial selection on codon usage and that single synonymous codon replacements within such genes can have a marked effect on bacterial fitness.
Keywords: codon usage, lowly expressed genes, evolution, competition experiments, fitness, natural selection
Introduction
The genetic code consists of 61 codons encoding only 20 amino acids. This results in the encoding of 18 of the amino acids by more than a single codon. Each of these 18 amino acids is encoded by 2–6 different codons, which are referred to as synonymous to each other. The synonymous codons encoding the same amino acid are sometimes referred to as a codon family. Codon families consisting of two or four members that differ only in their third nucleotide are referred to, respectively, as 2-fold- or 4-fold-degenerate codon families. Codon usage is biased, meaning that within codon families some synonymous codons are more frequently used than others (Grantham et al. 1980). These biases result from the background substitution biases of a genome as well as from natural selection that favors the use of specific synonymous codons over others (Shields and Sharp 1987; Hershberg and Petrov 2008).
Natural selection favors the use of certain synonymous codons over others due to a variety of reasons. The major and most well studied of reasons is variation in the abundance of different tRNA molecules within the cell. The usage of codons recognized by more abundant tRNAs (referred to as preferred codons) enables faster recognition of the codons by their tRNA molecules (Varenne et al. 1984; Hershberg and Petrov 2008; Gingold and Pilpel 2011). This in turn facilitates more efficient translation elongation resulting in less ribosomal delay and less protein misfolding (Zhou et al. 2009; Tuller et al. 2010). Usage of preferred codons also decreases the chance of errors in the incorporation of amino acids during translation (Akashi 1994). When the proteome as a whole is translated in an efficient and accurate manner, this results in a reduction in global translation costs.
The codon usage of highly expressed genes is expected to affect global translation costs much more than the codon usage of more lowly expressed genes (Hershberg and Petrov 2008; Gingold and Pilpel 2011). A highly expressed gene that is translated in an inefficient manner should lead to a much stronger sequestering of ribosomes, greatly reducing the pool of free ribosomes available for the translation of other genes. At the same time, the inaccurate translation of a highly expressed gene may result in a higher number of mistranslated and/or misfolded proteins, which could overload the cell’s degradation mechanisms or aggregate and disrupt cell functions (Gregersen 2006). Fitting with this, changes in the codon usage of several highly expressed genes were shown to affect cellular fitness (Carlini 2004; Agashe et al. 2013; Firnberg et al. 2014; Brandis and Hughes 2016; Hauber et al. 2016; Knöppel et al. 2016). As expected from the stronger effect of codon usage in highly expressed genes on global translation costs, highly expressed genes tend to display much higher codon bias than lowly expressed genes (Bennetzen and Hall 1982; Gouy and Gautier 1982; Hershberg and Petrov 2008).
Although it is reasonable to predict that selection on codon usage is stronger on highly expressed genes, some evidence indicates that selection on codon usage may also affect genes with lower expression levels (dos Reis and Wernisch 2009; Zhou et al. 2009). Zhou et al. have found a relationship between protein structure and codon usage (Zhou et al. 2009). Specifically, they found that preferred codons are more frequently used at buried protein residues and at protein sites at which mutations cause larger changes in free energy. Such an association may be a signal of selection on translation accuracy, as it demonstrates that preferred codons are more frequently used at sites that are more important for protein folding. Zhou et al. found this association between preferred codon usage and protein structure is true for both highly and lowly expressed gene, although it is stronger for highly expressed genes.
Here, we add to the evidence that selection on translation accuracy affects the codon usage of genes that are lowly expressed, by carrying out the Akashi test (Akashi 1994) and demonstrating higher usage of preferred codons at positions encoding conserved amino acids. We then further demonstrate that both positions encoding conserved and positions encoding less conserved amino acids are subject to selection on codon usage, even in the 10% of genes expressed at the lowest levels within the E. coli genome. Finally, we experimentally demonstrate, for the first time, that single synonymous codon replacements within an essential yet lowly expressed gene can significantly affect bacterial fitness.
Materials and Methods
Data Sets
All genomes used in this study were downloaded from the NCBI RefSeq genomes FTP: ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/ with the accession numbers: GCA_000005845.2 (Escherichia coli str. K-12 substr. MG1655), GCA_000474015.1 (Klebsiella pneumoniae CG43), GCA_000493535.1 (Salmonella enterica subsp. enterica serovar Typhimurium str. DT2), and GCA_000834315.1 (Yersinia pestis Pestoides F).
The gene expression data were combined from two publically available, normalized expression data sets (Faith et al. 2008; Hu et al. 2009). Expression data for genes for which data was available from both sources was averaged across both sources. In cases in which only one database provided data of expression for a gene, expression was estimated based on that database.
Test for Selection on Translation Accuracy (Akashi Test)
Each protein coding sequence from the E. coli genome was compared against the genomes of K. pneumoniae, S. typhimurium, and Y. pestis, using FASTA (Pearson and Lipman 1988). The best-obtained matching sequence was then compared against the E. coli genome in the same manner. Only the 2000 best-aligned protein-coding sequences from the E. coli genome that had a reciprocal best match in each of the three other genomes were taken for the following step.
