

Abstract
Although the catalog of cancer-associated mutations in protein-coding regions is nearly complete for all major cancer types, an assessment of regulatory changes in cancer genomes and their clinical significance remain largely preliminary. Adopting bottom-up approach, we quantify the effects of different sources of gene expression variation in a cohort of 3899 samples from 10 cancer types. We find that copy number alterations, epigenetic changes, transcription factors and microRNAs collectively explain, on average, only 31–38% and 18–26% expression variation for cancer-associated and other genes, respectively, and that among these factors copy number alteration has the highest effect. We show that the genes with systematic, large expression variation that could not be attributed to these factors are enriched for pathways related to cancer hallmarks. Integrating whole genome sequencing data and focusing on genes with systematic expression variation we identify novel, recurrent regulatory mutations affecting known cancer genes such as NKX2-1 and GRIN2D in multiple cancer types. Nonetheless, at a genome-wide scale proportions of gene expression variation attributed to recurrent point mutations appear to be modest so far, especially when compared to that attributed to copy number changes – a pattern different from that observed for other complex diseases and traits. We suspect that, owing to plasticity and redundancy in biological pathways, regulatory alterations show complex combinatorial patterns, modulating gene expression in cancer genomes at a finer scale.
INTRODUCTION
Increasing evidence suggests that regulatory alterations leading to gene expression changes play critical roles in complex traits and diseases, and it is anticipated that genomic alterations with regulatory consequences have important roles in cancer as well (1). Gene expression in mammalian genomes is regulated at different levels; point mutations, copy number alterations, epigenetic modifications, and post-transcriptional (and post-translational) modifications can potentially regulate abundance of gene products. Most major cancer types have been systematically profiled for copy number, CpG methylation and transcriptomic changes. Even though detection of driver alterations in protein coding regions (reviewed in (2)) has reached near saturation in all major types of cancer (3), our understanding of the prevalence of regulatory alterations and their significance remain largely preliminary (1), primarily due to lack of whole genome sequencing (WGS) data for cohorts of samples until recently.
Genome-wide patterns of genomic alterations, including potential regulatory mutations are emerging from the WGS studies including the International Cancer Genome Consortium (ICGC) (4). Early findings presented two polarizing viewpoints regarding the prevalence and significance of regulatory point mutations. The discovery of recurrent mutations in TERT promoter in multiple cancer types including melanoma advocated for a genome-wide scans to identify recurrent noncoding regulatory mutations (5,6). In contrast, two studies reported relatively few additional novel recurrent promoter mutations (e.g. CLPTM1L, SDHD) beyond the classic TERT promoter mutations after analyzing thousands of samples (7,8). Smith et al. (9) identified a signature of accelerated somatic evolution marked by clusters of non-recurrent mutations in the promoters of cancer genes that had pathway-level consequences. Several recent reports (4,6,10–14) identified hotspots of mutations and InDels (e.g. in BCL2 promoter in lymphoma); but, in some cases, recurrent mutations had no apparent regulatory effects on downstream genes, and in some other cases, mutations with regulatory significance did not necessarily have a base-pair level recurrence (14). It is argued that systematic identification of all ‘driver’ noncoding mutations, similar to that used for coding mutations, using a top-down WGS-guided approach would require very large cohorts (14). Nonetheless, ICGC studies, when published will provide important insights. Other modes of gene expression modulation such as epigenetic deregulation and chromatin-level changes appear to be pervasive in multiple cancer types (15). Complex regulation involving miRNA regulatory network was also shown to be common in cancer (16). Oncogenic copy number alterations are major drivers behind activity of cancer pathways (17). There are other examples as well. Previous studies have assessed effects of these factors or their limited combinations on gene expression (18–21). However, it remains unclear to what extent gene expression variation in cancer genomes could be attributed to these factors, including point mutations, and their combinations. Here we adopt a novel approach, complementary to the ongoing genome-centric efforts, to dissect the sources of gene expression variations in a cohort of 3899 samples from 10 cancer types, and then integrate whole genome sequencing data for a subset of the cases to identify novel, recurrent somatic mutations associated with altered expression of genes in cancer genomes.
MATERIALS AND METHODS
Data sources and preprocessing
TCGA data for gene expression (polyA+ IlluminaHiSeq), methylation (HumanMethylation450), somatic copy number (segmented copy number measured by Affymetrix Genome-Wide Human SNP Array 6.0), tumor purity estimates, and miRNA expression (IlluminaHiseq) for 10 cancer types was obtained from UCSC Xena (http://xena.ucsc.edu). Supplementary Table S1 tabulates total number of samples in each cancer type for which data was downloaded and analyzed. All data was mapped to the human reference genome hg19. Log2(x + 1) transformed RSEM normalized count gene expression data was used. In case of methylation, TSS of a gene was defined as the start of the longest transcript of a gene, and the probe closest to the TSS was considered for further analysis. The miRNA-mRNA pairs were identified based on the experimentally validated sets in the miRTarBase (Release 6.1) (22) (http://mirtarbase.mbc.nctu.edu.tw). Similarly for transcription factor-target gene (TF-TG) pairs, only validated targets were considered for analysis (http://www.grnpedia.org/trrust/;version 2). The cancer samples with no tumor purity or SCNA (Somatic Copy Number Alterations) data were removed. If a gene spanned multiple SCNA segments, the longest one was considered; such cases were exceedingly rare affecting <1% of cases. Missing values for covariates and potential regulators were replaced with NA. eQTL (Expression quantitative trait loci) data was obtained from the GTEx consortium (v6) (23).
somExVar framework
A total of ∼17, 000 RefSeq, known protein-coding genes that passed the selection criterion defined above and had available expression profiles were analyzed using the pipeline. Low gene expression estimates were floored at 0.001 for downstream statistical analyes. The genes for which data for at least four potential regulators of expression (CpG methylation, SCNA, miRNA expression and TF expression) was available for at least 30 samples were selected for further analysis. For each gene, in each sample we also obtained copy number estimates, promoter methylation status, expression levels of known regulating transcription factors and miRNAs, and also additional attributes such as tumor purity estimates to model gene expression as a function of these features.
