

Abstract
Objective
To assess temporal trends in familial amyotrophic lateral sclerosis (FALS) incidence rates in an Irish population and to determine factors influencing FALS ascertainment.
Methods
Population-based data collected over 23 years, using the Irish amyotrophic lateral sclerosis (ALS) register and DNA biobank, were analyzed and age-standardized rates of FALS and associated familial neuropsychiatric endophenotypes were identified.
Results
Between 1994 and 2016, 269 patients with a family history of ALS from 197 unique families were included on the register. Using stringent diagnostic criteria for FALS, the mean age-standardized FALS incidence rate for the study period was 11.1% (95% confidence interval [CI], 8.8–13.4). The FALS incidence rate increased steadily from 5.2% in 1994 to 19.1% in 2016, an annual increase of 0.7% (95% CI, 0.5–0.9, p < 0.0001). Inclusion of the presence of neuropsychiatric endophenotypes within kindreds increased the FALS incidence rate to 30%. The incidence of FALS in newly diagnosed individuals from known families increased significantly with time, accounting for 50% of all FALS diagnoses by 2016. The mean annual rate of recategorization from “sporadic ALS” to “FALS” was 3% (95% CI, 2.6–3.8).
Conclusions
The true population-based rate of FALS is at least 20%. Inclusion of extended endophenotypes within kindreds increases the rate of FALS to 30%. Cross-sectional analysis of clinic-based cohorts and stringent definitions of FALS underestimate the true rate of familial disease. This has implications for genetic counseling and in the recognition of presymptomatic stages of ALS.
Amyotrophic lateral sclerosis (ALS) is recognized as a primary motor system degeneration of complex genetic origin. The condition is usually distinguished into “familial ALS” (FALS), in which other family members are reported to have had ALS, and “sporadic ALS” (SALS), in which there is no discernible family history. At least 30 mendelian-inherited genes have been implicated in the familial form of ALS.1 However, variants in the majority of mendelian-inherited genes have also been identified in apparently sporadic cases.2–5
Recent modeling suggests that the interaction between the genetic and environmental risk factors varies depending on the specific genetic variants involved, implying that most genetic causes of ALS are likely to be incompletely penetrant, and that most apparently, SALS occurs in the context of variable genomic risk.6,7 Moreover, higher rates of neuropsychiatric conditions have been described among first- and second-degree relatives of ALS probands,8,9 and 14% polygenic overlap has been reported between ALS and schizophrenia.10 Whether the presence of these neuropsychiatric phenotypes within relatives should be considered in the definition of familial disease remains to be determined.
In this study, we have used previously proposed criteria with varying levels of stringency to assess temporal trends in FALS incidence rates in an Irish population over a 23-year period. We have taken into account the evolution of thinking around the definition of FALS and have sought to evaluate the impact of recently identified genetic and phenotypic representations of ALS and related conditions on how we define FALS.
Methods
Data collection
An Irish population-based register for patients with ALS has been in operation since 1994 with an associated DNA bank operating since 1999.11–13 Details of case ascertainment and validation methodologies have been described extensively elsewhere.11,12 Briefly, individuals confirmed to have possible, probable, or definite ALS according to the El Escorial criteria14 are enrolled on the register. Following confirmation of diagnosis, a semistructured telephone interview is conducted using a standardized questionnaire. All informants are asked specifically about the occurrence of any neurodegenerative or neuropsychiatric conditions among their first- and second-degree relatives. A diagnosis of frontotemporal lobar dementia (FTD) in a relative is accepted if (1) they were diagnosed by a physician with expertise in cognitive disorders or (2) the description of the relative was deemed to meet the Neary criteria15 by a neurologist experienced in diagnosing patients with FTD. For those identified posthumously, a direct chart review and interview with a family member is conducted where possible. The ALS DNA biobank has been used to identify established pathogenic variants in genes implicated in ALS pathogenesis.16 These DNA samples are stored to allow for retrospective assessment of new genes.
Standard protocol approvals, registrations, and patient consents
All patients included in the Irish ALS register between January 1, 1994, and December 31, 2016, were invited to participate in the study. Informed written consent for the study was obtained from all participants. This study was approved by the Beaumont Hospital Research Ethics Committee (15/40).
