

Abstract
In this work, the dissolution profiles of nine meloxicam tablet brands marketed in Argentina have been evaluated. As meloxicam is a Class 2 Biopharmaceutical Classification System (BSC) drug, interchangeability between commercial products must be demonstrated through in vivo bioequivalence studies. However, in our country, such studies remain to be performed.
Dissolution studies have been performed according to USP 38 and evaluated by fitting experimental data to the zero and first-order, the Hixson-Crowell, the Higuchi, and the Weibull model-dependent methods. To test the pertinence of these release models, the Akaike Information Criteria (AIC) were used.
All brands satisfied the dissolution profiles (phosphate buffer, pH 7.5) established in the USP. The comparison between the dissolution profiles was carried out by model-dependent and model-independent methods. The Weibull model provided the best kinetic curve adjustment. Brands I, II, IV and VI had the best fitting, with the maximum determination coefficient and the smallest AIC values. Model-independent methods included ratio test and the fit factors. The Dissolution Efficiency (DE) and Mean Dissolution Time (MDT) were analysed with ANOVA and the DGC method. In both cases, brand I did not show similarity with the rest of the brands. Using fit factors, only brands I, II and V were similar to each other.
Significant differences were found among the in vitro dissolution profiles of meloxicam tablets belonging to the nine brands. As meloxicam is a class 2 BCS drug, interchangeability between commercial products must be demonstrated through in vivo bioequivalence studies. However, in Argentina, such studies remain to be performed. Our results demonstrate that caution must be exercised as regards interchangeability of generic products.
Keywords: Meloxicam, Tablets, Dissolution profiles, Commercial products, Model-dependent method, Model-independent method
1. Introduction
It is known that low water solubility causes a decrease in the release rate of a drug. Thus, the formulation of oral delivery forms of these drugs is a permanent challenge in the pharmaceutical industry. In many cases, the dissolution rate of these drugs is a limiting factor affecting the therapeutic activity. To enhance dissolution several techniques have been developed, such as salt formation, and the development of inclusion complexes and pro-drugs (Jafar et al., 2010, Noolkar et al., 2013).
Three categories of dissolution test specifications for immediate release products are described in the guide provided by the Centre for Drug Evaluation and Research at the Food and Drug Administration (Food and Drug Administration, 1997): (a) single-point specifications, (b) two-points specifications and (c) dissolution profile comparison. Although this point estimate approach is suitable for drug products containing API with high solubility-high permeability, it may not be adequate for low solubility drugs or for products with modified release characteristics. According to the guide: “for highly soluble and rapidly dissolving drug products (BCS classes 1 and 3), a single-point dissolution test specification of 85% (Q = 80%) in 60 min or less is sufficient as a routine quality control test for batch-to-batch uniformity”. For class 2 BSC APIs, it is recommended that a two-point dissolution specification be established, one at 15 min and the other at 30, 45, or 60 min, in order to ensure 85% dissolution (Food and Drug Administration,1997).
Although weight, hardness, content uniformity, friability and disintegration are tests often employed for the analysis of an immediate release solid dosage form, the most important are the quantitation of the API, the determination of impurities and the dissolution test (Al Ameri et al., 2012). Once the dissolution specifications are set for a new drug, based on availability studies, such specifications become official for the development of all subsequent drug products having the same active ingredients. According to good laboratory practices, dissolution is considered one of the most important tests that is to be performed on a solid pharmaceutical dosage form (Al Ameri et al., 2012). This test is also used to control stability of the drug product and batch-to-batch consistency (Food and Drug Administration, 1997, Administración Nacional de Medicamentos, Alimentos y Tecnología Médica, 1999).
Economic reasons account for the widespread use of generic medicines. Thus, in vivo bioequivalence studies are necessary to ensure the existence of therapeutic equivalence between the generic medicine and the original drug product containing the same active drug, except when the drug is highly soluble and highly permeable, i.e., class I BCS compounds.
