

Abstract
We explored arsenic-gene interactions influencing pancreatic beta-cell activity in the Strong Heart Family Study (SHFS). We considered 42 variants selected for associations with either beta-cell function (31 variants) or arsenic metabolism (11 variants) in the SHFS. Beta-cell function was calculated as homeostatic model - beta corrected for insulin resistance (cHOMA-B) by regressing homeostatic model - insulin resistance (HOMA-IR) on HOMA-B and adding mean HOMA-B. Arsenic exposure was dichotomized at the median of the sum of creatinine-corrected inorganic and organic arsenic species measured by high performance liquid chromatography-inductively coupled plasma mass spectrometry (HPLC-ICPMS). Additive GxE models for cHOMA-B were adjusted for age and ancestry, and accounted for family relationships. Models were stratified by center (Arizona, Oklahoma, North Dakota and South Dakota) and meta-analyzed. The two interactions between higher vs. lower arsenic and SNPs for cHOMA-B that were nominally significant at P < 0.05 were with rs10738708 (SNP overall effect −3.91, P = 0.56; interaction effect with arsenic −31.14, P = 0.02) and rs4607517 (SNP overall effect +16.61, P = 0.03; interaction effect with arsenic +27.02, P = 0.03). The corresponding genes GCK and TUSC1 suggest oxidative stress and apoptosis as possible mechanisms for arsenic impacts on beta-cell function. No interactions were Bonferroni-significant (1.16 × 10−3). Our findings are suggestive of oligogenic moderation of arsenic impacts on pancreatic β-cell endocrine function, but were not Bonferroni-significant.
Keywords: genetic epidemiology, environmental epidemiology, susceptibility, arsenicals, diabetes, pancreas
INTRODUCTION
Genetic determinants of pancreatic beta cells’ dysfunction and contribution to diabetes pathophysiology are drawing increasing attention (Lawlor et al., 2017). Arsenic is a potential predictor of diabetes (Maull et al., 2012) and related traits including pancreatic beta cell function (Grau-Perez et al., 2017), and has genome-wide epigenetic effects (Bailey and Fry, 2014) which may moderate genetic risk by affecting the regulation of genes relevant to diabetes pathogenesis (Martin et al., 2017). Genetic variation such as in the arsenic (III) methyltransferase gene influences arsenic metabolism (Wood et al., 2006), altering the dose available to exert toxic effects on the pancreas. Limited evidence exists regarding the combined influence of genetic variants and arsenic exposure on pancreatic function. The objective of this study was to explore the joint association of SNPs and arsenic exposure with diabetes-related traits in the Strong Heart Family Study (SHFS).
METHODS
Study Population
The Strong Heart Study [SHS] is a cohort of American Indians from participating tribes and communities in Arizona, Oklahoma, and North Dakota and South Dakota (Lee et al., 1990). Its genetic component, the SHFS, recruited from extended families of SHS participants (North et al., 2003). Details on participant recruitment and information obtained in clinical visits have been published (Lee et al., 1990; North et al., 2003). Baseline recruitment for this study was conducted in two phases [533 participants in 1998–99; 1,941 in 2001–03]. Follow-up visits were conducted in 2001–03, 2005–06 and 2014–15. Inclusion criteria for this analysis were urine arsenic measures, genetic data passing quality control, and absence of diabetes at baseline [n=1,923].
Protocols were approved by the Indian Health Service Institutional Review Board, Institutional Review Boards of the participating Institutions, and participating communities. One of the communities [n=488] recently opted out of participation in future studies and therefore were excluded from this analysis. All participants provided written informed consent and tribal consent.
Beta-Cell Function, Insulin Resistance, and Incident Diabetes
During follow-up, 256 participants developed incident diabetes defined as fasting blood glucose ≥ 6.99 mmol/L [126 mg/dL], or use of insulin or oral hypoglycemic medications at follow-up visits. Among participants without diabetes, we calculated the homeostatic model - beta corrected for insulin resistance (HOMA-B) using [20 * fasting insulin in mU/L]/[fasting glucose in mmol/L − 3.5], and homeostatic model – insulin resistance (HOMA-IR) [mmol/L] using [fasting insulin in mU/L * fasting glucose in mmol/L]/22.5 (Matthews et al., 1985). Since HOMA-B scores are difficult to interpret without taking HOMA-IR into account (Pfutzner et al., 2010; Yang et al., 2010), we regressed HOMA-B on HOMA-IR and added the mean HOMA-B to the model residual for interpretability based on Willett’s nutrient correction method (Willett and Stampfer, 1986). These scores calculated per the method of Willett and Stampfer were called corrected HOMA-B (cHOMA-B) and were used as the primary outcome (S1 Figure).
Measurement of Arsenic Exposure
Spot urine samples were collected in the morning of the baseline visit and frozen and stored at −70°C. Methodology of arsenic measurements have been detailed elsewhere (Scheer et al., 2012). Briefly, concentrations were determined using high performance liquid chromatography-inductively coupled plasma mass spectrometry [HPLC-ICPMS]. Arsenic species levels below limit of quantitation [0.10 μg/L] were imputed as the limit of quantitation divided by the square root of 2 [0.07 μg/L]. Concentrations of urine inorganic and methylated arsenic species were corrected for creatinine and the sum was taken as a biomarker of inorganic arsenic exposure.