Diagnostic criteria for FALS
The presence and classification of FALS was determined using our previously defined criteria (figure 1).17
Figure 1. Byrne criteria for familial amyotrophic lateral sclerosis (FALS).

Analysis of register data
Data from the Irish ALS register from 1994 to 2016 were interrogated. All cases reporting a history of suspected or confirmed ALS or FTD in at least 1 relative were collated. Where necessary, genealogical records were reviewed. The DNA database was cross-referenced with the clinical database, and the genetic status was determined in all cases for whom DNA was available. Additional individuals with a known gene variant, not previously identified by a family history of ALS or FTD, were collated. Cases with a diagnosis of Kennedy disease were excluded. Individuals who had a family history suspicious for ALS (e.g., relative died of “muscle wasting disease”), in whom we could not confirm the diagnosis, were excluded. Similarly, individuals with a relative with dementia, in whom the nature of the dementia could not be accurately determined, were excluded.
Our previously reported criteria were applied to all identified FALS cases. Annual age-standardized incidence rates for FALS and FALS subclassifications were calculated as a proportion of the total number of ALS cases diagnosed annually, using the pooled Irish ALS register population from 1994 to 2016 and considering the following age bands: ≤39, 40–49, 50–59, 60–69, 70–79 and 80+ years.
Where multiple members of the same kindred were identified, pedigrees were constructed to include first-, second-, and third-degree relatives where possible. All newly diagnosed individuals from previously identified kindreds were identified. Crude incidence rates of newly diagnosed individuals from known families were calculated as a proportion of total number of ALS families presenting annually. Dates of diagnosis of FALS for all cases were obtained from the ALS register and cross-referenced against medical records where applicable. All cases were grouped by whether they were recategorized from SALS to FALS or identified as FALS at the time of diagnosis. Annual rates of recategorization were calculated by dividing the number of recategorized FALS individuals by the number of individuals diagnosed with ALS annually.
Analysis by additional phenotype and endophenotype
All cases with a confirmed family history of FTD were identified. Secondary analysis identified all probands with a confirmed family history of all-type dementia and/or schizophrenia/psychosis. The crude incidence rates of probands with a confirmed family history of FTD, all-type dementia, and possible familial endophenotypes (e.g., schizophrenia/psychosis13) were calculated by dividing by the number of patients diagnosed with ALS annually.
Genetic screening and analysis
All patients were tested for established high-penetrance ALS-associated variants. Patients were screened for the presence of the pathogenic GGGGCC hexanucleotide repeat expansion in C9orf72 by repeat-primed PCR as described previously.18 This methodology has previously been validated with positive and negative controls using reverse transcriptase PCR and Southern blots. Amplified fragments were measured by capillary electrophoresis on an Applied Biosystems 3500 Series Genetic Analyzer and visualized using Gene Mapper v.4.0. Patients with 30 hexanucleotide repeats or above were deemed positive for the expansion. Next-generation DNA sequencing was performed through Illumina paired-end, PCR-free, whole-genome sequencing and Illumina paired-end, target-enriched sequencing. Sequence data were generated for 676 patients and 446 population-matched controls. Participants were screened for exonic and splice-site variants in the exons of 30 genes considered to be linked to ALS (ALS2, ANG, CHCHD10, CHMP2B, DAO, DCTN1, ELP3, ERBB4, FIG4, FUS, hnRNPA1, LMNB1, MATR3, NEFH, OPTN, PFN1, PRPH, SETX, SIGMAR1, SOD1, SPAST, SPG11, SQSTM1, TAF15, TARDBP, TBK1, UBQLN2, UNC13A, VAPB, and VCP). To screen for high penetrance, variants that were present in any controls or at a maximum allele frequency exceeding 0.05 in reference population data sets were filtered. Variants that were present in the ALSonline genetics database (ALSoD)19 or the Human Gene Mutation Database V.2017.220 and are reported in the literature as being familial or highly penetrant were considered to be mendelian causes of ALS. Patients for whom DNA was not available and without confirmed family history of ALS or FTD were categorized as nonfamilial cases. All cases with an established mendelian-inherited ALS gene variant were identified, and the crude incidence rate of mendelian-inherited ALS was calculated by dividing by the number of patients diagnosed with ALS annually.
