

Abstract
Background
Clinical trials of therapies directed against nodes of the signaling axis of phosphatidylinositol-3 kinase/Akt/mammalian target of rapamycin (mTOR) in glioblastoma (GBM) have had disappointing results. Resistance to mTOR inhibitors limits their efficacy.
Methods
To determine mechanisms of resistance to chronic mTOR inhibition, we performed tandem screens on patient-derived GBM cultures.
Results
An unbiased phosphoproteomic screen quantified phosphorylation changes associated with chronic exposure to the mTOR inhibitor rapamycin, and our analysis implicated a role for glycogen synthase kinase (GSK)3B attenuation in mediating resistance that was confirmed by functional studies. A targeted short hairpin RNA screen and further functional studies both in vitro and in vivo demonstrated that microtubule-associated protein (MAP)1B, previously associated predominantly with neurons, is a downstream effector of GSK3B-mediated resistance. Furthermore, we provide evidence that chronic rapamycin induces microtubule stability in a MAP1B-dependent manner in GBM cells. Additional experiments explicate a signaling pathway wherein combinatorial extracellular signal-regulated kinase (ERK)/mTOR targeting abrogates inhibitory phosphorylation of GSK3B, leads to phosphorylation of MAP1B, and confers sensitization.
Conclusions
These data portray a compensatory molecular signaling network that imparts resistance to chronic mTOR inhibition in primary, human GBM cell cultures and points toward new therapeutic strategies.
Keywords: cancer, ERK, glioma, glioblastoma, GSK3B, MAP1B, MEK, microtubule, mTOR, tubulin
Importance of the study
This study presents a novel molecular mechanism of resistance to chronic mTOR inhibition that delineates posttranslational modifications from the kinases ERK and GSK3B to the microtubule interacting protein MAP1B. Aside from the basic insights into molecular signaling, this study also informs therapeutic intervention. We found that inhibition of both mTOR complexes 1 and 2 may trigger inhibitory phosphorylation of GSK3B and, surprisingly, a MAP1B-mediated mode of resistance to chronic mTOR inhibition. Although mTOR inhibitors have not been as effective as hoped in the treatment of GBM, new-generation inhibitors of mTOR and ERK in combination with MEK inhibitors offer new possibilities for treatment which must be informed by knowledge of molecular signaling involved in resistance to treatment. Furthermore, the identification of MAP1B-regulated microtubule stabilization as a means of resistance provides a set of therapeutic targets. This study provides compelling new knowledge in our understanding of the molecular response and resistance to chronic mTOR inhibition and indicates a direction for future combinatorial therapy in the treatment of GBM.
Mammalian (or mechanistic) target of rapamycin (mTOR) is one downstream effector of the Akt pathway that, as mTOR complex 1 (mTORC1), activates protein translation through phosphorylation of ribosomal protein S6 (pRPS6; also known as p70S6 or pS6) and eukaryotic initiation factor 4E binding protein 1 (4EBP1; see review1). Rapamycin, the mTOR pathway specific inhibitor originally used to identify mTOR,2–5 primarily exerts its effects via mTORC1. However, mTORC2 assembly has also been found to be impaired, in certain cell cultures, by chronic rapamycin treatment.6
Mammalian TOR signaling is critical for the promotion of cellular growth and proliferation.1 The mTOR signaling pathway can be activated by genetic alteration of the upstream epidermal growth factor receptor (EGFR) signaling pathway, which is frequently mutated and amplified in glioblastoma (GBM).7 Moreover, inhibition of protein kinase B (Akt), a kinase that activates mTOR signaling, has been indicated to deplete brain tumor stem cells, the putative cell type responsible for malignancy and recurrence in GBM.8 As a result of upstream activation, mTOR signaling is frequently activated in GBM. For these reasons, targeting of the mTOR signaling pathway has been an area of intense therapeutic interest.
In a clinical trial for glioma,9 half of the patients treated with rapamycin responded. However, many of the patients did not show evidence of mTOR inhibition, and in fact had elevated mortality associated with increased Akt activation. While there are divergent views on possible modes of rapamycin resistance,9–17 one theory is that feedback activation of mTORC2 activates that pathway. In fact, mixed kinase inhibitors such as BEZ235 were developed, in part, to circumvent these modes of resistance by targeting mTORC1, mTORC2, and phosphatidylinositol-3 kinase (PI3K). Another mode of resistance to mTOR inhibition is through activated extracellular signal-regulated kinase (ERK), a molecular response to mTOR inhibition in GBM, and inhibition of ERK has been shown, in vivo, to sensitize GBM to mTOR inhibition.18,19 However, other theories of resistance have also been developed, compelling us to perform a comprehensive characterization of the mechanisms that modulate resistance to chronic mTOR inhibition within GBM cells.
Here, we utilized phosphoproteomic analysis of primary GBM cultures under chronic mTOR inhibition followed by a targeted short hairpin (sh)RNA screen and functional studies to elucidate an ERK-modulated, glycogen synthase kinase (GSK)3B–mediated, microtubule-associated protein (MAP)1B–dependent mechanism of resistance.
Materials and Methods
See the Supplementary material for a complete methods description.
Tumor Collection
Human GBM (World Health Organization [WHO] grade IV) brain tumor samples were collected following surgical resection under institutional review board–approved protocols with patient consent and graded by a neuropathologist in accordance with the WHO established guidelines.20
Primary GBM Cell Cultures
All cultures used were a subset of those previously described by Laks et al21 (Supplementary Table S1). Each culture was analyzed initially by microarray at low passage number. Key features of the lines, including EGFR status and phosphatase and tensin homolog status were ascertained at low passage number and compared with the initial pathology report. If the sample did not agree with the pathology report, the sample was not used for this study. This procedure was repeated at irregular intervals. All experiments were performed at as low a passage number as possible from frozen stocks, and for those experiments that required higher passaging, characteristics of the cells were evaluated at irregular intervals, including phosphatase and tensin homolog and EGFR status, proliferation rate, general morphology of the cultures, and mycoplasma status. If any of these parameters became discordant, the culture was not used. For some cultures, as described by Laks et al,21 microarrays were repeated at low and high passage numbers, which showed a general maintenance of characteristics (accession #GSE98995). Cells were tested annually for mycoplasma with the LookOut Mycoplasma PCR Detection Kit (Sigma). We chose cultures that were expedient for the experimental setting. HK296, originally chosen for the phosphoproteomics experiment, took too long (2 wk) to become rapamycin resistant (proliferate) and was thus not an expedient in vitro model. In contrast, HK301 took only 2 days to begin to proliferate under rapamycin treatment. HK374 was chosen for in vivo models, as it had already been demonstrated by our lab to form rapid subcutaneous tumors.
Exposure Groups
Three exposure groups were devised for the purpose of the phosphoproteomics study: (i) dimethyl sulfoxide (DMSO) as control; (ii) chronic rapamycin treatment (the rapamycin-resistant population after 100 nM rapamycin, twice weekly; and (iii) acute rapamycin (100 nM rapamycin for 4 h). The latter 2 exposure groups were compared with the DMSO-control to normalize changes and serve as a baseline.