Some of the features in the model that potentially affect gene expression might be correlated, and thus we implemented a Principal Component Regression with Gamma distribution to model the residuals. As a first step, the available features were resolved along the principal axes, and then the principal components were used to construct a generalized linear model with log2 expression estimates as a response variable, where error-terms could be modeled using the Gamma distribution. In such a model, it is not possible to directly estimate the variance explained by R2. Instead, we used pseudo variable R2 (denoted as D2) of the generalized linear model as a proxy for total gene expression variation explained by the model. Downstream analyses were performed based on the results of the Principal Component Regression with Gamma distribution to model the residuals. Genes with low proportion of variance explained by somatic copy number alterations, CpG methylation, regulating transcription factor and miRNA expression in the model were prioritized for downstream analyses. Since gene expression estimates have also been modeled often using a lognormal distribution (24), as an alternate approach we constructed a linear model with log2 expression estimates as a response variable, and error-terms modeled using a Gaussian distribution. Overlap of genes between two models for each cancer has been shown in Supplementary Figure S1. The Jaccard Index ranged from 15–57% across different cancers and the overall Jaccard index was 27% with 55 overlapping calls out of 204 total calls from the two models.
Genes with recurrent sources of expression variation often show bimodal expression due to systematic expression variation between the samples with and without that regulatory variant (25,26). We used Hartigans' dip test while identifying genes for which the distribution of model residuals showed significant departure from unimodality (i.e. bimodal or multimodal). For the genes with significant dip-test P-value, we used two component mixture models to distinguish different clusters (or populations) in the residual distribution. The parameters of the mixture model were estimated using expectation maximization. Bimodal separation score (S) (Equation 1) was calculated to assess the separation of two populations. Genes with significant dip test P-value and/or high bimodal separation score are attractive targets for search for recurrent, potential regulatory alterations.
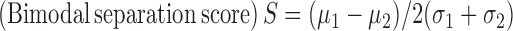 |
(1) |
The model framework has been named as somExVar. R code and example data for somExVar has been provided as Supplementary File 2 (zip file).
Somatic mutation analysis
Somatic mutations were identified from whole genome sequencing data for a subset of the samples from above cohorts by the PCAWGS initiative within ICGC (4). Variant calls were obtained from the ICGC data portal, upstream and downstream regions of the genes with high and low proportion of variance explained by somatic copy number alterations, CpG methylation, regulating transcription factor and miRNA expression identified by somExVar were scanned for potential regulatory somatic mutations (27). For each gene locus, somatic point mutations and small InDels within ±1 Mb flanking regions of the genes were identified; those were then classified as adjacent (gene overlapping – 1 kb), proximal (1–10 kb), intermediate (10–100 kb) and distal (100 kb–1 Mb), depending on their location relative to the gene locus. This identified 5164 point mutations and 17, 339 InDels across all cancer cohorts. The mutations were annotated for their regulatory potential using RegulomeDB (Version 1.1) (28), which assigns a score for each variant by integrating known motifs, transcription factor binding data, evolutionary conservation etc. as obtained from the UCSC Genome Browser (29). Additionally, regulatory potential of variations was also checked with FunSeq2 (v2.10) (30) as an alternate approach. Eukaryotic Promoter Database (v.005) (31) was used to check for mutation burden in alternate promoter sequences.
Somatic mutation patterns were analyzed at a genome-wide scale, but a particular emphasis was laid to assess somatic mutation burden in regulatory regions for the bottom 5% of genes in terms of explained variance by the model (D2) for each cancer type, for which features such as copy number etc did not substantially explain expression variance. For each such gene, samples were categorized into two groups based on their model residual values. The top 10 percentile samples with large model residual values (LR), and the 25–75 percentile samples that were representative of the distribution and had relatively small model residual values (SR) were scanned for SNVs and InDels within 10 kb upstream of their TSS regions. Finally, genes with significant dip test P-value and/or high bimodal separation score were scanned for recurrent regulatory mutations following criteria similar to that described above. A flowchart representing overall analysis strategy used in the study is shown in Supplementary Figure S2.
Statistical analysis
All analysis was performed in R. Principal component analysis was performed using ‘prcomp’ function, and then ‘glm’ function was used to construct the generalized linear model with ‘gamma’ distribution chosen to model the error-terms. The ‘Mixtool’ package (version 1.1.0) was used to estimate the parameters of the populations in the mixture model using expectation maximization. The Mann–Whitney U test (or Chi square test where applicable) was used to identify differential survivals, treatment outcome, metastasis status and extent of regional lymph node involvement between two populations. The Chi-sq trend test was used to compare the burden of somatic mutations or clinical features such as metastatic status, lymph node metastasis, or clinical response between groups of samples. Kaplan-Meier analysis was used to assess survival difference among classes of samples. Kaplan–Meier analyses were performed on overall survival data for these cohorts downloaded from UCSC Xena (http://xena.ucsc.edu/survival-plots/). FDR correction for multiple testing was used where applicable. Five percent cut-off was used for FDR correction for all analyses.
RESULTS
SCNA - highest contributor in gene expression variation
Integrating somatic copy number alteration (SCNA), CpG methylation, mRNA and miRNA expression data for 3899 samples from 10 cancer types, we first examined to what extent copy number alterations, epigenetic changes, and modulation of expression of transcription factors and miRNAs explain variation in expression of known genes in cancer genomes. To this end, first using a univariate analysis in each cohort for each gene, we estimate the extent of shared variance in rank between gene expression and its potential regulators using R2 (Spearman coefficient) as a proxy for variation explained (PVE). Figure 1B represents heatmap of R2 (PVE) for all cancer related genes in 10 different cancer types, where major cancer genes are highlighted. Supplementary Table S2 shows R2 values for all four potential regulators in different cancer types.
