


Abstract
Non-coding RNA regulatory elements are important for viral replication, making them promising targets for therapeutic intervention. However, regulatory RNA is challenging to detect and characterise using classical structure-function assays. Here, we present in cell Mutational Interference Mapping Experiment (in cell MIME) as a way to define RNA regulatory landscapes at single nucleotide resolution under native conditions. In cell MIME is based on (i) random mutation of an RNA target, (ii) expression of mutated RNA in cells, (iii) physical separation of RNA into functional and non-functional populations, and (iv) high-throughput sequencing to identify mutations affecting function. We used in cell MIME to define RNA elements within the 5′ region of the HIV-1 genomic RNA (gRNA) that are important for viral replication in cells. We identified three distinct RNA motifs controlling intracellular gRNA production, and two distinct motifs required for gRNA packaging into virions. Our analysis reveals the 73AAUAAA78 polyadenylation motif within the 5′ PolyA domain as a dual regulator of gRNA production and gRNA packaging, and demonstrates that a functional polyadenylation signal is required for viral packaging even though it negatively affects gRNA production.
INTRODUCTION
Once thought to be a passive carrier of genetic information between the DNA and the protein world, RNA is now appreciated to play a central role in the regulation of almost all cellular activity (1). RNA is unique in that it encodes information in both its sequence and its structure. Like its counterpart DNA, the order of nucleotides in RNA represents the sequence of amino acids during protein synthesis. However, unlike the regular double stranded DNA helix, RNA molecules can fold into complex and elaborate three-dimensional structures that impart functionality by serving as recognition sites for proteins, small molecules, and other nucleic acids.
RNA viruses, with their compact and efficiently encoded genomes, are perfect models of complex RNA function. The genomic RNA (gRNA) of HIV-1 encodes nine proteins: the major structural proteins, Gag, Pol and Env; the regulatory proteins Tat and Rev; and the accessory proteins Vpu, Vpr, Vif and Nef. In addition to its coding capacity, the HIV-1 gRNA is replete with cis-acting regulatory sequences that interact in complex ways to modulate gene expression through effects on RNA processing, stability, export and translation. These regulatory sequences are especially concentrated within the 5′ untranslated region (UTR) and the beginning of the Gag coding sequence (2–7). This region of the gRNA is highly structured, and folds into a series of relatively independent functional domains (Figure 1A): the Trans-Activating Response (TAR) for transcription; PolyA for polyadenylation; the primer binding site (PBS) for reverse transcription; SL1 or the dimerization initiation site (DIS) for gRNA dimerization; SL2 contains the major splice donor (SD) site; and SL3 is historically considered the major packaging signal (Psi). Together, these functional domains regulate key steps of the HIV-1 life cycle (8–11), and serve to highlight the fact that the gRNA sustains not only protein synthesis, but is an active participant in the viral infection process.
Figure 1.

(A) The HIV-1 5′UTR folds into a series of structural domains that control key steps of the HIV-1 life cycle including transcription, translation, export, packaging and reverse transcription. From 5′ to 3′ these structural domains are: transactivation response (TAR) for transcription; PolyA stem loop for polyadenylation; the primer binding site (PBS) for reverse transcription; SL1 promotes gRNA dimerization; SL2 contains the major splice donor (SD) site; SL3 has historically been considered the major packaging signal (Psi); the sequences surrounding the AUG start codon are thought to be involved in a base-pairing interaction with the upstream U5 region. (B) In cell Mutational Interference Mapping Experiment (in cell MIME). The proviral genome is randomly mutated using error prone PCR, and subsequently cloned into a gRNA expression vector. The structural and enzymatic proteins, Gag and Gag-Pol are expressed from a separate expression plasmid. Co-transfection of the mutant library and Gag/Gag-Pol expression plasmid into 293T cells leads to the transcription of mutant RNAs and subsequent sorting of functional and non-functional RNA populations by the viral and cellular machinery. Viral RNA present in cells and virus is reverse transcribed. Viral cDNA and the input DNA plasmid is amplified, fragmented, barcoded, sequenced on an Illumina HiSeq2500, and analysed using the MIMEAnTo software.
Because regulatory elements are essential for viral replication, they represent promising, yet still underexplored antiviral targets (12). Indeed, pioneering work in Hepatitis C virus (HCV) demonstrates that non-coding RNA can be targeted therapeutically with high barriers to resistance, providing impetus for the systematic discovery of functional RNA motifs in viral genomes (13). To date, regulatory RNA is most often identified using truncation or deletion mutants in individual assays to define regions of functionality. However, regulatory regions often overlap in complex RNAs, making these laborious experiments difficult to interpret. Indeed, attempts to define the minimal HIV-1 packaging signal have led to largely conflicting results, and evidence can be found in the literature that almost all regions of the 5′UTR are required for packaging (14), including TAR (15,16), the poly-A stem loop (17,18), PBS (14,18), SL1 (14,19–21), SL2 (22), SL3 (23–25), as well as the first nucleotides of gag (6,26,27). Many of these studies used large and imprecise deletions that likely compromised the global folding of the RNA, and some of these studies may not be correctly interpreted. For example, TAR was once considered part of the HIV-1 packaging signal, until work by the Berkhout lab revealed that packaging defects were caused by TAR mutation induced misfolding of the HIV-1 leader RNA (28–30). Finally, truncation and deletion mutagenesis experiments are rarely able to define regions of functionality at single nucleotide resolution, nor do they provide enough information to mechanistically understand RNA function. Thus, there is an urgent need for new high-resolution and quantitative methods to analyse RNA function, especially within the native cellular environment.
We have recently developed Mutational Interference Mapping Experiment (MIME) as a powerful and high resolution method to identify functional regions within long RNA molecules in vitro (31). We previously used MIME to precisely map the binding site of the HIV-1 Pr55Gag protein on the viral gRNA in vitro, finding that Pr55Gag recognises the region encompassing nucleotides 227 to 337 (31). Whilst Pr55Gag binding to the gRNA is presumed to be the major determinant of gRNA packaging into viral particles, it is currently unclear whether Pr55Gag recognises this same site within cells (32), nor whether there are additional regulatory or packaging signals that may define binding sites for cellular (33) and viral proteins, or even nucleic acids (34). Additionally, the minimal signal required to direct HIV-1 gRNA into viral particles has yet to be precisely determined, with the packaging signal possibly comprising the entire 5′UTR and up to half of the Gag coding sequence (35,36). How such an extended packaging signal interconnects with other regulatory motifs situated in the same region is an open question, whose answer would undoubtedly help with the engineering of safe HIV-1 lentiviral vectors for gene therapy purposes.
Here, we have adapted MIME to identify RNA regulatory sequences within the HIV-1 genome during its replication in cells (in cell MIME) (Figure 1B). By varying the functional selection criteria, we obtained two distinct and high-resolution maps of regulatory RNA controlling intracellular gRNA production and gRNA packaging, respectively. We found three RNA motifs regulating intracellular gRNA production and two motifs regulating genome packaging. Strikingly, a 73AAUAAA78 hexamer sequence within 5′ PolyA regulated both gRNA production and packaging, revealing the cellular polyadenylation machinery as a dual regulator of HIV-1 replication.
MATERIALS AND METHODS
Molecular clones
Mutant libraries were cloned into pDRNL43 NotI ATGaag Tat(–) ΔEnv which is a derivative of pDRNL43ΔEnv (37) modified to contain (i) NotI 431GCgGCcGC439 and NgoMIV 958GccGgC964 restriction sites for the cloning of the mutant library (positions based on pNL43 proviral DNA), (ii) a substitution in the initiation codon of gag to prevent Gag expression (27), (iii) a stop codon preventing Tat protein expression, (iv) a deletion in env (for biosafety). Gag and GagPol, and accessory proteins Tat and Rev, were expressed from the packaging vector pCMVΔR8.9 (38). PolyA and SL2 mutants were introduced into pDRNL43ΔEnv. Site directed mutagenesis was carried out utilising standard molecular biology techniques using the oligonucleotides listed in Supplementary Table S1.
Cell culture
Human embryonic kidney 293 (HEK 293T) cells were maintained at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with glutamine, penicillin, streptomycin and 10% (v/v) heat-inactivated fetal calf serum.
In cell mutational interference mapping experiment (MIME)
Mutagenesis
RNA expression vector (pDRNL43 NotI ATGaag NgoMIV Tat(–) ΔEnv) was mutated by error-prone PCR using the Mutazyme II DNA polymerase (Agilent) and the primers NL43_NotI_Fw and NgoMIV_Rv (Supplementary Table S1). We chose Mutazyme II as it is reported to produce a more uniform mutational spectrum than traditional error-prone PCR. The PCR reaction volume was 50 μl and consisted of 100 ng of template DNA, 1× buffer, 200 μM dNTPs, 0.5 μM of each primer, 2.5 U of Mutazyme II DNA polymerase. PCR cycling conditions were 95°C for 2 min followed by 35 cycles of 95°C for 30 s, 55°C for 30 s and 72°C for 1 min. We performed two or three rounds of PCR mutagenesis in duplicate. Mutated amplicon libraries were further amplified with the same primers used for mutagenesis using Phusion polymerase (NEB). PCR reaction volume was 50 μl and consisted of ∼50 ng of mutated DNA, 1× HF buffer, 200 μM dNTPs, 0.5 μM of each primer, 1 U of Phusion polymerase. Eight PCR amplifications were performed using the PCR cycling conditions 98°C for 30 s, followed by 30 cycles of 98°C for 10 s and 72°C for 1 min. Amplified libraries were column purified (Macherey-Nagel) and stored at –20°C until further use.
Cloning of library
Column purified amplicon libraries and the RNA expression vector pDRNL43 NotI NgoMIV Tat(–) ΔEnv were digested with NotI and NgoMIV. Vector and inserts were gel purified on a 1% agarose gel, and ligated overnight at an approximate molar ratio of 1 (vector):5 (insert) using a temperature cycling protocol of 30 s at 10°C followed by 30 s at 30°C. Overnight ligations were column purified using Nucleospin Gel and PCR Clean-up columns (Macherey-Nagel) and stored at –20°C until further use.