Candidate SNPs
We selected 42 candidate SNPs based on associations with either cHOMA-B [31 SNPs, S1 Table] or arsenic metabolism [11 SNPs, S2 Table] (Balakrishnan et al., 2017) in the SHFS. Genotyping was done with DNA from blood collected at baseline using Illumina Infinium Cardio-Metabo DNA Analysis BeadChip [MetaboChip (Voight et al., 2012) supplemented with a Golden Gate genotyping panel (Illumina) containing 670 candidate SNPs for arsenic metabolism and toxicity. Family-based imputation of genotyped SNPs was done with a PEDSYS-compatible version of Merlin using human genome build 18 [NCBI36/hg18] and 1000 Genomes as the reference panel (Abecasis et al., 2002). Quality control of genetic data excluded participants with call rate <95%, outliers in identity-by-descent [IBD] clustering, or outliers in principal components analysis. We excluded SNPs with minor allele frequency [MAF] <1%, call rate <98%, that were not autosomal, that were not polymorphic, or that violated Hardy-Weinberg equilibrium [HWE] P < 10−5.
Statistical Analysis
We tested the hypotheses that there would be heterogeneous pancreatic β-cell function across persons with additive doses of the candidate SNPs and arsenic exposure dichotomized at the median [5.93 μg/L] using interaction models. Modeling interactions and describing subgroup effects is appropriate when there are differences in the effects of one variable (e.g., arsenic) according to another variable (e.g., SNPs) (Berrington de Gonzalez and Cox, 2007). The main outcome of interest was cHOMA-B. Additive genetic models were fit for each candidate SNP, adjusting for age and principal components of genome-wide markers. Models accounted for pedigree structures and stratified by center (Arizona, Oklahoma, and North/South Dakota). Center-specific associations were combined using inverse-variance-weighted meta-analysis. Center-specific analysis was done using SOLAR (Almasy and Blangero, 1998), and meta-analysis using METAL (Willer et al., 2010). We used a nominal P-value threshold of 0.05. The effect Bonferroni-corrected alpha was 1.16 × 10−3. Among variants with a significant meta-analyzed interaction effect, we also report the main genetic effect from the model without interaction terms. In secondary analyses, we considered log-transformed HOMA-IR scores and incident diabetes as outcomes. For incident diabetes, we performed sensitivity analyses where case definition included glycated hemoglobin thresholds, in addition to analyses excluding controls with impaired fasting glucose.
RESULTS
The median arsenic exposure among participants was 5.93 μg/L (IQR 3.56–9.96); 962 participants were at or below median arsenic exposure and 961 were above median. Median HOMA-B score was 155.2 (IQR 104.9–246) and after correction for HOMA-IR, median cHOMA-B was 186.3 [IQR 153.3–236.2] (Table 1). Among 42 candidate variants from 31 genes, we found suggestive interactions although no associations passed the Bonferroni-corrected significance threshold [S3-S5 Tables]. In particular, two SNPs had nominal interactions with arsenic exposure for c-HOMAB meta-analysis (Table 2): an intergenic SNP rs4607517 [G>A, MAF 0.26], localized to GCK and YKT6 on chromosome 7, and rs10738708 [A>C, MAF 0.43], localized to TUSC1.
Table 1.
Baseline Characteristics by Median Arsenic Exposure During Follow-Up
| At or Below Median (n=962) | Above Median (n=961) | Total (n=1,923) | P-value | |
|---|---|---|---|---|
| Mean age, yrs (SD) | 38.2 (16.1) | 34.7 (14.8) | 36.5 (15.6) | <0.01 |
| No. female (%) | 591 (61.4) | 566 (58.9) | 1,157 (60.2) | 0.26 |
| Center (%) | <0.01 | |||
| Arizona | 43 (4.5) | 155 (16.1) | 198 (10.3) | |
| Oklahoma | 466 (48.4) | 353 (36.7) | 819 (42.6) | |
| North/South Dakota | 453 (47.1) | 453 (47.1) | 906 (47.1) | |
| Median arsenic, μg/L (IQR) | 3.6 (2.4–4.7) | 10.0 (7.6–15.0) | 5.9 (3.6–10.0) | <0.01 |
| Mean BMI, kg/m2 (SD) | 29.8 (6.9) | 31.0 (7.7) | 30.4 (7.3) | <0.01 |
| Mean fasting glucose, mmol/L (SD) | 93.2 (9.8) | 93.6 (10.7) | 93.4 (10.3) | 0.39 |
| Mean fasting insulin, pmol/L (SD) | 15.6 (15.5) | 17.6 (17.2) | 16.6 (16.4) | <0.01 |
| Median HOMA-IR (IQR) | 2.6 (1.6–4.4) | 2.9 (1.8–4.8) | 2.7 (1.7–4.6) | <0.01 |
| Median HOMA-B (IQR) | 148.4 (100.4–224.2) | 165.5 (107.9–264.7) | 155.2 (104.9–246) | <0.01 |
| Median cHOMA-Ba (IQR) | 184.7 (153.3–230.1) | 187.3 (153.4–240.7) | 186.3 (153.3–236.2) | 0.35 |
Abbreviations: BMI, body mass index; HOMA-B, homeostatic model assessment-beta cell function; HOMA-IR, homeostatic model assessment-insulin resistance; yrs, years.