Figure 1.

(A) A schematic diagram showing different genomic and epigenomic features involved in the regulation of gene expression in cancer genomes: Somatic Copy Number Alterations (SCNA), CpG methylation, microRNA (miRNA) expression, and Transcription factor (TF) expression. (B) The proportion of gene expression variation (R2) explained by individual potential regulators in 10 different cancer cohorts is shown. Each row represents a gene and each column represents one cancer type. For each gene the feature with highest R2 was shown in the heatmap, as indicated by color. Intensity of respective color indicates extent of proportion of variation explained (PVE) for a given feature. Selected cancer-associated genes are highlighted. Also see Supplementary Figure S3 and Supplementary Table S3 for additional details. BLCA: Bladder Urothelial Carcinoma, BRCA: Breast invasive carcinoma, CESC: Cervical squamous cell carcinoma and endocervical adenocarcinoma, COAD: Colon adenocarcinoma, HNSC: Head and neck Squamous Cell Carcinoma, KIRC: Kidney renal clear cell carcinoma, LUAD: Lung adenocarcinoma, LUSC: Lung squamous cell carcinoma, PRAD: Prostate adenocarcinoma, SKCM: Skin Cutaneous Melanoma.
We find that the SCNA log2 ratio approximately explained an average of 4–15% (up to 87% for selected genes) variation in gene expression across different cancer types (Supplementary Figure S3A). In some cancer types such as LUSC, LUAD, HNSC and SKCM, it explained even higher expression variation. We observed similar trends when cancer associated genes (Supplementary Table S3 shows list of cancer associated genes from COSMIC database) were analyzed (Supplementary Figure S3). Within individual cancer types, the extent of expression variation explained by SCNA varied over a wide range, and for certain genes like DDX3Y (in HNSC) and STAU1 (in COAD); the extent of variance explained by CNVs was high (>85%). Also, for some of the known cancer genes such as AKT1 (LUSC), RAF1 (BLCA), PCM1 (COAD) and WHSC1L1 (LUSC), SCNAs explained 67–72% variation in gene expression. In general, in all cohorts SCNA explained a significantly higher proportion of expression variation for known cancer genes compared to other genes (Mann Whitney U test; FDR adjusted P-value < 5e–04).
In contrast, the extent of shared variance (PVE) in rank between gene expression and promoter methylation was, on average, relatively low (2.5–3.5% average, up to 70% for some genes: Supplementary Figure S3B). However, there were notable exceptions, such as EF1AY expression in HNSC, for which expression variation across samples was predominantly explained by promoter methylation. Similarly, for known cancer genes such as RANBP17 (HNSC) and HOXA9 (BLCA), methylation could explain 53–60% of gene expression variation (Supplementary Table S2). Transcription factors and miRNA expression explained on average 3.8–12% expression variation across cancer types. For WAS and ITGB2 in LUSC and SKCM, respectively, expression variation was predominantly explained by their regulating transcription factors. Notwithstanding the genes such as TP53, ERG, BCL6 that directly or indirectly auto-regulate their expression via feedback loops, the extent of expression variation explained by known transcription factor and miRNAs was modest (Supplementary Figure S3C and D respectively). One possible caveat could be that the genome-wide regulator-target gene correspondence is incomplete and context-dependent for a majority of transcription factors and microRNA. Overall, among the above factors, copy number alteration contributed a significantly greater proportion of PVE (Wilcoxon test; FDR adjusted P-value < 5e–2) in gene expression in cancer genomes across all major cancer types, except COAD. Thus, it appears that SCNA explains gene expression variation to a greater extent for a majority of genes followed by miRNA and transcription factor expression.
A framework to detect genes with unexplained expression variation - somExVar
Some sources of gene expression variation in cancer genomes may be partly correlated. For instance, E-box methylation in promoter of EGFR and CASP8 influences binding of the N-Myc transcription factor (32). To study these effects, we developed a computational framework, somExVar, which performs a multivariate analysis and estimates the extent of expression variation, for each gene in each cancer type, explained by a combination of factors, after adjusting for tumor purity (Figure 2A; Materials and Methods). The extent of proportion of expression variance explained was calculated using D2 (PVE; similar to R2 in the linear regression models) and is shown in Figure 2B. Therefore, 1 – D2 (Supplementary Figure S4A) indicates the extent of gene expression variability that could not be explained by the features included in the model, and might be attributed to other factors. Many cancer genes such as BCL2, MYC, CDK4 etc. had most of the expression variance already explained by a combination of known features across all cancer types (Supplementary Figure S4B). In addition, some genes such as BTK and WAS had most of their expression variation explained by a single mechanism across all cancer types: for example, the variation in gene expression of BTK was primarily explained by its correlation with the expression of the transcription factor (SP1/SP3/SPI1) across all cancer types. The average explained variance in gene expression was 18–26% across most cancer types, except for KIRC and PRAD where it was lower. It was also noted that on average, copy number alteration is a major determinant (higher D2) of gene expression variation in all major cancer types, and in contrast, those genes for which overall expression variation explained is lower, tend to have miRNA as major contributor of expression variation, as seen in Figure 1B and separately shown in Supplementary Figure S5. In some cases, gene expression variation could arise due to molecular subtypes. In breast cancer, where such subtypes are well defined, D2 for all the genes in three subtypes viz. Luminal A, Luminal B and basal breast cancer was computed individually. Supplementary Figure S6 shows distribution of D2 values for all genes across different subtypes of breast cancer, separately as well as all subtypes merged together (labeled as BRCA). Average variation explained across two subtypes viz. Luminal B and Basal (average D2: 0.29 and 0.36, respectively) was significantly higher as compared to the combined BRCA dataset (average D2: 0.23) (Wilcoxon test; FDR corrected P-value < 5e – 12). Further, for SKCM cohort, we classified the samples as primary tumor (64 samples) and metastatic tumors (259 samples) and repeated the analyses. As shown in Supplementary Figure S7, D2 was found to be significantly higher in case of primary tumors as compared to metastatic tumors (Wilcoxon test; FDR corrected P-value < 5e–12). Further, in case of COAD, CIMP status was used for classifying samples as CIMP+ or CIMP–. D2 was calculated using somExVar for CIMP+ and CIMP– samples (69 and 78 samples respectively) and was compared to PVE for mixed group of samples. After classification, a significant increase in D2 was observed especially for CIMP– samples (Wilcoxon test FDR corrected P-value < 3.46e–12) (Supplementary Figure S8). Therefore, subtype or context can contribute to systematic gene expression, and subtype-aware assessment may improve overall PVE in some cases.