Isobaric Tags for Relative and Absolute Quantitation
Cells were pelleted and run through the protocol for isobaric tags for relative and absolute quantitation (iTRAQ), which was used according to standard methods to determine changes in phosphorylation from rapamycin to DMSO conditions.22 See the Supplementary material.
Kinase Enrichment Analysis
KEA (http://amp.pharm.mssm.edu/lib/kea.jsp; last accessed 12 August 2017)23 was utilized to determine candidate kinases that were associated with our list of proteins with phosphorylation changes under chronic rapamycin conditions.
Chronic Rapamycin Treatment
Chronic rapamycin (LC Laboratories, http://www.LCLabs.com; last accessed 12 August 2017) was administered twice weekly at 100 nM for at least 7 days.
Acute Drug Treatments
Five thousand cells were plated in each well of a 96-well plate and treated with serial dilutions of each drug and after 7 days of proliferation, cell number was assessed (see Supplementary Methods for more information).
Sensitization Selumetinib (MEK Inhibitor)+Rapamycin or BEZ235
Five thousand cells were plated in each well of a 96-well plate and treated with a serial dilution of selumetinib with either DMSO, 100 nM rapamycin, or 10 nM BEZ235. After 7 days, cell number was assessed by relative fluorescence of Hoechst labeled cells as detected by the Acumen 3 plate reader (TTPLabtech). Relative half-maximal inhibitory concentration (IC50) values were generated using GraphPad Prism software.
Western Blots
Western blots were performed according to previously described methods.21
CDK4 Inhibition
A serial dilution of PD0332991 (Selleckchem) was used in combination with 100 nM rapamycin or with DMSO (at 0.1% final concentration) as a control.
Targeted shRNA Screen
We performed a targeted shRNA screen against 50 of 52 candidates who underwent phosphorylation changes in chronic rapamycin and were associated with GSK3B. HK301 GBM cells were grown on 384-well plates (1000 cells/well) for 3 days with each shRNA. After 3 days, cells were treated with and without 100 nM rapamycin. Cells were re-treated with agent after another 3 days. After the initial treatment, cells were grown for 7 days. Cell number was determined using CellTiter-Glo 3D (Promega), which estimates cell number based on measured ATP levels.
In Vivo Subcutaneous Xenotransplantation of shMAP1B Cells
All animal experiments were done with protocol approval by the Office of Animal Research Oversight at UCLA and the UCLA institutional animal care and use committee. Five hundred thousand HK374 cells with either control or MAP1B knockdown (KD) were injected subcutaneously into the flanks of NSG mice. After 24 days, all tumors were established and we started daily intraperitoneal injection treatment with either DMSO or rapamycin in a final volume of 100 μL as follows—Group 1: control KD + DMSO; Group 2: control KD + 5 mg/kg rapamycin; Group 3: MAP1B KD + DMSO; and Group 4: MAP1B KD + 5 mg/kg rapamycin (LC Laboratories #R-5000). Tumors were measured with calipers every 2 days for 14 days. At day 15 post daily i.p. treatment, mice were euthanized and tumors were collected and lysed in radioimmunoprecipitation assay buffer (Thermo) with phosphatase and protease inhibitors and homogenized with a syringe plunger through a cell strainer (100 μm).
Tumor Volume Measurements
The tumor volume was calculated for the volume of a sphere with the formula Volume = 4/3*3.1415*(Diameter/2)^3. The mean diameter of the tumors for each mouse was utilized. Tumors that failed to form significant sizes before the onset of treatment (within the 95th percentiles) were excluded from further analysis.
Western Blots of Subcutaneous Tumors of shMAP1B Cells
Protein from subcutaneous tumors was collected individually and then by treatment category. Protein concentration was determined by Bradford assay. Protein was boiled in an equal volume of Laemmli Sample Buffer (Bio-Rad #161-0737) with 5% beta mercaptoethanol for 5 minutes. Western blots were carried out to validate inhibition of therapeutic targets, with beta actin (Abcam #8277) as a loading control. The antibodies used were the same as the antibodies used in the “Western Blots” section above.
In Vivo Subcutaneous Xenotransplantation for Combinatorial Treatment
Injected into both flanks of 24 male, 2-month-old, NOD/SCID-gamma null mice was 1 × 106 HK374 firefly luciferase–green fluorescent protein GBM cells. Each of the 4 treatment groups (DMSO, rapamycin [Rapa], selumetinib [Sel], and Rapa+Sel) consisted of 6 mice. Daily i.p. injection treatment in 1 of 4 groups was initiated after 14 days of tumor proliferation—Group 1: DMSO; Group 2: rapamycin 5 mg/kg; Group 3: selumetinib 35 mg/kg; Group 4: Rapa (5 mg/kg) + Sel (35 mg/kg). Each treatment was delivered in DMSO as a vehicle + 12.5% Carbowax (Polyethylene Glycol 400, Fisher Scientific) in a final volume of 100 μL. Rapamycin was from LC Laboratories (#R-5000). Selumetinib was from Selleckchem (#S1008). Tumors were measured with calipers every 2 days. A similar in vivo study was performed in the same manner in a separate trial but with 75 mg/kg selumetinib.
Western Blots of Subcutaneous Tumors for Combinatorial Treatment
Protein from subcutaneous tumors was collected and pooled by treatment category. Protein concentration was determined by Bradford assay.
Microtubule Staining and Quantification
Immunofluorescence microscopy was carried out essentially as described previously.24 See Supplementary Methods for details.
Statistics
Summary statistics were performed using ANOVA followed by post hoc Student’s t-tests as implemented by Stata 8.0 or GraphPad Prism 5 software. In certain cases, Welch’s correction for unequal variances was utilized. In other cases, the nonparametric Mann–Whitney test was employed. IC50 values were generated and compared using GraphPad Prism. A P-value < 0.01 was used to determine significance and adjust for multiple comparisons.
Results
GSK3B Is Associated with Phosphorylation Changes Under Chronic mTOR Inhibition
We compared the phosphoproteome of HK296 GBM cells treated with vehicle control (DMSO), acute rapamycin, or chronic rapamycin. Many proteins demonstrated inverse changes in acute compared with chronic rapamycin conditions (Fig. 1A). We applied stringent criteria (see Supplementary Methods) and identified 425 proteins with phosphorylation changes after chronic rapamycin treatment. Validation of 2 of these phosphorylation changes via western blot is shown in Fig. 1B.
Fig. 1.

