


Abstract
microRNAs (miRNAs) play key roles in cancer, but their propensity to couple their targets as competing endogenous RNAs (ceRNAs) has only recently emerged. Multiple models have studied ceRNA regulation, but these models did not account for the effects of co-regulation by miRNAs with many targets. We modeled ceRNA and simulated its effects using established parameters for miRNA/mRNA interaction kinetics while accounting for co-regulation by multiple miRNAs with many targets. Our simulations suggested that co-regulation by many miRNA species is more likely to produce physiologically relevant context-independent couplings. To test this, we studied the overlap of inferred ceRNA networks from four tumor contexts—our proposed pan-cancer ceRNA interactome (PCI). PCI was composed of interactions between genes that were co-regulated by nearly three-times as many miRNAs as other inferred ceRNA interactions. Evidence from expression-profiling datasets suggested that PCI interactions are predictive of gene expression in 12 independent tumor- and non-tumor contexts. Biochemical assays confirmed ceRNA couplings for two PCI subnetworks, including oncogenes CCND1, HIF1A and HMGA2, and tumor suppressors PTEN, RB1 and TP53. Our results suggest that PCI is enriched for context-independent interactions that are coupled by many miRNA species and are more likely to be context independent.
INTRODUCTION
MicroRNAs (miRNAs) are endogenous, small, noncoding, single-stranded RNA molecules that are evolutionarily conserved. They act as post-transcriptional regulators that modulate the rates of both protein translation and mRNA decay (1–3). miRNAs are involved in virtually every cellular process, from early development (4–6) to organ function (7,8). Their dysregulated expression is associated with human diseases, including diabetes (9), cancer (10) and infection (11). Conversely, more than half of all human mRNAs contain likely miRNA target sites and are post-transcriptionally regulated. Collectively, miRNAs regulate nearly all developmental pathways, including in the context of oncogenesis (12).
Numerous studies have shown that miRNA target abundance can alter miRNA activity (13–18). The corresponding regulatory mechanism has been named competing endogenous RNA (ceRNA). We call miRNAs that potentially co-regulate RNAs shared miRNAs, and note that two RNA species may become coupled through their shared miRNAs (19). For simplicity, we refer to shared miRNAs responsible for inferred interactions between ceRNAs as ceRNA mediators and networks of inferred ceRNA interactions as ceRNETs. When dysregulation of one mRNA species—e.g. via copy number alterations (CNAs) or mutations that change its transcriptional regulation—modulates the expression of another mRNA through ceRNA regulation, we call the former a ceRNA regulator and the latter a ceRNA target, respectively. Notably, however, ceRNA interactions are mostly bidirectional, and, thus, depending on which one of the two is independently dysregulated, either one can be considered the regulator or the target.
Mathematical models for ceRNA regulation have been developed by multiple groups (20,21), and include models for ceRNA co-regulation (22), for the effects of miRNA–target binding strength on ceRNA regulation (23,24), for the effects of the number of miRNA binding sites per ceRNA (25), and for the interplay between ceRNA regulation and other types of regulatory interactions (26–29). Biochemically validated models that focus on a specific ceRNA interaction, mediated by a single shared miRNA, suggest that high miRNA abundance levels and low miRNA-to-target abundance ratios are required for physiologically relevant ceRNA regulation (14,15,30). While these models helped improve our understanding of ceRNA regulation, they did not account for miRNAs that have hundreds of other targets or for ceRNA regulation by multiple shared miRNA species.
We propose a model that directly accounts for (i) regulation of hundreds of targets by each miRNA that mediates the interaction of a specific ceRNA-pair, (ii) existence of multiple distinct ceRNA mediators for the same ceRNA pair and (iii) participation of each ceRNA in multiple ceRNA interactions that are mediated by distinct sets of miRNAs. Modeling these properties addresses a key gap in our understanding of ceRNA regulation, and suggests that multiple ceRNA regulators can act in concert to induce profound dysregulation of common target genes involved in human pathophysiology. We describe the regulatory potential and limitations of ceRNA regulation according to our model and present specific conclusions drawn from its analysis that are supported by evidence from biochemical assays.
Simulations of ceRNA regulation using established parameter values suggested that ceRNA interactions that are mediated by many (∼16 in our simulations) miRNA species become virtually independent of the abundance of the individual shared miRNAs and nearly omnipresent. Because of the variability of the expression of their many shared miRNAs across contexts, we expect these interactions to be implemented by a minority of the potential miRNA mediators in each context. To test these conclusions, we studied the overlap between predicted ceRNETs from four highly distinct tumor contexts. Our analysis suggested that predicted ceRNA interactions across the four contexts are mediated by many miRNAs—17 on average—and that these miRNAs often have unique expression signatures in each tumor context. Consequently, these interactions were supported by non-overlapping sets of mediating miRNAs in each context.
MATERIALS AND METHODS
A kinetic model for of ceRNA regulation
Our steady-state formulation for RNA abundance and miRNA–ceRNA complex formation is supported by the observations that the half-life of most mRNAs (hours) (31), is much longer than both the diffusion time of a regulator (32) and the balance between miRNA–target binding and their disassembly (33). We note that because miRNAs have a long half-life (up to 72 h) (34–36), even relatively low rates of miRNA synthesis can produce high abundance and consistent regulation.
We first used the model to study interactions between two arbitrary RNA species—RNA1 and RNA2—as a function of the count and abundance of their shared miRNA regulators at steady state. We then extended this model to account for other miRNAs and other targets of the shared miRNAs (Supplementary Figure S1). We accounted for up to hundreds of additional targets for each shared miRNA species and for up to dozens of additional miRNA regulators for each target RNA. Our Monte Carlo analysis suggested that the ceRNA regulation efficiency depends both on the number of shared miRNA species and on the abundance of their high-affinity targets.
Single interaction model
We begin by describing a naïve single-interaction model that accounts for interactions between miRNAs and their target. ceRNA regulation is a consequence of changes in the abundance of free miRNAs, which in turn is a function of changes to miRNA degradation rates and the abundance of miRNA–target complexes—increases in miRNA–target complexes amount to reductions in the abundance of miRNAs that are free to regulate other targets. To model both, we describe the binding of miRNAs to their targets, as well changes to their abundance, using Michaelis–Menten kinetics, represented schematically below.
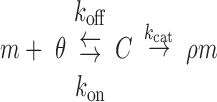 |
Here, 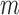 and
and 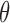 represent the abundances of a miRNA and its target, respectively, and
represent the abundances of a miRNA and its target, respectively, and
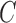 represents the miRNA–target complex.
The parameters
represents the miRNA–target complex.
The parameters 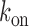 ,
,
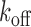 and
and 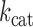 are chemical
rate constants with units of molecule/second. We assume that miRNAs and targets that form
complexes will be degraded asymmetrically. To model this, we use the dimensionless rescue
parameter
are chemical
rate constants with units of molecule/second. We assume that miRNAs and targets that form
complexes will be degraded asymmetrically. To model this, we use the dimensionless rescue
parameter 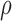 to allow for a proportion of
bound miRNAs to escape degradation and be reused (recycled) by the RNAi machinery (23,36–38). While
to allow for a proportion of
bound miRNAs to escape degradation and be reused (recycled) by the RNAi machinery (23,36–38). While 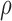 can take values between 0 and
1, we set
can take values between 0 and
1, we set 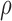 closer to 1 so that most bound
miRNAs will escape degradation and could be reused to target other genes; higher
closer to 1 so that most bound
miRNAs will escape degradation and could be reused to target other genes; higher
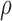 leads to weaker ceRNA effects.
The abundance of
leads to weaker ceRNA effects.
The abundance of 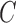 is dependent on both miRNA and Ago
abundances, and the latter may have a dominant modulatory effect on miRNA activity and
target coupling (38,39). Our model produces the following equations, which describe changes in free
target abundance (Equation 1), free miRNA
abundance (Equation 2), and miRNA–target
complex abundance (Equation 3) as a function
of their synthesis rates (
is dependent on both miRNA and Ago
abundances, and the latter may have a dominant modulatory effect on miRNA activity and
target coupling (38,39). Our model produces the following equations, which describe changes in free
target abundance (Equation 1), free miRNA
abundance (Equation 2), and miRNA–target
complex abundance (Equation 3) as a function
of their synthesis rates (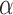 ) and degradation rates
(
) and degradation rates
(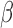 )—both assumed to be
constant—as well as the chemical rate constants that dictate complex formation and
disassembly.
)—both assumed to be
constant—as well as the chemical rate constants that dictate complex formation and
disassembly.
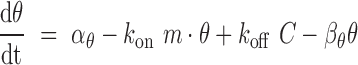 |
(1) |
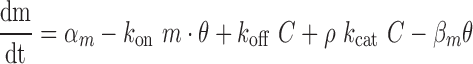 |
(2) |
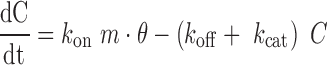 |
(3) |
Typically, the complex binds and unbinds much faster than RNA transcription and
degradation (40,41): with unbinding expected ∼10 min after binding (42), whereas transcription and degradation times have been estimated at
over 100 min for the average length mRNA (42,43). Consequently, the complex is expected to be in
quasi-equilibrium with respect to the abundance of miRNAs and their targets, and the rate
of change to complex abundance can be approximated as zero, i.e. 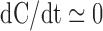 . This suggests
that target-dependent miRNA degradation is the predominant force associated with changes
in free miRNA abundance at steady state.
. This suggests
that target-dependent miRNA degradation is the predominant force associated with changes
in free miRNA abundance at steady state.