Statistical analysis
We fitted linear regression models to estimate the annual mean change in incidence rates with calendar year as the predictor variable and dependent variables: total FALS, definite FALS, probable FALS, possible FALS, FALS from previously identified families, recategorized FALS, mendelian-inherited ALS and probands with a positive family history of FTD, all-type dementia, and schizophrenia/psychosis, respectively. To determine whether increasing rates of FALS were a function of higher rates of “possible FALS” diagnoses, we tested the hypothesis that the total FALS (b1) and combined “definite” and “probable FALS” (b2) beta coefficients were not statistically different from each other. A simple linear regression model with total FALS and combined “definite” and “probable FALS” as main effects with joint interaction term (b1*b2) was fitted via bias corrected bootstrap (1,000 resamples). SPSS Statistics Version 24 was used to identify and estimate the parameters of the linear models and to test for statistical significance.
Results
Demographics
A total of 2173 individuals, diagnosed with ALS between 1994 and 2016, were recorded on the Irish ALS register. Of these, 313 individuals had potential FALS based on a family history of suspected or confirmed ALS or FTD in at least 1 relative or the presence of the mendelian-inherited ALS gene mutation in the proband (figure 2A). Thirty-five individuals with a family history suspicious for ALS or FTD in whom it was not possible to confirm the relatives' diagnoses and 9 individuals with proven Kennedy disease were excluded. Two hundred sixty-nine registered patients with ALS comprising 197 unique families were included in the final analysis (figure 2B). Ninety-four individuals carried a known ALS-causative gene mutation (C9orf72 [89], TARDBP [1], FUS [2], SOD1 [1], and SQSTM1 [1]). Fifty-one patients carrying the C9orf72 variant reported a family history of FTD (figure e1, links.lww.com/NXG/A49).
Figure 2. Flow chart of patients who met the inclusion/exclusion criteria for the study.
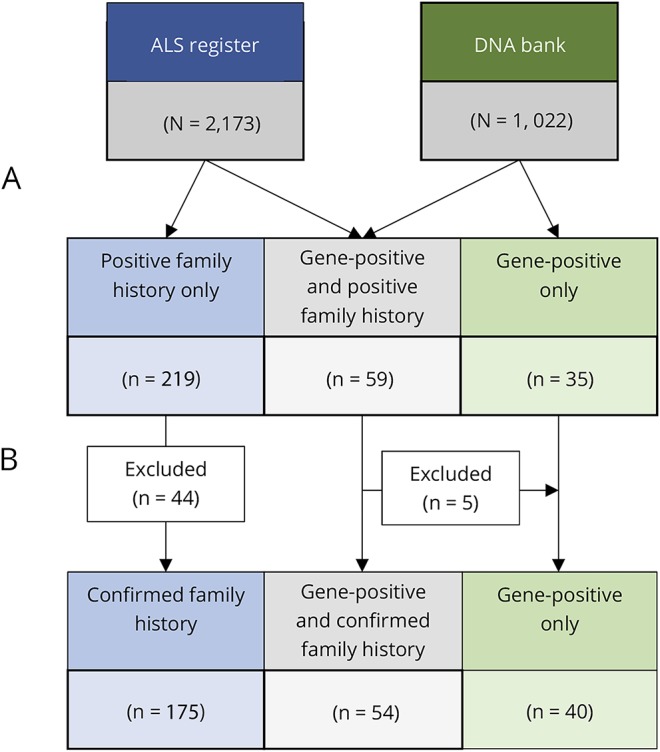
(A) All patients with ALS registered with the Irish ALS register from 1994 to 2016 who reported a history of suspected or confirmed ALS or FTD in at least 1 relative were identified. All patients with an established, highly penetrant ALS variant (C9orf72 89, TARDBP 1, FUS 2, SOD1 1, and SQSTM1 1) were identified from the DNA database. (B) Thirty-five patients with a family history suspicious for ALS or FTD in whom it was not possible to confirm the relatives' diagnoses and 9 patients with Kennedy disease were excluded. Five C9orf72-positive patients with a family history suspicious for ALS or FTD in whom it was not possible to confirm the relatives' diagnoses were recategorized into gene-positive only category. ALS = amyotrophic lateral sclerosis; FTD = frontotemporal lobar dementia.