Corrected HOMA-B; Sum of residuals adjusted for HOMA-IR and mean HOMA-B.
Table 2.
Strongest Interactions of Candidate SNPs with Arsenic for cHOMA-B.
| SNP rsID | Chr | Position | Genes | Alleles | MAF | SNP effect among persons with low arsenic | Arsenic effect among major allele homozygote | Interaction | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Beta | P-value | Beta | P-value | Beta | P-value | ||||||
| rs4607517 | 7 | 44202193 | GCK, YKT6 | G/A | 0.26 | −35.90 | 2.59e-3 | −6.10 | 0.50 | +27.02 | 0.03 |
| rs10738708 | 9 | 25231316 | TUSC1 | A/C | 0.43 | +30.47 | 2.74e-3 | +35.48 | 0.02 | −31.14 | 0.02 |
Abbreviations: Chr, chromosome; cHOMA-B, corrected homeostatic model assessment-beta cell function; MAF, minor allele frequency; SNP, single nucleotide polymorphism.
The “Beta” in table refers to estimated difference in means. The notation “e-3” is concise for “x 10−3 ”.
Among participants with lower than median arsenic exposure, each copy of allele A of rs4607517 was associated with a decrease in cHOMA-B of 35.9 [P 2.59 × 10−3]. Among participants with higher than median arsenic exposure, the genotype G/G was associated with a decrease in cHOMA-B of 6.1, G/A with a decrease of 14.98, and A/A with a decrease of 23.86. Stratified by center, the expected effect for cHOMA-B scores below median arsenic exposure for allele A was −31.76 for Arizona, −50.59 for Oklahoma, and −32.27 for North/South Dakota (Figure 1, Panel A; S3 Table). The difference in expected cHOMA-B scores comparing above vs. below median arsenic exposure with genotype G/G was +11.94 for Arizona, −22.83 for Oklahoma, and −32.58 for North/South Dakota; with genotype G/A was +29.93, +10.36, and +3.96; and with genotype A/A was +47.92, +43.55, and +40.50 [Figure 1, Panel B; S3 Table]. The overall association of rs4607517 with cHOMA-B, without considering arsenic, was +16.61 per increasing dose of G allele (P = 0.03).
Figure 1. Predicted cHOMA-B by rs4607517 Genotype.
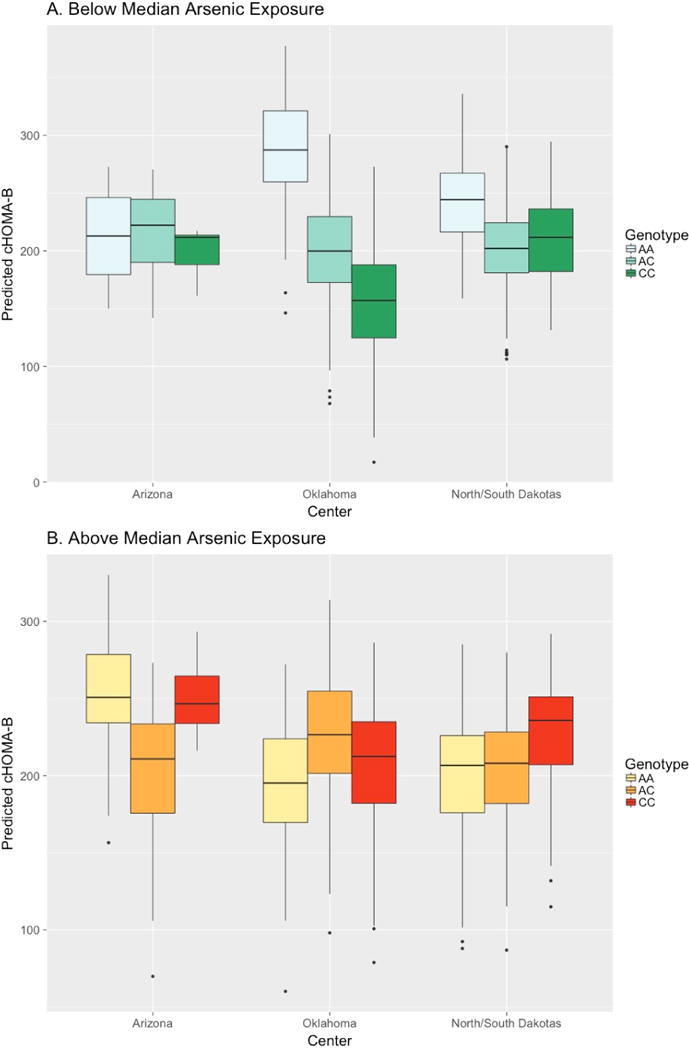
Expected effect by center for cHOMA-B scores of rs4607517 genotypes (G>A, MAF 0.26) for arsenic exposure below (Panel A) and above (Panel B) median.