A multiple sequence alignment (MSA) was subsequently generated for each of the obtained groups of orthologous protein-coding sequences using Muscle (Edgar 2004). Based on these alignments, the amino acids of the E. coli sequence were divided into two categories—conserved, if they were fully conserved across the four species, and variable if not.
Three methods were used to assign E. coli preferred codons:
The correlation method: For each codon family, the preferred codon is assigned as the one whose frequency within a gene most significantly positively correlates with the overall codon bias of the gene. If the frequency of neither codon within a codon family significantly correlates with overall levels of codon bias, no preferred codon is assigned for that codon family. We previously assigned preferred codons of hundreds of bacterial genomes using this method (Hershberg and Petrov 2009). In the current study, we used our preferred codon assignments for E. coli str. K-12 substr. MG1655 from this previous study. We were able to assign preferred codons for this bacterium for all 18 amino acids encoded by more than a single codon.
Ribosomal enrichment method: For each codon family, the preferred codon is assigned as the codon within that codon family that is most frequently used within ribosomal genes. Ribosomal gene sequences were extracted based on E. coli str. K-12 substr. MG1655 genome annotations.
tRNA copy number method: According to this method the preferred codon of each codon family is the one that is recognized by the tRNA present within the genome at the highest copy number. tRNA copy numbers for the E. coli str. K-12 substr. MG1655 genome were extracted from (Chan and Lowe 2009). The tRNA copy number method cannot always be used to identify a single preferred codon, as several codons may be recognized by tRNAs with equal copy numbers. In the case of E. coli str. K-12 substr. MG1655 we were able to assign preferred codons, using this method, for only 15 of the 18 amino acids for which there is more than one synonymous codon.
Preferred codons identified according to each of these three methods are summarized in supplementary table S1, Supplementary Material online.
Of the 2,000 reciprocally matched E. coli protein-coding sequences used in our analyses only 1,850 had expression data. These 1,850 protein-coding sequences were divided into ten equally sized groups according to their expression levels. For each of the 18 amino acids that are encoded by more than one codon, in each of the 1,850 gene sequences, a contingency table was generated, with amino acid conservation and codon preference as variables.
Finally, the Mantel–Haenszel estimate for the common odds ratio ( (Mantel and Haenszel 1959; Mantel 1963) was calculated according to the following formula:
Where, is the calculated odds ratio for a group of genes; the index j represents each of the genes, contained within this group and the index i represents each of the 18 amino acids that are encoded by more than a single codon. a is the number of times an amino acid is conserved and encoded by preferred codon, b is the number of times an amino acid is conserved and encoded by nonpreferred codon, c is the number of times an amino acid is variable and encoded by preferred codon and d is the number of times an amino acid is variable and encoded by nonpreferred codon. Finally, n is the sum of a–d.
In order to control for differences in the number of conserved and variable sites between expression groups, we repeated the above test after randomly drawing 5,000 conserved and 5,000 variable amino acid positions from each of the ten expression bins. Contingency tables were generated based on each such draw and the Mantel–Haenszel estimate and its significance was calculated for each draw and expression bin, based on its contingency tables as described above. Each random draw was repeated 10,000 times and the odds ratios were averaged across these draws. Figure 1b depicts these averages, across expression bins, with their standard deviations.
Fig. 1.

—Selection on translation accuracy affects both lowly and highly expressed genes in E. coli. Presented are the Akashi test odds ratios calculated for the association between conserved protein positions and the usage of preferred codons at those positions. (a) Akashi test odds ratio was calculated using the entire protein-coding gene sequences. (b) Average Akashi test odds ratio calculated from 10,000 random samplings of 5,000 conserved and 5,000 variable codons from each expression bin. Error bars represent the standard deviation across the mean for the 10,000 samplings.
Testing for Selection on Variable Amino Acid Sites
In order to examine whether variable positions of protein-coding sequences use a higher fraction of preferred codons than expected based on background substitution biases alone, we sought to generate randomized gene sequences in which codon usage was determined based on the composition of adjacent intergenic sequences. To generate these randomized sequences, we followed the following procedure as described in Hershberg and Petrov (2009). 1) The first 100 4-fold degenerate and 2-fold degenerate codons of each protein-coding gene were extracted. Genes that had <100 2-fold and 4-fold degenerate codons were removed from consideration (This was done in order to weigh genes equally). 2) For each protein-coding gene its two adjacent intergenic sequences were extracted and concatenated. From this concatenated sequence a 100 base-pair segment was selected at random and randomly shuffled. intergenic regions shorter than 50 bases were removed from consideration, and if for a gene there was not at least 100 bases of adjacent intergenic region, the gene in its entirety was removed from the analysis. 3) Random gene sequences were generated using the real coding sequences as a backbone and replacing the third codon positions, based on the shuffled adjacent intergenic sequences, whereas maintaining the encoded protein sequence. In the case of a 2-fold degenerate codon family such as the Tyrosine, TA(T/C), a TAC was selected if the corresponding intergenic position contained either a G or a C and TAT if the corresponding intergenic position contains an A or a T. As with the Akashi test above, we divided protein-coding genes into ten equally sized bins, based on their gene expression levels. Each bin contained 67 gene sequences. For each of the 14 2-fold and 4-fold amino acids in each expression level group, the amino acid sites were separated to conserved and variable, and separate contingency tables were generated, with sequence origin (real or randomized) and codon preference as variables. The Mantel–Haenszel estimate for the common odds ratio and its significance was calculated, as described above in the Akashi test section, based on the contingency tables of each expression level group for variable and for conserved amino acid sites separately. Generation of random protein-coding sequences and subsequent statistical testing was repeated 10,000 times. Figure 2 depicts the average odds ratio across these 10,000 tests as well as its standard deviation, for each expression bin, separately for conserved and variable protein positions.