Setting 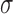 as shown in Equation (4) simplifies Equations (1)–(3) and reduces the number of system parameters, producing Equations (5) and (6). The parameter
as shown in Equation (4) simplifies Equations (1)–(3) and reduces the number of system parameters, producing Equations (5) and (6). The parameter 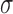 can now be interpreted as
the regulatory efficiency of the miRNA–target interaction, which depends on miRNA-binding
affinity.
can now be interpreted as
the regulatory efficiency of the miRNA–target interaction, which depends on miRNA-binding
affinity.
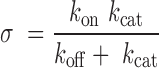 |
(4) |
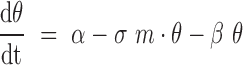 |
(5) |
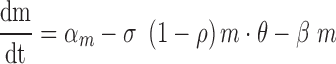 |
(6) |
The first derivative of 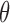 (
(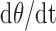 in Equation 5), represents the balance between the synthesis
rate
in Equation 5), represents the balance between the synthesis
rate 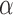 of the miRNA target, its
miRNA-mediated degradation rate
of the miRNA target, its
miRNA-mediated degradation rate 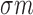 and its miRNA-independent
degradation rate
and its miRNA-independent
degradation rate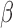 . Similarly, the first
derivative of
. Similarly, the first
derivative of 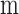 (
(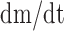 , Equation 6) represents the balance between the miRNA’s
synthesis rate
, Equation 6) represents the balance between the miRNA’s
synthesis rate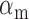 and its
degradation rate, which is a linear combination of its target-independent degradation
and its
degradation rate, which is a linear combination of its target-independent degradation
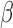 and its target-mediated
degradation
and its target-mediated
degradation 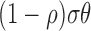 .
This, in turn, is guided by the rescue parameter
.
This, in turn, is guided by the rescue parameter 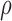 (23,36,37), which depends on the quality of its 3′ pairing
(18,30,36,38,44)—a common feature of miRNA binding
(45)—as well as on target abundance (16).
(23,36,37), which depends on the quality of its 3′ pairing
(18,30,36,38,44)—a common feature of miRNA binding
(45)—as well as on target abundance (16).
Model extension to multiple shared miRNAs and targets
In a physiologic setting, both shared miRNAs and their targets are embedded in a network of densely-connected interacting RNAs. Each shared miRNA may have hundreds of targets, and each target may be regulated by dozens of additional miRNAs. The kinetic model presented in Equations (4)–(6) can be thus extended to study miRNA-mediated regulation in the presence of N competing targets and M additional regulating miRNAs, as given in Equations (7) and (8) below.
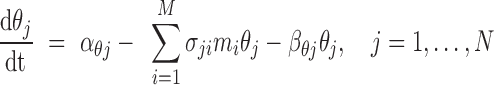 |
(7) |
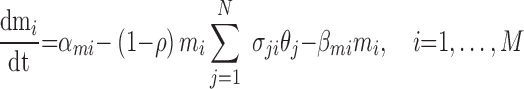 |
(8) |
Moreover, miRNAs and their targets cooperate as a part of a large network of interactions, with only a subset of the genes being regulated by any given miRNA. This has the effect of selectively depleting the miRNA pool that is available for binding and regulation.
Mean field approximation
Our model describes the kinetic dependence between RNAs regulated by a group of miRNAs, assuming that each RNA interacts with each miRNA. In a more realistic cellular context, however, miRNAs and their targets are immersed in a large network of interactions, and only a subset of genes are regulated by any given miRNA. This has the effect of selectively depleting the number of miRNAs that are available for binding and regulation.
While explicitly considering all possible miRNA–target interactions in the cell is infeasible—computational predictions estimate that there might be as many of 1200 miRNAs regulating 14 600 target RNAs—we are not interested in the detailed description of each interaction but on their average effect on genes of interest, and therefore the systems can be modeled with a mean field approximation. In our simulations, we accounted for a context-independent regulatory network with 1220 miRNAs—of which only 15% were expressed in each context—that targeted 14 576 genes through 490 000 interactions (Supplementary Table S1). Interestingly, simulation results with up to 5× more or fewer predicted miRNA–target interactions had almost no effect on predicted target fold changes—average fold changes for interactions mediated by 20 miRNAs ranged from 1.61 to 1.62 and by 10 miRNAs from 1.21 to 1.22. As a point of reference, Cupid miRNA–target predictions are based on a majority vote within a bootstrapping-classification scheme; increasing the requirement from 50% to 95% of the votes lead to less than a 0.5-fold change in the number of predicted miRNA–target interactions. Consequently, we concluded that, under proposed parameters, the mean-field approximation is robust to changes to the stringency of miRNA–target prediction methods.
We used a detailed kinetic description to model a small subset of miRNAs and their targets and replaced all additional interactions by the average values of miRNA and target abundances. These were computed by averaging the kinetic equations of all molecular species at steady state:
 |
(9) |
where 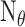 ,
,
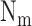 and
and 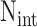 are the
total number of targets, miRNAs, and miRNA-mediated interactions. The equations can be
solved and the average abundances of miRNAs and their targets,
are the
total number of targets, miRNAs, and miRNA-mediated interactions. The equations can be
solved and the average abundances of miRNAs and their targets, 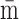 and
and 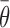 , can be obtained as a
function of the average transcription rates, degradation rates, and regulatory efficiency,
, can be obtained as a
function of the average transcription rates, degradation rates, and regulatory efficiency,
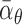 ,
,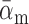 ,
,
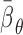 ,
,
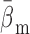 and
and
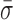 . Provided with these
average values, we can now return to the kinetic model and include all additional
interactions described by their mean field abundances:
. Provided with these
average values, we can now return to the kinetic model and include all additional
interactions described by their mean field abundances:
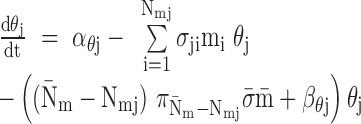 |
(10) |
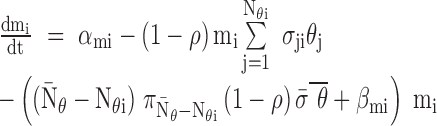 |
(11) |
where
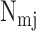 is the number of
additional miRNAs that regulate target
is the number of
additional miRNAs that regulate target 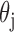 ,
,
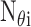 is the number
of targets regulated by
is the number
of targets regulated by 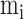 ,
,
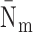 and
and
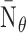 are
respectively the average number of miRNAs that regulate a single RNA and the average
number of RNA targets per miRNA;
are
respectively the average number of miRNAs that regulate a single RNA and the average
number of RNA targets per miRNA; 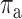 takes the
value 1 if
takes the
value 1 if 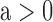 , 0 otherwise. For RNA targets
of interest, this factor allows us to scale the effects of mean-field interactions as a
function of the number of miRNAs explicitly considered. Namely, studying a particular gene
and its k miRNA regulators, the mean field approximation of miRNA effects
on this target is altered to acknowledge that the effects of k miRNAs
that regulate this target have already been accounted for.
, 0 otherwise. For RNA targets
of interest, this factor allows us to scale the effects of mean-field interactions as a
function of the number of miRNAs explicitly considered. Namely, studying a particular gene
and its k miRNA regulators, the mean field approximation of miRNA effects
on this target is altered to acknowledge that the effects of k miRNAs
that regulate this target have already been accounted for.
Using this modified system of equations, we were able to model the coupling effect between two genes with consideration for some of the indirect regulation that may affect their coupling activity. Namely, we considered the miRNAs that target both these genes and modulate their regulation, additional miRNAs that target one of these genes but not the other and thus may influence the dynamic range of its abundance, additional targets T of all said miRNA regulators that may affect the abundance of free miRNAs, and the miRNAs that target T.
Model parameters
We used the kinetic model (Equations 7
and 8) to investigate how changes to the
abundance of RNA2 affect the abundance of RNA1. We set model
coefficients, including the miRNA-independent degradation rate 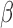 ,
regulatory efficiency
,
regulatory efficiency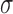 , RNA synthesis rates,
miRNA–target binding affinities, and the rescue parameter
, RNA synthesis rates,
miRNA–target binding affinities, and the rescue parameter 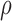 according to published studies, as given in Table 1 (31,33,36,46); typically, when using these parameters we obtained
according to published studies, as given in Table 1 (31,33,36,46); typically, when using these parameters we obtained
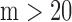 and
and 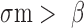 . Published
coefficients were adopted to achieve more physiological conditions. To study relative
target fold changes, we fit generalized extreme value distributions using MathWorks
Statistics Toolbox Distribution Fitting to simulation data. Each fit is reproduced in
Supplementary Figure S5.
. Published
coefficients were adopted to achieve more physiological conditions. To study relative
target fold changes, we fit generalized extreme value distributions using MathWorks
Statistics Toolbox Distribution Fitting to simulation data. Each fit is reproduced in
Supplementary Figure S5.
Table 1. Parameters used in the analytical and numerical simulations of the model to produce Figure 1, with corresponding references from the literature.