For rs10738708, each copy of allele C was associated with an increase in cHOMA-B of 30.47 [P = 2.74 × 10−3] among participants exposed to below median arsenic. Among participants exposed to above median arsenic, genotype AA of rs10738708 was associated with an increase in cHOMA-B of 35.48, AC with an increase of 34.81, and CC with an increase of 34.14. By center, the expected effect for cHOMA-B scores below median arsenic exposure for allele A was −33.20 for Arizona, −60.38 for Oklahoma, and −21.37 for North/South Dakota [Figure 2, Panel A; S3 Table]. The difference in expected cHOMA-B scores comparing above vs. below median arsenic exposure with genotype G/G was −6.49 for Arizona, −67.22 for Oklahoma, and −39.14 for North/South Dakotas; with genotype G/A was +8.44, −7.66, and −9.86; and with genotype A/A was +23.37, +51.90, and +19.42 (Figure 2, Panel B; S3 Table). The overall association of rs10738708 with cHOMA-B, without considering arsenic, was −3.91 per increasing dose of C allele (P = 0.56).
Figure 2. Predicted cHOMA-B by rs10738708 Genotype.
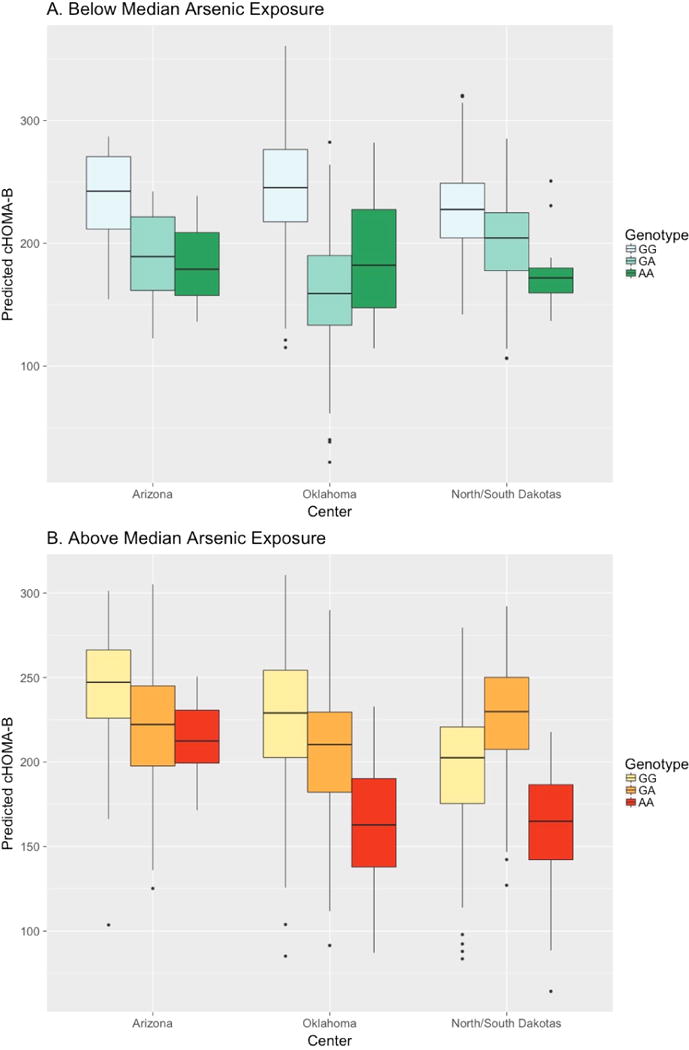
Expected effect by center for cHOMA-B scores of rs10738708 genotypes (A>C, MAF 0.43) for arsenic exposure below (Panel A) and above (Panel B) median.
There were two interactions in Arizona and one in the Dakotas that reached nominal significance. In Arizona, rs4139 [A>G, MAF 0.35] localized to FSTL5 on chromosome 4 and associated with antagonistic interaction effect of +53.44 [P = 0.03] [S3 Table]. In addition, rs3740393 [G>C, MAF 0.21], an index SNP for arsenic metabolism localized to AS3MT on chromosome 10, had an antagonistic interaction effect of −90.00 [P = 0.01] [S3 Table]. In North and South Dakota, there was a nominal antagonistic interaction association for the intergenic SNP rs12642615 [G>A, MAF 0.38] located between FLJ16686 and KIAA1239 [32.94, P = 0.05].
The interactions between arsenic and SNPs for HOMA-IR scores [S4 Table] and for incident diabetes [S5 Table] were not Bonferroni-significant.
DISCUSSION
We identified several suggestive interactions between arsenic exposure and genetic variants for beta-cell function, although none were significant after accounting for multiple comparisons. We highlight two variants, rs4607517 [GCK, YKT6] and rs10738708 [TUSC1], that had suggestive interactions and discuss possible mechanisms below.