Fig. 2.
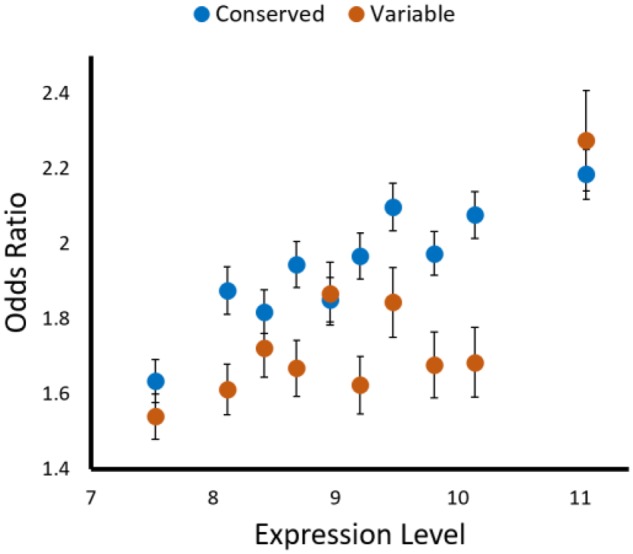
—For both lowly and highly expressed genes, preferred codons are enriched relative neutral expectations at both conserved and variable protein sites. For each protein-coding gene sequence, a set of 10,000 randomized gene sequences in which codon usage was determined based on the composition of adjacent intergenic sequences was generated. These randomized sequences maintained the amino acid sequence of the original gene, but had a frequency of preferred codons that was determined by background substitution biases alone (see Materials and Methods). Presented are the average odds ratio for the association between the frequencies of preferred or nonpreferred codons and the sequence being real or randomized, as a function of expression level. Error bars represent the standard deviation across the mean for the 10,000 sequence randomizations.
Material Sources
All primers were purchased from Integrated DNA Technologies (IDT).
Molecular biology plasmids and kits were purchased from New England Biolabs (NEB) unless otherwise mentioned.
Strain Constructions
Replacement of the Native Promoter with a Weak Promoter
The natural P3 promoter located upstream to the bla gene in the plasmid pBR322 was replaced with the weak promoter J23103 that was chosen from the Anderson promoter collection (Anderson 2009). A pair of primers each containing half of the promoter were designed (supplementary table S2, Supplementary Material online). PCR was carried out with a Q5 polymerase and the PCR products were then ligated with a T4 DNA ligase.
Site-Directed Mutagenesis
Primers with partial overlap were designed for the two synonymous replacements (supplementary table S2, Supplementary Material online). PCR was carried out with a Q5 polymerase while the pBR322 plasmid with the modified promoter was used as the template.
Linear Transformation
A DNA fragment containing the weak promotor upstream the WT bla gene or the synonymous mutated bla gene, was linearly transformed (Datsenko and Wanner 2000) into the chromosomal region 3635970–3636206 of previously generated E. coli K12 MG1655 strains, containing antibiotic resistance cassettes (either kanamycin or chloramphenicol) (Katz and Hershberg 2013). Primers with their sequence partially complementary to the MG1655 genome were designed (supplementary table S2, Supplementary Material online) and the induction of the lambda red system was carried out with the plasmid pKD119 (Coli Genetic Stock Center).
Following each step of the procedure and at the procedures end, Sanger sequencing was used to verify that the desired sequence was indeed obtained.
Western Blot Analysis
To verify that indeed the bla gene controlled by the weak promoter is expressed at much lower levels than the gene controlled by its native promoter, Western blot analyses were carried out. This enabled us to compare protein levels obtained when the chromosomally residing gene was controlled by the weak promoter to the levels obtained when the gene was controlled by its native promoter (supplementary fig. S4, Supplementary Material online). Periplasmatic beta-lactamase proteins were extracted from E. coli cells as previously described (BÜDeyrİ GÖKgÖZ et al. 2015). The proteins were run on 12% SDS PAGE and transformed onto a nitrocellulose membrane or used for Comassie staining. Following transformation the membrane was blotted with 5% nonfat milk and then probed with 1: 200 mouse monoclonal anti beta-lactamase antibody (Santa Cruz Biotechnology, Inc). Goat antimouse HRP was used as the second antibody (Jackson ImmunoResearch Laboratories, Inc).