Figure 2.

(A) somExVar workflow: multiple genomic and epigenomic features (potential regulators), as mentioned in Figure 1 are integrated for 10 different cancer types, and a generalized principal component regression is implemented with normalized gene expression as the response variable. Model residuals are modeled using Gamma or log-normal distributions, and genes with high proportions of unexplained variance by the features included in model are prioritized, and investigated for pathway enrichment and burden of potential regulatory somatic mutations. As a special case, genes showing systematic expression variation i.e. bimodality in residual distribution are scanned for recurrent somatic mutations in their regulatory regions and tested for association with clinical features in the available samples. B) Gene expression variation explained (PVE; here represented by D2) by all features in the model for all genes across different cancers. Each point is a gene and higher value represents higher gene expression variation explained. PVE (D2) for cancer genes across all cancer types is shown in Supplementary Figure S4B.
We hypothesize that other regulatory changes (e.g. promoter mutations) might explain a proportion of unexplained expression variation, and the genes with high systematic unexplained variation in expression may be rational targets for potential regulatory alterations with large effect sizes. We first demonstrated the utility of this approach using two well-characterized examples. SCNAs affecting BRCA1 expression are common in multiple cancers including ovarian cancer and breast cancer (33). In our dataset for BRCA samples, BRCA1 expression was significantly different among the samples with deletion, duplication compared to wild type BRCA1 (Wilcoxon test P-value: 5e–03/2.2e–16, Supplementary Figure S9A). We first used the somExVar pipeline to identify sources of variation in BRCA1 expression without including SCNA in the model and identified large systematic variation in the model residuals. This was evident when the samples were grouped according to their BRCA1 copy number status and model residuals were compared (deletion/ duplication; Wilcoxon test P-value: 3.8e–16/2.5e–13, Supplementary Figure S9B). Subsequently, when SCNA status was included in the model, the systematic differences disappeared (Wilcoxon test P-value > 0.05; Supplementary Figure S9C), and the proportion of PVE in BRCA1 expression explained increased by 13% (generalized linear model R2 increased from 0.68 [without CNV] to 0.81 [with CNV]). In fact, among the known features included in the model, SCNA explained the largest proportion of PVE. This suggests that copy number alteration is by far the leading mechanism driving expression variation of BRCA1. As a second example, we analyzed SDHD, which has been reported to carry recurrent regulatory mutations driving its decreased expression in melanoma (Wilcoxon test P-value < 1e–03, Supplementary Figure S10A, (8)). It is noteworthy that copy number alteration at the SDHD locus is also common. In case of SDHD promoter mutation, 7 out of 13 samples carrying the mutant allele also had copy number loss (CN log2 ratio ←0.1) of SDHD locus (remaining were copy neutral, CN log2 ratio: −0.1 to 0.1). But in the cohort, of the 40 samples that had whole genome sequencing data, 3 and 1 sample had copy number deletion and amplification, respectively. Including only promoter methylation, tumor purity, transcription factor and miRNA expression, somExVar identified systematic difference in residuals among the samples carrying promoter mutations/SCNA and those that were wild type (Supplementary Figure S10B). Once again, after inclusion of promoter mutation /SCNA status (Supplementary Figure S10C and D respectively) in the model, overall variation in residuals was considerably reduced, and there was no significant difference between the two groups (Wilcoxon test P-value > 0.05). In this case, SCNA explained proportionally more PVE than the recurrent promoter mutation at chr11: 111 957 523, 111 957 541 and 111 957 544. Taken together, somExVar presented a rational approach to prioritize candidate gene sets.
Genes with large, unexplained expression variation enriched for cancer related pathways
We then applied the somExVar approach to 3899 samples from 10 cancer types integrating SCNA, CpG methylation, TF expression and miRNA expression data. In each cohort, 83–94% genes had >50% unexplained expression variation (Supplementary Figure S4A). Unexplained variance in gene expression was relatively high across different cancer types (70–80%), suggesting that attributes other than those included in the model have the potential to modulate gene expression at different levels. It is noteworthy that some cancer types such as prostate cancer (PRAD), that have a relatively limited number of identified driver mutations in coding regions (2,34–37) on average show large expression variation, potentially suggestive of regulatory abnormalities. At the gene level, known oncogenes such as OLIG2, ROS1 and LMO1 had large proportions of unexplained variance in the expression in all cancer types.
The genes with high proportions of unexplained expression variance had non-random pathway preferences. The top 5% of genes with high unexplained expression variance (1 – D2) in each cancer type were analyzed for canonical pathways. Such genes were significantly enriched for Matrisome (Extracellular Matrix proteins and associated factors) and signaling by GPCR (G Protein Coupled receptors) related pathways consistently across all cancers (Figure 3). Similar pathways were enriched in case of middle 50–60% genes also. Dysregulation of ECM composition and structure is known to play a role in invasive cancer (38). Similarly, there is evidence of GPCRs controlling processes like proliferation and invasion in tumorigenesis (39). On the other hand, bottom 5% genes with low unexplained variance showed almost exclusive enrichment for cell cycle related pathways.