Transfection
Transfections of HEK 293T cells were carried out using the X-tremeGENE-9 DNA Transfection Reagent (Roche) according to the manufacturer's instructions. Briefly, cells were seeded at 70% confluence in 100 mm cell culture dishes and co-transfected with 2.5 μg mutant library, 2.5 μg of pCMVΔR8.9 packaging vector, 1 μg of pCMV RFP with 1 μl of X-tremeGENE-9 per μg DNA. 36 h post-transfection, virus containing media was harvested for storage at 4°C and cells were replenished with fresh media to allow for a second virus harvest 24 h later. Virus containing supernatant was pooled and clarified by centrifugation at 1462 g for 30 min, then passed through a 0.22 μm filter to remove cellular debris. Purified virus was concentrated by ultracentrifugation at 100 000 g through a 20% sucrose cushion.
RNA extraction
RNA was extracted from viral or cellular pellets using TriReagent (MRC) according to the manufacturer's instructions. Briefly, cells or virions were lysed in 1 ml of TriReagent and incubated at room temperature for 5 min. 0.2 ml of chloroform was added, followed by vigorous mixing, and a further incubation at room temperature for 15 min. After centrifugation at 12 000 g for 15 min at 4°C, the upper aqueous phase was transferred into a new tube. RNA was precipitated by the addition of 0.5 ml of isopropanol and 1 μg of glycogen followed by centrifugation at 12 000 g for 15 min at 4°C. RNA pellets were washed once in 500 μl of 70% EtOH, air dried, and resuspended in 200 μl of RNase free H2O. Viral and cellular RNA was then treated to remove contaminating plasmid DNA with 5 μl of RNase free DNase I (Roche), 5 μl RNasin (Promega) in 1× buffer (40 mM Tris–HCl, 10 mM NaCl, 6 mM MgCl2, 1 mM CaCl2, pH 7.9) for 2 h at 37°C. RNA was then extracted with phenol–chloroform, chloroform and precipitated with EtOH, washed with 70% EtOH and dissolved in ultra-pure water. Cellular and viral RNA pellets were dissolved in 200 μl and 20 μl of RNase free H2O, respectively.
Reverse transcription
Four microliter of RNA was mixed with 1 μl of a 10 μM stock of primer NL43_544_Rv (Supplementary Table S1), denatured at 90°C for 2 min and then chilled on ice. Reverse transcription was carried out in a total volume of 10 μl by adding 1× buffer (50 mM Tris–HCl pH 8.3, 6 mM MgCl2, 40 mM KCl), 200 nM dNTPs and 2 U of RNasin (Promega) and 2 U of AMV RT (MP Biomedicals). Samples were incubated for 5 min at 42°C, 30 min at 50°C and 10 min at 60°C and diluted 1/10 with 90 μl H2O before use. Negative reverse transcription controls were carried out in the absence of AMV RT to check for the absence of contaminating plasmid DNA. cDNA was quantified by qPCR using primers NL43_C1_seq and NL43_NgoMIV_seq (Supplementary Table S1) and Brilliant II SYBR master mix (Agilent). cDNA was normalized and amplified with primers NL43_C1_seq and NL43_NgoMIV_Seq (Supplementary Table S1) using Phusion polymerase. PCR reaction volume was 50 μl and consisted of 105 copies of DNA, 1× HF buffer, 200 μM dNTPs, 0.5 μM of each primer, 1 U of Phusion polymerase. PCR amplifications were performed in duplicate using the PCR cycling conditions 98°C for 10 s, followed by 30 cycles of 98°C for 10 s, 55°C for 15 s and 72°C for 1 min. Pooled PCR products were isolated on a 1% agarose gel and purified using Nucleospin Gel and PCR Clean-up columns (Macherey-Nagel).
Fragmentation
As 2 × 100 nt Illumina sequencing does not completely cover the ∼500 bp fragment analysed in this study, we randomly fragmented 500 ng of gel purified dsDNA with 2.5 μl 10× buffer, 2.5 μl of 10× BSA and 2.5 μl of NEBNext dsDNA fragmentase in a total volume of 25 μl for 45 min at 37°C. Samples were verified on a 1% agarose gel, and digestion was confirmed as a smear on the gel. Fragmented samples were purified using Nucleospin Gel and PCR Clean-up columns, according to the manufacturer's instructions (Macherey-Nagel).
Library preparation
Fragmented DNA was first repaired using 1× T4 DNA ligase buffer (NEB), 0.4 mM each dNTPs, 1 mM ATP, 0.5 μl of Escherichia coli DNA ligase (from NEB NEBNext dsDNA fragmentase kit), 4.5 U of T4 DNA polymerase (NEB) and 25 U of T4 polynucleotide kinase (NEB) in 50 μl total volume for 1 h at 20°C. Enzymes were then heat inactivated by incubating samples for 30 min at 75°C. DNA was A-tailed by adding 12.5 U of Klenow fragment (3′-5′ Exo-) and 1.25 μl of 100 μM dATP and incubating for 45 min at 37°C. Following a second round of enzyme heat inactivation for 30 min at 75°C, adaptor ligation was performed by adding 9 μl of fresh 10× T4 DNA ligase buffer (NEB), 28 μl of 24% PEG 600 (NEB), 1 μl of 12.5 μM pre-annealed adaptors, and 2.5 μl of T4 DNA ligase (NEB) followed by incubation at 20°C for 1 h. Adaptor sequences IlluminaMAs and IlluminaMAa (Supplementary Table S1) were annealed by mixing in 1× ligase buffer (NEB), heating to 95°C for 1 min and slow cooling to room temperature. Samples were purified using Nucleospin Gel and PCR Clean-up columns. Y-shaped Illumina adaptors were converted into dsDNA using the PCR cycling conditions 98°C for 30 s followed by 5 cycles of 98°C for 15 s, 63°C for 30 s and 72°C for 30 s using the Illumina_1.0 and Illumina Index (Supplementary Table S1) with Phusion polymerase. PCR reaction volume was 50 μl and consisted of adaptor ligated DNA, 1× HF buffer, 200 μM dNTPs, 0.5 μM of each primer, 1 U of Phusion polymerase. Samples were then run on a 1% agarose gel and the range corresponding to 200–600 bp range was isolated, and purified using Nucleospin Gel and PCR Clean-up columns. DNA libraries were quantified by qPCR using Illumina PE PCR primer 1.0 and one of the Illumina Index primers (for multiplexing) with Brilliant II SYBR master mix. Samples were normalized and then re-amplified by PCR Illumina PE PCR primer 1.0 and one of the Illumina Index primers using the PCR cycling conditions 98°C for 30 s; followed by 6 cycles of 98°C for 15 s, 63°C for 30 s and 72°C for 30 s with Phusion polymerase. Samples were then pooled, and a final size selection was performed on a 1% agarose gel to re-isolate the range 200–600 bp ensuring the removal of Illumina adaptor dimers. Samples were sequenced on a single lane of a HiSeq 2500 instrument in 100 bp paired end mode, according to established procedures (IGBMC sequencing platform, Strasbourg, France).
RT-qPCR
Packaging efficiency of wild-type and mutant HIV-1 were carried out by transfecting 106 293T cells with 250 ng of plasmid using 4.5 μl of polyethylenimine per μg of DNA (1 mg/ml; Polysciences). Thirty six hours post-transfection, viral supernatant was clarified by centrifugation, syringe filtered through 0.22 μm pores, and pelleted through a 20% sucrose cushion, as outlined above. 293T cells were washed once in PBS. Viral and cellular RNA was then extracted using TriReagent (MRC), treated with DNase I, phenol/chloroform extracted, chloroform extracted, and EtOH precipitated as outlined above. Cellular and viral RNA pellets were dissolved in 200 μl and 20 μl of RNase free H2O, respectively. Three microliters of RNA were mixed with 2 μl of a 5× mix of random hexamer and anchored oligodT (5× mix; 12.5 μM dT20VN; 17.5 μM N6) denatured at 90°C for 2 min and then chilled on ice. Reverse transcription was carried out in a total volume of 10 μl by adding 1× buffer (50 mM Tris–HCl pH 8.3, 6 mM MgCl2, 40 mM KCl), 200 nM dNTPs and 2 U of RNasin and 2 U of AMV RT (MP Biomedicals). Samples were incubated for 5 min at 42°C, 30 min at 50°C and 10 min at 60°C and diluted 1/10 with 90 μl H2O before use. Negative reverse transcription controls were carried out in the absence of AMV RT to check for the absence of contaminating plasmid DNA. gRNA, spliced viral RNA, and GAPDH mRNA were quantified by TaqMan qPCR assay using the primers listed in Supplementary Table S1. A standard curve was generated from 109 to 103 copies of plasmid containing the relevant target. Negative controls demonstrated the DNA contamination levels were present at <1% in all samples. Packaging efficiency was determined by calculating the ratio of the total amount of each RNA present in the supernatant by the amount present in the cells.
Analysis of in cell MIME data
Relation between nucleotide frequencies and the effect on intracellular gRNA production
Employing the derivation outlined in the Supplementary Text Equations (S1)–(S12), we can deduce the effect of a mutation 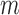 at position
at position 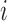 in the RNA from the frequency of that mutation in the DNA library relative to the frequency in the cells
in the RNA from the frequency of that mutation in the DNA library relative to the frequency in the cells 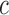 , i.e.
, i.e.
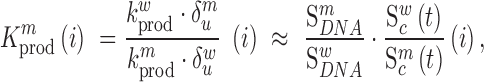 |
(1) |
where 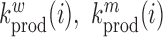 denote the rate of intracellular production of the wild type viral RNA and the viral RNA that carries a particular mutation (i.e. A→C, A→G, A→U, if the wild type base is adenosine) at position
denote the rate of intracellular production of the wild type viral RNA and the viral RNA that carries a particular mutation (i.e. A→C, A→G, A→U, if the wild type base is adenosine) at position 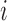 and
and 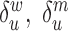 are the corresponding rates of RNA degradation. The ratios
are the corresponding rates of RNA degradation. The ratios 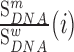 and
and 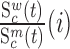 denote the frequency of mutations in the DNA library and in the pool of viral RNA located in the cell. Whenever the measure above is larger than 1, mutations decrease HIV-1 RNA levels. In order to identify regions that are important for gRNA production, one may also depict the impact of the mutation mmax(i) that has a maximal impact at position i only, as shown in Figure 2A; where
denote the frequency of mutations in the DNA library and in the pool of viral RNA located in the cell. Whenever the measure above is larger than 1, mutations decrease HIV-1 RNA levels. In order to identify regions that are important for gRNA production, one may also depict the impact of the mutation mmax(i) that has a maximal impact at position i only, as shown in Figure 2A; where 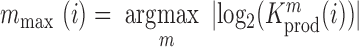 and where m denotes all those mutations that have a significant impact on binding at nucleotide position i (if any), or all possible mutations otherwise.
and where m denotes all those mutations that have a significant impact on binding at nucleotide position i (if any), or all possible mutations otherwise.