Competition Experiments
In order to measure relative fitness, we carried out competition experiments. For these experiments, two strains were competed: a strain with the WT bla gene and an additional antibiotic resistance cassette (either kanamycin or chloramphenicol), and a strain having a synonymously mutated bla gene as well as the other additional antibiotic resistance cassette. Reciprocal competitions with the two antibiotic resistance cassettes between the two strains were carried out in order to eliminate potential fitness effects of the marker kan and chl cassettes. In each competition experiment, the two competing strains were grown separately to an O.D of 0.2, and were then diluted 1:100 into the same 2 ml well containing Mueller–Hinton (MH) medium and 50 mg/l ampicillin and incubated for 7 h at 37°C. To determine the initial relative frequencies of each strain, a sample from each well, prior to the incubation, was serially diluted and plated in duplicates on Luria Broth (LB) agar plates supplemented with either kanamycin (50 µg/ml) or chloramphenicol (25 µg/ml). Plates were incubated overnight allowing for the growth of visible colonies and colony-forming units (CFU) were then quantified by colony counting. To minimize counting errors and noise, plates containing <30 colonies or over 400 colonies were discarded. Following the 7 h incubation of the competing bacteria, the final relative abundances of each strain were determined as was done prior to the incubation, through plating on plates containing kan or chl and calculating CFU.
From the resulting initial and final CFU counts, the relative fitness was calculated according to:
Where W is the relative fitness and N is the CFU number. The i and f subscripts denote the initial and final measurements and the superscripts denote the W.T and mutated genes. W values for each separate experiment are summarized in supplementary table S7, Supplementary Material online. To test whether W is significantly higher than 1 across a group of experiments, we used a Mann–Whitney paired, one-tailed test. The mean values of W for each group of experiments, the number of independent experiments carried out and the resulting P-values are summarized in table 1.
Table 1.
Single Synonymous Codon Replacements within a Lowly Expressed Beta Lactamase Gene Significantly Affect Relative Fitness in the Presence of Ampicillin
| mut kan/W.T chl |
mut chl/W.T kan |
mut/W.T |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Relative fitnessa | P valueb | nc | Relative fitnessa | P valueb | nc | Relative fitnessa | P valueb | nc | |
| S68 (AGC→TCC) | 1.021 | 0.0375 | 17 | 1.073 | 0.0004 | 17 | 1.047 | <0.0001 | 34 |
| S128 (AGT→TCT) | 1.065 | 0.015 | 16 | 1.063 | 0.0174 | 18 | 1.064 | 0.0005 | 34 |
Relative fitness of mutant versus wildtype, calculated as specified in the Materials and Methods.
P values according to a Mann–Whitney paired one-sided test.
n = number of independent experiments carried out.
Results
Both Lowly and Highly Expressed Genes are Subject to Selection on Translation Accuracy in E. coli
The most well accepted test for natural selection on translation accuracy was developed by Hiroshi Akashi (Akashi 1994). This test examines whether preferred codons are used more frequently at sites encoding amino acids that are more conserved in evolution. The assumption of Akashi’s test is that amino acids that are more evolutionarily conserved are more often important for protein folding and/or function. Hence, it would be more crucial to translate such important sites accurately. Therefore, if selection indeed exists for translation accuracy, conserved amino acid sites would more frequently be encoded by preferred codons. Akashi’s test was previously applied on many different genomes, demonstrating the effects of selection for translation accuracy in mammals, flies and bacteria (Akashi 1994; Stoletzki and Eyre-Walker 2007; Drummond and Wilke 2008).
In order to examine whether E. coli genes, expressed at different levels, are subject to selection on translation accuracy, we applied the Akashi test on E. coli gene sequences (Materials and Methods). To perform the Akashi test, amino acid positions need to be classified according to their evolutionary conservation. We therefore focused on 2000 E. coli protein-coding genes that best aligned to their orthologs in three other Enterobacteria genomes (Salmonella enterica, Klebsiella pneumonia and Yersinia pestis, see Materials and Methods). Based on the alignments of these protein-coding genes we classified each amino acid as evolutionarily conserved, if it was identical across all four species or variable, if it was not.
To carry out the Akashi test the codons encoding each amino acid residue must also be classified as preferred or nonpreferred. We considered three major methods of identification of preferred codons. The first method is referred to as the “correlation method.” According to the “correlation method”, preferred codons of each codon family are those whose frequency of usage within genes most significantly positively correlates with the overall levels of codon bias of the gene (Hershberg and Petrov 2009). The second method (“ribosomal enrichment method”) examines which codons are most frequently used within highly expressed ribosomal genes. The third method we considered for preferred codon assignment is the “tRNA copy number method.” According to this method, the preferred codon of each codon family is the one that is recognized by the tRNA present within the genome at the highest copy number. If indeed tRNA copy number correlates with the abundance of a tRNA molecule within the cell (Gingold and Pilpel 2011; Quax et al. 2015), this may be the most reliable method of all. However, sometimes this method cannot be used to identify a single preferred codon, as several codons may be recognized by tRNAs with equal copy numbers. Indeed, in E. coli we are unable to use the “tRNA copy number method” to assign codons for three out of the 18 amino acids for which there is more than one synonymous codon.