Figure 3.

Pathway enrichment analysis for genes with low (bottom 5% genes), intermediate (mid 50–60%), and high (top 5% genes) unexplained variance in gene expression across all cancers. ECM remodeling and GPCR signaling pathways were consistently significant across almost all cancer types in top 5% genes (low PVE) genes, whereas bottom 5% genes (high PVE) show almost exclusive enrichment for cell cycle related pathways. Color intensity represents –log10(FDR-p.value). Dark blue color represents significance.
Patterns of somatic mutations associated with high unexplained expression variation
Focusing on the genes with large, unexplained variation in gene expression residuals in the model, we investigated whether the burden of somatic mutations (SNVs and InDels) in the regulatory regions correlated with the extent of expression variation i.e. for those genes, whether the samples with high model residuals had more somatic mutations compared to those with better model fits and smaller residuals (Figure 4A). When the top 5% of genes with high unexplained variation (D2<0.02) were considered, overall there was no systematic difference in somatic mutation burden in their proximal noncoding regions, but in 3–15% of the cases, the candidate genes had higher somatic mutation burden within −10 kb to +1 kb of the TSS (Transcription Start Site) in the samples with large model residuals (LR) compared to the samples with small model residuals (SR) (Supplementary Table S4). In a majority of these cases, the number of samples with WGS data available was inadequate for statistically meaningful comparisons. Limiting the analysis to those cases with an adequate number of samples, we found examples of two such genes, MUCL1 in CESC and NMBR in SKCM (Independence test; P-value < 1e–04 for each; Figure 4B), which showed significantly different expression (MUCL1: Wilcoxon pval < 1e–04, NMBR: Wilcoxon pval < 1e–04) in LR versus SR samples (Figure 4C and D). Supplementary Figure S11 shows burden of somatic mutations between SR and LR samples for all genes across different cancer types. It was observed that for the genes with low PVE in the model, there was a relative and significant increase (FDR adjusted P-value < 0.05) in the mutation burden in the LR group, when compared to the high PVE group. Further, for genes which showed high somatic mutation burden in LR samples as compared to SR samples, we analyzed mutational signatures for somatic SNVs upto 10 kb upstream of TSS for genes, Although, no consistent difference in enrichment of signatures was observed between SR and LR groups, but some cancers e.g. lung (LUAD and LUSC) showed enrichment for smoking related signature; Signature 4 (Supplementary Figure S12). Further, integrating clinical data we noted that several genes, with significant mutation burden, had systematic difference in clinical outcome between samples that have large and small model residuals. For example, in case of ENTPD2 (LUAD) and TMC6 (SKCM), LR samples had poor survival compared to SR samples Figure 4E and F. But the number of samples was small, and information regarding molecular subtype and covariates was limited, so we cautiously interpret the results.
Figure 4.

(A) A schematic representation for analysis of somatic mutation burden in samples with small residuals (SR) versus large residual (LR) in low PVE genes. Genes with low explained variation in gene expression (D2 < 0.02; bottom 5 percentile) were prioritized, and for each gene, the samples were categorized into two groups; samples with SR (small model residual values) and LR (large residual values)—which were compared for somatic mutation burden in regulatory regions of the respective genes, changes in gene expression and clinical features. (B) Representative examples: for MUCL1 and NMBR genes (genes with low explained variance or low PVE in gene expression) the LR samples had high mutation burden as compared to the SR samples. Each row represents one sample in each of the two groups; red bars indicate somatic mutations in −10 kb and +1 kb of TSS in each sample. (C and D) Boxplot indicating significantly differential expression between SR and LR samples for MUCL1 in CESC and NMBR in SKCM. (E and F) KM plot indicating significant survival rate differences between SR and LR samples for ENTPD2 in LUAD and TMC6 in SKCM.
Detection of recurrent somatic mutations in regulatory regions of candidate genes
Recurrent regulatory alterations are expected to result in systematic variation in gene expression. Therefore, we prioritized those genes for which the model residuals had large variance and bimodal (or multimodal) patterns suggesting potential non-random (genetic or non-genetic) sources of variation. We present the pan cancer view of all genes that show bimodal patterns of residual in expression detected in the model in Supplementary Figure S13. A significant proportion of those cases (55 out of 138 genes) were also detected by an alternative approach (Gaussian distribution) (Supplementary Figure S14). To further prioritize the candidate genes for their clinical relevance, we overlaid clinical data and investigated whether their gene expression patterns were associated with metastasis status, the extent of regional lymph node involvement, treatment success, and survival. 52 genes showed significant Chi-square P-values for the above-mentioned parameters (Supplementary Table S5). One of such examples is SLC1A6 in BLCA. When we grouped the samples from the BLCA cancer cohort (n = 144) into two modal populations by the model residuals of SLC1A6 expression, they had significant difference in N0 status for the extent of spread to lymph node (Chi-square test FDR adjusted P-value < 5e–03) and survival (P-value < 1e–03, Supplementary Figure S15) so that the patients with large SLC1A6 expression residuals had greater spread to lymph node as well as consistently poor survival.