Figure 2.

In cell Mutational Interference Mapping Experiment (in cell MIME) discovery of RNA motifs regulating HIV-1 gRNA production (A) Log2 Kprod showing the maximal effect of mutations on RNA production in cells with the HIV-1 5′ UTR and Gag coding region (smoothed with a linear, two-sided convolution filter of width 2). Functional domains are indicated with coloured boxes below the graph. Positions with significant effects on RNA production are indicated by black triangles above the graph. Three regions with significant (P < 0.05) and strong (log2 Kprod ≥ 1 or ≤ –1; gray dotted line) effects on gRNA production are highlighted with red circles. (B to D) Mutations with maximal effect on log2 Kprod mapped on RNA structure. Positions impairing RNA production are shown in red. Positions improving RNA production shown in blue. Box and whisker plots show effect of each class of mutation on log2 Kprod. Black dot shows median, box shows quartiles (25% and 75%) and whiskers show extremes (excluding outliers beyond 1.5× IQR). Mutation classes are colour coded: red mutated to A; green mutated to C; blue mutated to G; yellow mutated to U. (B) Effect of mutations on gRNA production (log2 Kprod) mapped to TAR. (C) Effect of mutations on gRNA production (log2 Kprod) mapped to 5′ PolyA. All mutations to AAUAAA sequence improve gRNA production except for a single A to U mutation. (D) Effect of mutations on gRNA production (log2 Kprod) mapped to SL2. Mutations impairing gRNA production cluster to the U1 snRNA binding site.
Relation between nucleotide frequencies and the effect on packaging
Similarly, using derivations (S1–S6) and (S13–S19) in the Supplementary Text, we can deduce the effect of any mutation 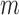 at position
at position 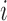 in the RNA on packaging from the frequency of mutation
in the RNA on packaging from the frequency of mutation 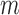 in the cell
in the cell 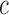 relative to the frequency in the virions
relative to the frequency in the virions 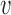 , i.e.
, i.e.
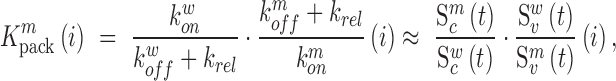 |
(2) |
where 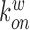 ,
, 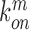 ,
, 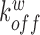 ,
, 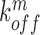 denote the binding- and dissociation rate of the RNA to/from the packaging complex and the rate
denote the binding- and dissociation rate of the RNA to/from the packaging complex and the rate 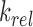 denotes the rate at which RNA bound to the packaging complex is released from the cell after packing into nascent virions. In order to identify regions that are important for packaging, one may also depict the impact of the mutation mmax(i) that has a maximal impact at position i only, as shown in Figure 3A.
denotes the rate at which RNA bound to the packaging complex is released from the cell after packing into nascent virions. In order to identify regions that are important for packaging, one may also depict the impact of the mutation mmax(i) that has a maximal impact at position i only, as shown in Figure 3A.
Figure 3.

In cell Mutational Interference Mapping Experiment (in cell MIME) discovery of RNA motifs regulating HIV-1 gRNA packaging. (A) Log2 Kpack showing the maximal effect of mutations on RNA packaging with the HIV-1 5′ UTR and Gag coding region (smoothed with a linear, two-sided convolution filter of width 2). Functional domains are indicated with coloured boxes. Positions with significant effects on gRNA packaging are indicated by black triangles. Two regions with significant (P < 0.05) and strong (log2 Kpack ≥ 1; gray dotted line) effects on gRNA packaging are highlighted with dot red line/circle. (B and C) Mutations with maximal effect on log2 Kpack represented on RNA structure. Positions impairing gRNA packaging are shown in red. Positions improving gRNA packaging are shown in blue. Box and whisker plots show effect of each class of mutation on log2 Kpack. Black dot shows median, box shows quartiles and whiskers show extremes (excluding outliers beyond 1.5× IQR). Mutation classes are colour coded: red mutated to A; green mutated to C; blue mutated to G; yellow mutated to U. (B) Effect of mutations on gRNA packaging expressed as Log2 Kpack mapped to 5′ PolyA. All mutations to AAUAAA sequence impair gRNA packaging except for a single A to U mutation. (C) Effect of mutations on gRNA packaging expressed as log2 Kpack mapped to RNA structure in the region SL1–SL3. (D) Qualitative comparison between the significant effects of mutations on Pr55Gag binding determined by in vitro MIME (upper portion, green) and the effects of mutations on gRNA packaging by in cell MIME (lower portion, blue). Sites significantly affecting both are pictured red. Color-coded arrows below (for in cell) and above (for in vitro) indicate the affected functional domain (colored boxes on the bottom). Filled arrows show significant effects at sites in both in vitro and in cell.
Error correction
The mutation frequencies 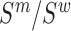 needed to evaluate Equations (1) and (2) are not known exactly, however, next generation sequencing (NGS) of the distinct RNA pools (DNA library, cellular RNA and RNA in virions) gives their frequencies in the NGS reads
needed to evaluate Equations (1) and (2) are not known exactly, however, next generation sequencing (NGS) of the distinct RNA pools (DNA library, cellular RNA and RNA in virions) gives their frequencies in the NGS reads 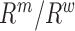 . These reads however contain substantial sequencing errors, which we have to correct for, akin to the method presented in the Supplementary Notes of (31,39). Error correction allows us then to directly estimate the effects of each mutation
. These reads however contain substantial sequencing errors, which we have to correct for, akin to the method presented in the Supplementary Notes of (31,39). Error correction allows us then to directly estimate the effects of each mutation 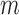 for all position
for all position  from the nucleotide frequencies observed in the NGS reads, provided we have a sufficient signal-to-noise ratio (see Supplementary Text):
from the nucleotide frequencies observed in the NGS reads, provided we have a sufficient signal-to-noise ratio (see Supplementary Text):
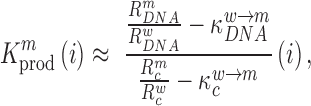 |
(3) |
where  denotes the probability to falsely detect a wild type nucleotide w as some mutant m at position i.
denotes the probability to falsely detect a wild type nucleotide w as some mutant m at position i.  is computed from experiments with wild type libraries for each type of mutation m and for each position i, akin to (31,39) and as exemplified in the Supplementary Text.
is computed from experiments with wild type libraries for each type of mutation m and for each position i, akin to (31,39) and as exemplified in the Supplementary Text.
Similarly, for packaging, we derive
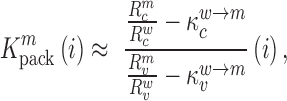 |
(4) |
Statistical assessment of effects
The above described method provides a single estimate of the relative effect for each nucleotide position and for each possible mutation, but it does not assess the confidence range of this estimate, or whether a mutation at position i has a significant impact on binding. In the following, we make use of a jacknife-like re-sampling procedure to estimate the confidence of each relative effect estimate, analogous to the methods in (31,39): In brief, if we are interested in the effect of a mutation m at position i, then for each pair of nucleotide positions (i,j), we can re-compute 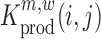 , respectively
, respectively 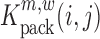 , N times (i.e. for each j ≠ i). Each of these estimates can be computed according to:
, N times (i.e. for each j ≠ i). Each of these estimates can be computed according to:
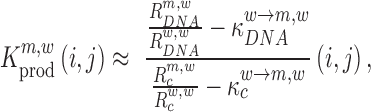 |
(5) |
and analogously,
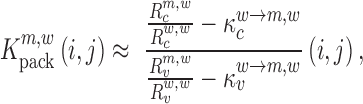 |
(6) |
where 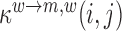 denotes the probability to falsely detect a wild type nucleotide w at position i as some mutant m and to correctly detect the wild type at position j ≠ i as wild type, with derivations provided in the Supplementary Text. To test whether a mutation at position i significantly increases/decreases gRNA production, i.e.
denotes the probability to falsely detect a wild type nucleotide w at position i as some mutant m and to correctly detect the wild type at position j ≠ i as wild type, with derivations provided in the Supplementary Text. To test whether a mutation at position i significantly increases/decreases gRNA production, i.e. 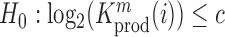 ,
, 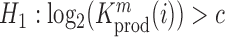 , the raw P-value can be computed according to
, the raw P-value can be computed according to
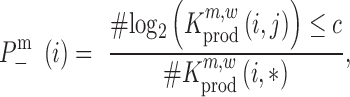 |
(7) |
where ‘#’ denotes the ‘number of estimates’ and * indicates that all positions j are evaluated. To test whether a mutation at position i significantly decreases 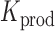 ,
,
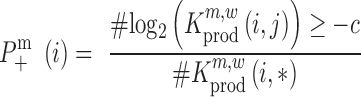 |
(8) |
We used P < 0.05 to detect significance. Note, that one can test any threshold 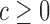 (e.g. 2-fold increase/decrease, etc.). Throughout the manuscript we chose
(e.g. 2-fold increase/decrease, etc.). Throughout the manuscript we chose 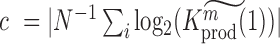 i.e. the average over all positions i, i.e.
i.e. the average over all positions i, i.e. 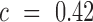 . An analogous scheme can be used to assess the effects on packaging, where we determined threshold
. An analogous scheme can be used to assess the effects on packaging, where we determined threshold 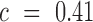 . All reported P-values were corrected by the false discovery rate (FDR)-based method of Benjamini-Hochberg.
. All reported P-values were corrected by the false discovery rate (FDR)-based method of Benjamini-Hochberg.