Secondary analysis of the Irish ALS register identified 392 patients with a confirmed relative with dementia (reported as Alzheimer disease [131], FTD [51], and unspecified [210]) and 57 patients with a confirmed family history of schizophrenia.
Incidence of FALS
The mean annual crude FALS incidence rate was 11.1% (95% confidence interval [CI], 8.9–13.3) for the study period, and the corresponding mean age-standardized FALS incidence rate was 11.1% (95% CI, 8.8–13.4). However, the age-standardized FALS incidence rate increased steadily from 5.2% in 1994 to 19.1% in 2016, representing an overall increase of 0.7% (95% CI, 0.5–0.9, p < 0.0001) per annum (figure 3A). Using our previously published criteria for “definite FALS,” the mean age-standardized incidence rate was 4% (95% CI, 2.9–5.0) for the entire study period. However, between 1994 and 2016, this increased at a rate of 0.2% (95% CI, 0.01–0.3, p = 0.007) annually (figure 3B). For “probable FALS,” the age-standardized incidence rate was 3.1% (95% CI, 2.3–3.9) but did not increase with time (p = 0.318) (figure 3C). Conversely, the age-standardized incidence for “possible FALS” increased by 0.4% (95% CI, 0.2–0.5, p < 0.0001) annually, with an overall mean rate of 4.6% (95% CI, 3.1–6.1) (figure 3D). There was no difference between the total FALS (b = 0.007) and combined “definite” and “probable FALS” (b = 0.003) beta coefficients (p = 0.671) (figure 3E).
Figure 3. Temporal trends in FALS incidence.

Temporal trends in age-standardized FALS incidence for total FALS (A), definite FALS (B), probable ALS (C), and possible FALS (D). Temporal trends in age-standardized FALS incidence for total FALS compared with definite and probable FALS (E). ALS = amyotrophic lateral sclerosis; FALS = familial ALS.
FALS from previously identified families and recategorized FALS
A mean of 2.9% (95% CI, 1.8–4.1) of individuals diagnosed with ALS each year were from known FALS families, increasing by 0.3% annually (95% CI, 0.2–0.4, p < 0.0001). The relative contribution of newly diagnosed individuals from known families increased annually (p = 0.001), accounting for 50% of all FALS diagnoses in 2016. To prevent an underestimation of effect size due to unrecognized FALS in SALS individuals (i.e., those in whom a second family member has not yet been affected), the final 3 years of data collection were excluded. For the remaining years, the overall mean rate of recategorization from sporadic to FALS was 3% (95% CI, 2.6–3.8) annually. This did not change with time (p = 0.177).
Mendelian-inherited ALS
From 1999 to 2016, the mean crude incidence rate for known mendelian-inherited ALS was 4.49% (95% CI, 2.8–6.2) annually. A temporal increase of 0.4% (95% CI, 0.2–0.6) annually (p = 0.02) was observed, driven by C9orf72-positive ALS patients, who accounted for 89 of 94 known mendelian-inherited forms. The other cases, all of which were sporadic, were associated with mutations in TARDBP (1), FUS (2), SQSTM1 (1), and a previously recognized rare SOD1 variant.
Impact of phenotype
From 1994 to 2016, a mean of 1.9% (95% CI, 1.1–2.9) of individuals diagnosed with ALS annually had a confirmed positive family history of FTD, increasing by 0.2% (95% CI, 0.01–0.39; p = 0.001) per year. The mean crude incidence rates of probands with a confirmed family history of any form of dementia or schizophrenia were 14.87% (95%% CI, 9.3–20.5) and 2.2% (95% CI, 1.4–3.0), respectively. Both demonstrated annual increases of 1.8% (95% CI, 1.4–2.2; p = 0.0001) and 0.2% (95% CI, 0.18–0.22; p = 0.0001), respectively.