Phosphoproteomics of mTOR inhibition. (A) Heatmap of phosphoproteomics results comparing chronic rapamycin treatment or acute rapamycin treatment with DMSO treated controls. The relative levels of phosphopeptides are expressed as log2 of the ratios relative to control (DMSO). The heatmap on the left depicts proteins with ≥2-fold differences and the one on the right shows those with ≥4-fold differences. (B) Western blot of phosphorylated 4EBP1-T37/46 (p4EBP1), phosphorylated RB1-S249 (pRB1), and phosphorylated RPS6-S240/244 (pS6) demonstrating changes after 14 days of chronic rapamycin treatment (14 days, 100 nM). Phosphoproteomics values (−2.28x and +2.17x) above blots indicate the fold changes for p4EBP1-T46 and pRB1-S249, respectively, in phosphorylation levels as detected by the phosphoproteomics screen. See also Supplementary Table S1.
KEA23 revealed that GSK3B is significantly associated with 52 out of 425 of these phosphorylated proteins (P < 0.001; Supplementary Table S1). GSK3B had the most substrates and the second lowest P-value on this list of kinases, while cell division cycle 2 (cyclin-dependent kinase 4 [CDK4]) had the lowest P-value (P < 0.0001; Supplementary Table S1). Preliminary experiments found no sensitization to rapamycin upon combinatorial treatment with a CDK4 inhibitor, PD0332991 (data not shown).
Attenuation of GSK3B Confers Resistance to Chronic mTOR Inhibition
Combinatorial treatment of GBM cultures with a serial dilution of rapamycin or BEZ235 in the presence of 1 μM CHIR99021, a selective GSK3B inhibitor, conferred resistance to rapamycin (Fig. 2A) and to BEZ235 (Fig. 2B). This was true in a variety of cell cultures tested (Supplementary Figure S1A–G). Western blot of the CHIR99021-treated GBM culture demonstrated that GSK3B activity was attenuated as its downstream target p-4EBP1-T46 was diminished (Fig. 2C). Furthermore, depletion of GSK3B via shRNA (Fig. 2D) did not affect GSK3 alpha and resulted in a dramatic increase in resistance to rapamycin (Fig. 2E) and to BEZ235 (Fig. 2F) in HK301 and in other cell cultures tested (Supplementary Figure S1H–K). These trends were validated with a second shGSK3B construct (Supplementary Figure S2). These data indicate that GSK3B modulates resistance to mTOR pathway specific inhibition, even when mTORC2 and PI3K are additionally targeted by the combinatorial inhibitor BEZ235.
Fig. 2.

GSK3B inhibition confers resistance to mTOR pathway inhibition. (A) Dose response to a serial dilution of rapamycin with co-treatment of the GSK3B inhibitor CHIR99021 (1 μM) (P = 0.0054, Mann–Whitney test). (B) Dose response to a serial dilution of BEZ235 with co-treatment of the GSK3B inhibitor CHIR99021 (1 μM), P < 0.0001 comparing IC50 values. (C) Western blot of HK301 cells after 2 hours treatment with DMSO, rapamycin (100 nM), CHIR99021 (4 μM), or rapamycin (100 nM) + CHIR99021 (4 μM). The top band of 3 bands in the phosphorylated 4EBP1, indicated by the arrow, is for threonine-46. (D) Western blot of GSK3B knockdown demonstrates specificity for GSK3B with no depletion of GSK3A. (E) Fitted curve of log-transformed values for a serial dilution of rapamycin in HK301 GBM cells with and without GSK3B knockdown, P < 0.0001 comparing IC50 values. (F) Fitted curve of log transformed values for a serial dilution of BEZ235 in HK301 GBM cells with and without GSK3B knockdown. P = 0.0007 comparing IC50 values. N = 3 independent experiments for A, B, E, and F. See also Supplementary Figures S1 and S2.
The Relative Roles of RICTOR and RAPTOR in Conferring Inhibitory Phosphorylation of GSK3B Vary Among GBM Cultures
Phosphorylation of GSK3B at serine 9 is known to inhibit its kinase activity.25 We discovered that GSK3B consistently becomes phosphorylated at serine 9 in response to prolonged rapamycin treatment in human GBM cell cultures (Fig. 4D). As mTOR exists in 2 distinct complexes, mTORC1, associated with regulatory associated protein of mTOR (RAPTOR), and mTORC2, associated with rapamycin-insensitive companion of mTOR (RICTOR),26 we sought to determine which mTOR complex was responsible for GSK3B phosphorylation. In HK157, shRNA-mediated knockdown of either RAPTOR or RICTOR resulted in phosphorylation of GSK3B (Fig. 3A). However, in HK301, RAPTOR knockdown resulted in enhanced phosphorylation of GSK3B while RICTOR knockdown did not.
Fig. 4.

Knockdown of MAP1B confers sensitivity to chronic mTOR inhibition. (A) Fluorescent images of wells from 384 well plates used in the targeted shRNA screen display green fluorescent protein–labeled cells in shMAP1B conditions at 10 days posttreatment (DMSO-control or 100 nM rapamycin [RAPA]). White scale bar represents 2.67 mm. (B) Fitted curve of log transformed values for a serial dilution of rapamycin in HK217 GBM cells, with and without MAP1B knockdown, after 6 days of proliferation, P = 0.0002 comparing IC50 values (N = 3). (C) Fitted curve of log transformed values for a serial dilution of BEZ235 in HK217 GBM cells, with and without MAP1B knockdown, after 6 days of proliferation, P = 0.0028 comparing IC50 values (N = 4). (D) Western blot of HK301 cells. Western blot columns are in order: 7 day chronic DMSO-control, 7 day rapamycin, shRNA-control (C), shMAP1B (M), shGSK3B (G). (E) Observed points, standard error bars, and fitted lines for normalized tumor volumes of HK374 subcutaneous tumors at different time intervals. By day 12 posttreatment, the shMAP1B under rapamycin treatment demonstrates increased sensitivity and reduced tumor growth, as indicated by a comparison of the slopes (P < 0.0001) as well as by comparison of the means (P = 0.0323, Kruskal‒Wallis test). (F) Western blot of the tumor lysates demonstrates that the shMAP1B transplanted tumors maintained depletion of MAP1B throughout the tumor growth. Phosphorylated RPS6 levels demonstrate that rapamycin inhibited its target pathway in the tumors. (G) Bar graph shows mean ± SEM for the corrected total cell fluorescence (CTCF) of alpha-tubulin in HK217 GBM cells after 7 days of chronic rapamycin treatment followed by acute exposure to 5 μM Nocodazole (30 min). There is a significant increase in the rapamycin conditions compared with the DMSO conditions for the pGIPZ-Ctrl vector (P = 0.001; Mann‒Whitney test). There is no significant difference between the DMSO and rapamycin conditions for the shMAP1B infected cells (P = 0.1431). Each condition represents the corrected total cell fluorescence of 10 individual cells. (H) Representative photographs of alpha tubulin staining of HK217 GBM cells in the different 7 day treatment conditions and after acute Nocodazole treatment. White scale bar indicates 10 μM. See also Supplementary Table S2, and Supplementary Figures S4, S5.
Fig. 3.