Supplementary table S1, Supplementary Material online summarizes the preferred codon assignments made by each of the three methods. As can be seen from this table, for 11 of the 18 codon families all three methods assign the same preferred codon. The “correlation method” and the “tRNA copy number method” disagree in the assignment of preferred codons for only two codon families, whereas the “tRNA copy number method” and the “ribosomal enrichment method” disagree when it comes to the same two codon families as well as an additional two. This, as well as our previous analyses (Hershberg and Petrov 2012), led us to have more confidence in the assignments made by the “correlation method” (compared with those made by the “ribosomal enrichment method”). Since the “tRNA copy number method” does not allow for the assignment of preferred codons for all 18 codon families, we first performed the Akashi test by classifying codons as preferred or nonpreferred using the “correlation method.” To make certain that the manner in which we chose to assign codons as preferred or nonpreferred does not significantly affect our conclusions we also repeated the analyses described below using only the 11 codon families for which all three methods assign the same preferred codon. Our results remained entirely consistent with what we report below (see Supplementary Material).
To examine whether genes with various expression levels show a signal of selection on translation accuracy, we performed the Akashi test on groups of protein-coding genes divided by their expression levels, based on microarray data (Faith et al. 2008; Hu et al. 2009). Of the 2,000 genes with the best sequence alignments, 1,850 had expression data. We divided these 1,850 genes into ten equally sized groups, based on their levels of expression. For each group, Akashi’s test odds ratio was calculated (Materials and Methods). When preferred codons are used significantly more frequently at sites encoding conserved amino acids, this would be reflected in an odds ratio value that is significantly higher than 1. We found that all groups, including those with the lowest expression level, significantly passed the Akashi test (had an odds ratio significantly >1, fig. 1a andsupplementary table S3, Supplementary Material online). At the same time groups with higher expression levels had larger, and more significant odds ratios compared with the lower expression level groups (fig. 1a andsupplementary table S3, Supplementary Material online). This indicates that selection on translation accuracy affects both highly expressed and lowly expressed genes, although it may tend to affect highly expressed genes more strongly.
Proteins belonging to the different expression groups differ in their length (supplementary fig. S2, Supplementary Material online), and in the proportion of evolutionary conserved and variable amino acids they contain. This might affect the extent to which the various groups of genes pass the Akashi test. Therefore, we repeated the Akashi test calculations after randomly drawing the same number (5,000) of conserved and variable amino acids for each of the ten expression level groups. The random sampling of conserved and variable amino acids and their codons was carried out 10,000 times, and the odds ratio for each sampling as well as the average odds ratio, across these 10,000 comparisons was calculated. This average odds ratio was higher than 1 across all bins (fig. 1b). The percentage of samplings for which a significantly higher than 1 odds ratio was obtained (at P < 0.05) varied across expression bins (supplementary table S5, Supplementary Material online). However, across expression bins, at the least 74% of samplings resulted in an odds ratio value that was significantly higher than 1. At random, only 5% of samplings would be expected to obtain such significant odds ratio values. Therefore, even when eliminating the difference in length and conservation of the various expression bins, a significant signal of selection on translation accuracy remains for both lowly and highly expressed genes.
Selection on Codon Usage Also Affects Less Conserved Protein Positions, Even in Lowly Expressed Genes
Next, we wanted to examine whether positions encoding less conserved amino acids within lowly expressed genes are also subject to selection on codon usage. Codon usage can be affected by both natural selection, favoring the usage of preferred codons, and by the background substitution biases of the genome, which drive towards using more GC rich or more AT rich codons (Knight et al. 2001; Chen et al. 2004; Hershberg and Petrov 2008, 2009). To examine whether there is selection on codon usage within positions encoding both conserved and variable amino acids, we asked whether such positions are enriched for preferred codons, relative what would be expected according to the background substitution biases of the E. coli genome. To do so, we generated for each E. coli protein-coding gene randomly perturbed synonymous sequences. In these perturbed sequences the 2- and 4-fold codon third positions were replaced with a randomly drawn nucleotide from the protein’s adjacent intergenic sequence (while maintaining the protein sequence of the gene, Materials and Methods). To make certain that genes are weighted equally, from each such synonymous sequence we extracted the first 100 2- and 4-fold codon positions. For each protein coding sequence, we generated 10,000 such randomly perturbed segments.
Protein-coding genes were divided into ten equally sized groups, according to their expression levels. For each expression level, we could then compare the fraction of preferred codons found within the “real” protein-coding segments to those found in each of the 10,000 sets of corresponding randomly perturbed segments. To examine whether preferred codons were used more frequently in the “real” segments, relative the perturbed ones, we calculated Mantel–Haenszel odds ratio values (see Materials and Methods). Prior to these calculations we further classified the amino acid positions contained within each gene segment based on conservation (as in our Akashi test classifications, Materials and Methods). This enabled us to examine whether preferred codons were used more frequently than expected based on background substitution biases alone, separately for conserved and variable amino acid positions. We found that for both conserved and variable amino acid positions the individual odds ratio values calculated for each of the 10,000 segment comparisons, for each expression bin were always significantly >1 (P ≪ 0.001). Figure 2 depicts the mean odd ratio values across all 10,000 comparisons, for each of the ten expression level bins, separately for conserved and variable amino acid positions. These results demonstrate, as we have previously shown using Akashi’s test that, across expression bins, there is selection for the usage of preferred codons at evolutionary conserved amino acid positions. These results further demonstrate that, across expression bins, there is also selection for the usage of preferred codons at positions that encode nonconserved amino acids. These findings hold even for the 10% of genes that are most lowly expressed within the E. coli genome.