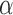
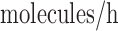
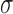
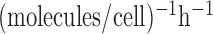
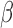
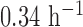
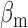
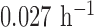
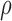
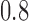
We used a fixed average recycling rate 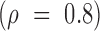 that
translate to a conservative (low) target-mediated degradation rate—equivalent to a miRNA
degradation event per five targeting events. We selected αm to match a miRNA
expression distribution observed in profiles of tumor samples. Namely, we created a
distribution of miRNA-basal transcription rates based on miRNA expression profiles from
255 TCGA prostate cancer samples (47) and our
average mRNA synthesis rates (48). Here, for each
given tumor sample, αm and miRNA and mRNA abundances
that
translate to a conservative (low) target-mediated degradation rate—equivalent to a miRNA
degradation event per five targeting events. We selected αm to match a miRNA
expression distribution observed in profiles of tumor samples. Namely, we created a
distribution of miRNA-basal transcription rates based on miRNA expression profiles from
255 TCGA prostate cancer samples (47) and our
average mRNA synthesis rates (48). Here, for each
given tumor sample, αm and miRNA and mRNA abundances 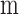 and
and
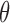 were estimated using Table
1 parameters together with their expression
estimates in the sample following Equation S12 in Schwanhäusser et al.
(31); the resulting αm distribution
was then normalized to produce a minimum αm of 0 and the average rate of 2
molecules/h (49). In our simulations,
αm values were sampled from the resulting distribution; we note that profiles
of TCGA cancer samples from each of fourteen tested tumor types produced nearly identical
αm distributions, suggesting that our αm distribution is not
context specific. Using these parameters, our simulations predicted that couplings that
are mediated by twenty miRNAs lead to a 1.5-fold change in the abundance of the target
gene in response to a 5-fold change in the abundance of the other gene—i.e. 80%
silencing—as shown in Figure 1.
were estimated using Table
1 parameters together with their expression
estimates in the sample following Equation S12 in Schwanhäusser et al.
(31); the resulting αm distribution
was then normalized to produce a minimum αm of 0 and the average rate of 2
molecules/h (49). In our simulations,
αm values were sampled from the resulting distribution; we note that profiles
of TCGA cancer samples from each of fourteen tested tumor types produced nearly identical
αm distributions, suggesting that our αm distribution is not
context specific. Using these parameters, our simulations predicted that couplings that
are mediated by twenty miRNAs lead to a 1.5-fold change in the abundance of the target
gene in response to a 5-fold change in the abundance of the other gene—i.e. 80%
silencing—as shown in Figure 1.
Figure 1.

Simulation: the ceRNA effect as a function of the number of mediating miRNA species. We simulated changes in steady-state abundance of RNA1 as a function of the steady-state abundance of RNA2 for varying synthesis rates αm of their common miRNA regulators when each of these miRNAs regulates 400 other targets. In repeated simulations, synthesis rates for each miRNA varied between 1 and 4000 molecules per hour and were selected based on a distribution derived from TCGA miRNA expression profiles in prostate cancer tumors. All other parameters were taken from published estimates (Table 1). We report curves fitted to RNA1 fold changes (Supplementary Figure S2) in 1000 repeated simulations following RNA2 5-fold upregulation—from 50 to 250 molecules per cell—and where the two ceRNAs are co-regulated by up to 20 miRNAs. Minimal changes were observed when the two ceRNAs were co-regulated by 1–2 miRNAs (inset), but co-regulation by 16 or more miRNAs consistently increased RNA1 abundance by ∼50%.
Using published parameters avoids optimization and training, and is less likely to be
biased, overfitted, or limited to specialized conditions. However, because these values
are only estimates and in order to better understand the relationship between the model's
parameters, we repeated the simulation using a wide range of parameters, including α from
0.5 to 8 molecules/h, σ from 0.005 to 0.05 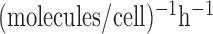 ,
β from 0.04 to 0.6 h–1, βm from 0.005 to 0.34 h–1, and
,
β from 0.04 to 0.6 h–1, βm from 0.005 to 0.34 h–1, and
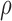 from 0.7 to 0.95 (50). Note that the range selection for α and the mean
expression for αm were motivated by observations made by Marzi et
al. (49). We extended the range for
from 0.7 to 0.95 (50). Note that the range selection for α and the mean
expression for αm were motivated by observations made by Marzi et
al. (49). We extended the range for
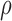 , which was previously set to
0.75 or 0.9 (38) to enable an investigation of the
effects of
, which was previously set to
0.75 or 0.9 (38) to enable an investigation of the
effects of 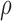 (Supplementary Figures S2 and S3).
The range for β and βm was chosen to span 10× around values given in Table
1 and used for generating Figure 1 simulations.
(Supplementary Figures S2 and S3).
The range for β and βm was chosen to span 10× around values given in Table
1 and used for generating Figure 1 simulations.
Constructing ceRNA interaction networks (ceRNETs)
ceRNETs were assembled from independently-predicted ceRNA interactions, with predictions
made by an algorithm based on the previously described Hermes and Cupid (22,51). Hermes
and Cupid predict coupling between miRNA targets based on the relative size of their
shared miRNA regulatory program and the conditional mutual information between these genes
and their mediating miRNAs. Namely, given genes Ti and Tj, and the
set of miRNAs that regulate them 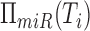 and
and
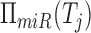 , their
shared program is identified by taking the intersection
, their
shared program is identified by taking the intersection 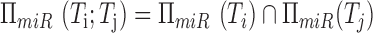 .
First, test that the size of
.
First, test that the size of 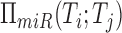 relative
to the sizes of the individual programs is statistically significant at FDR < 1E–2 by
Fisher's exact test. Then, evaluate the statistical significance
relative
to the sizes of the individual programs is statistically significant at FDR < 1E–2 by
Fisher's exact test. Then, evaluate the statistical significance 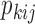 (P-value) of the
test
(P-value) of the
test 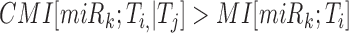 ,
where CMI and MI stand for conditional mutual information and mutual information
respectively, and the variables indicate the expression of the corresponding RNA species
(51). The CMI is estimated using an adaptive
partitioning algorithm (52) by first iteratively
partitioning the 3D expression space evenly into 8 partitions per iteration until
partitions are balanced (P > 0.05 by Chi-squared test), and then
summing up CMI across partitions. P-values for each triplet are computed
using a null-hypothesis where the candidate modulator's expression
(
,
where CMI and MI stand for conditional mutual information and mutual information
respectively, and the variables indicate the expression of the corresponding RNA species
(51). The CMI is estimated using an adaptive
partitioning algorithm (52) by first iteratively
partitioning the 3D expression space evenly into 8 partitions per iteration until
partitions are balanced (P > 0.05 by Chi-squared test), and then
summing up CMI across partitions. P-values for each triplet are computed
using a null-hypothesis where the candidate modulator's expression
(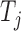 ) is shuffled 1000 times, thus
preserving the pairwise mutual information between miRNA and target. Final significance
across the entire set of miRNA mediators is computed using Fisher's method to integrate
both regulatory directions, i.e. Ti affecting
miRk regulation of Tj as well
as Tj affecting miRk regulation of
Ti, for all miRNA mediators
) is shuffled 1000 times, thus
preserving the pairwise mutual information between miRNA and target. Final significance
across the entire set of miRNA mediators is computed using Fisher's method to integrate
both regulatory directions, i.e. Ti affecting
miRk regulation of Tj as well
as Tj affecting miRk regulation of
Ti, for all miRNA mediators 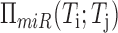 .
Specifically, the value
.
Specifically, the value 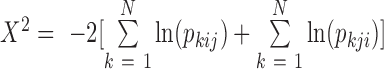 was computed and used to estimate a significance P-value for the entire
program. Note that
was computed and used to estimate a significance P-value for the entire
program. Note that 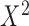 follows a Chi-square distribution,
with
follows a Chi-square distribution,
with 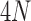 degrees of freedom, where
degrees of freedom, where
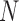 is the number of miRNAs in the shared
program. Finally, only prediction passing significance of FDR < 1E–3 were selected.
Note that selected predictions by Hermes have been validated in glioblastoma cell lines
(51).
is the number of miRNAs in the shared
program. Finally, only prediction passing significance of FDR < 1E–3 were selected.
Note that selected predictions by Hermes have been validated in glioblastoma cell lines
(51).
In order to identify miRNA mediators in addition to ceRNA interactions, we modified our prediction algorithms to perform greedy addition of miRNA mediators and to optimize the combined P-value for each predicted interaction. Namely, for each candidate interaction, we searched for the minimum combined P-value through the greedy forward inclusion of individual miRNAs: mediators were included only if they improved the combined P-value as estimated using Fisher's method. Those that failed to improve the combined P-value lack functional evidence for mediating regulation. We constructed interaction networks for glioblastoma (53) (423 samples, 12 032 genes, 469 miRNAs profiled), ovarian cancer (54) (583 samples, 12 032 genes, 713 miRNAs profiled), prostate cancer (47) (140 samples, 23 614 genes, 367 miRNAs profiled) and breast cancer (55) (207 samples, 18 748 genes, 524 miRNAs profiled). The resulting predicted networks had 527 430 (glioblastoma), 532 869 (ovarian), 476 456 (prostate) and 447 011 (breast) predicted interactions, and are given in Supplementary Tables S2–S5.
Constructing the pan-cancer ceRNA interactome (PCI)
We studied the extent, overlap, and potential relevance of predicted ceRNA interactions across four tumor types, including glioblastoma (GBM) (53), ovarian (OV) (54), prostate (PRAD) (47) and breast (BRCA) (55) adenocarcinoma. Cupid predicts miRNA targets by integrating sequence-based predictions made by multiple methods (Supplementary Table S1). We note that ceRNA inference is not based on ceRNA co-expression, and is instead based on assessing whether the abundance of one RNA affects regulation of the other by their shared miRNA program (and vice-versa) based on the statistical significance of their conditional mutual information (22,51). As such, we did not make ceRNA inferences based on pairwise coexpression or co-regulation but rather based on evidence for a more complex, three-way interaction model; see details in Methods.