Depletion of mTORC1 and, in certain GBM cultures, mTORC2 can promote inhibitory phosphorylation of GSK3B. (A) Western blot of GBM cultures HK157 and HK301 demonstrating efficacy of knockdown and phosphorylation status of GSK3B, ERK, and Akt. (B) Mean relative cell number and standard error bars for a serial dilution of rapamycin treatments in HK157 in the presence or absence of shRICTOR, P = 0.0004 comparing mean IC50 values (N = 3, log scale). (C) Mean relative cell number and standard error bars for a serial dilution of BEZ235 treatments in HK157 in the presence and absence of shRICTOR, P < 0.001 comparing IC50 values (N = 3, log scale). (D) Mean relative cell number and standard error bars for a serial dilution of rapamycin treatments in HK301 in the presence and absence of shRICTOR (P = 0.8618) is depicted comparing IC50 values (N = 2, log scale). (E) Mean relative cell number and standard error bars for a serial dilution of BEZ235 treatments in HK301 in the presence and absence of shRICTOR, P = 0.1012 comparing IC50 values (N = 2, log scale). See also Supplementary Figure S3.
GSK3B is known to be phosphorylated by Akt25,27 and through ERK-activated P90-RSK1.28 In HK157, RAPTOR knockdown resulted in a minor elevation of phosphorylated ERK, while RICTOR knockdown did so more convincingly (Fig. 3A), suggesting that phosphorylated ERK could mediate the induction of phosphorylated GSK3B resulting from RICTOR knockdown. No such effects were detectable in HK301.
We then sought to determine the impact of these findings on rapamycin sensitivity. While RICTOR knockdown did not influence sensitivity to either drug in HK301, it did protect HK157 cells against both rapamycin and BEZ235 (Fig. 3D, E). RAPTOR knockdown prevented the cells from proliferating and was therefore not suitable for further in vitro study.
Taken together, the data indicate that the relative roles of mTORC1 and mTORC2 inhibition in mediating GSK3B phosphorylation can vary from tumor to tumor. Moreover, these results provide a bridge between mTORC2 inhibition, ERK activation, and the GSK3B-mediated mechanism of resistance.
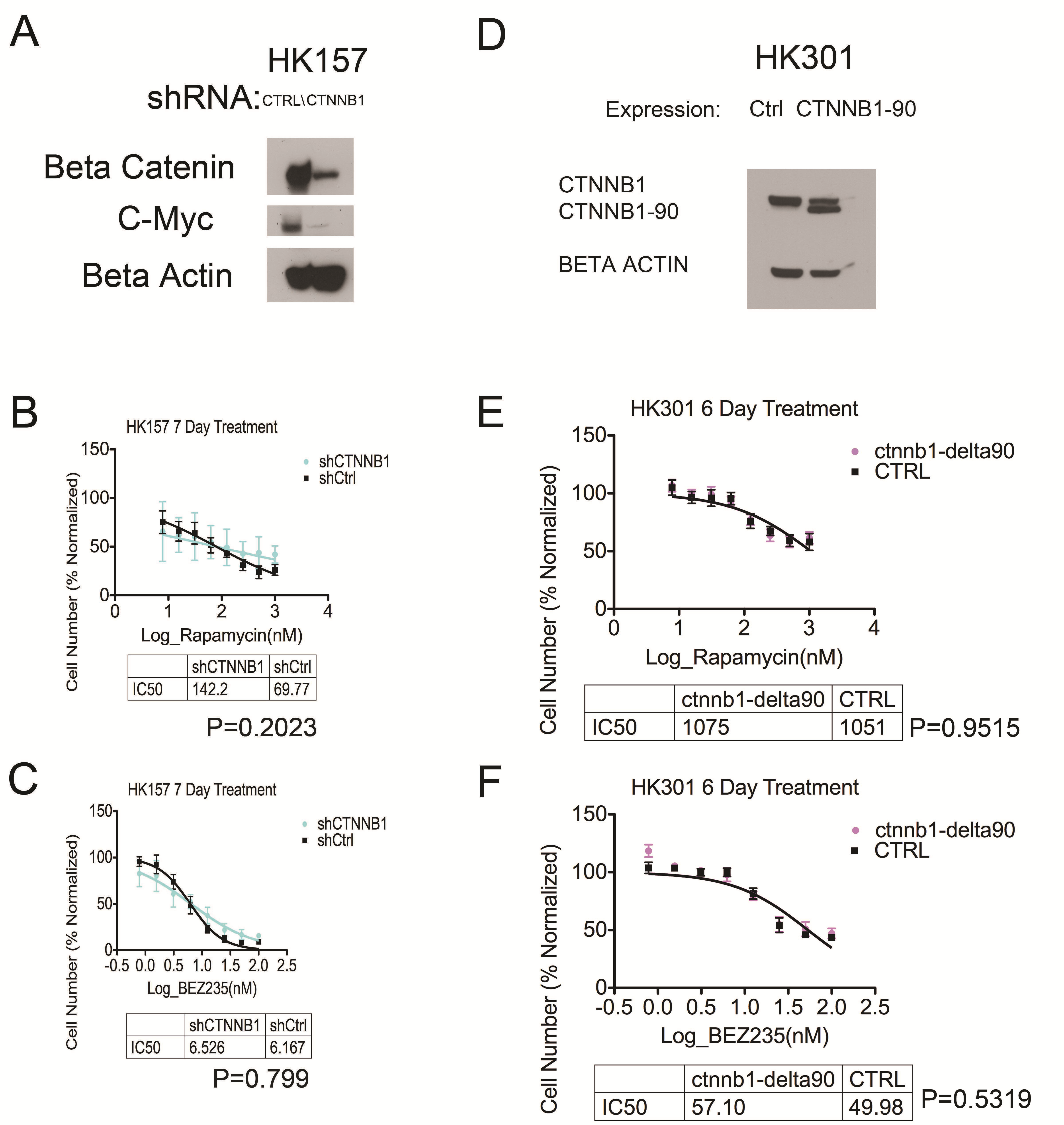
Resistance to mTOR Inhibition by Phosphorylation of GSK3B Is Not Mediated by an Effect on β-Catenin
GSK3B phosphorylates β-catenin, marking it for degradation.29–31 We therefore studied whether activated β-catenin mediates the observed attenuation of GSK3B-mediated resistance to rapamycin. Depletion by shRNA of β-catenin mRNA, catenin beta-1 (CTNNB1), did not confer sensitivity to either rapamycin (Supplementary Figure S3B) or BEZ235 (Supplementary Figure S3C). Likewise, overexpression of constitutively active β-catenin (CTNNB1-delta90) did not confer resistance to chronic mTOR inhibition by either rapamycin or BEZ235 (Supplementary Figure S3E, F).