Quality criteria
For each pair of reads Rm,w(i,j) / Rw,w(i,j), we assessed its respective signal-to-noise ratio in the corresponding RNA pool (DNA, cell and virion) according to:
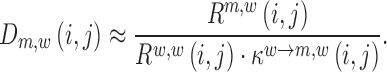 |
(9) |
If the ratio was below the user-supplied threshold of 2 in both samples (DNA library versus cell and cell versus virion), the corresponding estimates in Equations (5) and (6) were discarded. If the signal was below the threshold in only one of the samples, the respective estimate was tagged as either being a lower- or upper estimate of the mutations' effect and assigned the value of the median effect estimate on RNA production or packaging respectively. This has the following reason: if a mutation strongly decreases RNA production, the frequency of that mutation in the cellular RNA may fall below the required signal-to-noise ratio (a multiple of the sequencing error) and the (negative) effect on RNA production may actually be higher than estimable. Conversely, if the frequency of that mutation was below the minimum signal-to-noise ratio in the DNA library, but HIV-1 RNA levels increase in the cellular RNA above the threshold, the (positive) effect on RNA production may actually be higher than estimable. Likewise, if a mutation strongly decreases packaging, the frequency of that mutation in the virions may also fall below the sequencing error, while the signal within the cells is sufficient. Again, the actual (negative) effect on packaging may be larger than estimable. We only evaluated Equations(5) and (6) for positions j where the total number of sequence fragments covering both i and j was at least 50% of the maximum coverage. For determining P-values, at least 300 estimates had to fulfil the quality criteria.
RESULTS
Mutational interference mapping experiment (MIME) in cells
The 5′UTR folds into a series of functional domains that regulate almost every stage of the HIV-1 life cycle (2,3,5), including intracellular gRNA production and packaging into viral particles. For the most part, the RNA sequences regulating these processes have been mapped to individual stem loops, but a complete nucleotide level understanding of their function is largely lacking. We recently developed Mutational Interference Mapping Experiment (MIME) for dissecting RNA structure and function at single nucleotide resolution (31). This technique is based on (i) the random mutation of the RNA of interest (ii) the physical separation of RNA into functional and non-functional populations and (iii) high-throughput sequencing to identify mutations affecting function. In theory, MIME can be applied to any process where functional and non-functional RNAs can be physically separated, including within cells during a native viral replication cycle. We reasoned that during replication, mutated viral RNA would be naturally segregated into functional and non-functional populations. That is, correctly transcribed and processed viral RNAs will accumulate in the cytoplasm over RNAs that are poorly transcribed or degraded due to defective processing. Similarly, viral RNAs that are efficiently selected for packaging will be more abundant in virions compared to packaging defective RNAs. By physically isolating and sequencing these mutant populations, regulatory RNA controlling two different stages of viral replication can be dissected in cells at unprecedented detail.
The entire 5′UTR and the beginning of the Gag coding region (6,26,27) is thought to contain RNA sequences regulating HIV-1 replication, therefore we targeted the first ∼500 nucleotides of the gRNA for functional analysis. Because mutating the Gag coding region could complicate the identification of non-coding regulatory RNA i.e. by introducing mutations that interfere with viral assembly, we first designed a conditional co-transfection system to separate the production of mutated gRNA from the expression of the viral structural proteins. gRNA was expressed from the lab adapted pNL4.3 HIV-1 vector modified to include (i) restriction sites for cloning of the mutant library, (ii) a substitution in the gag ATG start codon preventing Gag/Gag-Pol expression, (iii) a stop codon preventing Tat expression and (iv) a deletion in env for biosafety purposes. Unaltered viral proteins Pr55Gag, Pr160GagPol and the accessory proteins Tat and Rev were expressed from a separate packaging vector. In this experimental setup, only co-transfected cells produce gRNA, ensuring that all gRNA is produced in the presence of the viral assembly machinery. The inclusion of restriction sites did not affect viral replication in single round assays (Supplementary Figure S1), and the ATG start codon mutation prevents Gag expression without significantly affecting encapsidation (27).
We performed in cell MIME (Figure 1B) using six mutant libraries tested in three independent experiments (two mutant libraries per experiment). Mutations were introduced using commercial PCR based mutagenesis technology. These libraries were then cloned into the gRNA expression vector and co-transfected into 293T cells together with the packaging vector. Viral and cellular gRNA were harvested, reverse transcribed, randomly fragmented, and sequenced using the Illumina HiSeq 2500 platform in 100 nt paired end mode. We also sequenced DNA derived from both the wild-type and mutant DNA libraries, with the non-mutated WT sequences used to control for errors introduced during library preparation and sequencing.
Altogether, we aligned 180 million sequences to the reference genome, finding 1.08 × 108 mutations from 2.15 × 1010 base pairs (Supplementary Table S2). Raw substitution rates were found to be significantly higher in the mutant library compared to the WT controls (Supplementary Figure S2), demonstrating that biologically relevant mutations could be clearly distinguished from the background errors introduced during library preparation and sequencing (P-value < 0.01). Importantly, we were able to use the substitution frequencies in the wild-type control to obtain error-corrected mutation frequencies, thus eliminating any biases from errors introduced during library preparation and sequencing (Materials and Methods section and Supplementary Text) (31,39). Error-corrected mutation rates were similar across all six independent libraries from the three independent experiments (Supplementary Figure S3) and were highly reproducible for all classes of mutations (Supplementary Figures S4 and S5). Importantly, error corrected mutation rates steadily decreased from DNA (median = 4.8 × 10−3), cellular gRNA (median = 4.2 × 10−3) to virion gRNA (median = 3.3 × 10−3) providing evidence for purifying selection as the viral life-cycle proceeds (Supplementary Figure S3). Interestingly, A–G mutations were found to be consistently more abundant in cellular gRNA (Supplementary Figure S6) compared to the input DNA (P-value < 0.01). These cellular A–G mutations were enriched at 5′AA3′ and 5′UA3′ dinucleotides and seemed to cluster at unpaired adenines near regions of double stranded RNA structure (Supplementary Figure S7). Although the biological basis for these abundant A-G mutations is unclear, their nature is suggestive of an editing activity by the dsRNA adenosine deaminases, ADAR1 or ADAR2 (40,41) (Supplementary Figure S7). Whilst intriguing, this phenomenon is likely unrelated to the processes of gRNA production and packaging investigated here, so we ignored this class of mutation in the following analysis.
Regulation of intracellular gRNA production
We first focused on identifying RNA sequences regulating gRNA production in infected cells by comparing the mutation rate in the DNA library with mutations found in the gRNA in cells. Intuitively, mutations impairing gRNA production should be depleted in cells compared to the input DNA. Conversely, mutations improving RNA production should be enriched in the cellular gRNA compared to the input DNA library. Indeed, formal modelling of this biological process revealed a direct relation between mutation frequency and effect on gRNA production (Supplementary Text). In other words, the frequency of a mutation 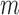 at position
at position 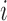 in the DNA library
in the DNA library 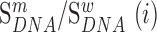 , divided by the mutation frequency in the cellular RNA
, divided by the mutation frequency in the cellular RNA 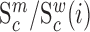 is directly proportional to the decrease/increase of intracellular viral RNA production caused by that mutation. By adapting a previously developed analytical framework (31,39), we were able to infer the mechanistic effects of all mutations m at all positions i on gRNA production and stability simultaneously, summarized as
is directly proportional to the decrease/increase of intracellular viral RNA production caused by that mutation. By adapting a previously developed analytical framework (31,39), we were able to infer the mechanistic effects of all mutations m at all positions i on gRNA production and stability simultaneously, summarized as 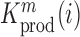 (see Equation (1) in Materials and Methods and Supplementary text). Moreover, we were also able to statistically ascertain mutation effects at each position.
(see Equation (1) in Materials and Methods and Supplementary text). Moreover, we were also able to statistically ascertain mutation effects at each position. 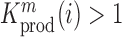 means that the mutation
means that the mutation 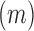 at position
at position 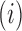 decreases gRNA production and stability. Conversely,
decreases gRNA production and stability. Conversely, 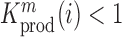 identifies mutations
identifies mutations 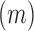 at position
at position 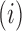 that increase gRNA production and stability. Upon performing this analysis, we found three distinct regulatory regions that strongly and significantly affected gRNA production, both positively and negatively (Figure 2A, Supplementary Data Files). These regions mapped to the domains TAR, PolyA and SL2, respectively.
that increase gRNA production and stability. Upon performing this analysis, we found three distinct regulatory regions that strongly and significantly affected gRNA production, both positively and negatively (Figure 2A, Supplementary Data Files). These regions mapped to the domains TAR, PolyA and SL2, respectively.
Unsurprisingly, TAR was identified as a positive regulator of gRNA production, consistent with its crucial role in enhancing viral transcription (42,43). This was seen as a strong depletion of mutations in TAR in the cellular gRNA when compared to the input DNA library (Figure 2A, 2B). Although we were not able to analyse the extreme 5′ part of the TAR (due to the binding of a specific primer to this region during sequencing library preparation), it was notable that mutations to the apical portion were more strongly depleted in cells compared to the distal portion of the stem-loop (Figure 2B). This apical region is known to be important for gRNA production by assembling with the HIV-1 Trans-Activator of Transcription (Tat) protein and the cellular factor P-TEFb (42–47). Furthermore, these results are in agreement with detailed mutant-revertant and phylogenetic studies showing that the distal portion of TAR is less important for gene expression compared to the apical portion (48,49). Altogether, these data evidence the ability of in cell MIME to discover regulatory RNA in an unbiased fashion.
The second regulatory motif was found to reside within the 5′ PolyA. Unexpectedly, mutations in the 5′ PolyA were enriched in cells compared to the input DNA, indicating that this motif plays a negative role in gRNA production. Strikingly, mutations improving gRNA production mapped precisely to the 73AAUAAA78 hexamer within the 5′ PolyA apical loop (Figure 2A, 2C). All mutations to this hexamer were enriched in cellular gRNA compared to DNA, except for a single 73AAUAAA78 to 73AUUAAA78 substitution (Figure 2C). As AAUAAA and AUUAAA are the most abundant cellular polyadenylation signals (50), these data imply a role for the cellular polyadenylation machinery in regulating intracellular gRNA levels.
The third regulatory motif mapped to the splice donor site within SL2. Here, mutations were strongly depleted in cellular gRNA compared to DNA (Figure 2A, D) demonstrating that sequences within SL2 are required for gRNA production. Interestingly, mutations disrupting gRNA production mapped precisely to the U1snRNA binding site 289GGUGAGU295 (Figure 2D), and all classes of mutation to this region disrupted gRNA production. This was somewhat surprising, as one might expect that disrupting the splice donor site would increase unspliced gRNA production by eliminating the splicing of viral RNAs. Nevertheless, the opposite effect is observed here, and our data argue that an interaction between U1snRNA and the splice donor site is required for gRNA production.