Next, overlaying mutation and genomic data, we asked whether such changes are frequently associated with common germ line SNPs or recurrent somatic mutations. Overlaying GTEx data, we found that 53% (86 out of 162 total unique genes across all cancers by both models) of the genes detected by somExVar (with significant bimodal patterns of model residuals) are associated with common SNPs with known, significant eQTL relationships. For instance, genes such as RPS28 (P-value < 4.0e–07), RPL9 (P-value < 2e–7), and ERAP2 (P-value < 3e–04) showed eQTL-linked expression variation (23). This observation demonstrates that somExVar rationally prioritizes the targets of genuine regulatory variations. We subsequently excluded all instances with known eQTLs while detecting candidate genes with somatic regulatory mutations. Supplementary Table S6 shows the details of all the genes that had a significant bimodal pattern in the distribution of residuals after excluding the eQTLs. For the subset of the samples where whole genome sequencing data was available (Supplementary Table S1), we scanned genomic regions for these genes up to ±1000 kb searching for somatic SNVs and InDels. The average mutation density in whole genome was ∼24 mutations/mb and 3.2 mutations/mb for substitutions and InDels respectively across the ten cancer types. Supplementary Figure S16 shows that mutation rate estimates for the promoter (10 kb upstream) regions are marginally lower than that estimated at the genome level, consistently across the cancer types. While transitions were more common in gene regions, transversions were more common in the upstream regions (Supplementary Figure S17). Further, mutation calls were prioritized based on their recurrence and regulatory impact on nearby gene. Towards this, we found 33 recurrent single nucleotide variations and 134 small InDels in 63 genes across all 10 cancers, which were predicted to have high regulatory potential (SNVs: Figure 5B, Indels: Supplementary Figure S18). List of prioritized SNVs and InDels is provided as Supplementary Table S7 and Supplementary Table S8 respectively. No additional candidates were found when alternate promoter sequences from Eukaryotic Promoter Database were considered.
Figure 5.

(A) A schematic representation for analyses of the genes that show bimodal pattern of model residuals indicating systematic patterns of expression variation among the samples in a cohort. Genes with high bimodal separation score for the model residual distribution were scanned for recurrent, variations with potential regulatory significance. (B) A plot showing total number of somatic regulatory variations (SNVs) identified in upstream regions of all the genes significant from somExVar (after removing eQTLs) across all cancers. Color represents score for potential regulatory impact (Red: high impact, Green: low impact). (C) Recurrent, potential regulatory mutations (SNVs and InDels) upstream of NKX2-1 in the HNSC samples disrupt the SP1/SP3 transcription factor binding site. (D) Boxplot showing significant gene expression changes for NKX2-1 in the samples carrying recurrent mutation at SP1/SP3 binding site in NKX2-1 promoter v/s those samples with no promoter mutations.
Integrating whole genome sequencing data, we detected recurrent somatic mutations with regulatory potential in the NKX2-1 promoter within 230bp upstream of the transcription start site in the HNSC cohort (Figure 5C). 25% (10/39) samples investigated had a somatic single nucleotide substitution and small InDels—two samples had SNV [chr14:36989660:A>C], while eight others had small 2–4 bp deletions [chr14: 36989645–36989649]. The genomic position was in the DNase hypersensitive region, overlapped with motifs for SP1, SP3, SP4, RREB1, SREBP, UF1H3BETA, CAC-binding protein, and KROX, and also carried ChIPSeq signal for RBBP5 and EZH2. SP3 is one of the known regulators of the NKX2-1 gene and can act as a repressor, thereby decreasing the expression of downstream genes. Samples with a mutation at this position disrupt the binding site for SP3, and thus had significantly higher expression compared to wild type (P-value = 0.03, Wilcoxon rank sum test), Figure 5D. According to our model, the inclusion of these somatic mutations led to a 7% increase in PVE for the expression level of the NKX2-1 gene. NKX2-1 is a thyroid specific transcription factor, which binds to the thyroglobulin promoter and regulates the expression of thyroid-specific genes (40). Also, it has been shown to play role in lung adenocarcinomas (41). Using a similar approach, we detected additional novel recurrent somatic mutations [chr19:48894669], SNVs and InDels, (in 11 samples of HNSC and 9 more samples across different cancers) upstream of the GRIN2D locus. The recurrent SNV overlaps with the binding motif for NFKB1, with the mutation disrupting the canonical motif. We also found small InDels at a recurrent position near the TLX2 gene in the HNSC cohort. It disrupted the predicted binding motif of the SP1 transcription factor, potentially altering TLX2 gene expression. Mutations in NKX2-1 and GRIN2D were predicted to have regulatory impact by both RegulomeDB (28), and Funseq2 (30), which highlight the utility of our approach to complement the classic top-down approach.
It was interesting to note that, even when recurrent regulatory mutations were associated with expression changes of target genes, in many cases, such mutations were not the only modulators of gene expression. For instance, SDHD expression was affected by promoter mutations in some samples and copy number alteration in some other samples (Supplementary Figure S4). Recently, Rheinbay et al. (14) reported genes, like LEPROTL1, FOXA1 and ALDOA1 etc. harboring regulatory mutations in their upstream regions with an effect on their gene expression. It is noteworthy that these genes, apart from having regulatory mutations, also harbor copy number alterations and missense mutations that are known to affect their gene expression levels (Supplementary Figure S19). somExVar could explain the variance in expression of these genes to a high degree (D2 = 0.60, 0.48 and 0.38 respectively), with SCNA as a major contributor (PVE by SCNA being 0.15–0.48 across three genes). Thus, for SDHD and other examples, point mutations are not the only modifiers of gene expression, and rather most of these genes experience expression changes via multiple mechanisms.
DISCUSSION
In this study, we present an integrative approach to identify the major sources of gene expression variation in cancer genomes, and identify candidates with large unexplained variation in gene expression after accounting for the effects of different modes of regulation such as copy number alterations, epigenetic changes, transcription factors, and miRNAs. Our initiative presents a rational strategy to prioritize targets for an investigation into potential regulatory alterations.