MAP1B Mediates Attenuated GSK3B-Conferred Resistance to mTOR Pathway Specific Inhibition
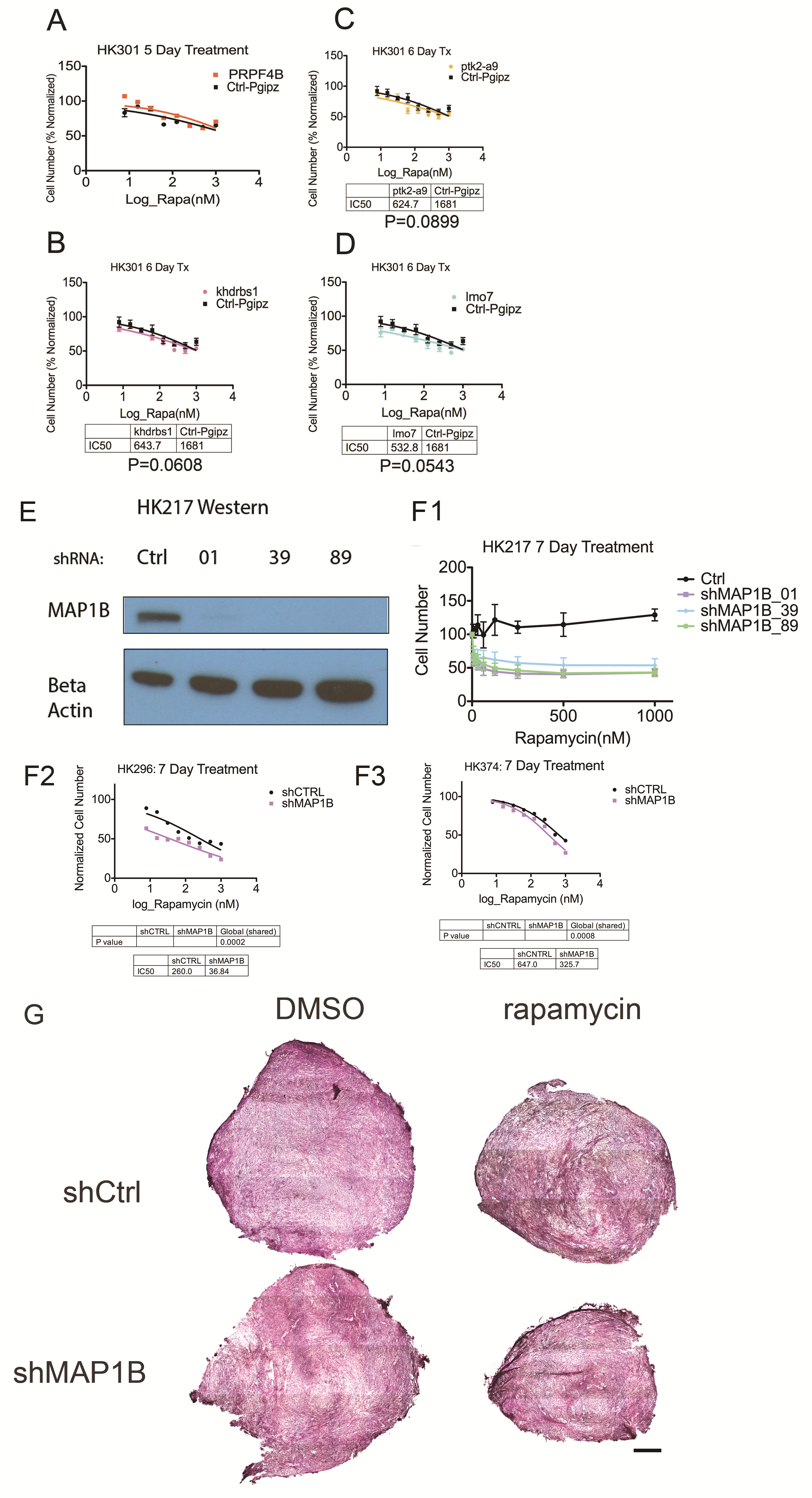
We performed a targeted shRNA screen to knock down expression of 50 out of 52 of the proteins that underwent phosphorylation changes under chronic rapamycin and were associated with GSK3B in our KEA analysis. Sixty-two percent of these proteins had decreased phosphorylation sites under chronic rapamycin, 38% had increased phosphorylation. From this screen, 5 sensitizing candidates were chosen (Z-score ≤ −2; Supplementary Table S2). Each of these candidates except for pre-mRNA processing factor 4B (PRPF4B) was hypophosphorylated under chronic rapamycin conditions, consistent with attenuated GSK3B function.
We found that sensitization effects were not consistently reproducible for knockdown of PRPF4B, K homology RNA binding domain containing signal transduction associated 1 (KHDRBS1), protein tyrosine kinase 2 (PTK2), and Lin11/Isl-1/Mec-3 (LIM) domain only protein 7 (LMO7) (Supplementary Figure S4). However, knockdown of MAP1B consistently conferred sensitization to mTOR inhibition (Fig. 4A–C). In GBM culture HK217, the IC50 of rapamycin for shMAP1B-treated cells was reduced by over 25-fold from the IC50 for shControl cells (P = 0.0002; Fig. 4B). These results were reproduced using 3 different constructs to knockdown MAP1B with the effects on sensitization roughly associated with the efficacy of MAP1B knockdown (Supplementary Figure S4E, F1). These associations of MAP1B depletion and sensitization to mTOR inhibition were also apparent in GBM cultures HK296 (Supplementary Figure S4F2), HK374 (Supplementary Figure S4F3), and HK301 (data not shown).
In HK217, the IC50 of BEZ235 for shMAP1B-treated cells was lower by 1.6-fold from the IC50 for shControl cells (P = 0.0028; Fig. 4C). These data indicate that for some GBM cultures, MAP1B promotes resistance to mTOR inhibition in a manner that is not completely abrogated by inhibition of mTORC2.