Synonymous Replacement of Single Codons within an Essential Lowly Expressed Gene Can Significantly Affect Fitness
Previous studies have directly demonstrated that synonymous codon replacements within highly expressed genes can substantially affect fitness. In order to examine whether this also extends to lowly expressed genes, we sought to test whether the synonymous replacement of a single codon, encoding a functionally important amino acid, within a lowly expressed yet essential protein can alter bacterial fitness. As a model we used the bla gene encoding the Beta-lactamase TEM-1 protein, which induces E. coli resistance to ampicillin (Heffron et al. 1975). In the presence of concentrations of ampicillin higher than the minimal inhibitory concentration (MIC) of the antibiotic, this gene becomes essential for E. coli growth. The bla gene is normally fairly highly expressed as it resides on a plasmid (pBR322) under the control of a relatively strong promoter. In order to artificially generate a weakly expressed version of the gene, we extracted the gene from its native plasmid and cloned it into the E. coli chromosome, under the control of a weak promoter (Materials and Methods). Since the manipulated bla gene is encoded chromosomally, rather than on a plasmid, we remove the possible effect of plasmid copy number on the expression of the gene. Using Western analysis, we validated that the chromosomally encoded bla gene, regulated by the weak promoter was relatively lowly expressed (supplementary fig. S4, Supplementary Material online). In order to examine how synonymous codon replacements within this now lowly expressed essential gene affect fitness, we introduced synonymous mutations into the gene prior to cloning it into the chromosome, using site-directed mutagenesis.
We focused on two serine residues (S68 and S128) that were previously shown to be important for protein function. Replacing these amino acids with any other amino acid tested was shown to severely reduce protein function (Firnberg et al. 2014). In the wildtype bla sequence, S68 is encoded by the codon AGC and S128 is encoded by the codon AGT. Both of these codons are recognized by the same single copy GCT-anticodon tRNA (Chan and Lowe 2009). We replaced the S68 AGC codon and the S128 AGT codons with TCC and TCT, respectively. Both TCC and TCT are recognized by the same GGA anticodon tRNA, which appears in two copies within E. coli (Chan and Lowe 2009). Because we are replacing codons recognized by a lower copy tRNA with codons recognized by a higher copy tRNA, we expected to observe an improvement in fitness in the mutants relative the wildtype.
To examine whether the synonymous replacement of codons at S68 and S128 affected bacterial fitness, we conducted competition experiments. In these experiments bacteria chromosomally carrying the wildtype bla gene were grown together with bacteria chromosomally carrying either the S68 or the S128 synonymous codon replacement (in both the wildtype and the mutants the bla gene was placed under the control of the same weak promoter). The growth media in which the strains were competed contained 50 mg/L ampicillin. This antibiotic concentration abolishes E. coli growth in the absence of the bla gene, but enables growth of strains containing the weakly expressed, chromosomal bla gene (data not shown). Prior to growth the relative abundance of each strain type was quantified. Following 7 h of growth (to mid log) bacteria were harvested and the relative abundance of each strain type was again quantified. This enabled us to determine whether the mutant or wildtype strains replicated faster during the first 7 h of growth (Materials and Methods). In order to be able to quantify the abundance of each strain, different strains grown together were marked with either a kanamycin (kan) or a chloramphenicol (chl) resistance cassette. Colony forming units (CFU) were then quantified by plating of colonies on either kan or chl. To make certain that the cassettes themselves did not drive observed differences, we carried out reciprocal experiments (wildtype marked by kan/mutant marked by chl and wildtype marked by chl/mutant marked by kan). For each of the mutants 34 independent experiments were carried out (table 1 andsupplementary table S7, Supplementary Material online).
In our competition experiments, both strains carrying the mutated bla genes grew significantly faster than the strain carrying the wildtype bla gene (P ≪ 0.001, table 1). Over the 7 h of competition, the two mutants grew ∼5–6% more than the wildtype. These results remained significant whether the wildtype was marked with kan and the mutant with chl or the opposite (P < 0.05 for all comparisons, table 1). Thus, our results demonstrate that synonymous replacements of single codons, encoding important amino acids within a lowly expressed essential gene can significantly affect bacterial fitness.
Discussion
It was originally thought that since synonymous codons encode the same amino acid, they were functionally indistinguishable from each other. For this reason, synonymous mutations from one codon to another, encoding the same amino acid are to this day often referred to as “silent” mutations. This notion has long been known to be false; at least when it came to highly expressed genes, where codon usage has been shown to be ubiquitously subject to selection from bacteria to mammals (Akashi 1994; Stoletzki and Eyre-Walker 2007; Drummond and Wilke 2008; Hershberg and Petrov 2008). Much less focus has been given to examining selection on codon usage within lowly expressed genes. Here, we support previous findings (Zhou et al. 2009) suggesting that selection on translation accuracy affects codon usage within lowly expressed genes. Furthermore, within lowly expressed genes, we found that selection affects codon usage both at sites encoding conserved amino acids and sites encoding less conserved amino acids. Our results therefore indicate that there may be very few sites within protein-coding genes that evolve in an entirely neutral, or “silent” manner.
We cannot provide a clear cause for the selection we observed to be affecting less conserved amino acid positions, in both highly and lowly expressed genes. Such selection could be due to the need to maintain translation accuracy, if some less conserved amino acids are also important for protein function and/or folding. On the other hand, such selection could also stem from the need to maintain translation efficiency, or for other yet unknown reasons.