In vitro dissolution profiles can be compared by three groups of methods: (A) methods based on analysis of variance, (B) model-dependent methods, (C) model-independent methods. Dissolution data can either be analysed in their crude form or they can be transformed using simple ANOVA-based methods. The ANOVA is a useful method to detect differences between the profiles in level and shape (Polli et al., 1997, Yuksel et al., 2000, Costa and Souza Lobo, 2001, Adams et al., 2001, LeBlond et al., 2016).
Model-dependent methods are based on an appropriate curve fitting procedure: the zero and first-order, the Hixson-Crowell, the Higuchi, the quadratic, the Weinbull, the Gompertz and the logistic (Yuksel et al., 2000, Costa et al., 2003, Khan et al., 2013). Model-dependent methods explore the mathematical equations governing the liberation profile as a function of certain parameters related to the pharmaceutical dosage form. These models allow an easy quantitative interpretation of data. These methods are always applied in the formulation-development stage.
Model-independent methods generate a single value from a dissolution profile, thus allowing data to be compared directly. Model-independent methods include fit factors and ratio tests. Ratio tests determine, at the same sampling time, the percentage of drug dissolved, the mean dissolution time (MDT) and the dissolution efficiency percentage (DE) of the test formulation in relation with a reference formulation (Khan and Rhodes, 1972, Aguilar et al., 2008). Fit factors are known as the difference factor (f1) and the similarity factor (f2) (The United States Pharmacopeia, 2015a, Moore and Flanner, 1996, European Medicines Agency, 2010, Administración Nacional de Medicamentos, Alimentos y Tecnología Médica, 2016).
A class 2 BCS compound is permeable but relatively insoluble. Various techniques have been developed to improve the dissolution rate of class 2 compounds, including micronization, inclusion complex formation, complexation, conversion into amorphous state and solid dispersion (Jafar et al., 2010).
According to the FDA, EMA and WHO, the comparison of pharmaceutical products containing class 2 BCS API must be performed by in vivo bioequivalence studies.
Meloxicam, 4-hydroxy-2-methyl-N-(5-methylthiazol-2-yl)-2H-1,2-benzothiazine-3-carboxamide-1,1-dioxide is an oxicam derivative with non-steroidal anti-inflammatory properties whose poor water solubility limits its therapeutic efficacy. According to its solubility and permeability, meloxicam belongs to class 2 BCS (Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the U.S. Food and Drug Administration, 2017, Pappich and Martinez, 2015, Sugihara et al., 2015).
The aim of the present study was to evaluate and compare the dissolution profile of nine commercial products containing Meloxicam 15 mg. marketed in Argentina, on the basis of their in vitro dissolution characteristics using the dissolution conditions in the USP monograph for Meloxicam Tablets (The United States Pharmacopeia, 2015b). Our results demonstrate that caution must be exercised as regards interchangeability of generic products.
2. Materials and methods
2.1. Reagents
Analytical grade potassium dihydrogen phosphate (Biopack, Argentina) and sodium hydroxide (Anedra, Argentina) were used.
Meloxicam was purchased in Unifarma (Argentina), with 99.9% purity, as calculated with reference to the dried substance (India).
2.2. Materials
Nine commercial brand tablets containing 15 mg meloxicam were purchased from pharmacies in Buenos Aires (Argentina). All tests were performed before the product expiration dates, which were similar among brands.
2.3. Apparatus and procedure
All dissolution studies were performed using USP Apparatus 2 (The United States Pharmacopeia, 2015b) in a Vankel, VK 7010 (Varian Inc., USA) manual-sampling dissolution bath. The meloxicam tablets dissolution test was performed at 75 ± 1 rpm in phosphate buffer pH = 7.5. The acceptance criterion was Q = 70% of the labeled amount of meloxicam released in 30 min.
The dissolution medium volume was 900 ml. In all experiments, 10 ml sample aliquots were withdrawn at 5, 15, 30, 45 and 60 min using micropipettes, without medium replacement. The withdrawn amounts were adjusted in the calculations. All samples were filtered through 10.0 µm filter paper (Whatman 91), which was properly validated with the standard solution comparing its performance with that obtained with membrane filters. The drug amount dissolved was determined spectrophotometrically in a Cary 1E Varian UV–VIS spectrophotometer (Victoria, Australia) at 362 nm. To prove peak purity, the second order derivative UV spectra was recorded against dissolution medium as reference solution. This procedure was performed with each product.