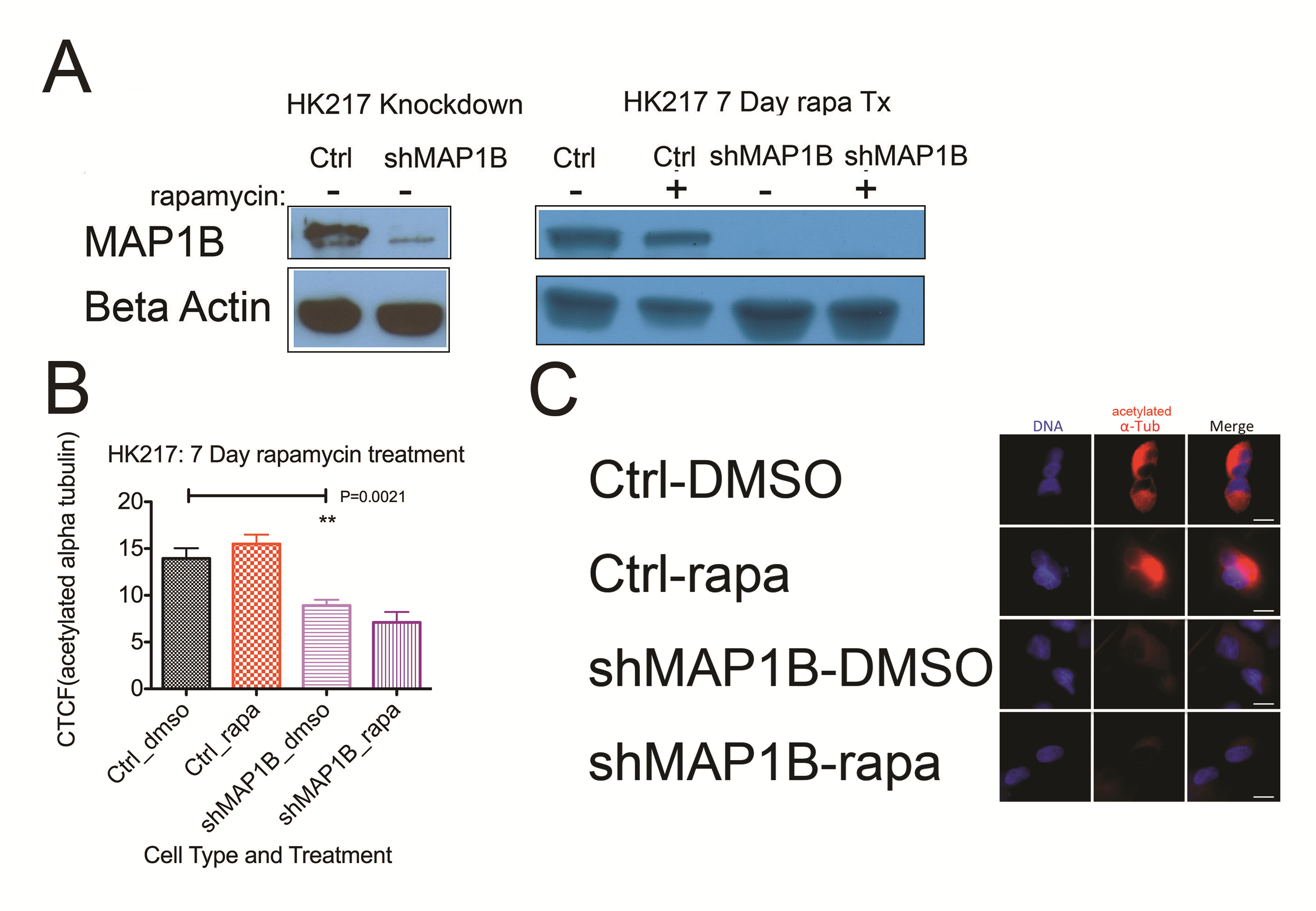
In our phosphoproteomics screen, MAP1B had decreased phosphorylated serine 1265 upon chronic rapamycin treatment (0.435 ratio of phosphorylated MAP1B for chronic rapamycin/DMSO treated controls). We found that in addition to increased phosphorylation of GSK3B, chronic rapamycin resulted in diminished phosphorylation of MAP1B at residue T1270 (Fig. 4D). Moreover, depletion of GSK3B diminished phosphorylation of MAP1B (Fig. 4D).
To determine the effects of MAP1B on rapamycin sensitivity in vivo, we performed subcutaneous xenografts of HK374 cells that were either control shRNA treated or depleted of MAP1B by shMAP1B lentivirus infection prior to transplantation. Incipient tumors were treated with either DMSO or rapamycin and tumor growth was measured over time. Strikingly, the shMAP1B cohort had a significant sensitization to mTOR inhibition by rapamycin, as evident by reduced tumor growth (Fig. 4E). These shMAP1B tumor cells maintained their depletion of MAP1B throughout the treatment until the endpoint as depicted in western blots of the resultant tumors (Fig. 4F). In addition, the western blot of tumor lysates for phosphorylated RPS6 demonstrates that rapamycin effectively inhibited its pathway in vivo (Fig. 4F). Tumors in each cohort had similar appearances on hematoxylin and eosin staining with a relatively high cellular density and somewhat diminished cellular density in the middle of the sections (Supplementary Figure S4G). These data indicate that depletion of MAP1B sensitizes GBM cells to mTOR inhibition in an in vivo model of GBM tumor growth by about 50%, which is an effect equivalent to the reduction of the IC50 for HK374 cells in vitro (Supplementary Figure S4F3).
As phosphorylated MAP1B is known to destabilize microtubules,32 we examined the effects of chronic rapamycin and MAP1B on microtubule stability. Depletion of MAP1B with shRNA resulted in effective knockdown of MAP1B even after 7 days of treatment with chronic rapamycin (Supplementary Figure S5A). In control-infected cells (shCtrl), chronic rapamycin induced increased resistance to the microtubule destabilizing agent Nocodazole compared with DMSO treated cells (Mann–Whitney test, P = 0.001; Fig. 4G, H) as measured by alpha tubulin immunocytochemistry. This resistance was abrogated by MAP1B depletion (Fig. 4G, H). Upon depletion of MAP1B (shMAP1B), the level of acetylated alpha tubulin, an indicator of stabilized microtubules, was significantly reduced compared with control cells (Mann–Whitney test, P = 0.0021, Supplementary Figure S5B, C). These data indicate that in primary human GBM cultured cells, chronic rapamycin treatment stabilizes microtubules in a MAP1B-dependent manner.
Combinatorial Therapy Targeting ERK and mTOR Abrogates the GSK3B/MAP1B-Dependent Mechanism of Resistance
As our mechanistic studies in vitro implicated ERK activation in the regulation of GSK3B phosphorylation, we next sought to determine whether MAP1B is modulated by ERK signaling to impart resistance to mTOR inhibition in an in vivo model of human GBM.
First, we demonstrated, in vitro, that HK374 GBM cells have a dramatic sensitization to mitogen-activated protein kinase kinase (MEK) (the immediate upstream modulator of ERK) inhibition by selumetinib upon additional mTOR inhibition by either rapamycin or BEZ235 (Fig. 5A). Next, we utilized a subcutaneous xenograft model to study whether MAP1B was modulated by ERK within the context of inhibition of mTOR and MEK. Caliper measurements of tumor size (mm3) over days of treatment depict the responses of tumors to combinatorial or single agent treatment with selumetinib (35 mg/kg) and rapamycin (5 mg/kg; Supplementary Figure S6A). Volume quantification of tumors at day 9 indicates a significant reduction in tumor size between combinatorial treated mice and single agent treated mice or controls (Fig. 5B).
Fig. 5.

In vivo support for our model of regulation of resistance to mTOR inhibition. (A) Representative sample of sensitization effect on HK374. Log scale of relative cell numbers following treatment with a serial dilution of doses of selumetinib (μM) in the presence of DMSO (black line), 100 nM rapamycin (red line), or 10 nM BEZ235 (green line). The calculated IC50 values are displayed for each combinatorial treatment (selumetinib + second inhibitor listed). (B) Quantification of tumor value at day 9 is depicted. Treatment groups (mean ± SEM) are compared with the DMSO treated control. P-values for the Mann‒Whitney statistical comparison test are indicated for each comparison. ANOVA comparing all groups had P = 0.0010. (C) Western blot of pooled xenograft tumors (6 animals/treatment group, 12 tumors/treatment group) depicts effective targeting of rapamycin and selumetinib after 9 days of treatment as well as phosphorylation status of MAP1B and GSK3B. (D) Schematic of proposed model of resistance to chronic mTOR inhibition. Dotted line represents indirect association. Red color indicates inactivation under chronic treatment, green color indicates activation. See also Supplementary Figure S6.