Each tumor-specific ceRNA-network included >5000 genes participating in ∼500K predicted interactions: each was defined by two RNAs and by the miRNAs that couple them. The four networks are given in Supplementary Tables S2–S5. Surprisingly, each pair of ceRNA networks had ∼300 000 ceRNA interactions in common or ∼43% on average of the total number of interactions inferred by the algorithm in each tumor context. If the true ceRNETs were completely tumor-context independent, and given an estimated false-negative rate of ∼30% (56), one would expect ∼49% of the inferred ceRNA interactions to be identical across tumor pairs—each interaction will be identified with a 70% chance in each network. Thus, this result suggests that ceRNA networks are largely tumor-context independent. In comparison, other regulatory networks inferred from the same data were dramatically more tumor-specific. For instance, only 1% of transcriptional interactions, as inferred by the ARACNe algorithm (57,58), were conserved between GBM and BRCA. Similarly, comparisons of protein-protein interactomes, assembled by identical high-throughput experimental assays such as yeast-2-hybrids, rarely contain >10% identical interactions (59).
Moreover, 164 623 ceRNA interactions between 3803 genes—about a third of those inferred in each tumor and 13% of the size of the union of the four ceRNETs—were common to all four ceRNETs (FDR<1E-3); these interactions form a pan-cancer ceRNA interactome (PCI) and are reported in Supplementary Table S6. Remarkably, if the actual ceRNETs—as opposed to our predicted networks that are incomplete and include false positives—were identical in all four tumor contexts, the expected overlap would be 24% (0.7 to the power of four, based on a 30% false negative rate). This observation supports the hypothesis that a majority of ceRNA interactions are cancer-context independent. In addition, a comparison of the number of miRNA co-regulators and predicted miRNA mediators for PCI interactions in each context suggested that, on average, PCI interactions have nearly three-times as many co-regulating miRNAs (Figure 2C) as other (more context-specific) ceRNA interactions. Note that co-regulating miRNAs are miRNAs that are predicted to co-regulate the coupled genes based on sequence analysis and cross-species conservation—this prediction is expression-independent—as opposed to miRNA mediators, which are predicted based on evidence for ceRNA regulation from RNA-expression profiles.
Figure 2.

The overlap of ceRNETs in four tumor datasets identified a context-independent ceRNET (PCI). ceRNETs were inferred based on RNA profile datasets from breast, glioblastoma, ovarian and prostate tumors. (A) A significantly large set of nearly 165,000 ceRNA interactions were common to all four networks (P < 1E–12 by permutation testing) and these comprise the pan-cancer ceRNA interactome (PCI). (B) Most miRNA mediators of PCI interactions are context specific. We used Krippendorff's alpha coefficients (α) to test the overlap between mediator sets of PCI interactions across networks; α>0.8 indicates good agreement. In total, 85% of conserved interactions had −0.15 < α < 0.15 (no relation); 9% had α < −0.15 (weak dissimilarity); 6% had α > 0.15 (weak similarity); no PCI interaction had α > 0.8. (C) Considering each context, coupled genes in PCI were co-regulated by nearly three times more miRNAs than other ceRNET interactions, respectively.
To test the statistical significance of the network overlap, we performed permutation tests by swapping one million ceRNA interactions (network edges) at random between genes with shared miRNA regulators in each of the four networks. Such an approach preserves node connectivity and network topology while ensuring that randomized interactions are supported by a wild-type-comparable set of miRNA regulators (51). Following 1E-12 such tests, we never observed a comparable number of common interactions across the four networks, suggesting an upper bound for the P-value, P < 1E–12 (Figure 2A). The result excludes the possibility that PCI is a by-product of network topology or co-regulation by miRNAs. In addition, to quantitatively address the potential contamination by transcriptional regulation events, we measured the overlap of breast and colon cancer transcriptional networks (60) with PCI. Our analysis revealed that less than 0.6% of the >185,000 candidate interactions in each of the two contexts are represented in PCI.
Tumor-specificity of PCI mediators
To study individual miRNAs that mediate PCI interactions in multiple cancer types, we focused on 247 miRNAs that could be detected in all of the four datasets. Surprisingly, only eleven of these were inferred to mediate interactions in all four networks (Cluster J in Supplementary Figure S7), suggesting that pan-cancer interactions may indeed be mediated by different miRNA in each context. This is consistent with the results of the kinetic model analysis, because a miRNA that is in the appropriate rate-limiting kinetic regime to mediate a PCI interaction in one context may be expressed in a non-rate-limiting regime (i.e. too high or too low) in a different context, thus failing to provide a significant contribution. Furthermore, systematic analysis of PCI interactions confirmed that miRNAs inferred to functionally mediate these interactions are highly context specific. Indeed, for 66% of the PCI interactions, no miRNA was inferred as mediating the interaction in all four networks. See Supplementary Table S7 for interactions that are similarly mediated across networks, and Supplementary methods for detail.
Identifying frequent miRNA mediators in each coupling network
When identifying miRNAs that were frequently predicted mediators of target coupling in each context, we considered only miRNAs that were predicted mediators of PCI interactions. miRNAs were evaluated for the significance of the odds ratio between the predicted frequency for that specific miRNA and the average predicted frequency for all other miRNA mediators across all interactions. We used a right-tailed Chi-square (χ2) test to examine if the odds ratio in a given context was significantly large, setting a P < 1E–5 cutoff for selection of frequent mediators. At this stringent threshold, the analysis identified 50–100 frequent miRNAs mediators within each context. In total, of the 247 miRNAs that were profiled across all four datasets, a total of 134 miRNAs were identified as frequent mediators of target coupling (Supplementary Figure S7). Interestingly, most of these miRNAs mediate coupling in tumor-specific fashion, i.e. in one tumor but not in the others.
Predicting the expression of PCI targets
We used a ridge-regression with Glmnet for Matlab within a 10-fold cross-validation
analysis scheme (61,62) to predict the expression of each PCI target from the expression of its
inferred regulators. Ridge regression was used to minimize over-fitting. For each PCI
target, in each 10-fold cross-validation step, Glmnet constructs a regression model using
training samples to fit an estimate 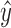 for the PCI-target expression
profile
for the PCI-target expression
profile 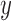 . The test-set residuals
(
. The test-set residuals
(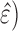 are then compiled
across the 10 testing sample sets by taking the difference between the target expression
profile
are then compiled
across the 10 testing sample sets by taking the difference between the target expression
profile  and the fitted estimate
and the fitted estimate
 , i.e.
, i.e. 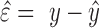 . To
calculate R2, we take the sum of the square of the residuals
across all samples,
. To
calculate R2, we take the sum of the square of the residuals
across all samples, 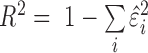 /
/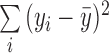 ,
where
,
where 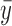 is the mean expression of the PCI
target across the dataset. To assign P-values to the predictive ability
we used bootstrapping. Namely, in bootstrap runs, the PCI-target expression profile
is the mean expression of the PCI
target across the dataset. To assign P-values to the predictive ability
we used bootstrapping. Namely, in bootstrap runs, the PCI-target expression profile
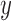 is replaced by
is replaced by 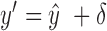 , where
, where
 and
and
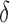 is populated by random selection
from
is populated by random selection
from 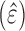 , with replacement
(63,64).
Glmnet regression was repeated for one thousand bootstrapping
, with replacement
(63,64).
Glmnet regression was repeated for one thousand bootstrapping 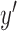 s,
estimating bootstrapping R2 using 10-fold cross validation
analysis to produce a null distribution and a P-value was assigned by
comparing the R2 to this distribution. Note that this approach
is virtually identical to the one used by Barretina et al. (65), with the only difference being that the more
conservative ridge-regression was used. When computing correlations between expression
profiles of ceRNA targets and the standardized totals of the expression profiles of their
predicted PCI regulators, the expression profile of each gene was quantile normalized and
standardized (51). Pearson correlations were
computed between the expression profiles of the ceRNA target and the total expression of
its regulators; no learning or optimization was performed.
s,
estimating bootstrapping R2 using 10-fold cross validation
analysis to produce a null distribution and a P-value was assigned by
comparing the R2 to this distribution. Note that this approach
is virtually identical to the one used by Barretina et al. (65), with the only difference being that the more
conservative ridge-regression was used. When computing correlations between expression
profiles of ceRNA targets and the standardized totals of the expression profiles of their
predicted PCI regulators, the expression profile of each gene was quantile normalized and
standardized (51). Pearson correlations were
computed between the expression profiles of the ceRNA target and the total expression of
its regulators; no learning or optimization was performed.
Protein activity inference
We used VIPER (66) to analyze high-throughput assays and test whether inferred target-ceRNA protein activity was altered following silencing of their ceRNA regulators. VIPER estimates changes to the activity of a protein based on changes to the expression profiles of its predicted downstream targets (22,67,68). Downstream targets were inferred by ARACNe (58,69) in each tumor context.
Biochemical experiments
Details on cell and culture conditions, RNA interference and reverse transfection, over-expression and forward transfection, mutagenesis, RT-PCR analysis, high-throughput quantitative RT-PCR analysis, and dual luciferase reporter assays are given in Supplemental methods.
RESULTS
We first present an analysis of our simulated ceRNA interactions based on a kinetic model for ceRNA regulation. Our simulations pointed to required conditions for effective ceRNA regulation and predicted that some ceRNA interactions are expected to be context independent. We then describe our efforts to computationally and biochemically test interactions that are predicted to be tumor-context independent.