To provide a balanced perspective, we also note potential limitations of our analysis. First, we model gene expression using the Gamma distribution, which may not be an optimal choice for some genes. As single cell digital gene expression data becomes available from multiple tissue types, it might be possible to choose the model more optimally. Second, our model includes only CNAs, CpG methylation, transcription factors and miRNAs, but other factors such as chromatin modifications, enhancers, splice variations, gene fusion etc. which can also be important modifiers of gene expression, could not be included in the model for lack of appropriate data. Although we considered major cancer types separately (e.g. lung adeno- and squamous cell carcinoma) in many other cancers molecular bases of subtypes and their clinical relevance are unclear, and could not be explicitly considered in the model. But we did perform a subtype wise analysis for breast cancer, for which molecular subtypes are well defined, and showed that subtype-aware analysis may further improve the estimates of explained variance in gene expression in some cases. Nonetheless, our method was able to identify the key sources of expression variation in known cases such as BRCA1 and SDHD, and also those associated with known eQTLs. Ultimately our analysis identified novel targets of recurrent regulatory alterations (e.g. NKX2-1).
Our Pan-cancer analysis indicates that copy number alterations, epigenetic changes, and transcriptional and post-transcriptional regulatory factors collectively explain, on average, 18–26% expression variation at a genome-wide scale. The proportion of expression variation of known cancer genes explained by the above factors was higher at 31–38%. In both cases, copy number alteration had the highest effect size. It is likely that factors such as tumor subtype contribute to expression variation, and the effective PVE after adjusting for these confounders could actually be higher. Genes with large unexplained expression variance were enriched for cancer related pathways such as matrisome and GPCR signaling. Unlike the oncogenic mutations in coding regions, regulatory mutations (associated with gene expression) with base-pair level recurrence appear to be relatively less common in non-coding regions and had relatively small effect sizes. For instance, the recurrent SDHD promoter mutation increased the variation explained in the model only by 1%. Also, when incorporated in somExVar workflow, promoter mutations lead to 2–5% improvement in average PVE in general across different cancer types (Supplementary Figure S20 and Supplementary Table S9). Nonetheless, it appears that even for the genes with known regulatory mutations (e.g. SDHD, FOXA1 etc.), point mutations are not the only modifiers of gene expression, and rather most of these genes experience expression changes via multiple mechanisms. These findings are in line with the emerging concept (42–45) that, owing to plasticity and redundancy in biological networks, complex patterns of noncoding regulatory mutations and non-genetic regulatory changes might be common in cancer genomes.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank other members of De laboratories and the Center for Systems and Computational Biology at Rutgers Cancer Institute for helpful discussions. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Lung Cancer Research Foundation (sponsored by Elliot's Legacy and Joan's Legacy); Boettcher Foundation. Funding for open access charge: Start up fund of Dr De.
Conflict of interest statement. None declared.
REFERENCES
- 1. Khurana E., Fu Y., Chakravarty D., Demichelis F., Rubin M.A., Gerstein M.. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016; 17:93–108. [DOI] [PubMed] [Google Scholar]
- 2. Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A. Jr., Kinzler K.W.. Cancer genome landscapes. Science. 2013; 339:1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., Meyerson M., Gabriel S.B., Lander E.S., Getz G.. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. International Cancer Genome, C. Hudson T.J., Anderson W., Artez A., Barker A.D., Bell C., Bernabe R.R., Bhan M.K., Calvo F., Eerola I. et al. International network of cancer genome projects. Nature. 2010; 464:993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heidenreich B., Rachakonda P.S., Hemminki K., Kumar R.. TERT promoter mutations in cancer development. Curr. Opin. Genet. Dev. 2014; 24:30–37. [DOI] [PubMed] [Google Scholar]
- 6. Melton C., Reuter J.A., Spacek D.V., Snyder M.. Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat. Genet. 2015; 47:710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fredriksson N.J., NY L., Nilsson J.A., Larsson E.. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat. Genet. 2014; 46:1258–1263. [DOI] [PubMed] [Google Scholar]
- 8. Weinhold N., Jacobsen A., Schultz N., Sander C., Lee W.. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014; 46:1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith K.S., Yadav V.K., Pedersen B.S., Shaknovich R., Geraci M.W., Pollard K.S., De S.. Signatures of accelerated somatic evolution in gene promoters in multiple cancer types. Nucleic Acids Res. 2015; 43:5307–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang M.T., Asthana S., Gao S.P., Lee B.H., Chapman J.S., Kandoth C., Gao J., Socci N.D., Solit D.B., Olshen A.B. et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016; 34:155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feigin M.E., Garvin T., Bailey P., Waddell N., Chang D.K., Kelley D.R., Shuai S., Gallinger S., McPherson J.D., Grimmond S.M. et al. Recurrent noncoding regulatory mutations in pancreatic ductal adenocarcinoma. Nat. Genet. 2017; 49:825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Imielinski M., Guo G., Meyerson M.. Insertions and deletions target lineage-defining genes in human cancers. Cell. 2017; 168:460–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Juul M., Bertl J., Guo Q., Nielsen M.M., Switnicki M., Hornshoj H., Madsen T., Hobolth A., Pedersen J.S.. Non-coding cancer driver candidates identified with a sample- and position-specific model of the somatic mutation rate. Elife. 2017; 6:e21778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rheinbay E., Parasuraman P., Grimsby J., Tiao G., Engreitz J.M., Kim J., Lawrence M.S., Taylor-Weiner A., Rodriguez-Cuevas S., Rosenberg M. et al. Recurrent and functional regulatory mutations in breast cancer. Nature. 2017; 547:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fiziev P., Akdemir K.C., Miller J.P., Keung E.Z., Samant N.S., Sharma S., Natale C.A., Terranova C.J., Maitituoheti M., Amin S.B. et al. Systematic epigenomic analysis reveals chromatin states associated with melanoma progression. Cell Rep. 2017; 19:875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sumazin P., Yang X., Chiu H.S., Chung W.J., Iyer A., Llobet-Navas D., Rajbhandari P., Bansal M., Guarnieri P., Silva J. et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011; 147:370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hanahan D., Weinberg R.A.. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 18. Fleischer T., Edvardsen H., Solvang H.K., Daviaud C., Naume B., Borresen-Dale A.L., Kristensen V.N., Tost J.. Integrated analysis of high-resolution DNA methylation profiles, gene expression, germline genotypes and clinical end points in breast cancer patients. Int. J. Cancer. 2014; 134:2615–2625. [DOI] [PubMed] [Google Scholar]