The in vivo experiment was repeated with a higher dose of selumetinib (75 mg/kg). Again, subcutaneous tumor sizes were significantly reduced in this combinatorial treatment compared with single agent treatment (Supplementary Figure S6B), but toxicity issues at this higher dose caused the loss of several mice.
Western blots (Fig. 5C) of protein recovered from the tumors in the lower dose cohort demonstrate that administered therapeutic treatments effectively inhibited their molecular targets. Rapamycin inhibited phosphorylated RPS6 (pRPS6), and selumetinib inhibited levels of phosphorylated ERK (pERK). Phosphorylated Akt-S473 (pAkt) is downregulated in the chronic rapamycin group, suggesting that chronic rapamycin inhibited mTORC2 as well as mTORC1 in these HK374 GBM cells. Importantly, the western blot also depicts activation of pERK in the chronic rapamycin group. These data indicate that abrogating the activated ERK response to mTOR inhibition can diminish tumor growth.
Our data indicate that GSK3B and MAP1B are signaling nodes downstream from the ERK activation response to mTOR inhibition. Chronic rapamycin conditions demonstrated increased inhibitory phosphorylation of GSK3B and diminished phosphorylation of MAP1B (Fig. 5C). With combinatorial mTOR and ERK inhibition (Rapa + Sel), these mechanisms of resistance were diminished: GSK3B had attenuated phosphorylation and MAP1B had greater phosphorylation compared with the rapamycin treated conditions (Fig. 5C). These data support a model of resistance wherein chronic mTOR inhibition leads to activated ERK, inhibitory phosphorylation of GSK3B, and diminished phosphorylation of MAP1B (Fig. 5D).
Discussion
Here, we find that inhibitory phosphorylation of GSK3B is an adaptive response to chronic mTOR inhibition. Our data contribute to and extend the literature supporting a role for GSK3B in the mTOR pathway.33–35 In certain GBM tumor cells, we discovered that upstream activation of ERK induces phosphorylation of GSK3B and the downstream activation of MAP1B. By first demonstrating a functional role of MAP1B in resistance to mTOR inhibition, and then elucidating the cascade of signaling between activated ERK and MAP1B, we infer that MAP1B is an effector of upstream ERK activation in its mechanism of resistance to mTOR inhibition, in at least some GBM.
Our biochemical study demonstrates that phosphorylation of MAP1B is modulated by GSK3B, a finding supported by previous studies.36–38 The identification of MAP1B as a functional protein in GBM is somewhat surprising on the surface. MAP1B is highly expressed in neurons, and most studies have investigated the role of MAP1B in neuronal growth. However, MAP1B is highly expressed in human glial progenitors (http://web.stanford.edu/group/barres_lab/brainseqMariko/brainseq2.html; last accessed 12 August 2017), although its role in these cells or in glioma is yet unknown.
The major function of MAP1B in neurons has been associated with stabilization of microtubules. A role for microtubule stability in mediating resistance to targeted therapy has not been described. Our study demonstrates that rapamycin induces microtubule stabilization that is abrogated by depletion of MAP1B. Microtubule stability may confer aspects of cellular protection, as microtubules function to coordinate the cellular stress response, promote cell survival under harsh environments, and confer chemoresistance.39 Furthermore, rapamycin-resistant cells are vulnerable to agents that induce further microtubule stability, as rapamycin sensitizes cells to Taxol in a GSK3B-dependent manner,40 and stabilized microtubules were shown to sensitize ovarian cancer cells to Taxol.41 On the other hand, if microtubule stability via MAP1B is a mode of resistance to mTOR inhibition, then one may surmise that microtubule destabilization may confer sensitivity to mTOR inhibition. Indeed, this has been shown to be the case in certain cancer cells that demonstrate a synergistic response to rapamycin and the microtubule destabilizing agent vinorelbine.42 The combination of rapalogs and vinorelbine has undergone clinical trials for the treatment of certain cancers, although not GBM due to the blood–brain barrier. Thus, molecular resistance to mTOR inhibition may be exploited by employing mTOR inhibition together with microtubule stabilization agents.
Our results have implications for therapeutic strategies in the treatment of GBM. In certain GBM cultures, inhibition of mTORC2 contributes to resistance by promoting phosphorylation of GSK3B. These findings suggest that treatment with BEZ235 or similar mixed kinase inhibitors, designed to overcome resistance, may instead result in activation of GSK3B-mediated resistance.
In clinical trials of solid tumors, combinatorial therapy targeting MEK and mTOR faced severe clinical issues due to drug toxicity.43 We found similar problems in mice. However, a novel ERK inhibitor has been shown to synergize with MEK inhibitors44,45 and may allow for lower doses of MEK inhibitor to be employed to reduce drug toxicity. Thus, despite previous setbacks with toxicity, present and future pharmacological advances may permit combinatorial therapy targeting MEK, ERK, and mTOR to provide an efficacious therapeutic strategy in the treatment of certain GBM. Our delineation of this molecular pathway for resistance to chronic mTOR inhibition reveals vulnerabilities that may be exploited by combinatorial targeting of mTOR, MEK/ERK, and microtubule stabilization in the treatment of GBM.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was supported by National Institute of Neurological Disorders and Stroke (NINDS) grant NS052563; the Dr Miriam and Sheldon G. Adelson Medical Research Foundation; California Institute of Regenerative Medicine (CIRM) training grant TG2-01169; and a Whitcome Fellowship. Mass spectrometry analysis was provided by the Bio-Organic Biomedical Mass Spectrometry Resource at UCSF (A. L. Burlingame, Director) supported by funding from the Biomedical Technology Research Centers program of the NIH National Institute of General Medical Sciences, NIH NIGMS 8P41GM103481.
Acknowledgments
We acknowledge the support of the NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691). We thank Dr Paul Mischel for helpful comments on the manuscript.
Conflict of interest statement. The authors disclose no potential conflicts of interest.