Although our results did show that selection in favor of preferred codon usage affects both lowly and highly expressed genes, it is important to note that the signals of selection do appear to be much stronger for highly expressed genes. This is not surprising since, as we mention in the introduction, inefficient and inaccurate translation is expected to more strongly increase global translation costs, when it affects genes that are highly expressed.
The expression data we used to classify genes into expression bins was extracted under specific growth-permissive conditions. It is quite possible that some of the genes we classified as lowly expressed, under these conditions are expressed at higher levels under other conditions. At the same time, as discussed above, we do observe a much weaker signal of selection affecting the genes we classified into the lower expression bins, compared with the genes classified into higher expression bins. This indicates that on average genes classified into the lower expression bins are indeed expressed at lower levels than genes classified into the higher expression bins.
Although in general, we found that higher expression bins displayed a stronger signal for selection on translation accuracy, compared with lower expression bins, we did observe a puzzling reduction in the Akashi odds ratio for the highest expression bin (fig. 1). The reason for this reduction seems to be that the genes contained within the highest expression bin tend to have a very high frequency of preferred codons encoding their less conserved amino acids (supplementary fig. S5A, Supplementary Material online). This leads to a reduction in the difference between the preferred codon usage at conserved versus nonconserved amino acid sites (supplementary fig. S5B, Supplementary Material online), which in turn reduces the Akashi odds ratio. We hypothesize that strong selection on translation efficiency is responsible for the increased frequency of preferred codons at less conserved amino acid sites, within the most highly expressed genes. After all, selection on translation efficiency is expected to be stronger on highly expressed genes and to affect both codons encoding conserved amino acids and codons encoding less conserved amino acids. Interestingly, ribosomal genes contained within the highest expression bin use far less preferred codons at less conserved amino acid sites, than nonribosomal genes contained within the same expression bin (supplementary fig. S5A, Supplementary Material online). Ribosomal genes are often used in studies in order to define patterns of codon usage among highly expressed genes. The fact that these genes behave so differently than other highly expressed genes demonstrates that it may be problematic to use these genes in such a manner.
Even when it became apparent that selection does indeed affect synonymous substitutions, it was often argued that such selection would be expected to be quite weak. However, recent studies have revealed that synonymous sites may in fact often be subject to strong selection (Lawrie et al. 2013; Machado et al. 2017). The fitness effects we observed for the single codon substitutions we introduced into the lowly expressed beta-lactamase gene were quite substantial (on the order of 5% difference in growth over 7 h). Such observable fitness effects should be quite strongly affected by natural selection. It is quite possible, however, that these are extreme cases. After all, we have specifically modified a gene that is entirely essential under the conditions investigated, at residues at which amino acid replacements greatly reduce function (Firnberg et al. 2014). Additionally, we examined the fitness effects of only two synonymous codon replacements, finding a pronounced fitness effect in the direction we expected in both cases. Further studies will need to be carried out in order to determine the distribution of fitness effects of synonymous substitutions, within lowly expressed genes.
In our computational analyses, we found that preferred codons are used more frequently at sites encoding conserved relative nonconserved amino acids. Additionally, we found a higher usage of preferred codons than expected from background substitution biases (estimated from intergenic sequence composition) at both conserved and variable amino acid positions. In our experimental analyses, we found that manipulating codon usage of a lowly expressed gene at a single site from a codon recognized by a lower copy number tRNA to a synonymous codon recognized by a higher copy number tRNA can lead to an observable improvement in fitness. These results fit well with a model by which selection on translation optimization affects codon usage in lowly expressed genes. However, it is important to note that we cannot be certain that other selective pressures, unrelated to translation optimization may be driving some of our observations. For example, it is possible that the fitness improvements we observed stem from changes to mRNA structure, rather than from changes to codon usage. It is also possible that preferred codons are enriched both at sites encoding conserved and nonconserved amino acids, due to reasons that are not directly related to translation optimization. Therefore, while our results clearly demonstrate selection affecting synonymous sites within lowly expressed genes, further studies will be needed to conclusively determine whether this stems directly from selection on codon usage and translation optimization.
To conclude, we find that substitution of even a single synonymous codon within an essential yet lowly expressed gene can carry substantial, observable effects on bacterial fitness. Fitting with this, signals of selection on synonymous sites can be demonstrated even for those 10% of genes that are most lowly expressed within the E. coli genome. Such signals of selection are not limited to sites encoding conserved amino acids, but are also present for sites encoding less conserved amino acids.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by a Leonard and Diane Sherman Interdisciplinary Graduate School Fellowship (to A.Y.), by an ISF grant (No. 756/17, to R.H.) and by the Rappaport Family Institute for Research in the Medical Sciences (to R.H.). The described research was carried out in the Rachel and Menachem Mendelovitch Evolutionary Process of Mutation and Natural Selection Research Laboratory.