- 19. Ng S., Collisson E.A., Sokolov A., Goldstein T., Gonzalez-Perez A., Lopez-Bigas N., Benz C., Haussler D., Stuart J.M.. PARADIGM-SHIFT predicts the function of mutations in multiple cancers using pathway impact analysis. Bioinformatics. 2012; 28:i640–i646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun Z., Asmann Y.W., Kalari K.R., Bot B., Eckel-Passow J.E., Baker T.R., Carr J.M., Khrebtukova I., Luo S., Zhang L. et al. Integrated analysis of gene expression, CpG island methylation, and gene copy number in breast cancer cells by deep sequencing. PLoS One. 2011; 6:e17490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thingholm L.B., Andersen L., Makalic E., Southey M.C., Thomassen M., Hansen L.L.. Strategies for integrated analysis of genetic, epigenetic, and gene expression variation in Cancer: Addressing the challenges. Front Genet. 2016; 7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chou C.H., Chang N.W., Shrestha S., Hsu S.D., Lin Y.L., Lee W.H., Yang C.D., Hong H.C., Wei T.Y., Tu S.J. et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016; 44:D239–D247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hebenstreit D., Fang M., Gu M., Charoensawan V., van Oudenaarden A., Teichmann S.A.. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol. Syst. Biol. 2011; 7:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Consortium, G.T., Laboratory, D.A., Coordinating Center -Analysis Working, G., Statistical Methods groups-Analysis Working, G., Enhancing, G.g., Fund, N.I.H.C., Nih/Nci, Nih/Nhgri, Nih/Nimh, Nih/Nida Genetic effects on gene expression across human tissues. Nature. 2017; 550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCall M.N., Illei P.B., Halushka M.K.. Complex sources of variation in tissue expression Data: Analysis of the GTEx lung transcriptome. Am. J. Hum. Genet. 2016; 99:624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cancer Genome Atlas Research, N. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The cancer genome atlas Pan-Cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012; 22:1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fu Y., Liu Z., Lou S., Bedford J., Mu X.J., Yip K.Y., Khurana E., Gerstein M.. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 2014; 15:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dreos R., Ambrosini G., Groux R., Cavin Perier R., Bucher P.. The eukaryotic promoter database in its 30th year: focus on non-vertebrate organisms. Nucleic Acids Res. 2017; 45:D51–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perini G., Diolaiti D., Porro A., Della Valle G.. In vivo transcriptional regulation of N-Myc target genes is controlled by E-box methylation. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:12117–12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walsh T., Casadei S., Coats K.H., Swisher E., Stray S.M., Higgins J., Roach K.C., Mandell J., Lee M.K., Ciernikova S. et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA. 2006; 295:1379–1388. [DOI] [PubMed] [Google Scholar]
- 34. Cancer Genome Atlas Research, N. The molecular taxonomy of primary prostate cancer. Cell. 2015; 163:1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kan Z., Jaiswal B.S., Stinson J., Janakiraman V., Bhatt D., Stern H.M., Yue P., Haverty P.M., Bourgon R., Zheng J. et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010; 466:869–873. [DOI] [PubMed] [Google Scholar]
- 36. Berger M.F., Lawrence M.S., Demichelis F., Drier Y., Cibulskis K., Sivachenko A.Y., Sboner A., Esgueva R., Pflueger D., Sougnez C. et al. The genomic complexity of primary human prostate cancer. Nature. 2011; 470:214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grasso C.S., Wu Y.M., Robinson D.R., Cao X., Dhanasekaran S.M., Khan A.P., Quist M.J., Jing X., Lonigro R.J., Brenner J.C. et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bonnans C., Chou J., Werb Z.. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014; 15:786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bar-Shavit R., Maoz M., Kancharla A., Nag J.K., Agranovich D., Grisaru-Granovsky S., Uziely B.. G protein-coupled receptors in cancer. Int. J. Mol. Sci. 2016; 17:E1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carre A., Szinnai G., Castanet M., Sura-Trueba S., Tron E., Broutin-L’Hermite I., Barat P., Goizet C., Lacombe D., Moutard M.L. et al. Five new TTF1/NKX2.1 mutations in brain-lung-thyroid syndrome: rescue by PAX8 synergism in one case. Hum. Mol. Genet. 2009; 18:2266–2276. [DOI] [PubMed] [Google Scholar]
- 41. Kang Y., Hebron H., Ozbun L., Mariano J., Minoo P., Jakowlew S.B.. Nkx2.1 transcription factor in lung cells and a transforming growth factor-beta1 heterozygous mouse model of lung carcinogenesis. Mol. Carcinog. 2004; 40:212–231. [DOI] [PubMed] [Google Scholar]
- 42. Araya C.L., Cenik C., Reuter J.A., Kiss G., Pande V.S., Snyder M.P., Greenleaf W.J.. Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations. Nat. Genet. 2016; 48:117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Colebatch A.J., Di Stefano L., Wong S.Q., Hannan R.D., Waring P.M., Dobrovic A., McArthur G.A., Papenfuss A.T.. Clustered somatic mutations are frequent in transcription factor binding motifs within proximal promoter regions in melanoma and other cutaneous malignancies. Oncotarget. 2016; 7:66569–66585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Denisova E., Heidenreich B., Nagore E., Rachakonda P.S., Hosen I., Akrap I., Traves V., Garcia-Casado Z., Lopez-Guerrero J.A., Requena C. et al. Frequent DPH3 promoter mutations in skin cancers. Oncotarget. 2015; 6:35922–35930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fredriksson N.J., Elliott K., Filges S., Van den Eynden J., Stahlberg A., Larsson E.. Recurrent promoter mutations in melanoma are defined by an extended context-specific mutational signature. PLoS Genet. 2017; 13:e1006773. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.