Discussion
The Irish ALS population-based register is flexibly designed to allow for both the categorization of FALS and the application of different levels of stringency of the definition of FALS. The availability of data from the register, combined with island status, low immigration rates, and comparatively large family sizes makes Ireland an ideal place to study FALS. By applying our previously reported criteria to the Irish ALS population-based register, we have shown that the mean annual age-standardized incidence rate for any definition of FALS over a 23-year period was 11.1%. This is consistent with the commonly quoted figure of 10% for the population rate of FALS observed in other populations of European ancestry.21,22 However, we have also identified an increasing trend in FALS ascertainment, ranging from 5.2% in 1994 to 19.1% in 2016, with over 50% of newly diagnosed FALS cases in 2016 coming from second generations within known FALS families. This in turn may drive the observed increasing trend in “definite” FALS as with increasing awareness of the prevalent FALS families, certainty of FALS classification increases.
The mean rate of recategorization from “SALS” to “FALS” was 3% annually. This is higher than previously reported crude incidence rates of ALS in relatives of patients with SALS of 1.2%.23 The difference is most likely a function of differences in classification criteria used. Indeed, there are numerous reasons why some FALS individuals are misclassified as SALS, including incomplete ascertainment of extended kindreds, and small family size.17 Less frequently, SALS may be misclassified as FALS.24 The methodological approach used in this study is consistent with standard approaches used in a clinical environment with only ALS cases with confirmed ALS and/or FTD cases in relatives included in our analysis. Overall, as the opportunities to misclassify FALS as SALS are greater than vice versa,17 the possibility of underestimating the true FALS rate within this study far exceeds the possibility an overestimation.
We have also found a mean incidence rate of known mendelian-inherited genes associated with ALS of 4.49% (95% CI, 2.8–6.2), increasing annually. This figure is driven by C9orf72-positive ALS, which accounts for the majority of known mendelian-inherited ALS in Ireland. In our data set, of the 35 C9orf72-positive ALS patients without a confirmed family history of ALS or FTD, 9 patients were unable to provide complete information on their relatives. The remaining patients reported at least 1 first- or second-degree relative with unspecified dementia, a neuropsychiatric disorder or unconfirmed ALS (figure e-2, links.lww.com/NXG/A49). Our findings are thus in keeping with our previous work suggesting that truly “sporadic” ALS associated with C9orf72 repeat expansions are rare and that the majority of those carrying this variant have a family history of either neurodegenerative or neuropsychiatric disease.
We have also demonstrated an increased trend in incidence of probands with confirmed positive family histories of FTD, all-type dementia, and schizophrenia reflecting the recognition of the importance of the extended phenotype within ALS kindreds. Our findings also demonstrate a systematic difference in the clinical phenotype between historical cases and those recently enrolled consistent with information creep, a common finding in longer standing registers.25 Our observations strongly suggest that FALS criteria should be expanded to incorporate the presence of these extended phenotypes within ALS kindreds. We have previously shown that the likelihood of FALS increases based on the number of patients with ALS within a kindred and the size of the extended kindred.17 Using the same analyses, we have now calculated that a kindred of 38 will introduce a 5% probability of having a relative with FTD26 by chance alone, but that the presence of 2 or more relatives with FTD is sufficiently unlikely by chance (p = 0.025) to provide a credible criterion for FALS. Similarly,27 we have calculated a 5% probability of having 1 relative with schizophrenia in the presence of at least 5 unaffected relatives. However, there is a diminishing probability of having 2 or more first- or second-degree relatives with schizophrenia within extended kindreds, rendering the presence of 3 or more family members with schizophrenia, a credible criterion by which to extend the definition of FALS (figure 4A). This calculation is consistent with our recent cluster analysis of ALS kindreds with extended neuropsychiatric endophenotypes.9 Conversely, assuming a lifetime risk of developing dementia in those over 65 of approximately 1 in 5,28 the presence of dementia in 2 relatives within an ALS kindred is insufficient to make a diagnosis of likely FALS, irrespective of the family size (figure 4B).
Figure 4. Binomial probability distribution.