Regulation of gRNA packaging
We next searched for RNA motifs regulating gRNA packaging into virions by comparing the mutation rate in the cellular RNA with that found in RNA extracted from viral particles. In cells, gRNA packaging comprises multiple molecular events, including the formation of a protein : RNA packaging complex, its transport to the cell surface, and its assembly into viral particles. Modelling of this process demonstrates that the frequency of a mutation 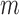 at position
at position 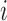 in the cells
in the cells 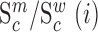 divided by the mutation frequency in the virion RNA
divided by the mutation frequency in the virion RNA 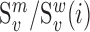 is proportional to the mutation's effect on packaging (see Equation (2) in Methods and Supplementary Text). We derived the term
is proportional to the mutation's effect on packaging (see Equation (2) in Methods and Supplementary Text). We derived the term 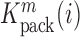 that summarizes the underlying processes (Equation 2). When
that summarizes the underlying processes (Equation 2). When 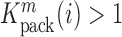 , the mutation
, the mutation 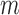 at position
at position 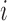 decreases gRNA packaging, when
decreases gRNA packaging, when 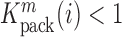 , it increases packaging. Analogous to the analysis of in cell MIME data for RNA production, we adopted the previously developed analytical framework for error correction and statistical analysis (31,39) (Materials and Methods section and Supplementary Text). Upon analysis, we identified two distinct regions that strongly and significantly affected gRNA packaging (Figure 3 A, Supplementary Data Files). These regulatory sequences mapped to the 5′ PolyA and the region SL1-SL3, respectively.
, it increases packaging. Analogous to the analysis of in cell MIME data for RNA production, we adopted the previously developed analytical framework for error correction and statistical analysis (31,39) (Materials and Methods section and Supplementary Text). Upon analysis, we identified two distinct regions that strongly and significantly affected gRNA packaging (Figure 3 A, Supplementary Data Files). These regulatory sequences mapped to the 5′ PolyA and the region SL1-SL3, respectively.
Strikingly, the packaging signal within 5′ PolyA mapped precisely to the same 5′ PolyA sequence 73AAUAAA78 that we identified as a strong regulator of gRNA production (Figure 3A and 3B). Indeed, mutations to this sequence have similar effects on gRNA packaging as mutations to the Psi region (Figure 3A). Like their effect on gRNA production, all mutations to this hexamer sequence impaired gRNA packaging into virions, except for a single 73AAUAAA78 to 73AUUAAA78 substitution (Figure 3B). Again, because 73AAUAAA78 and 73AUUAAA78 sequences function as canonical polyadenylation signals, our data provide evidence that the cellular polyadenylation machinery plays an important role in regulating not only gRNA production, but also its incorporation into viral particles (50).
The second packaging signal overlapped the domains SL1 to SL3 (Figure 3C) that we have previously identified as the Pr55Gag binding site in vitro (31). These data therefore confirm the idea that Pr55Gag is a central player in the selection of the gRNA (Figure 3D). However, we did find some differences between the sequences required for Pr55Gag binding in vitro and those directing gRNA packaging into virions. First, and most remarkably, the 257GCGCGC262 palindromic sequence within the SL1 apical loop seen as crucial for Pr55Gag binding to gRNA in vitro, was not required for gRNA packaging in cells (Figure 3C and 3D, Supplementary Figure S8). Second, SL2 was slightly more important for gRNA packaging in cells compared to Pr55Gag binding in vitro. However, it remained relatively minor when compared to SL1 and SL3 (Figure 3D). Finally, mutations to the stem of SL1 had comparable effects on packaging in cells as mutations to the stem of SL3, in contrast to the situation in vitro where SL1 stem mutations were much more deleterious than mutations to the SL3 stem (Supplementary Figure S8) (4,31). Altogether, the region SL1-SL3 is a major gRNA packaging determinant, with SL1 shown to be the most important stem-loop given that is over 2.5 times larger than SL3 (2,3).
Role of the AAUAAA PolyA motif in gRNA production and packaging
To confirm the role of the 5′ PolyA and the U1snRNA binding site on gRNA production, we introduced mutants into these two regions and tested their impact on viral replication in transfected cells by reverse transcription quantitative PCR (RT-qPCR). We inhibited the 5′ polyadenylation signal either by its complete deletion (73ΔAAUAAA78) or its mutation to 73AAUGAA78 (Figure 4A). We also included a 73AUUAAA78 mutation to serve as a canonical polyadenylation control (Figure 4A). Whilst disruption of 5′ polyadenylation did not lead to a detectible increase in the quantity of gRNA in the cell compared to wild-type (99.0% 73ΔAAUAAA78; 100.2% 73AAUGAA78), we did observe a significant increase in the quantity of spliced RNA produced in the 5′polyadenylation defective mutants compared to wild-type (266.4% 73ΔAAUAAA78P<0.05; 292.8% 73AAUGAA78P < 0.01) (Figure 4B). On the other hand, the 73AUUAAA78 mutant produced both gRNA and spliced RNA at levels comparable to wild-type (107.6% gRNA; 131.7% spliced RNA) (Figure 4B). Together, these data confirm that the 5′PolyA canonical polyadenylation signal regulates gRNA production.
Figure 4.

Role of the AAUAAA polyA motif in gRNA production and packaging. (A) 5′PolyA mutants contain point mutations or deletions to the AAUAAA sequence. SL2 mutant containing substitutions within the U1snRNA binding site. (B) Production of gRNA and spliced viral RNA (mRNA Tat) for 5′ polyA and SL2 mutants. Bar charts represent six independent experiments. (C) Relative packaging efficiency of gRNA and spliced viral RNA into viral particles, expressed as a virus/cellular RNA (36). Bar charts represent 3 independent experiments. Statistical tests were carried out using ANOVA corrected for multiple comparisons.
We next assessed the role of the 289GGUGAGU295 U1snRNA binding site by designing a mutation predicted to disrupt 289UACGAGU295 U1snRNA binding (Figure 4A). Disruption of this binding site led to a thousand-fold reduction in cellular levels of gRNA and spliced RNA (0.3% genomic; 0.15% spliced) (Figure 4B) whereas combining the U1snRNA binding site mutant 289UACGAGU295 with a deletion of the 5′ PolyA 73ΔAAUAAA78 signal caused levels of gRNA to return to wild-type (110.9%). These data demonstrate a functional interaction between the 5′PolyA and the U1snRNA binding site in gRNA production in agreement with a model that U1snRNA binding is required to inhibit 5′ premature polyadenylation (51,52). To our surprise, spliced viral RNA could also be detected at nearly wild-type levels in this double mutant (118.3%) despite disruption of the U1snRNA binding site. Sequencing of the PCR products revealed that splicing still occurred within SL2, even in the absence of a canonical splice donor sequence, but the splice site was shifted by four nucleotides in the 3′ direction (Supplementary Figure S9). Activation of cryptic splice donor sites has been observed upon mutation of the HIV-1 gRNA (52,53). This highlights that splice site selection is extremely complex, and likely balanced by RNA structure (54,55) as well as interactions between positive and negative splicing elements (53).
To further assess the role of the PolyA sequence on gRNA packaging, we next quantified by RT-qPCR the relative packaging efficiency of gRNA and spliced viral RNAs into viral particles expressed as the ratio of RNA found in cells compared to virus (56). We found a significant reduction (P<0.05) in packaging efficiency to 43% and 48% of wild-type, for the 73ΔAAUAAA78 and 73AAUGAA78 mutants respectively, whereas the packaging efficiency of the 78AUUAAA78 mutant remained at 92% of wild-type (Figure 4C). Conversely, we found that spliced viral RNA was incorporated with much greater efficiency in the 73ΔAAUAAA78 and 73AAUGAA78 mutant, at 266% and 293% of wild-type, respectively (Figure 4C). This compared to a non-significant 131% of wild-type spliced gRNA incorporation for the 73AUUAAA78 mutant (Figure 4C). Altogether our results demonstrate that a canonical polyadenylation motif in the 5′ PolyA is required for efficient gRNA packaging, even though it is ordinarily repressed during HIV-1 replication.
DISCUSSION
RNA molecules are important regulators of biological activity (1). They play key roles in bacterial (57) or viral infection processes (58,59), and defects in RNA regulation have been implicated in human disease (60). Although this makes RNA an extremely attractive medical target, RNA-based treatments have so far been challenging to develop.
The first step in exploiting RNA as a drug target is to identify RNA motifs with the most potential for therapeutic intervention. Unfortunately, the functional flexibility of RNA means that the same stretches of RNA often perform multiple roles, which greatly complicates the identification of regulatory RNA by traditional truncation and deletion mutagenesis. This problem is especially evident within RNA viral genomes, where fierce evolutionary pressure for ‘data compression’ means that regulatory and coding regions overlap in complex ways that impede the understanding of their function. Here, we have implemented in cell MIME to pinpoint regulatory RNA in an unbiased fashion at single nucleotide resolution (31,39). Using different methods of functional selection, we could dissect a complex regulatory network controlling gRNA production and packaging into virions (Figure 5). Surprisingly, a common sequence within 5′ PolyA both negatively and positively regulated these respective processes.
Figure 5.

Five regulatory elements controlling HIV-1 replication. gRNA production is positively regulated by sequences within TAR and by the U1snRNP binding site within SL2. gRNA production is negatively regulated by the AAUAAA motif in 5′ polyA. The U1snRNP binding site is required for repression of 5′ polyadenylation. gRNA packaging into virions requires both the Pr55Gag binding site (SL1-SL3), and the AAUAAA motif in 5′ PolyA. Positive regulatory elements are highlighted in green. Negative regulatory elements are highlighted in red.