The intergenic SNP rs4607517 had an additive allelic effect on cHOMA-B among persons with low arsenic of −35.9 [P = 2.59 × 10−3] and an antagonistic interactive effect (difference in effect for persons with high arsenic) of +27.2 [P = 0.03]. Located between GCK and YKT6, genetic variation in rs4607517 has been associated with BMI and fasting glucose (Dupuis et al., 2010; Manning et al., 2012). Human and animal studies have shown that GCK is highly correlated to glucose metabolism, i.e., it is responsible for upregulating glucose-6-phosphatase production via hexokinase in pancreatic beta cells (Osbak et al., 2009). Other variants involved in pancreas development and insulin secretion adversely influence the response to arsenic exposure, with oxidative stress and apoptosis or other cell death mechanisms being top candidate pathways (Hectors et al., 2011).
We found an antagonistic interaction between rs10738708, upstream of TUSC1, and arsenic exposure on cHOMA-B although the estimated effects vary by center. The influence of TUSC1 on pancreatic beta cells or arsenic metabolism is not well established. One possible explanation for this finding relates to apoptosis and the rate of cell growth. When TUSC1 is introduced, tumor cell lines grow slower (Shan et al., 2013); perhaps it also has a role in beta cell development and proliferation. Research in hepatocellular carcinoma cell lines and surgical samples (paired cancer and non-cancer) has suggested that DNA methylation is likely important for regulation of TUSC1 (Shimizu et al., 2014). The primary rationale for studying arsenic interactions with genes in this study was in part that arsenic is known to affect genome-wide epigenetics (Bailey and Fry, 2014). In this study, among the participants with higher arsenic there were less dramatic differences by TUSC1 genotype in cHOMA-B, especially in Oklahoma (Figure 2). However, especially given the limited prior evidence for TUSC1 in diabetes pathophysiology, and the lack of statistical significance, these findings are tentative.
Our study has several strengths. First, the SHFS has rigorous and consistent data collection among communities affected by both diabetes and arsenic. Second, the complex pedigrees provide increased power to detect genetic effects and consequently interaction effects. Third, correcting HOMA-B scores for insulin resistance allows for a more independent, accurate investigation of beta-cell function compared to raw HOMA-B scores (Pfutzner et al., 2010; Yang et al., 2010). Lastly, although exposure to arsenic is low to moderate, the distribution of arsenic exposure across various centers provides a heterogeneous sample to assess GxE associations.
Our study has limitations. This cross-sectional analysis cannot establish the cause- and-effect relation of the combined influence of genetic variants and arsenic exposure on beta-cell function. Another limitation was the number of participants that developed diabetes over follow-up as well as the number of participants in each genotypic subgroup. To understand the effect of arsenic exposure among different genotypic subgroups, either larger sample sizes or experimental trials reducing exposure to arsenic are required. Finally, we cannot rule out the possibility of unmeasured confounders rather than genotypic subgroups within arsenic exposure categories accounting for observed differences in beta-cell function. for example, if there are other environmental exposures differences that correlate with SNP allele frequencies or with arsenic across these populations, some of the interaction attributed to these SNPs and arsenic could be due to or masked by unmeasured environmental variables. The observed heterogeneity in associations therefore might reflect confounding of the interacting variables. However, it is also possible that unmeasured variables differing between sites, including genetic features, could be higher-order effect modifiers if they impact expression of TUSC1, GCK, YKT6 or other biologically important genes, or the biologically effective dose of arsenic.
These exploratory associations may be consistent with oligogenic moderation of arsenic pancreatic endocrine function, but the lack of Bonferroni-corrected statistical significance precludes strong inferences. Since we used a candidate SNP approach focused on common variants, we did not assess the role of rare variation within these pathways or the many other pathways by which arsenic and genes hypothetically could jointly impact pancreatic beta-cell function. Replication in other epidemiological studies and validation through toxicological experiments would strengthen the tentative findings that oxidative stress and apoptosis could relevant be toxicological mechanisms by which arsenic may impact pancreatic function.
Supplementary Material
Highlights.
Genetic variants near tumor suppressor TUSC1 may alter arsenic pancreatic toxicity.
Genetic variation near glucokinase GCK and secretory protein YKT6 genes may alter arsenic pancreatic toxicity.
Additional research on apoptosis and oxidative stress mechanisms is warranted.
Acknowledgments
The authors would like to thank the participants and staff of the SHS and the SHFS for their important contributions. MOG is guarantor of the manuscript.
FUNDING
This work was funded by the National Institute of Environmental Health Sciences (NIEHS) (R01ES021367, 1R01ES025216, 5P30ES009089, P30ES019776, 5T32ES007141, and P42ES010349) and the National Heart Lung and Blood Institute (NHLBI) grants supporting the Strong Heart Study: U01-HL41642, U01-HL41652, U01-HL41654, U01-HL65520, U01-HL65521, R01-HL109315, R01HL109301, R01HL109284, R01HL109282 and R01HL109319).