Analysis of our ceRNA model suggests that effective regulation requires many miRNA mediators
Models for ceRNA regulation suggest that effective regulation is only possible when miRNA mediators operate in a rate-limiting regime (15,25). To test this assertion, we extended an established kinetic interaction model to describe the miRNA–target complex formation and its effects on the abundance of both miRNAs and targets, Supplementary Figure S1 (38,39,70). The resulting model (see Methods) accounts for stoichiometric degradation of miRNA–target complexes and supports the assertion that ceRNA regulation is unlikely to result from purely catalytic titration of miRNAs or from perturbations of their transcription rates (12,27,33).
We used this model to study miRNA–target couplings that may be mediated by multiple miRNAs (1 ≤ nm ≤ 20), where each miRNA regulated over 400 other targets. Namely, we computed the probability of observing a given RNA1 fold-change as a function of RNA2 changes over a 5-fold range according to our model using parameters that were motivated by published assays and with miRNA expression assigned based on expression distributions obtained from TCGA profiles of primary tumors (47). Simulation results using Table 1 parameters are given in Figure 1. Our analysis suggested that RNA1 fold-changes became virtually independent of the expression of each individual miRNA when the interaction was mediated by many randomly selected miRNAs. The average RNA1 fold-change was modest when the interaction was mediated by few shared miRNAs (1 ≤ nm ≤ 10), but increasing the number of miRNA mediators (to nm > 10) led to rapid asymptotic convergence to a 1.5-fold change in RNA1 abundance (under Table 1 parameters). RNA-target coupling efficiency was highly variable and depended on individual miRNA expression when the number of miRNA mediators was 10 ≤ nm ≤ 16, but appeared independent of miRNA expression for ceRNA interactions with nm > 16.
Varying Table 1 parameters produced
qualitatively similar results but with varying nm cutoffs and
RNA1 fold changes. Most importantly, for all parameter settings tested,
nm = 1 and nm = 6 produced
negligible expectation for ceRNA effects, while nm = 16 and
nm = 20 produced effects with expected physiological
relevance (Supplementary Figure
S2); e.g. simulations with nm = 16,
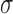 = 0.022, and
= 0.022, and
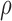 = 0.8, and setting
= 0.8, and setting
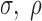 as in Table
1, produced fold changes ranging from 1.3 to
2.8 (Supplementary Figure S3).
Every distribution in Figure 1 is given as a single
data point in Supplementary Figures S2A
and S3. We also note that 7% of genes that are potentially co-regulated by at
least one miRNA (Supplementary Table
S1) are potentially co-regulated by at least 16 miRNAs, and even fewer are
expected to be expressed in each context. We emphasize that our conclusions about the
number of miRNA mediators that are expected for effective ceRNA regulation are dependent
on the parameters used to populate the proposed model. Our parameter selection was
motivated by estimates from published data, but these may not be appropriate for all
contexts. However, it is important to note that, independently of the set of parameters,
our model suggests that interactions with a greater number of miRNA mediators are expected
to produce greater and more consistent ceRNA effects.
as in Table
1, produced fold changes ranging from 1.3 to
2.8 (Supplementary Figure S3).
Every distribution in Figure 1 is given as a single
data point in Supplementary Figures S2A
and S3. We also note that 7% of genes that are potentially co-regulated by at
least one miRNA (Supplementary Table
S1) are potentially co-regulated by at least 16 miRNAs, and even fewer are
expected to be expressed in each context. We emphasize that our conclusions about the
number of miRNA mediators that are expected for effective ceRNA regulation are dependent
on the parameters used to populate the proposed model. Our parameter selection was
motivated by estimates from published data, but these may not be appropriate for all
contexts. However, it is important to note that, independently of the set of parameters,
our model suggests that interactions with a greater number of miRNA mediators are expected
to produce greater and more consistent ceRNA effects.
Our simulations suggested that the ceRNA effect strongly depends on the number of miRNA
mediators and the stability of miRNAs—including miRNA recycling rates—as well as their
relative binding affinity. In general, as clearly shown in Figure 1 and Supplementary
Figure S2A, ceRNA effects increased with the number of mediating miRNAs. However,
increasing the recycling rate 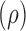 for interactions that were
mediated by fewer miRNAs was compensatory and increased the ceRNA effect in our
simulations (Supplementary Figure
S3B). In all cases, higher binding affinities (
for interactions that were
mediated by fewer miRNAs was compensatory and increased the ceRNA effect in our
simulations (Supplementary Figure
S3B). In all cases, higher binding affinities (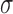 ) and lower target-independent
miRNA degradation rates (βm) lead to stronger effects (see Methods, Supplementary Figure S3). In
addition, our simulations suggested that the number of targets for each miRNA mediator
affected their ability to mediate interactions, and that cooperative regulation of a
target by multiple ceRNA regulators increased the overall expected target fold change
(Supplementary Figure S4).
) and lower target-independent
miRNA degradation rates (βm) lead to stronger effects (see Methods, Supplementary Figure S3). In
addition, our simulations suggested that the number of targets for each miRNA mediator
affected their ability to mediate interactions, and that cooperative regulation of a
target by multiple ceRNA regulators increased the overall expected target fold change
(Supplementary Figure S4).
In conclusion, analyses results, together with the observation that the distribution of miRNA expression profiles is nearly identical across tumor contexts (71,72), Supplementary Figures S2 and S3, suggested that genes that are co-regulated by many miRNAs are more likely to be coupled as ceRNAs, irrespective of tumor type or the abundance of individual species of shared miRNAs. Such ceRNA interactions are expected to be tumor-context independent.
PCI interactions are implemented in a context-specific manner
Our model predicts that, while effective ceRNA regulation is dependent on many factors, RNAs that are potentially co-regulated by many shared miRNAs are more likely to result in ceRNA interactions, independent of individual miRNA abundance (10,73). Details on the construction of PCI are given in Materials and Methods, but we note that PCI interactions were inferred independently in each of the four tumor contexts. This, however, was not the case for the specific miRNAs that mediate them. While PCI interactions were predicted to regulate gene expression in all context, the sets of their mediating miRNAs were virtually non-overlapping across contexts (Figures 2B and Supplementary Figure S3). This observation is in line with predictions by ceRNA models: PCI interactions are generally supported by many miRNAs; miRNA expression distributions are similar across contexts, but individual miRNA expression is context specific; while miRNA species that do not operate in a rate-limiting regime are expected to have minimal contributions to ceRNA couplings (23,74), a sufficient number of mediators are expected to support PCI interactions in many contexts.
Our own simulations suggested that each RNA-miRNA-RNA triplet has its own dynamics, depending on expression, degradation, and binding affinity parameters. Thus, the contribution of each miRNA mediator to each ceRNA interaction is dependent on many parameters and only those mediators that are expressed within their rate-limiting regime for this interaction will effectively contribute to its implementation. For ceRNA pairs that share many miRNAs—i.e. 17 miRNA species or 10 miRNA families, on average (Supplementary Figure S6)—this all but guarantees that at least some expressed miRNA species will operate in the right kinetic regime to mediate ceRNA coupling. Indeed, each inferred PCI interaction was predicted to be mediated by only 7 of the 17 shared miRNAs, on average (or five miRNA families). Consequently, considering evidence from tumor profiles, it is not surprising that the set of miRNA mediators for a given coupling were tumor-context specific (Figure 2B), and that the PCI is mostly composed of ceRNAs that are co-regulated by many miRNA species (Figure 2C). An example for the miRNAs predicted to mediate interactions in a small network clique is given in Figure 3; only 2 of the 109 miRNAs predicted to mediate these PCI ceRNA interactions were predicted in all four contexts.
Figure 3.

A densely connected subnetwork of oncogenes in PCI is mediated by varying populations of miRNAs across the four ceRNETs. CCND1, HMGA2 and HIF1a are predicted to regulate each other as ceRNAs in PCI. As an illustrating example of context-dependent mediation for PCI interactions, we identified the miRNAs that are predicted to mediate each of the three interactions in each of the four ceRNETs. For example, Cupid predicted that CCND1 and HIF1a have 38 shared miRNAs, but their ceRNA interaction was predicted to be mediated by only 16, 13, 12 and 11 miRNAs in BRCA, GBM, OV, and PRAD, respectively. miR-19a was the only mediator predicted to mediate this interaction in all four ceRNETs, resulting in no cross-ceRNET similarity: Krippendorff's alpha coefficient of −0.03 and Jaccard index of 0.03.
Biochemical assays support PCI’s tumor-context independence
The PCI was inferred by analyzing independent TCGA datasets for GBM, BRCA, OV and PRAD. We selected two small PCI subnetworks for experimental follow-up in multiple tumor contexts: one includes three oncogenes (CCND1, HIF1A, and HMGA2) and the other includes three tumor suppressors (PTEN, RB1 and TP53). These ceRNA interactions were tested in A549 (lung cancer, LUAD), C3A (liver cancer, LIHC), HT-29 (colon cancer, COAD), SK-MEL-28 (melanoma, SKCA), MCF7 (BRCA), U2-OS (osteosarcoma, OSCA), PC3 (PRAD) and SK-OV-3 (OV) cell lines, representing eight distinct tumor contexts, including five that were not used for ceRNA-interaction inference; GBM was excluded from the validation effort because the interaction between RB1 and PTEN has been previously reported in GBM cell lines (51).