With regards to gRNA production, we were able to identify three distinct RNA motifs (Figure 2). Reassuringly, mutations to TAR impaired gRNA production, with the apical portion proving to be more important for gRNA production than the distal portion. These results are in agreement with the deletion and mutagenesis studies pointing to the minimal sequences required for transactivation as spanning residues 19 to 43 (61,62). Although we were not able to analyse the U-rich bulge (nucleotides 23–25) constituting the binding site for the viral Tat protein (63,64), we could see the importance of the loop sequence (nucleotides 29–34) serving as the binding site for P-TEFb (65,66). Interestingly, TAR is assumed to be the most important motif for gRNA production, but we were able to identify several point mutations that were more detrimental to viral RNA levels in cells than those mapping to TAR. These mutations clustered within the U1 snRNA binding site within SL2 suggesting a crucial role for the splicing factor U1snRNA in gRNA production. Indeed, binding of U1 snRNP, and in particular the U1 snRNP protein 70K, to the HIV-1 gRNA is important for the repression of polyadenylation at the 5′ PolyA site (51,52,67). Without this repression, only short prematurely polyadenylated transcripts would be generated, preventing production of the full-length gRNA. In agreement, our mutations designed to impair U1 snRNA binding strongly repress gRNA production, and that this phenotype could be counteracted by deletion of the 73AAUAAA78 (Figure 4). We also observed that individual mutants to 73AAUAAA78 were enriched in cells compared to the wild-type sequence. Presumably, disruption of the 5′ canonical polyadenylation signal enhances viral RNA production by eliminating low levels of premature cleavage and polyadenylation. The general assumption is that 5′ PolyA is efficiently repressed in wild-type HIV-1 through inhibitory RNA structure (68–71), proximity to the 5′ cap (72,73), the presence of downstream inhibitory sequences (51,52), and the absence of upstream activating sequences that are only present at the 3′ end of the gRNA (72,73). Our data argue that some level of cleavage and polyadenylation still occurs within the wild-type 5′ PolyA despite these inhibitory mechanisms (71).
With regards to gRNA packaging, we identified two distinct regions required for incorporation of gRNA into viral particles (Figure 3). The central packaging signal spanned SL1 to SL3 and closely corresponds to the Pr55Gag binding site previously defined in vitro (4,31) and a region found to bind Pr55Gag in PAR-CLIP crosslinking experiments in cells (32). Mutations to the 257GCGCGC262 sequence within the apical loop of SL1 did not impair gRNA packaging in cells. This palindromic sequence initiates gRNA dimerization via a kissing loop interaction – a conserved phenomenon within the retroviridae family (10,74,75). gRNA dimerization is required for viral replication (19,20,76–78) and presumed to be mechanistically linked to gRNA packaging (20,21,79). Given that identical mutations severely compromised Pr55Gag binding in a similar MIME assay conducted in vitro (31), it was surprising to see that mutations to this sequence did not impair gRNA packaging. Nevertheless, our in cell data is consistent with modest effects on gRNA packaging seen with SL1 apical loop mutants in a variety of studies (19,21,76,80,81). For example, in one study a single G to U mutation 257GCGCUC262 lead to a roughly two fold reduction in gRNA packaging (82), whereas our in cell MIME data shows that this same mutation packages 76% of wild-type (68–86% are the 5 and 95 percent confidence intervals) (Supplementary Data Files). SL1 apical loop mutants may also have less impact in cells compared to Pr55Gag binding assays conducted in vitro (76) due to the presence of yet unidentified redundant dimerization sites within the full length HIV-1 genome that were not present on the short RNA fragment tested in vitro. Alternatively, primary T-lymphocytes can partially rescue defects in reverse transcription induced by deletion or mutation of SL1, implying that cellular factors can also compensate for SL1 defects (76). Regardless of the role of the SL1 apical loop sequence, the SL1 stem and internal loop itself is a bona fide packaging signal, consistent with the fact that its deletion leads to severe packaging defects (18,56,80).
Finally, we make the discovery that 73AAUAAA78 within the 5′PolyA is an exceptionally strong packaging signal in cells. Previous studies have shown that destabilizing the PolyA hairpin decreases gRNA packaging (83,84) and that complete deletion of PolyA reduces gRNA packaging by 70% (18,84), similar to a combined deletion of SL1 and SL3 (56). Until now, the best explanation for why PolyA acts as a packaging determinant is that it binds to Pr55Gag directly during viral assembly (56). Although a truncated version of the Gag protein bound 5′ PolyA in an in vitro footprinting assay (85), this binding site was not seen by in vitro MIME using full length Pr55Gag protein (31). Furthermore, PAR-CLIP experiments conducted in cells also did not identify 5′ PolyA as a Pr55Gag binding site (32). We therefore find a mechanism involving the direct binding of Pr55Gag to the 5′ PolyA unlikely. Instead, our data suggests that the cellular polyadenylation machinery and gRNA packaging are mechanistically linked. Our evidence is two-fold: first, we localized the 5′ PolyA packaging signal, at single nucleotide resolution, to the 73AAUAAA78 canonical polyadenylation signal; second, we showed that all mutations to this sequence impair gRNA packaging except for a single A to U mutation (73AUUAAA78) forming the second most frequent polyadenylation signal found in mammalian cells (86). Together, these facts argue that a functional polyadenylation signal is required for gRNA packaging into virions. This would explain why HIV-1 conserves the 5′ polyadenylation signal, even though its presence is detrimental for gRNA production. Other retroviruses, such as mouse mammary tumor virus (MMTV) and avian leukosis-sarcoma virus (ALSV), encode a single copy of the AAUAAA polyadenylation signal in the 3′ end of the gRNA, demonstrating that different (seemingly more logical) gRNA organisations are possible.
How might polyadenylation facilitate gRNA packaging? Given that the gRNA packaging is thought to be mainly determined by Pr55Gag, one simple explanation could be that there exists a direct or indirect interaction between Pr55Gag and the cellular polyadenylation machinery. This interaction could help recruit or stabilize Pr55Gag on the gRNA to ensure that viral assembly occurs preferentially on the gRNA, rather than cellular RNAs. Interestingly, cellular RNAs that are preferentially packaged into viral particles exhibit particularly long 3′UTRs (87), possibly because these mRNAs are more likely to contain inhibited upstream polyadenylation sequences than the equivalent cellular RNAs with short UTRs. Which component(s) of the polyadenylation machinery are involved in gRNA packaging? The polyadenylation machinery comprises cleavage polyadenylation specificity factor (CPSF), cleavage factors Im and IIm (CFIm and CFIIm), cleavage stimulatory factor (CstF), poly(A) polymerase (PAP), and poly(A) binding protein II (88). At least some of these cellular polyadenylation factors bind to the 5′ polyadenylation signal even when cleavage and polyadenylation is repressed (51,52,68). Amongst these factors, CPSF6—a subunit of CFIm—stands out. Although it does not directly recognize the AAUAAA sequence (88,89), it is a key player in mRNA 3′ end processing and is involved in the repression of 5′ proximal polyadenylation sites (90,91). It also interacts with the CA domain of Pr55Gag providing a possible link between polyadenylation and HIV-1 biology (92–96). Delineating the mechanistic contributions of the cellular polyadenylation machinery to gRNA packaging could provide a new window of therapeutic opportunity not currently exploited by antiretroviral therapy. However, further work will be required to define its potential role in HIV-1 gRNA packaging.
In summary, we have used in cell MIME to identify at single nucleotide resolution RNA motifs regulating gRNA production and packaging into HIV-1 virions. One of the major advantages of the in cell MIME method is that low level random mutagenesis can pinpoint functional RNA motifs whilst reducing the risk of RNA misfolding that often occurs when large and imprecise deletion mutants are used. Although we have not done so here, in principle, in cell MIME data can also be used to identify RNA secondary structure important for regulatory function through the identification of compatible co-varying nucleotide positions. Thus, in cell MIME is a flexible and powerful methodology should help to identify novel regulatory RNA motifs in a wide range of pathogens, as well as lead to a better understanding of non-coding RNA molecules in eukaryotic cells.
DATA AVAILABILITY
Software for analysing MIME data is available from GitHub https://github.com/maureensmith/MIMEAnTo/. Processed data can be found in Supplementary Data Files. Raw sequencing reads are available through NCBI Gene Expression Omnibus number GSE109386.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Sidaction and ANRS (Agence Nationale de Recherches sur le SIDA et les Hépatites Virales) to R.M.; ANRS [ECTZ35556 to R.P.S.] and IDEX: Par delà les frontières, l’Université de Strasbourg to R.P.S.; German Ministry for Education & Science (BMBF) [031A307 to M.S. and M.v.K.]; Partenariat Hubert Curien (PHC) programme franco-allemand PROCOPE [37709NJ to R.P.S. and M.v.K.]; IGBMC Microarray and Sequencing platform, a member of the ‘France Génomique’ consortium [ANR-10-INBS-0009]. Funding for open access charge: CNRS.
Author contributions: R.P.S. designed the study. R.P.S. performed the MIME experiments including libraries for sequencing. R.P.S., M.R.S., L.D, F.J., P.C. and M.v.K. developed bioinformatic tools. M.v.K. and M.R.S. developed binding models and statistical tools. G.L., T.D. and C.M. performed replication studies of modified HIV-1. R.P.S., A-C.J. and M.M. tested PolyA and SL2 mutants. R.P.S., M.R.S., M.M., J.-C.P., M.v.K. and R.M. analyzed the data. R.P.S. and M.v.K. wrote the paper with contributions from the other authors.
Conflict of interest statement. None declared.