Footnotes
DESCRIPTION OF SUPPLEMENTAL DATA
Supplemental data contain 5 tables.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin - rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey K, Fry R. Arsenic-Associated Changes to the Epigenome: What Are the Functional Consequences? Curr Envir Health Rpt. 2014;1:22–34. doi: 10.1007/s40572-013-0002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan P, Vaidya D, Franceschini N, Voruganti VS, Gribble MO, Haack K, Laston S, Umans JG, Francesconi KA, Goessler W, North KE, Lee E, Yracheta J, Best LG, MacCluer JW, Kent J, Cole SA, Navas-Acien A. Association of Cardiometabolic Genes with Arsenic Metabolism Biomarkers in American Indian Communities: The Strong Heart Family Study (SHFS) Environ Health Perspect. 2017;125:15–22. doi: 10.1289/EHP251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrington de Gonzalez A, Cox DR. Interpretation of Interaction: A Review. The Annals of Applied Statistics. 2007;1:371–385. [Google Scholar]
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, Lindgren CM, Magi R, Morris AP, Randall J, Johnson T, Elliott P, Rybin D, Thorleifsson G, Steinthorsdottir V, Henneman P, Grallert H, Dehghan A, Hottenga JJ, Franklin CS, Navarro P, Song K, Goel A, Perry JR, Egan JM, Lajunen T, Grarup N, Sparso T, Doney A, Voight BF, Stringham HM, Li M, Kanoni S, Shrader P, Cavalcanti-Proenca C, Kumari M, Qi L, Timpson NJ, Gieger C, Zabena C, Rocheleau G, Ingelsson E, An P, O’Connell J, Luan J, Elliott A, McCarroll SA, Payne F, Roccasecca RM, Pattou F, Sethupathy P, Ardlie K, Ariyurek Y, Balkau B, Barter P, Beilby JP, Ben-Shlomo Y, Benediktsson R, Bennett AJ, Bergmann S, Bochud M, Boerwinkle E, Bonnefond A, Bonnycastle LL, Borch-Johnsen K, Bottcher Y, Brunner E, Bumpstead SJ, Charpentier G, Chen YD, Chines P, Clarke R, Coin LJ, Cooper MN, Cornelis M, Crawford G, Crisponi L, Day IN, de Geus EJ, Delplanque J, Dina C, Erdos MR, Fedson AC, Fischer-Rosinsky A, Forouhi NG, Fox CS, Frants R, Franzosi MG, Galan P, Goodarzi MO, Graessler J, Groves CJ, Grundy S, Gwilliam R, Gyllensten U, Hadjadj S, Hallmans G, Hammond N, Han X, Hartikainen AL, Hassanali N, Hayward C, Heath SC, Hercberg S, Herder C, Hicks AA, Hillman DR, Hingorani AD, Hofman A, Hui J, Hung J, Isomaa B, Johnson PR, Jorgensen T, Jula A, Kaakinen M, Kaprio J, Kesaniemi YA, Kivimaki M, Knight B, Koskinen S, Kovacs P, Kyvik KO, Lathrop GM, Lawlor DA, Le Bacquer O, Lecoeur C, Li Y, Lyssenko V, Mahley R, Mangino M, Manning AK, Martinez-Larrad MT, McAteer JB, McCulloch LJ, McPherson R, Meisinger C, Melzer D, Meyre D, Mitchell BD, Morken MA, Mukherjee S, Naitza S, Narisu N, Neville MJ, Oostra BA, Orru M, Pakyz R, Palmer CN, Paolisso G, Pattaro C, Pearson D, Peden JF, Pedersen NL, Perola M, Pfeiffer AF, Pichler I, Polasek O, Posthuma D, Potter SC, Pouta A, Province MA, Psaty BM, Rathmann W, Rayner NW, Rice K, Ripatti S, Rivadeneira F, Roden M, Rolandsson O, Sandbaek A, Sandhu M, Sanna S, Sayer AA, Scheet P, Scott LJ, Seedorf U, Sharp SJ, Shields B, Sigurethsson G, Sijbrands EJ, Silveira A, Simpson L, Singleton A, Smith NL, Sovio U, Swift A, Syddall H, Syvanen AC, Tanaka T, Thorand B, Tichet J, Tonjes A, Tuomi T, Uitterlinden AG, van Dijk KW, van Hoek M, Varma D, Visvikis-Siest S, Vitart V, Vogelzangs N, Waeber G, Wagner PJ, Walley A, Walters GB, Ward KL, Watkins H, Weedon MN, Wild SH, Willemsen G, Witteman JC, Yarnell JW, Zeggini E, Zelenika D, Zethelius B, Zhai G, Zhao JH, Zillikens MC, Borecki IB, Loos RJ, Meneton P, Magnusson PK, Nathan DM, Williams GH, Hattersley AT, Silander K, Salomaa V, Smith GD, Bornstein SR, Schwarz P, Spranger J, Karpe F, Shuldiner AR, Cooper C, Dedoussis GV, Serrano-Rios M, Morris AD, Lind L, Palmer LJ, Hu FB, Franks PW, Ebrahim S, Marmot M, Kao WH, Pankow JS, Sampson MJ, Kuusisto J, Laakso M, Hansen T, Pedersen O, Pramstaller PP, Wichmann HE, Illig T, Rudan I, Wright AF, Stumvoll M, Campbell H, Wilson JF, Bergman RN, Buchanan TA, Collins FS, Mohlke KL, Tuomilehto J, Valle TT, Altshuler D, Rotter JI, Siscovick DS, Penninx BW, Boomsma DI, Deloukas P, Spector TD, Frayling TM, Ferrucci L, Kong A, Thorsteinsdottir U, Stefansson K, van Duijn CM, Aulchenko YS, Cao A, Scuteri A, Schlessinger D, Uda M, Ruokonen A, Jarvelin MR, Waterworth DM, Vollenweider P, Peltonen L, Mooser V, Abecasis GR, Wareham NJ, Sladek R, Froguel P, Watanabe RM, Meigs JB, Groop L, Boehnke M, McCarthy MI, Florez JC, Barroso I. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau-Perez M, Kuo CC, Gribble MO, Balakrishnan P, Jones Spratlen M, Vaidya D, Francesconi KA, Goessler W, Guallar E, Silbergeld EK, Umans JG, Best LG, Lee ET, Howard BV, Cole SA, Navas-Acien A. Association of Low-Moderate Arsenic Exposure and Arsenic Metabolism with Incident Diabetes and Insulin Resistance in the Strong Heart Family Study. Environ Health Perspect. 2017;125:127004. doi: 10.1289/EHP2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hectors TL, Vanparys C, van der Ven K, Martens GA, Jorens PG, Van Gaal LF, Covaci A, De Coen W, Blust R. Environmental pollutants and type 2 diabetes: a review of mechanisms that can disrupt beta cell function. Diabetologia. 2011;54:1273–1290. doi: 10.1007/s00125-011-2109-5. [DOI] [PubMed] [Google Scholar]
- Lawlor N, Khetan S, Ucar D, Stitzel ML. Genomics of Islet (Dys)function and Type 2 Diabetes. Trends Genet. 2017;33:244–255. doi: 10.1016/j.tig.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ET, Welty TK, Fabsitz R, Cowan LD, Le NA, Oopik AJ, Cucchiara AJ, Savage PJ, Howard BV. The Strong Heart Study. A study of cardiovascular disease in American Indians: design and methods. Am J Epidemiol. 1990;132:1141–1155. doi: 10.1093/oxfordjournals.aje.a115757. [DOI] [PubMed] [Google Scholar]
- Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, Rybin D, Liu CT, Bielak LF, Prokopenko I, Amin N, Barnes D, Cadby G, Hottenga JJ, Ingelsson E, Jackson AU, Johnson T, Kanoni S, Ladenvall C, Lagou V, Lahti J, Lecoeur C, Liu Y, Martinez-Larrad MT, Montasser ME, Navarro P, Perry JR, Rasmussen-Torvik LJ, Salo P, Sattar N, Shungin D, Strawbridge RJ, Tanaka T, van Duijn CM, An P, de Andrade M, Andrews JS, Aspelund T, Atalay M, Aulchenko Y, Balkau B, Bandinelli S, Beckmann JS, Beilby JP, Bellis C, Bergman RN, Blangero J, Boban M, Boehnke M, Boerwinkle E, Bonnycastle LL, Boomsma DI, Borecki IB, Bottcher Y, Bouchard C, Brunner E, Budimir D, Campbell H, Carlson O, Chines PS, Clarke R, Collins FS, Corbaton-Anchuelo A, Couper D, de Faire U, Dedoussis GV, Deloukas P, Dimitriou M, Egan JM, Eiriksdottir G, Erdos MR, Eriksson JG, Eury E, Ferrucci L, Ford I, Forouhi NG, Fox CS, Franzosi MG, Franks PW, Frayling TM, Froguel P, Galan P, de Geus E, Gigante B, Glazer NL, Goel A, Groop L, Gudnason V, Hallmans G, Hamsten A, Hansson O, Harris TB, Hayward C, Heath S, Hercberg S, Hicks AA, Hingorani A, Hofman A, Hui J, Hung J, Jarvelin MR, Jhun MA, Johnson PC, Jukema JW, Jula A, Kao WH, Kaprio J, Kardia SL, Keinanen-Kiukaanniemi S, Kivimaki M, Kolcic I, Kovacs P, Kumari M, Kuusisto J, Kyvik KO, Laakso M, Lakka T, Lannfelt L, Lathrop GM, Launer LJ, Leander K, Li G, Lind L, Lindstrom J, Lobbens S, Loos RJ, Luan J, Lyssenko V, Magi R, Magnusson PK, Marmot M, Meneton P, Mohlke KL, Mooser V, Morken MA, Miljkovic I, Narisu N, O'Connell J, Ong KK, Oostra BA, Palmer LJ, Palotie A, Pankow JS, Peden JF, Pedersen NL, Pehlic M, Peltonen L, Penninx B, Pericic M, Perola M, Perusse L, Peyser PA, Polasek O, Pramstaller PP, Province MA, Raikkonen K, Rauramaa R, Rehnberg E, Rice K, Rotter JI, Rudan I, Ruokonen A, Saaristo T, Sabater-Lleal M, Salomaa V, Savage DB, Saxena R, Schwarz P, Seedorf U, Sennblad B, Serrano-Rios M, Shuldiner AR, Sijbrands EJ, Siscovick DS, Smit JH, Small KS, Smith NL, Smith AV, Stancakova A, Stirrups K, Stumvoll M, Sun YV, Swift AJ, Tonjes A, Tuomilehto J, Trompet S, Uitterlinden AG, Uusitupa M, Vikstrom M, Vitart V, Vohl MC, Voight BF, Vollenweider P, Waeber G, Waterworth DM, Watkins H, Wheeler E, Widen E, Wild SH, Willems SM, Willemsen G, Wilson JF, Witteman JC, Wright AF, Yaghootkar H, Zelenika D, Zemunik T, Zgaga L, Wareham NJ, McCarthy MI, Barroso I, Watanabe RM, Florez JC, Dupuis J, Meigs JB, Langenberg C. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–669. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EM, Styblo M, Fry RC. Genetic and epigenetic mechanisms underlying arsenic-associated diabetes mellitus: a perspective of the current evidence. Epigenomics. 2017;9:701–710. doi: 10.2217/epi-2016-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, Pi J, Silbergeld EK, Styblo M, Tseng CH, Thayer KA, Loomis D. Evaluation of the association between arsenic and diabetes: a National Toxicology Program workshop review. Environ Health Perspect. 2012;120:1658–1670. doi: 10.1289/ehp.1104579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North KE, Howard BV, Welty TK, Best LG, Lee ET, Yeh JL, Fabsitz RR, Roman MJ, MacCluer JW. Genetic and environmental contributions to cardiovascular disease risk in American Indians: the Strong Heart Family Study. Am J Epidemiol. 2003;157:303–314. doi: 10.1093/aje/kwf208. [DOI] [PubMed] [Google Scholar]
- Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanne-Chantelot C, Ellard S, Gloyn AL. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30:1512–1526. doi: 10.1002/humu.21110. [DOI] [PubMed] [Google Scholar]
- Pfutzner A, Derwahl M, Jacob S, Hohberg C, Blumner E, Lehmann U, Fuchs W, Forst T. Limitations of the HOMA-B score for assessment of beta-cell functionality in interventional trials-results from the PIOglim study. Diabetes Technology & Therapeutics. 2010;12:599–604. doi: 10.1089/dia.2010.0019. [DOI] [PubMed] [Google Scholar]
- Scheer J, Findenig S, Goessler W, Francesconi K, Howard B, Umans J, Pollak J, Tellez-Plaza M, Silbergeld E, Guallar E, Navas-Acien A. Arsenic species and selected metals in human urine: validation of HPLC/ICPMS and ICPMS procedures for a long-term population-based epidemiological study. Analytical Methods. 2012;4:406–413. doi: 10.1039/C2AY05638K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Z, Shakoori A, Bodaghi S, Goldsmith P, Jin J, Wiest JS. TUSC1, a putative tumor suppressor gene, reduces tumor cell growth in vitro and tumor growth in vivo. PLoS One. 2013;8:e66114. doi: 10.1371/journal.pone.0066114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu D, Kanda M, Nomoto S, Oya H, Takami H, Hibino S, Suenaga M, Inokawa Y, Hishida M, Takano N, Nishikawa Y, Yamada S, Fujii T, Nakayama G, Sugimoto H, Koike M, Fujiwara M, Kodera Y. Identification of intragenic methylation in the TUSC1 gene as a novel prognostic marker of hepatocellular carcinoma. Oncol Rep. 2014;31:1305–1313. doi: 10.3892/or.2013.2939. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kang HM, Ding J, Palmer CD, Sidore C, Chines PS, Burtt NP, Fuchsberger C, Li Y, Erdmann J, Frayling TM, Heid IM, Jackson AU, Johnson T, Kilpelainen TO, Lindgren CM, Morris AP, Prokopenko I, Randall JC, Saxena R, Soranzo N, Speliotes EK, Teslovich TM, Wheeler E, Maguire J, Parkin M, Potter S, Rayner NW, Robertson N, Stirrups K, Winckler W, Sanna S, Mulas A, Nagaraja R, Cucca F, Barroso I, Deloukas P, Loos RJ, Kathiresan S, Munroe PB, Newton-Cheh C, Pfeufer A, Samani NJ, Schunkert H, Hirschhorn JN, Altshuler D, McCarthy MI, Abecasis GR, Boehnke M. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793. doi: 10.1371/journal.pgen.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett W, Stampfer MJ. Total energy intake: implications for epidemiologic analyses. Am J Epidemiol. 1986;124:17–27. doi: 10.1093/oxfordjournals.aje.a114366. [DOI] [PubMed] [Google Scholar]
- Wood TC, Salavagionne OE, Mukherjee B, Wang L, Klumpp AF, Thomae BA, Eckloff BW, Schaid DJ, Wieben ED, Weinshilboum RM. Human arsenic methyltransferase (AS3MT) pharmacogenetics: gene resequencing and functional genomics studies. J Biol Chem. 2006;281:7364–7373. doi: 10.1074/jbc.M512227200. [DOI] [PubMed] [Google Scholar]
- Yang Q, Liu T, Shrader P, Yesupriya A, Chang MH, Dowling NF, Ned RM, Dupuis J, Florez JC, Khoury MJ, Meigs JB. Racial/ethnic differences in association of fasting glucose-associated genomic loci with fasting glucose, HOMA-B, and impaired fasting glucose in the U.S. adult population. Diabetes Care. 2010;33:2370–2377. doi: 10.2337/dc10-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.