For the number of relatives with schizophrenia (A) or dementia (B) against the kindred size. p < 0.05 is highlighted in blue.
Using stringent diagnostic criteria and excluding the presence of a neuropsychiatric endophenotype within kindreds, our data suggest that at least 20% of ALS cases in Ireland have FALS. Of these, over 40% of Irish ALS families carry the C9orf72 repeat expansion. However, by including a wider neuropsychiatric endophenotype among relatives as an additional criterion, the rate of FALS is likely to be in the region of 30%, although definitive estimates cannot yet be generated using our population-based register data, as historical ascertainment is incomplete.
This study has limitations. The presence of the C9orf72 repeat expansion was determined by repeat-primed PCR plus amplicon length analysis in blood samples. Although confirmation of each repeat expansion length using Southern blotting is recommended, and although there is acknowledged variation in amplicon length across the tissues, the approach used in this study was validated using positive and negative controls confirmed using Southern blot and is consistent with that adopted in the setting of diagnostic screening.
The definition and utility of the concept of FALS remains a matter of debate. Our data demonstrate that the estimated frequency of FALS within a population can be biased by both ascertainment methods, the level of stringency applied to the definition, and the inclusion or exclusion of extended phenotypes and endophenotypes that are biologically associated with ALS. Our population-based longitudinal data indicate that at least 20% of ALS is familial using stringent criteria. However, our data also suggest that a wider diagnostic categorization, to include FTD and neuropsychiatric conditions, is warranted.
Glossary
- ALS
amyotrophic lateral sclerosis
- CI
confidence interval
- FALS
familial ALS
- FTD
frontotemporal lobar dementia
- SALS
sporadic ALS
Author contributions
M. Ryan: study concept and design, analysis and interpretation of data, and manuscript composition. M. Heverin: study concept and design, analysis and interpretation of data, and revision of the manuscript for intellectual content. M.A. Doherty: acquisition of data, analysis and interpretation of data, and revision of the manuscript for intellectual content. N. Davis and E.M. Corr: acquisition of data. A. Vajda and N. Pender: study concept and design. R. McLaughlin: analysis and interpretation of data and revision of the manuscript for intellectual content. O. Hardiman: study concept and design, analysis and interpretation of data, and revision of the manuscript for intellectual content.
Study funding
The project is supported through the funding provided by Science Foundation Ireland (15/SPP/3244 and 16/ERCD/3854). O.H. receives support from the Health Research Board including Joint Programme in Neurodegeneration, Research Motor Neurone, and Science Foundation Ireland (FutureNeuro Centre). R.L.M. receives support from the Motor Neurone Disease Association (MNDA).
Disclosure
M. Ryan has received research support from Science Foundation Ireland. M. Heverin, M.A. Doherty, N. Davis, and E.M. Corr report no disclosures. A. Vajda has received research support from the Irish Health Research Board. N. Pender has received a speaker honorarium from Biogen; has served on the editorial board of the International Journal of Neuroscience; and has received research support from the Monkstown Hospital Foundation. R. McLaughlin has received research support from the Motor Neurone Disease Association of England and Science Foundation Ireland. O. Hardiman has received speaker honoraria/travel funding from Janssen Cilag, Biogen Idec, Sanofi Aventis, Novartis, and Merck-Serono; has been a member of advisory panels for Biogen Idec, Allergan, Ono Pharmaceuticals, Novartis, Cytokinetics, Treeway, Wave, NINDS CDE Team for ALS/MND, and Sanofi Aventis; serves as Editor-in-Chief of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia; serves on the editorial board of the Journal of Neurology Neurosurgery; coholds patents for Treatment of Central Nervous System Injury Inventors (RCSI); consults for Biogen Idec and Cytokinetics; and has received research support from Science Foundation Ireland. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol 2017;13:96–104. [DOI] [PubMed] [Google Scholar]