REFERENCES
- 1. Morris K.V., Mattick J.S.. The rise of regulatory RNA. Nat. Rev. Genet. 2014; 15:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilkinson K.A, Gorelick R.J., Vasa S.M., Guex N., Rein A., Mathews D.H., Giddings M.C., Weeks K.M.. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 2008; 6:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paillart J.C., Dettenhofer M., Yu X.F., Ehresmann C., Ehresmann B., Marquet R.. First snapshots of the HIV-1 RNA structure in infected cells and in virions. J. Biol. Chem. 2004; 279:48397–48403. [DOI] [PubMed] [Google Scholar]
- 4. Abd El-Wahab E.W., Smyth R.P., Mailler E., Bernacchi S., Vivet-Boudou V., Hijnen M., Jossinet F., Mak J., Paillart J.-C., Marquet R.. Specific recognition of the HIV-1 genomic RNA by the Gag precursor. Nat. Commun. 2014; 5:4304. [DOI] [PubMed] [Google Scholar]
- 5. Watts J.M., Dang K.K., Gorelick R.J., Leonard C.W., Bess J.W., Swanstrom R., Burch C.L., Weeks K.M.. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009; 460:711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lu K., Heng X., Garyu L., Monti S., Garcia E.L., Kharytonchyk S., Dorjsuren B., Kulandaivel G., Jones S., Hiremath A. et al. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science. 2011; 334:242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baudin F., Marquet R., Isel C., Darlix J.L., Ehresmann B., Ehresmann C.. Functional sites in the 5′ region of human immunodeficiency virus type 1 RNA form defined structural domains. J. Mol. Biol. 1993; 229:382–397. [DOI] [PubMed] [Google Scholar]
- 8. Mailler E., Bernacchi S., Marquet R., Paillart J.-C., Vivet-Boudou V., Smyth R.. The life-cycle of the HIV-1 Gag–RNA complex. Viruses. 2016; 8:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karn J., Stoltzfus C.M.. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2012; 2:a006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paillart J.-C., Shehu-Xhilaga M., Marquet R., Mak J.. Dimerization of retroviral RNA genomes: an inseparable pair. Nat. Rev. Microbiol. 2004; 2:461–472. [DOI] [PubMed] [Google Scholar]
- 11. Isel C., Ehresmann C., Marquet R.. Initiation of HIV reverse transcription. Viruses. 2010; 2:213–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Le Grice S.F.J. Targeting the HIV RNA genome: high-hanging fruit only needs a longer ladder. Curr. Top. Microbiol. Immunol. 2015; 389:147–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Janssen H.L.A., Reesink H.W., Lawitz E.J., Zeuzem S., Rodriguez-Torres M., Patel K., van der Meer A.J., Patick A.K., Chen A., Zhou Y. et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013; 368:1685–1694. [DOI] [PubMed] [Google Scholar]
- 14. Clever J.L., Miranda D., Parslow T.G., Miranda D. Jr, Parslow T.G.. RNA structure and packaging signals in the 5′ leader region of the human immunodeficiency virus type 1 genome. J. Virol. 2002; 76:12381–12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Helga-Maria C., Hammarskjöld M.-L.L., Rekosh D.. An intact TAR element and cytoplasmic localization are necessary for efficient packaging of human immunodeficiency virus type 1 genomic RNA. J. Virol. 1999; 73:4127–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harrich D., Hooker C.W., Parry E.. The human immunodeficiency virus type 1 TAR RNA upper stem-loop plays distinct roles in reverse transcription and RNA packaging. J. Virol. 2000; 74:5639–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Russell R.S., Hu J., Laughrea M., Wainberg M.A., Liang C.. Deficient dimerization of human immunodeficiency virus type 1 RNA caused by mutations of the u5 RNA sequences. Virology. 2002; 303:152–163. [DOI] [PubMed] [Google Scholar]
- 18. Didierlaurent L., Racine P.J., Houzet L., Chamontin C., Berkhout B., Mougel M.. Role of HIV-1 RNA and protein determinants for the selective packaging of spliced and unspliced viral RNA and host U6 and 7SL RNA in virus particles. Nucleic Acids Res. 2011; 39:8915–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Berkhout B., van Wamel J.L.. Role of the DIS hairpin in replication of human immunodeficiency virus type 1. J. Virol. 1996; 70:6723–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laughrea M., Jetté L., Mak J., Kleiman L., Liang C., Wainberg M.A.. Mutations in the kissing-loop hairpin of human immunodeficiency virus type 1 reduce viral infectivity as well as genomic RNA packaging and dimerization. J. Virol. 1997; 71:3397–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paillart J.C., Berthoux L., Ottmann M., Darlix J.L., Marquet R., Ehresmann B., Ehresmann C.. A dual role of the putative RNA dimerization initiation site of human immunodeficiency virus type 1 in genomic RNA packaging and proviral DNA synthesis. J. Virol. 1996; 70:8348–8354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keane S.C., Heng X., Lu K., Kharytonchyk S., Ramakrishnan V., Carter G., Barton S., Hosic A., Florwick A., Santos J. et al. RNA structure. Structure of the HIV-1 RNA packaging signal. Science. 2015; 348:917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aldovini A., Young R.A.. Mutations of RNA and protein sequences involved in human immunodeficiency virus type 1 packaging result in production of noninfectious virus. J. Virol. 1990; 64:1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clavel F., Orenstein J.M.. A mutant of human immunodeficiency virus with reduced RNA packaging and abnormal particle morphology. J. Virol. 1990; 64:5230–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lever A., Gottlinger H., Haseltine W., Sodroski J.. Identification of a sequence required for efficient packaging of human immunodeficiency virus type 1 RNA into virions. J. Virol. 1989; 63:4085–4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luban J., Goff S.P.. Mutational analysis of cis-acting packaging signals in human immunodeficiency virus type 1 RNA. J. Virol. 1994; 68:3784–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nikolaitchik O.A., Rhodes T.D., Ott D., Hu W.-S.. Effects of mutations in the human immunodeficiency virus type 1 Gag gene on RNA packaging and recombination. J. Virol. 2006; 80:4691–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vrolijk M.M., Ooms M., Harwig A., Das A.T., Berkhout B.. Destabilization of the TAR hairpin affects the structure and function of the HIV-1 leader RNA. Nucleic Acids Res. 2008; 36:4352–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Das A.T., Vrolijk M.M., Harwig A., Berkhout B.. Opening of the TAR hairpin in the HIV-1 genome causes aberrant RNA dimerization and packaging. Retrovirology. 2012; 9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Das A.T., Harwig A., Vrolijk M.M., Berkhout B.. The TAR hairpin of human immunodeficiency virus type 1 can be deleted when not required for Tat-mediated activation of transcription. J. Virol. 2007; 81:7742–7748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smyth R.P., Despons L., Huili G., Bernacchi S., Hijnen M., Mak J., Jossinet F., Weixi L., Paillart J., von Kleist M. et al. Mutational interference mapping experiment (MIME) for studying RNA structure and function. Nat. Methods. 2015; 12:866–872. [DOI] [PubMed] [Google Scholar]
- 32. Kutluay S.B., Zang T., Blanco-Melo D., Powell C., Jannain D., Errando M., Bieniasz P.D.. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell. 2014; 159:1096–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mouland A.J., Mercier J., Luo M., Bernier L., DesGroseillers L., Cohen E.A.. The double-stranded RNA-binding protein Staufen is incorporated in human immunodeficiency virus type 1: evidence for a role in genomic RNA encapsidation. J. Virol. 2000; 74:5441–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eckwahl M.J., Arnion H., Kharytonchyk S., Zang T., Bieniasz P.D., Telesnitsky A., Wolin S.L.. Analysis of the human immunodeficiency virus-1 RNA packageome. RNA. 2016; 22:1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y., Nikolaitchik O.A., Rahman S.A., Chen J., Pathak V.K., Hu W.S.. HIV-1 sequence necessary and sufficient to package non-viral RNAs into HIV-1 particles. J. Mol. Biol. 2017; 429:2542–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McBride M.S., Schwartz M.D., Panganiban A.T.. Efficient encapsidation of human immunodeficiency virus type 1 vectors and further characterization of cis elements required for encapsidation. J. Virol. 1997; 71:4544–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gibbs J.S., Regier D.A., Desrosiers R.C.. Construction and In Vitro Properties of HIV-1 Mutants with Deletions in ‘Nonessential’ Genes. AIDS Res. Hum. Retroviruses. 1994; 10:343–350. [DOI] [PubMed] [Google Scholar]
- 38. Zufferey R., Nagy D., Mandel R.J., Naldini L., Trono D.. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997; 15:871–875. [DOI] [PubMed] [Google Scholar]
- 39. Smith M.R., Smyth R.P., Marquet R., von Kleist M.. MIMEAnTo—profiling functional RNA in mutational interference mapping experiments. Bioinformatics. 2016; 32:3369–3370. [DOI] [PubMed] [Google Scholar]
- 40. Lehmann K.A., Bass B.L.. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000; 39:12875–12884. [DOI] [PubMed] [Google Scholar]
- 41. Suspène R., Petit V., Puyraimond-Zemmour D., Aynaud M.-M., Henry M., Guétard D., Rusniok C., Wain-Hobson S., Vartanian J.-P.. Double-stranded RNA adenosine deaminase ADAR-1-induced hypermutated genomes among inactivated seasonal influenza and live attenuated measles virus vaccines. J. Virol. 2011; 85:2458–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sodroski J., Rosen C., Wong-Staal F., Salahuddin S.Z., Popovic M., Arya S., Gallo R.C., Haseltine W.A.. Trans-acting transcriptional regulation of human T-cell leukemia virus type III long terminal repeat. Science. 1985; 227:171–173. [DOI] [PubMed] [Google Scholar]
- 43. Sodroski J., Patarca R., Rosen C., Wong-Staal F., Haseltine W.. Location of the trans-activating region on the genome of human T-cell lymphotropic virus type III. Science. 1985; 229:74–77. [DOI] [PubMed] [Google Scholar]
- 44. Herrmann C.H., Rice A.P.. Lentivirus Tat proteins specifically associate with a cellular protein kinase, TAK, that hyperphosphorylates the carboxyl-terminal domain of the large subunit of RNA polymerase II: candidate for a Tat cofactor. J. Virol. 1995; 69:1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herrmann C.H., Gold M.O., Rice A.P.. Viral transactivators specifically target distinct cellular protein kinases that phosphorylate the RNA polymerase II C-terminal domain. Nucleic Acids Res. 1996; 24:501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bardaro M.F., Shajani Z., Patora-Komisarska K., Robinson J.A., Varani G.. How binding of small molecule and peptide ligands to HIV-1 TAR alters the RNA motional landscape. Nucleic Acids Res. 2009; 37:1529–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Richter S., Cao H., Rana T.M.. Specific HIV-1 TAR RNA loop sequence and functional groups are required for human cyclin T1-Tat-TAR ternary complex formation. Biochemistry. 2002; 41:6391–6397. [DOI] [PubMed] [Google Scholar]