Literature Cited
- Agashe D, Martinez-Gomez NC, Drummond DA, Marx CJ.. 2013. Good codons, bad transcript: large reductions in gene expression and fitness arising from synonymous mutations in a key enzyme. Mol Bio Evol. 30(3):549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H. 1994. Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. Genetics 136(3):927–935. doi: 10.1007/s12033-011-9383-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson promoter collection [Internet]. Registry of standard biological parts; 2009. Available from: http://parts.igem.org/Promoters/Catalog/Anderson
- Bennetzen JL, Hall BD.. 1982. Codon selection in yeast. J. Biol. Chem. 257(6):3026–3031. [PubMed] [Google Scholar]
- Brandis G, Hughes D.. 2016. The selective advantage of synonymous codon usage bias in Salmonella. PLoS Genet. 12(3):e1005926–e1005916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BÜDeyrİ GÖKgÖZ N, et al. , 2015. Investigation of the in vivo interaction between β-lactamase and its inhibitor protein. Turk J Biol. 39:485–492. [Google Scholar]
- Carlini DB. 2004. Experimental reduction of codon bias in the Drosophila alcohol dehydrogenase gene results in decreased ethanol tolerance of adult flies. J Evol Biol. 17(4):779–785. [DOI] [PubMed] [Google Scholar]
- Chan PP, Lowe TM.. 2009. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37(Database):D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SL, Lee W, Hottes AK, Shapiro L, McAdams HH.. 2004. Codon usage between genomes is constrained by genome-wide mutational processes. Proc Natl Acad Sci USA. 101(10):3480–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL.. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 97(12):6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M, Wernisch L.. 2009. Estimating translational selection in eukaryotic genomes. Mol Biol Evol. 26(2):451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO.. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134(2):341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, et al. , 2008. Many microbe microarrays database: uniformly normalized Affymetrix compendia with structured experimental metadata. Nucleic Acids Res. 36(Database):D866–D870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firnberg E, Labonte JW, Gray JJ, Ostermeier M.. 2014. A comprehensive, high-resolution map of a Gene’s fitness landscape. Mol Biol Evol. 31(6):1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Pilpel Y.. 2011. Determinants of translation efficiency and accuracy. Mol Syst Biol. 7(1):481–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Gautier C.. 1982. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 10(22):7055–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M, Mercier R, Pavé A.. 1980. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 8(1):r49–r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen N. 2006. Protein misfolding disorders: pathogenesis and intervention. J Inherit Metab Dis. 29(2-3):456–470. [DOI] [PubMed] [Google Scholar]
- Hauber DJ, Grogan DW, DeBry RW.. 2016. Mutations to less-preferred synonymous codons in a highly expressed gene of Escherichia coli: fitness and epistatic interactions. PLoS One 11(1):e0146375–e0146316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffron F, Sublett R, Hedges RW, Jacob A, Falkow S.. 1975. Origin of the TEM-beta-lactamase gene found on plasmids. J Bacteriol. 122(1):250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2009. General rules for optimal codon choice. PLoS Genet. 5(7):e1000556.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2012. On the limitations of using ribosomal genes as references for the study of codon usage: a rebuttal. PLoS One 7(12):e49060.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2008. Selection on codon bias. Annu Rev Genet. 42(1):287–299. [DOI] [PubMed] [Google Scholar]
- Hu P, et al. , 2009. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 7(4):e1000096–e1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz S, Hershberg R.. 2013. Elevated mutagenesis does not explain the increased frequency of antibiotic resistant mutants in starved aging colonies. PLoS Genet. 9(11):e1003968–e1003968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight RD, Freeland SJ, Landweber LF.. 2001. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2:research0010.0011–research0010.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöppel A, Näsvall J, Andersson DI.. 2016. Compensating the fitness costs of synonymous mutations. Mol Biol Evol. 33(6):1461–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrie DS, Messer PW, Hershberg R, Petrov DA.. 2013. Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet. 9(5):e1003527.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado HE, Lawrie DS, Petrov DA.. 2017. Strong selection at the level of codon usage bias: evidence against the Li-Bulmer model. bioRxiv. doi: 10.1101/106476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel N. 1963. Chi-square tests with one degree of freedom; extensions of the Mantel–Haenszel procedure. J Am Stat Assoc. 58(303):690–700. [Google Scholar]
- Mantel N, Haenszel W.. 1959. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 22(4):719–748. [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ.. 1988. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 85(8):2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quax TEF, Claassens NJ, Söll D, van der Oost J.. 2015. Codon bias as a means to fine-tune gene expression. Mol. Cell 59(2):149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields DC, Sharp PM.. 1987. Synonymous codon usage in Bacillus subtilis reflects both translational selection and mutational biases. Nucleic Acids Res. 15(19):8023–8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A.. 2007. Synonymous codon usage in Escherichia coli: selection for translational accuracy. Mol Biol. Evol. 24(2):374–381. [DOI] [PubMed] [Google Scholar]
- Tuller T, et al. , 2010. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141(2):344–354. [DOI] [PubMed] [Google Scholar]
- Varenne S, Buc J, Lloubes R, Lazdunski C.. 1984. Translation is a non-uniform process: effect of tRNA availability on the rate of elongation of nascent polypeptide chains. J Mol Biol. 180(3):549–576. [DOI] [PubMed] [Google Scholar]
- Zhou T, Weems M, Wilke CO.. 2009. Translationally optimal codons associate with structurally sensitive sites in proteins. Mol Biol Evol. 26(7):1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.