In each cell line, we measured expression fold changes of each gene, by qRT-PCR, following transfection of the 3′ UTR of each gene in its triplet, including their own 3′ UTR as a positive control; 3′ UTRs of MAPK13, RSAD1 and THNSL1 were used as negative controls. qRT-PCR assays were performed on Fluidigm BioMark HD system in 3 biological replicates and with three technical replicates per biological replicate. Results are summarized in Figure 4A–B, and detailed fold-change responses to 3′ UTR transfections are given in Supplementary Figures S8-S9; negative controls in A549 and U2-OS are shown in Figure 4C and Supplementary Figure S10. In total, in 80% of the experiments, predicted targets were significantly upregulated (P < 0.05 by t-test). While significant and highly consistent, individual responses were relatively small, with an average 1.4-fold increase in gene expression and only 29 interactions showing >50% increase; this is consistent with previously reported results (51,56,74) and with our model's predictions (Figure 1, a 5-fold increase of an RNA lead to a 1.5 increase of its ceRNA partner).
Figure 4.

Validation of oncogene and tumor-suppressor PCI subnetworks. Significant mRNA upregulation in response to 3′ UTR transfection of candidate PCI regulators in eight cell lines is shown for (A) a three-oncogene subnetwork and for (B) a three-tumor-suppressor subnetwork, and (C) negative controls. For each transfected 3′ UTR and each target gene, the panels identify cell lines where ceRNA targets were significantly (P < 0.05) upregulated in response to transfection (filled dots); missing dots designate cell lines where target expression was too low to be measured. Fold changes are depicted by a green-to-red log-scale gradient, with red showing a 2-fold change; fold change values are given in Supplementary Figures S8-S10. (D) Deletions of 7-base predicted miRNA binding sites were used to generate HIF1A 3′ UTR (WT) mutants (Ψ1 and Ψ2); deletions at Ψ1 and Ψ2 start at positions 3844 and 3964 of NM_001530, respectively. (E) Transfections of constructs containing Ψ1 and Ψ2 had weaker HMGA2 upregulation and rescue by HIF1A silencing, compared to WT constructs. Negative controls include perturbations of MAPK13, RSAD1 and THNSL1. (F) Luciferase activity fold changes of constructs containing WT, Ψ1, and Ψ2 after transfecting miRNAs predicted to modulate HMGA2 regulation and target deleted sites. The region deleted in Ψ1 was predicted to bind miR-17-5p, miR-18a-5p, miR-20a-5p and miR-106a-5p; Ψ2 lost a predicted binding site for let-7g-3p, miR-493-5p and let-7a-2-3p; miR-301a-3p was predicted to target WT, Ψ1 and Ψ2; and miR-557 was not predicted to regulate HIF1A. Notations ** and *** denote P< 0.01 and P< 0.001, respectively. Data are represented as mean ± SEM.
To confirm that observed interactions are mediated by miRNA binding sites in 3′ UTRs, we tested the effects of miRNA binding-site mutagenesis on regulation of HMGA2 by the HIF1A 3′ UTR in A549 cells (Figure 4D–F), compared regulation of PTEN cDNA and 3′ UTRs in PC3 (Figure 5A), and rescued the effects of siRB1 on PTEN and TP53 expression by transfecting RB1 3′ UTRs in U2OS (Figure 5B and C). We selected two HIF1A 3′ UTR miRNA binding sites for deletion. These sites, and associated miRNAs, had high confidence scores for regulating the two genes and mediating their interaction by members of multiple miRNA families. Each site, corresponding to a 7-base seed match of the regulating miRNAs, was independently deleted to produce two 3′ UTR mutants, Ψ1 and Ψ2 (Figure 4D). Transfections of mutants showed weaker HMGA2 up-regulation and siHIF1A rescue when compared with wildtype 3′ UTR (Figure 4E). Ψ1 and Ψ2 also showed reduced sensitivity to miRNAs that are predicted to target the deleted sites (Figure 4F).
Figure 5.

Validation of tumor-suppressor PCI interactions. (A) Perturbations using PTEN 3′ UTR and cDNA in PC3, where PTEN is –/–, suggest that RB1 and TP53 regulation by the PTEN 3′ UTR is independent of PTEN protein expression. (B) Silencing RB1 and transfecting its 3′ UTR in U2-OS cells down and upregulated PTEN and TP53 mRNA expression, respectively. Transfecting RB1 3′ UTR rescued the effects of siRB1 on PTEN and TP53 mRNA expression (P < 0.1 for PTEN and P < 1E–4 for TP53). Negative controls include perturbations of MAPK13, RSAD1 and THNSL1. (C) siRNA-mediated silencing of RB1 in U2-OS, SK-MEL-28 and A549 reduced PTEN and TP53 luciferase activity. Data are represented as mean ± SEM.
To disentangle the effects of RNA and protein-mediated regulation, we transfected PC3, a PTEN –/– cell line, with PTEN cDNA and PTEN 3′ UTR. PTEN 3′ UTR, but not cDNA transfections lead to significant upregulation of RB1 and TP53 in PC3 (Figure 5A). Moreover, in U2OS, siRNA-mediated silencing of RB1 downregulated PTEN and TP53, while RB1 3′ UTR transfection upregulated PTEN and TP53 expression and rescued the effects of siRB1 transfection (Figure 5B and C). These results suggest that observed effects are independent of protein activity and dependent on regulation by 3′ UTRs and the miRNA binding sites embedded in them.
To further test the effects of perturbations targeting ceRNA regulators on the activity of their protein targets, we used VIPER to analyze 13 high-throughput assays where each of the six genes was targeted by RNAi and genome-wide expression was measured by microarrays or RNA-Seq (75–85). To determine whether VIPER could be used to evaluate these assays, we first tested whether silencing of each gene resulted in a significant (P < 0.05, VIPER) dysregulation to its inferred activity; e.g. siPTEN was expected to alter PTEN activity. In total, four assays failed this control and were eliminated from further consideration. Results of 18 tests based on the remaining 9 assays are given in Supplementary Figure S11. In total, 16 of the 18 tests in osteosarcoma, breast, lung, ovary, prostate, and colon cancer cell lines showed significant reductions in the activity of our tested cancer genes following silencing of their ceRNA regulators (P < 0.05). Taken together, an investigation of six predicted ceRNA interactions from two networks of key cancer genes suggests that these interactions are present in every tested context, lead to changes to ceRNA–target protein activity, and are dependent on regulator 3′ UTR expression—and particularly on miRNA binding sites in 3′ UTR—but not on ceRNA-regulator protein expression.
PCI interactions predict expression in multiple contexts
To test whether PCI interactions may be present in contexts not used for its construction, we used PCI to predict expression in eight additional datasets. A unique property of ceRNETs is an increase in correlation between the expression of a specific target and the total expression of its ceRNA partners as a function of the number of partners (51). This property has been attributed to combinatorial regulation by multiple ceRNA regulators that compete for common miRNAs. To evaluate the predictive power of PCI, we report on (a) an evaluation of the predictive ability of PCI on the expression of targets in both tumor-related and non-tumor contexts and (b) median correlations between expression profiles of genes and the standardized totals of the expression profiles of their predicted PCI regulators.
We used a ridge-regression within a 10-fold cross-validation analysis scheme to predict the expression of each PCI target from the expression of its inferred regulators; see Methods for details. Our results, given in Figure 6, confirmed that PCI interactions are predictive of ceRNA-target expression in the contexts used for its construction—GBM, OV, PRAD and BRCA tumor profiles—as well as in other tumor contexts, disease contexts, and pools of expression profiles from healthy cells from Gene Expression Atlas (86). In every context tested, using both ridge regression and Pearson correlation, the expression of genes with few regulators in PCI could not be predicted, while profiles of genes with many regulators could be significantly predicted using the expression of their regulators. This observation is consistent with previous results based on ceRNA networks that were constructed in a context-specific manner (51).
Figure 6.

The predictive ability of PCI is context-independent. A unique property of ceRNETs is the increase in predictive ability and correlation between expression profiles of targets and their regulators as a function of the number of regulators (given in the x-axis in log2 scale) (51). (A) The predictive significance of PCI interactions as a function of the number of regulators, as estimated using ridge regression analysis, assigning significance by bootstrapping residuals. ceRNA targets with the same number of predicted regulators were binned together, and median P values are reported for each group. (B) Pearson correlation between the expression profiles of each target and the average expression of their regulators. Pearson correlation obtained from shuffled networks shown using dashed lines, P-values estimated using K-S test. As shown, in every context, predictive ability improved with the number of ceRNA regulators. For both (A) and (B), the top, middle, and bottom rows summarize analyses of TCGA datasets used to build the PCI, TCGA datasets for other tumor types, and both cancer and non-cancer datasets from Gene Expression Atlas, respectively.
DISCUSSION
Recently, multiple kinetic models for ceRNA regulation have provided a mechanistic rationale for the coupling of RNA species that are regulated by the same miRNAs (15,23,25,74). However, these models do not account for the effects of mediation by multiple miRNA species with many other targets. We proposed a model for ceRNA regulation that accounts for the effects of mediation by multiple miRNA species with hundreds of targets and for regulation by multiple ceRNAs. Our analyses suggested that effective ceRNA regulation is strongly influenced by the abundance of its miRNA mediators and by the number their targets and that interactions that are mediated by many miRNA species are more likely to produce measurable effects. As observed by others, our model predicted that one miRNA regulator is not likely to mediate physiologically relevant ceRNA regulation (30).