- 48. Klaver B., Berkhout B.. Evolution of a disrupted TAR RNA hairpin structure in the HIV-1 virus. EMBO J. 1994; 13:2650–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harrich D., Hsu C., Race E., Gaynor R.B.. Differential growth kinetics are exhibited by human immunodeficiency virus type 1 TAR mutants. J. Virol. 1994; 68:5899–5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tian B., Hu J., Zhang H., Lutz C.S.. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005; 33:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ashe M.P., Griffin P., James W., Proudfoot N.J.. Poly(A) site selection in the HIV-1 provirus: inhibition of promoter-proximal polyadenylation by the downstream major splice donor site. Genes Dev. 1995; 9:3008–3025. [DOI] [PubMed] [Google Scholar]
- 52. Ashe M.P., Pearson L.H., Proudfoot N.J.. The HIV-1 5′ LTR poly (A) site is inactivated by U1 snRNP interaction with the downstream major splice donor site. EMBO J. 1997; 16:5752–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Takata M., Soll S.J., Emery A., Blanco-Melo D., Swanstrom R., Bieniasz P.D.. Global synonymous mutagenesis identifies cis-acting RNA elements that regulate HIV-1 splicing and replication. PLoS Pathog. 2018; 14:e1006824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mueller N., van Bel N., Berkhout B., Das A.T.. HIV-1 splicing at the major splice donor site is restricted by RNA structure. Virology. 2014; 468:609–620. [DOI] [PubMed] [Google Scholar]
- 55. Mueller N., Berkhout B., Das A.T.. HIV-1 splicing is controlled by local RNA structure and binding of splicing regulatory proteins at the major 5′ splice site. J. Gen. Virol. 2015; 96:1906–1917. [DOI] [PubMed] [Google Scholar]
- 56. Houzet L., Paillart J.C., Smagulova F., Maurel S., Morichaud Z., Marquet R., Mougel M.. HIV controls the selective packaging of genomic, spliced viral and cellular RNAs into virions through different mechanisms. Nucleic Acids Res. 2007; 35:2695–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Duval M., Cossart P., Lebreton A.. Mammalian microRNAs and long noncoding RNAs in the host-bacterial pathogen crosstalk. Semin. Cell Dev. Biol. 2017; 65:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang Z., Zhao Y., Zhang Y.. Viral lncRNA: A regulatory molecule for controlling virus life cycle. Non-coding RNA Res. 2017; 2:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tycowski K.T., Guo Y.E., Lee N., Moss W.N., Vallery T.K., Xie M., Steitz J.A.. Viral noncoding RNAs: more surprises. Genes Dev. 2015; 29:567–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cooper T.A., Wan L., Dreyfuss G.. RNA and disease. Cell. 2009; 136:777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jakobovits A., Smith D.H., Jakobovits E.B., Capon D.J.. A discrete element 3′ of human immunodeficiency virus 1 (HIV-1) and HIV- 2 mRNA initiation sites mediates transcriptional activation by an HIV trans activator. Mol. Cell. Biol. 1988; 8:2555–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Berkhout B., Silverman R.H., Jeang K.T.. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell. 1989; 59:273–282. [DOI] [PubMed] [Google Scholar]
- 63. Dingwall C., Ernberg I., Gait M.J., Green S.M., Heaphy S., Karn J., Lowe A.D., Singh M., Skinner M.A., Valerio R.. Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (TAR) RNA in vitro. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:6925–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dingwall C., Ernberg I., Gait M.J., Green S.M., Heaphy S., Karn J., Lowe A.D., Singh M., Skinner M.A.. HIV-1 tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. EMBO J. 1990; 9:4145–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Feng S., Holland E.C.. HIV-1 tat trans-activation requires the loop sequence within tar. Nature. 1988; 334:165–167. [DOI] [PubMed] [Google Scholar]
- 66. Wei P., Garber M.E., Fang S.M., Fischer W.H., Jones K.A.. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998; 92:451–462. [DOI] [PubMed] [Google Scholar]
- 67. Ashe M.P., Furger A., Proudfoot N.J.. Stem-loop 1 of the U1 snRNP plays a critical role in the suppression of HIV-1 polyadenylation. RNA. 2000; 6:170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Klasens B.I., Thiesen M., Virtanen A., Berkhout B.. The ability of the HIV-1 AAUAAA signal to bind polyadenylation factors is controlled by local RNA structure. Nucleic Acids Res. 1999; 27:446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gee A.H., Kasprzak W., Shapiro B.A.. Structural differentiation of the HIV-1 polyA signals. J. Biomol. Struct. Dyn. 2006; 23:417–428. [DOI] [PubMed] [Google Scholar]
- 70. Graveley B.R., Fleming E.S., Gilmartin G.M.. RNA structure is a critical determinant of poly(A) site recognition by cleavage and polyadenylation specificity factor. Mol. Cell. Biol. 1996; 16:4942–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Das A.T., Klaver B., Berkhout B.. A hairpin structure in the R region of the human immunodeficiency virus type 1 RNA genome is instrumental in polyadenylation site selection. J. Virol. 1999; 73:81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cherrington J., Ganem D.. Regulation of polyadenylation in human immunodeficiency virus (HIV): contributions of promoter proximity and upstream sequences. EMBO J. 1992; 11:1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Valsamakis A., Schek N., Alwine J.C.. Elements upstream of the AAUAAA within the human immunodeficiency virus polyadenylation signal are required for efficient polyadenylation in vitro. Mol. Cell. Biol. 1992; 12:3699–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Paillart J.C., Marquet R., Skripkin E., Ehresmann B., Ehresmann C.. Mutational analysis of the bipartite dimer linkage structure of human immunodeficiency virus type 1 genomic RNA. J. Biol. Chem. 1994; 269:27486–27493. [PubMed] [Google Scholar]
- 75. Paillart J.C., Skripkin E., Ehresmann B., Ehresmann C., Marquet R.. A loop-loop ‘kissing’ complex is the essential part of the dimer linkage of genomic HIV-1 RNA. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:5572–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jones K.L., Sonza S., Mak J.. Primary T-lymphocytes rescue the replication of HIV-1 DIS RNA mutants in part by facilitating reverse transcription. Nucleic Acids Res. 2008; 36:1578–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hill M.K., Shehu-Xhilaga M., Campbell S.M., Poumbourios P., Crowe S.M., Mak J.. The dimer initiation sequence stem-loop of human immunodeficiency virus type 1 is dispensable for viral replication in peripheral blood mononuclear cells. J. Virol. 2003; 77:8329–8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sakuragi J.-I., Sakuragi S., Shioda T.. Minimal region sufficient for genome dimerization in the human immunodeficiency virus type 1 virion and its potential roles in the early stages of viral replication. J. Virol. 2007; 81:7985–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Russell R.S., Liang C., Wainberg M.A.. Is HIV-1 RNA dimerization a prerequisite for packaging? Yes, no, probably?. Retrovirology. 2004; 1:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Clever J.L., Parslow T.G.. Mutant human immunodeficiency virus type 1 genomes with defects in RNA dimerization or encapsidation. J. Virol. 1997; 71:3407–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. St Louis D.C., Gotte D., Sanders-Buell E., Ritchey D.W., Salminen M.O., Carr J.K., McCutchan F.E.. Infectious molecular clones with the nonhomologous dimer initiation sequences found in different subtypes of human immunodeficiency virus type 1 can recombine and initiate a spreading infection in vitro. J. Virol. 1998; 72:3991–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Clever J., Sassetti C., Parslow T.G.. RNA secondary structure and binding sites for gag gene products in the 5′ packaging signal of human immunodeficiency virus type 1. J. Virol. 1995; 69:2101–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Das A.T., Klaver B., Klasens B.I., van Wamel J.L., Berkhout B.. A conserved hairpin motif in the R-U5 region of the human immunodeficiency virus type 1 RNA genome is essential for replication. J. Virol. 1997; 71:2346–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Clever J.L., Eckstein D.A., Parslow T.G.. Genetic dissociation of the encapsidation and reverse transcription functions in the 5′ R region of human immunodeficiency virus type 1. J. Virol. 1999; 73:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kenyon J.C., Prestwood L.J., Lever A.M.L.. A novel combined RNA-protein interaction analysis distinguishes HIV-1 Gag protein binding sites from structural change in the viral RNA leader. Sci. Rep. 2015; 5:14369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Beaudoing E., Freier S., Wyatt J.R., Claverie J.M., Gautheret D.. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000; 10:1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Comas-Garcia M., Davis S.R., Rein A.. On the Selective Packaging of Genomic RNA by HIV-1. Viruses. 2016; 8:E246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sun Y., Zhang Y., Hamilton K., Manley J.L., Shi Y., Walz T., Tong L.. Molecular basis for the recognition of the human AAUAAA polyadenylation signal. Proc. Natl. Acad. Sci. U.S.A. 2017; 115:E1419–E1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Clerici M., Faini M., Aebersold R., Jinek M.. Structural insights into the assembly and polyA signal recognition mechanism of the human CPSF complex. Elife. 2017; 6:e33111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Martin G., Gruber A.R., Keller W., Zavolan M.. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012; 1:753–763. [DOI] [PubMed] [Google Scholar]
- 91. Shi Y., Di Giammartino D.C., Taylor D., Sarkeshik A., Rice W.J., Yates J.R., Frank J., Manley J.L.. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell. 2009; 33:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Engeland C.E., Brown N.P., Börner K., Schümann M., Krause E., Kaderali L., Müller G.A., Kräusslich H.-G.. Proteome analysis of the HIV-1 Gag interactome. Virology. 2014; 460–461C:194–206. [DOI] [PubMed] [Google Scholar]
- 93. Saito A., Henning M.S., Serrao E., Dubose B.N., Teng S., Huang J., Li X., Saito N., Roy S.P., Siddiqui M.A. et al. Capsid-CPSF6 interaction is dispensable for HIV-1 replication in primary cells but is selected during virus passage in vivo. J. Virol. 2016; 90:6918–6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lee K., Ambrose Z., Martin T.D., Oztop I., Mulky A., Julias J.G., Vandegraaff N., Baumann J.G., Wang R., Yuen W. et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe. 2010; 7:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rasheedi S., Shun M.-C., Serrao E., Sowd G.A., Qian J., Hao C., Dasgupta T., Engelman A.N., Skowronski J.. The cleavage and polyadenylation specificity factor 6 (CPSF6) subunit of the capsid-recruited pre-messenger RNA cleavage factor I (CFIm) complex mediates HIV-1 integration into genes. J. Biol. Chem. 2016; 291:11809–11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Price A.J., Fletcher A.J., Schaller T., Elliott T., Lee K., KewalRamani V.N., Chin J.W., Towers G.J., James L.C.. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 2012; 8:e1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Software for analysing MIME data is available from GitHub https://github.com/maureensmith/MIMEAnTo/. Processed data can be found in Supplementary Data Files. Raw sequencing reads are available through NCBI Gene Expression Omnibus number GSE109386.