References
- 1. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown EJ, Albers MW, Shin TB, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369(6483):756–758. [DOI] [PubMed] [Google Scholar]
- 3. Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4(5):335–348. [DOI] [PubMed] [Google Scholar]
- 4. Chiu MI, Katz H, Berlin V. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc Natl Acad Sci U S A. 1994;91(26):12574–12578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78(1):35–43. [DOI] [PubMed] [Google Scholar]
- 6. Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–168. [DOI] [PubMed] [Google Scholar]
- 7. Verhaak RG, Hoadley KA, Purdom E, et al. ; Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gallia GL, Tyler BM, Hann CL, et al. Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol Cancer Ther. 2009;8(2):386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cloughesy TF, Yoshimoto K, Nghiemphu P, et al. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5(1):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hosoi H, Dilling MB, Liu LN, et al. Studies on the mechanism of resistance to rapamycin in human cancer cells. Mol Pharmacol. 1998;54(5):815–824. [DOI] [PubMed] [Google Scholar]
- 11. Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009;5(5):725–726. [DOI] [PubMed] [Google Scholar]
- 12. Wang BT, Ducker GS, Barczak AJ, Barbeau R, Erle DJ, Shokat KM. The mammalian target of rapamycin regulates cholesterol biosynthetic gene expression and exhibits a rapamycin-resistant transcriptional profile. Proc Natl Acad Sci U S A. 2011;108(37):15201–15206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Y, Sun SY, Owonikoko TK, et al. Rapamycin induces Bad phosphorylation in association with its resistance to human lung cancer cells. Mol Cancer Ther. 2012;11(1):45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoang B, Benavides A, Shi Y, et al. The PP242 mammalian target of rapamycin (mTOR) inhibitor activates extracellular signal-regulated kinase (ERK) in multiple myeloma cells via a target of rapamycin complex 1 (TORC1)/eukaryotic translation initiation factor 4E (eIF-4E)/RAF pathway and activation is a mechanism of resistance. J Biol Chem. 2012;287(26):21796–21805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Totary-Jain H, Sanoudou D, Dautriche CN, Schneller H, Zambrana L, Marks AR. Rapamycin resistance is linked to defective regulation of Skp2. Cancer Res. 2012;72(7):1836–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Juengel E, Dauselt A, Makarević J, et al. Acetylation of histone H3 prevents resistance development caused by chronic mTOR inhibition in renal cell carcinoma cells. Cancer Lett. 2012;324(1):83–90. [DOI] [PubMed] [Google Scholar]
- 17. Akhavan D, Cloughesy TF, Mischel PS. mTOR signaling in glioblastoma: lessons learned from bench to bedside. Neuro Oncol. 2010;12(8):882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Albert L, Karsy M, Murali R, Jhanwar-Uniyal M. Inhibition of mTOR activates the MAPK pathway in glioblastoma multiforme. Cancer Genomics Proteomics. 2009;6(5):255–261. [PubMed] [Google Scholar]
- 19. Wei W, Shin YS, Xue M, et al. Single-cell phosphoproteomics resolves adaptive signaling dynamics and informs targeted combination therapy in glioblastoma. Cancer Cell. 2016;29(4):563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61(3):215–225; discussion 226–229. [DOI] [PubMed] [Google Scholar]
- 21. Laks DR, Crisman TJ, Shih MY, et al. Large-scale assessment of the gliomasphere model system. Neuro Oncol. 2016;18(10):1367–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jones AM, Nühse TS. Phosphoproteomics using iTRAQ. Methods Mol Biol. 2011;779:287–302. [DOI] [PubMed] [Google Scholar]
- 23. Lachmann A, Ma’ayan A. KEA: kinase enrichment analysis. Bioinformatics. 2009;25(5):684–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Torres JZ, Ban KH, Jackson PK. A specific form of phospho protein phosphatase 2 regulates anaphase-promoting complex/cyclosome association with spindle poles. Mol Biol Cell. 2010;21(6):897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–789. [DOI] [PubMed] [Google Scholar]
- 26. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saito Y, Vandenheede JR, Cohen P. The mechanism by which epidermal growth factor inhibits glycogen synthase kinase 3 in A431 cells. Biochem J. 1994;303(Pt 1):27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J. 1994;303(Pt 3):701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272(5264):1023–1026. [DOI] [PubMed] [Google Scholar]
- 30. Sakanaka C, Weiss JB, Williams LT. Bridging of beta-catenin and glycogen synthase kinase-3beta by axin and inhibition of beta-catenin-mediated transcription. Proc Natl Acad Sci U S A. 1998;95(6):3020–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu C, Li Y, Semenov M, et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108(6):837–847. [DOI] [PubMed] [Google Scholar]
- 32. Trivedi N, Marsh P, Goold RG, Wood-Kaczmar A, Gordon-Weeks PR. Glycogen synthase kinase-3beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J Cell Sci. 2005;118(Pt 5):993–1005. [DOI] [PubMed] [Google Scholar]
- 33. Sokolosky M, Chappell WH, Stadelman K, et al. Inhibition of GSK-3β activity can result in drug and hormonal resistance and alter sensitivity to targeted therapy in MCF-7 breast cancer cells. Cell Cycle. 2014;13(5):820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koo J, Wang X, Owonikoko TK, Ramalingam SS, Khuri FR, Sun SY. GSK3 is required for rapalogs to induce degradation of some oncogenic proteins and to suppress cancer cell growth. Oncotarget. 2015;6(11):8974–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koo J, Yue P, Gal AA, Khuri FR, Sun SY. Maintaining glycogen synthase kinase-3 activity is critical for mTOR kinase inhibitors to inhibit cancer cell growth. Cancer Res. 2014;74(9):2555–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lucas FR, Goold RG, Gordon-Weeks PR, Salinas PC. Inhibition of GSK-3beta leading to the loss of phosphorylated MAP-1B is an early event in axonal remodelling induced by WNT-7a or lithium. J Cell Sci. 1998;111 (Pt 10):1351–1361. [DOI] [PubMed] [Google Scholar]
- 37. García-Pérez J, Avila J, Díaz-Nido J. Implication of cyclin-dependent kinases and glycogen synthase kinase 3 in the phosphorylation of microtubule-associated protein 1B in developing neuronal cells. J Neurosci Res. 1998;52(4):445–452. [DOI] [PubMed] [Google Scholar]
- 38. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65(4):391–426. [DOI] [PubMed] [Google Scholar]
- 39. Parker AL, Kavallaris M, McCarroll JA. Microtubules and their role in cellular stress in cancer. Front Oncol. 2014;4:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dong J, Peng J, Zhang H, et al. Role of glycogen synthase kinase 3beta in rapamycin-mediated cell cycle regulation and chemosensitivity. Cancer Res. 2005;65(5):1961–1972. [DOI] [PubMed] [Google Scholar]
- 41. Yu Y, Gaillard S, Phillip JM, et al. Inhibition of spleen tyrosine kinase potentiates paclitaxel-induced cytotoxicity in ovarian cancer cells by stabilizing microtubules. Cancer Cell. 2015;28(1):82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mondesire WH, Jian W, Zhang H, et al. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res. 2004;10(20):7031–7042. [DOI] [PubMed] [Google Scholar]
- 43. Tolcher AW, Bendell JC, Papadopoulos KP, et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann Oncol. 2015;26(1):58–64. [DOI] [PubMed] [Google Scholar]
- 44. Hatzivassiliou G, Liu B, O’Brien C, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11(5):1143–1154. [DOI] [PubMed] [Google Scholar]
- 45. Hayes TK, Neel NF, Hu C, et al. Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell. 2016;29(1):75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.