Based on our model, we hypothesized that ceRNAs that are co-regulated by many miRNAs are expected to be context-independent. Interestingly, an analysis of the overlap between ceRNETs that were independently reconstructed from four tumor contexts—the PCI—suggested that surprisingly many ceRNA interactions were predicted in each of the four contexts and that these interactions were enriched for genes that are co-regulated by many miRNAs. Further analysis showed that PCI interactions are predictive of gene expression in each of twelve contexts, including tumor- and non-tumor cells. Moreover, biochemical assays supported PCI’s context-independent nature. An important consequence of this work is that oncogenic drivers that were previously thought to be completely uncoupled present strong crosstalk, including CCND1, HIF1A and HMGA2 and PTEN, TP53 and RB1. This suggests that aberrant up or downregulation of any of these genes may produce an effect greater than what may be accounted for in isolation. We plan to explore the full coupling of oncogenes and tumor suppressors in a follow-up manuscript.
Our analysis suggests that ceRNA interactions that are mediated by many miRNAs are expected to have physiologically relevant effects and that models for ceRNA regulation must make an effort to account for all RNAs that influence ceRNA regulation. Unexpectedly, our model predicted that thousands of ceRNA interactions are expected to regulate gene expression in context-independent fashion and that these interactions form a unique regulatory network that may be regulating and altering gene expression in virtually all cellular contexts.
Supplementary Material
ACKNOWLEDGEMENTS
The results published here are in part based upon data generated by The Cancer Genome Atlas pilot project established by the NCI and NHGRI as of January 2011. Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at http://cancergenome.nih.gov/. The dbGaP Accession number for the data analyzed in this paper is phs000178.v4.p4, dated 24 January 2011.
Author Contributions: H.S.C., M.R.M., X.Y., P.S. and A.C. conceived and contributed to modeling ceRNA regulation. M.R.M. and P.S. conducted simulation and analyzed the kinetic model. H.S.C., M.B. and P.S. analyzed TCGA data. E.V.K., D.L.N. and J.S. carried out and analyzed experiments. H.S.C., M.R.M., P.S. and A.C. wrote the manuscript. All authors read and approved the final manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) grant awards: The Outstanding NCI Investigator Award [1R35CA197745 to A.C.]; Centers for Cancer Systems Biology Consortium [1U54CA209997]; instrumentation grant to AC supporting high-performance computing [1S10OD012351]; instrumentation grant to AC supporting biological data storage [1S10OD021764]. Funding for open access charge: NCI.
Conflict of interest statement. Andrea Califano is founder and equity holder ofDarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. Columbia University is also an equity holder in DarwinHealth Inc.The other authors declare that they have no competing interests.
REFERENCES
- 1. Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fabian M.R., Sonenberg N., Filipowicz W.. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010; 79:351–379. [DOI] [PubMed] [Google Scholar]
- 3. Huntzinger E., Izaurralde E.. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011; 12:99–110. [DOI] [PubMed] [Google Scholar]
- 4. Wightman B., Ha I., Ruvkun G.. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993; 75:855–862. [DOI] [PubMed] [Google Scholar]
- 5. Kanellopoulou C., Muljo S.A., Kung A.L., Ganesan S., Drapkin R., Jenuwein T., Livingston D.M., Rajewsky K.. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005; 19:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Song J.L., Stoeckius M., Maaskola J., Friedländer M., Stepicheva N., Juliano C., Lebedeva S., Thompson W., Rajewsky N., Wessel G.M.. Select microRNAs are essential for early development in the sea urchin. Dev. Biol. 2012; 362:104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Amin N.D., Bai G., Klug J.R., Bonanomi D., Pankratz M.T., Gifford W.D., Hinckley C.A., Sternfeld M.J., Driscoll S.P., Dominguez B. et al. Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science. 2015; 350:1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen J.F., Murchison E.P., Tang R., Callis T.E., Tatsuguchi M., Deng Z., Rojas M., Hammond S.M., Schneider M.D., Selzman C.H. et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trajkovski M., Hausser J., Soutschek J., Bhat B., Akin A., Zavolan M., Heim M.H., Stoffel M.. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. 2011; 474:649–653. [DOI] [PubMed] [Google Scholar]
- 10. Lu J., Getz G., Miska E.A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B.L., Mak R.H., Ferrando A.A. et al. MicroRNA expression profiles classify human cancers. Nature. 2005; 435:834–838. [DOI] [PubMed] [Google Scholar]
- 11. Pfeffer S., Zavolan M., Grässer F.A., Chien M., Russo J.J., Ju J., John B., Enright A.J., Marks D., Sander C. et al. Identification of virus-encoded microRNAs. Science. 2004; 304:734–736. [DOI] [PubMed] [Google Scholar]
- 12. Broderick J.A., Zamore P.D.. Competitive endogenous RNAs cannot alter microRNA function in vivo. Mol. Cell. 2014; 54:711–713. [DOI] [PubMed] [Google Scholar]
- 13. Arvey A., Larsson E., Sander C., Leslie C.S., Marks D.S.. Target mRNA abundance dilutes microRNA and siRNA activity. Mol. Syst. Biol. 2010; 6:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bosson A.D., Zamudio J.R., Sharp P.A.. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol. Cell. 2014; 56:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luna J.M., Scheel T.K., Danino T., Shaw K.S., Mele A., Fak J.J., Nishiuchi E., Takacs C.N., Catanese M.T., de Jong Y.P. et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell. 2015; 160:1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cazalla D., Yario T., Steitz J.A.. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science. 2010; 328:1563–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M.. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013; 495:333–338. [DOI] [PubMed] [Google Scholar]
- 18. Powers J.T., Tsanov K.M., Pearson D.S., Roels F., Spina C.S., Ebright R., Seligson M., de Soysa Y., Cahan P., Theißen J.. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature. 2016; 535:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P.. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language. Cell. 2011; 146:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H.. starBase v2.0: decoding miRNA–ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yip D.K., Pang I.K., Yip K.Y.. Systematic exploration of autonomous modules in noisy microRNA-target networks for testing the generality of the ceRNA hypothesis. BMC Genomics. 2014; 15:1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chiu H.S., Llobet-Navas D., Yang X., Chung W.J., Ambesi-Impiombato A., Iyer A., Kim H.R., Seviour E.G., Luo Z., Sehgal V. et al. Cupid: simultaneous reconstruction of microRNA-target and ceRNA networks. Genome Res. 2015; 25:257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yuan Y., Liu B., Xie P., Zhang M.Q., Li Y., Xie Z., Wang X.. Model-guided quantitative analysis of microRNA-mediated regulation on competing endogenous RNAs using a synthetic gene circuit. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:3158–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yuan Y., Ren X., Xie Z., Wang X.. A quantitative understanding of microRNA-mediated competing endogenous RNA regulation. Quant. Biol. 2016; 4:47–57. [Google Scholar]
- 25. Ala U., Karreth F.A., Bosia C., Pagnani A., Taulli R., Léopold V., Tay Y., Provero P., Zecchina R., Pandolfi P.P.. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:7154–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bosia C., Pagnani A., Zecchina R.. Modelling competing endogenous RNA networks. PLoS One. 2013; 8:e66609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Figliuzzi M., Marinari E., De Martino A.. MicroRNAs as a selective channel of communication between competing RNAs: a steady-state theory. Biophys. J. 2013; 104:1203–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang P., Ning S., Zhang Y., Li R., Ye J., Zhao Z., Zhi H., Wang T., Guo Z., Li X.. Identification of lncRNA-associated competing triplets reveals global patterns and prognostic markers for cancer. Nucleic Acids Res. 2015; 43:3478–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paci P., Colombo T., Farina L.. Computational analysis identifies a sponge interaction network between long non-coding RNAs and messenger RNAs in human breast cancer. BMC Syst. Biol. 2014; 8:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Denzler R., McGeary S.E., Title A.C., Agarwal V., Bartel D.P., Stoffel M.. Impact of MicroRNA levels, target-site complementarity, and cooperativity on competing endogenous RNA-Regulated gene expression. Mol. Cell. 2016; 64:565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M.. Global quantification of mammalian gene expression control. Nature. 2011; 473:337–342. [DOI] [PubMed] [Google Scholar]
- 32. Jens M., Rajewsky N.. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat. Rev. Genet. 2015; 16:113–126. [DOI] [PubMed] [Google Scholar]
- 33. Wee L.M., Flores-Jasso C.F., Salomon W.E., Zamore P.D.. Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell. 2012; 151:1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krol J., Loedige I., Filipowicz W.. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010; 11:597–610. [DOI] [PubMed] [Google Scholar]
- 35. Hwang H.W., Wentzel E.A., Mendell J.T.. A hexanucleotide element directs microRNA nuclear import. Science. 2007; 315:97–100. [DOI] [PubMed] [Google Scholar]
- 36. Baccarini A., Chauhan H., Gardner T.J., Jayaprakash A.D., Sachidanandam R., Brown B.D.. Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr. Biol.: CB. 2011; 21:369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chatterjee S., Großhans H.. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature. 2009; 461:546–549. [DOI] [PubMed] [Google Scholar]