- 2.Jackson M, Al-Chalabi A, Enayat ZE, Chioza B, Leigh PN, Morrison KE. Copper/zinc superoxide dismutase 1 and sporadic amyotrophic lateral sclerosis: analysis of 155 cases and identification of a novel insertion mutation. Ann Neurol 1997;42:803–807. [DOI] [PubMed] [Google Scholar]
- 3.Kabashi E, Valdmanis PN, Dion P, et al. . TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008;40:572–574. [DOI] [PubMed] [Google Scholar]
- 4.Hubers A, Just W, Rosenbohm A, et al. . De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol Aging 2015;36:3117.e1–3117.e6. [DOI] [PubMed] [Google Scholar]
- 5.Gibson SB, Downie JM, Tsetsou S, et al. . The evolving genetic risk for sporadic ALS. Neurology 2017;89:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Chalabi A, Calvo A, Chio A, et al. . Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol 2014;13:1108–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chio A, Calvo A, Mazzini L, et al. . Genetic mutations shorten the multistep process in ALS. Amyotroph Lateral Scler Frontotemporal Degener 2017;18:1–73. [Google Scholar]
- 8.Byrne S, Heverin M, Elamin M, et al. . Aggregation of neurologic and neuropsychiatric disease in amyotrophic lateral sclerosis kindreds: a population-based case-control cohort study of familial and sporadic amyotrophic lateral sclerosis. Ann Neurol 2013;74:699–708. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien M, Burke T, Heverin M, et al. . Clustering of neuropsychiatric disease in first-degree and second-degree relatives of patients with amyotrophic lateral sclerosis. JAMA Neurol 2017;74:1425–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLaughlin RL, Schijven D, van Rheenen W, et al. . Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nat Commun 2017;8:14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Incidence and prevalence of ALS in Ireland, 1995–1997: a population-based study. Neurology 1999;52:504–509. [DOI] [PubMed] [Google Scholar]
- 12.O'Toole O, Traynor BJ, Brennan P, et al. . Epidemiology and clinical features of amyotrophic lateral sclerosis in Ireland between 1995 and 2004. J Neurol Neurosurg Psychiatry 2008;79:30–32. [DOI] [PubMed] [Google Scholar]
- 13.Byrne S, Elamin M, Bede P, et al. . Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol 2012;11:232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 15.Neary D, Snowden JS, Gustafson L, et al. . Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 16.Kenna KP, McLaughlin RL, Byrne S, et al. . Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet 2013;50:776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrne S, Bede P, Elamin M, et al. . Proposed criteria for familial amyotrophic lateral sclerosis Amyotroph Lateral scler 2011;12:157–159. [DOI] [PubMed] [Google Scholar]
- 18.Renton AE, Majounie E, Waite A, et al. . A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abel O, Shatunov A, Jones AR, Andersen PM, Powell JF, Al-Chalabi A. Development of a smartphone app for a genetics website: the amyotrophic lateral sclerosis online genetics database (ALSoD). JMIR Mhealth and Uhealth 2013;1:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stenson PD, Mort M, Ball EV, et al. . The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 2017;136:665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrne S, Walsh C, Lynch C, et al. . Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2011;82:623–627. [DOI] [PubMed] [Google Scholar]
- 22.Andersen PM. Genetic factors in the early diagnosis of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1(suppl 1):S31–S42. [DOI] [PubMed] [Google Scholar]
- 23.Hanby MF, Scott KM, Scotton W, et al. . The risk to relatives of patients with sporadic amyotrophic lateral sclerosis. Brain 2011;134:3454–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rabe M, Felbecker A, Waibel S, et al. . The epidemiology of CuZn-SOD mutations in Germany: a study of 217 families. J Neurol 2010;257:1298–1302. [DOI] [PubMed] [Google Scholar]
- 25.Rooney JPK, Brayne C, Tobin K, Logroscino G, Glymour MM, Hardiman O. Benefits, pitfalls, and future design of population-based registers in neurodegenerative disease. Neurology 2017;88:23212329. [DOI] [PubMed] [Google Scholar]
- 26.Coyle-Gilchrist IT, Dick KM, Patterson K, et al. . Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 2016;86:1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS Med 2005;2:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seshadri S, Wolf PA. Lifetime risk of stroke and dementia: current concepts, and estimates from the Framingham Study. Lancet Neurol 2007;6:1106–1114. [DOI] [PubMed] [Google Scholar]