- 38. Loinger A., Shemla Y., Simon I., Margalit H., Biham O.. Competition between small RNAs: a quantitative view. Biophys. J. 2012; 102:1712–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Levine E., Zhang Z., Kuhlman T., Hwa T.. Quantitative characteristics of gene regulation by small RNA. PLoS Biol. 2007; 5:e229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Briggs G.E., Haldane J.B.. A note on the kinetics of enzyme action. Biochem. J. 1925; 19:338–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morozova N., Zinovyev A., Nonne N., Pritchard L.-L., Gorban A.N., Harel-Bellan A.. Kinetic signatures of microRNA modes of action. RNA. 2012; 18:1635–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mathonnet G., Fabian M.R., Svitkin Y.V., Parsyan A., Huck L., Murata T., Biffo S., Merrick W.C., Darzynkiewicz E., Pillai R.S.. MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science. 2007; 317:1764–1767. [DOI] [PubMed] [Google Scholar]
- 43. Hartl D.L., Ruvolo M.. Genetics: Analysis of Genes and Genomes. 2011; 8th ednBoston, MA: Jones & Bartlett Learning [Google Scholar]
- 44. Ameres S.L., Horwich M.D., Hung J.H., Xu J., Ghildiyal M., Weng Z., Zamore P.D.. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010; 328:1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Helwak A., Kudla G., Dudnakova T., Tollervey D.. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013; 153:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang E., van Nimwegen E., Zavolan M., Rajewsky N., Schroeder M., Magnasco M., Darnell J.E. Jr. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 2003; 13:1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taylor B.S., Schultz N., Hieronymus H., Gopalan A., Xiao Y., Carver B.S., Arora V.K., Kaushik P., Cerami E., Reva B. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim Y.K., Kim V.N.. Processing of intronic microRNAs. EMBO J. 2007; 26:775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marzi M.J., Ghini F., Cerruti B., de Pretis S., Bonetti P., Giacomelli C., Gorski M.M., Kress T., Pelizzola M., Muller H.. Degradation dynamics of microRNAs revealed by a novel pulse-chase approach. Genome Res. 2016; 26:554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Horvathova I., Voigt F., Kotrys A.V., Zhan Y., Artus-Revel C.G., Eglinger J., Stadler M.B., Giorgetti L., Chao J.A.. The dynamics of mRNA turnover revealed by single-molecule imaging in single cells. Mol. Cell. 2017; 68:615–625. [DOI] [PubMed] [Google Scholar]
- 51. Sumazin P., Yang X., Chiu H.S., Chung W.J., Iyer A., Llobet-Navas D., Rajbhandari P., Bansal M., Guarnieri P., Silva J. et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011; 147:370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Darbellay G., Vajda I.. Estimation of the information by an adaptive partitioning of the observation space. IEEE Trans. Inform. Theory. 1999; 45:1315–1321. [Google Scholar]
- 53. TCGA-Consortium Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Buffa F.M., Camps C., Winchester L., Snell C.E., Gee H.E., Sheldon H., Taylor M., Harris A.L., Ragoussis J.. microRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer Res. 2011; 71:5635–5645. [DOI] [PubMed] [Google Scholar]
- 56. Chiu H.-S., Martínez M.R., Bansal M., Subramanian A., Golub T.R., Yang X., Sumazin P., Califano A.. High-throughput validation of ceRNA regulatory networks. BMC Genomics. 2017; 18:418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Basso K., Margolin A.A., Stolovitzky G., Klein U., Dalla-Favera R., Califano A.. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 2005; 37:382–390. [DOI] [PubMed] [Google Scholar]
- 58. Margolin A., Nemenman I., Basso K., Wiggins C., Stolovitzky G., Favera R., Califano A.. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006; 7:S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jansen R., Gerstein M.. Analyzing protein function on a genomic scale: the importance of gold-standard positives and negatives for network prediction. Curr. Opin. Microbiol. 2004; 7:535–545. [DOI] [PubMed] [Google Scholar]
- 60. Dutkowski J., Kramer M., Surma M.A., Balakrishnan R., Cherry J.M., Krogan N.J., Ideker T.. A gene ontology inferred from molecular networks. Nat. Biotechnol. 2013; 31:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zou H., Hastie T.. Regularization and variable selection via the elastic net. J. R. Stat. Soc. B. 2005; 67:301–320. [Google Scholar]
- 62. Tibshirani R. Regression shrinkage and selection via the lasso. J. Royal. Statist. Soc B. 1996; 58:267–288. [Google Scholar]
- 63. Parmigiani G., Garrett E.S., Irizarry R.A., Zeger S.L.. The Analysis of Gene Expression Data. 2003; NY: Springer; 1–45. [Google Scholar]
- 64. Anderson M.J., Robinson J.. Permutation tests for linear models. Australian & New Zealand Journal of Statistics. 2001; 43:75–88. [Google Scholar]
- 65. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehar J., Kryukov G.V., Sonkin D. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alvarez M.J., Shen Y., Giorgi F.M., Lachmann A., Ding B.B., Ye B.H., Califano A.. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nature Genetics. 2016; 48:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chiu H-S., Somvanshi S., Patel E., Chen T-W., Singh V.P., Zorman B., Patil S.L., Pan Y., Chatterjee S.S. TCGA et al. Pan-cancer analysis of lncRNA regulation supports their targeting of cancer genes in each tumor context. Cell reports. 2018; 23:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sumazin P., Chen Y., Treviño L.R., Sarabia S.F., Hampton O.A., Patel K., Mistretta T.A., Zorman B., Thompson P., Heczey A.. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology. 2017; 65:104–121. [DOI] [PubMed] [Google Scholar]
- 69. Lefebvre C., Rajbhandari P., Alvarez M.J., Bandaru P., Lim W.K., Sato M., Wang K., Sumazin P., Kustagi M., Bisikirska B.C. et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol. 2010; 6:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shimoni Y., Friedlander G., Hetzroni G., Niv G., Altuvia S., Biham O., Margalit H.. Regulation of gene expression by small non-coding RNAs: a quantitative view. Mol. Syst. Biol. 2007; 3:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ludwig N., Leidinger P., Becker K., Backes C., Fehlmann T., Pallasch C., Rheinheimer S., Meder B., Stähler C., Meese E.. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016; 44:3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Landgraf P., Rusu M., Sheridan R., Sewer A., Iovino N., Aravin A., Pfeffer S., Rice A., Kamphorst A.O., Landthaler M.. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007; 129:1401–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Volinia S., Calin G.A., Liu C.G., Ambs S., Cimmino A., Petrocca F., Visone R., Iorio M., Roldo C., Ferracin M. et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bosson Andrew D., Zamudio Jesse R., Sharp Phillip A. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol. Cell. 56:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Janus P., Szołtysek K., Zając G., Stokowy T., Walaszczyk A., Widłak W., Wojtaś B., Gielniewski B., Iwanaszko M., Brown R.. Pro-inflammatory cytokine and high doses of ionizing radiation have similar effects on the expression of NF-kappaB-dependent genes. Cell. Signal. 2018; 46:23–31. [DOI] [PubMed] [Google Scholar]
- 76. Peng G., Lin C.C.-J., Mo W., Dai H., Park Y.-Y., Kim S.M., Peng Y., Mo Q., Siwko S., Hu R.. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 2014; 5:3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dorr C., Janik C., Weg M., Been R.A., Bader J., Kang R., Ng B., Foran L., Landman S.R., O'Sullivan M.G.. Transposon mutagenesis screen identifies potential lung cancer drivers and CUL3 as a tumor suppressor. Mol. Cancer Res. 2015; 13:1238–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vivanco I., Palaskas N., Tran C., Finn S.P., Getz G., Kennedy N.J., Jiao J., Rose J., Xie W., Loda M.. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell. 2007; 11:555–569. [DOI] [PubMed] [Google Scholar]
- 79. Comisso E., Scarola M., Rosso M., Piazza S., Marzinotto S., Ciani Y., Orsaria M., Mariuzzi L., Schneider C., Schoeftner S.. OCT4 controls mitotic stability and inactivates the RB tumor suppressor pathway to enhance ovarian cancer aggressiveness. Oncogene. 2017; 36:4253. [DOI] [PubMed] [Google Scholar]
- 80. Labrecque M.P., Takhar M.K., Nason R., Santacruz S., Tam K.J., Massah S., Haegert A., Bell R.H., Altamirano-Dimas M., Collins C.C.. The retinoblastoma protein regulates hypoxia-inducible genetic programs, tumor cell invasiveness and neuroendocrine differentiation in prostate cancer cells. Oncotarget. 2016; 7:24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wu J., Liu Z., Shao C., Gong Y., Hernando E., Lee P., Narita M., Muller W., Liu J., Wei J.-J.. HMGA2 overexpression-induced ovarian surface epithelial transformation is mediated through regulation of EMT genes. Cancer Res. 2011; 71:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tobin N.P., Sims A.H., Lundgren K.L., Lehn S., Landberg G.. Cyclin D1, Id1 and EMT in breast cancer. BMC cancer. 2011; 11:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sun M., Song C.-X., Huang H., Frankenberger C.A., Sankarasharma D., Gomes S., Chen P., Chen J., Chada K.K., He C.. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:9920–9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Casimiro M.C., Wang C., Li Z., Di Sante G., Willmart N.E., Addya S., Chen L., Liu Y., Lisanti M.P., Pestell R.G.. Cyclin D1 determines estrogen signaling in the mammary gland in vivo. Mol. Endocrinol. 2013; 27:1415–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Perez-Perri J.I., Dengler V.L., Audetat K.A., Pandey A., Bonner E.A., Urh M., Mendez J., Daniels D.L., Wappner P., Galbraith M.D.. The TIP60 complex is a conserved coactivator of HIF1A. Cell Rep. 2016; 16:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Petryszak R., Keays M., Tang Y.A., Fonseca N.A., Barrera E., Burdett T., Füllgrabe A., Fuentes A.M.-P., Jupp S., Koskinen S.. Expression Atlas update—an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2015; 44:D746–D752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.