Twelve tablets of each preparation were studied to obtain statistically significant results.
2.4. Model-dependent methods
Five model-dependent approaches were used to compare the meloxicam dissolution profiles. The model-dependent approaches included the zero order, the first order, the Hixson-Crowell, the Higuchi and the Weibull models.
2.4.1. Zero order kinetics
The drug dissolution from pharmaceutical dosage forms that do not disaggregate and deliver the API slowly (assuming that the area is not modified and that no equilibrium conditions are reached) can be represented by the following equation:
| (1) |
where Q0 is the initial amount of drug in the pharmaceutical dosage form, Qt is the amount of drug in the pharmaceutical dosage form at time t and K0t is the proportionality constant.
2.4.2. First order kinetics
The amount of API delivered from pharmaceutical dosage forms following this dissolution profile are delivered with a rate that is comparable to the amount of API remaining within, in such way that the amount of API liberated per unit of time diminish. First order dissolutions can be represented by the following equation:
| (2) |
where Q0 is the initial amount of API in the pharmaceutical dosage form, Qt is the amount of API in the pharmaceutical dosage form at time t and K0 is the proportionality constant
2.4.3. Hixson-Crowell model
The Hixson-Crowell model assumes that the particle normal area is comparable to the cubic root of its volume. The following equation represents the dissolution profile:
| (3) |
where Q0 is the initial amount of API in the pharmaceutical dosage form, Qt is the amount of API in the pharmaceutical dosage form at time t and Kd is the dissolution constant.
2.4.4. Higuchi model
The Higuchi equation is a “square root of time” release kinetics. This kinetic model gives good experimental fitting data in API dissolution processes formulated as modified liberation systems or semisolid dosage forms. This model can be represented by the following equation:
| (4) |
where Kd is the dissolution constant.
2.4.5. Weibull model
The Weibull function is a mathematical model lacking physicochemical fundament and can be used to study the dissolution rate. The following equation represents the model:
| (5) |
where Q is the amount of API dissolved at time t, Q∞ is the amount of API dissolved at time infinite, also called “total dissolution”, t0 is the lag time and td is the time interval necessary to dissolve or release 63.2% of the API present in the pharmaceutical dosage form. The shape parameter β is obtained from the slope.
To test the pertinence of the release models employed, the Akaike Information Criteria (AIC) (Aguilar et al., 2008) were used. The AIC are a measure of the best fit based on maximum probability. When comparing data sets, the model associated with the smallest AIC value is considered the best fit. The AIC is only applicable when specimens with equal weighting scheme are compared.
| (6) |
where n is the number of dissolution data points, p is the number of parameters of the model, WSSR is the weight sum of square of residues.
2.5. Model-independent methods
2.5.1. Fit factors
Moore and Flanner (Moore and Flanner, 1996) have developed a simple model for independent approximation using fit factors. Fit factors are the difference factor f1, and the similarity factor f2. These fit factors contrast the difference between the percent API dissolved per unit time of a test with that of a reference formulation. Fit factors have been accepted by FDA Centre for Drug Evaluation and Research (CDER) (Food and Drug Administration, 1997) and the similarity factor has also been adopted by the European Medicines Agency (EMA) Committee for Medicinal Products for Human Use (CHMP) (European Medicines Agency, 2010) and World Health Organization (WHO) (World Health Organization, 2006) as a rating criterion of similarity between two in vitro dissolution profiles. In Argentina, regulations establish that changes in the post marketing stage as regards dissolution profiles must be evaluated using the similarity factor f2 (Administración Nacional de Medicamentos, Alimentos y Tecnología Médica, 2009).
The fit factors are denoted f1 (difference factor) and f2 (similarity factor) and are defined by Eqs. (7), (8).
| (7) |
| (8) |
(7) Difference factor (8) Similarity factor
where n is the number of dissolution sampling times, and Rt and Tt are the individual or mean percent dissolved at each time point, t, for the reference and test dissolution profiles, respectively. The parameter f1, whose values range from 0 to 15, and f2, whose values range from 50 to 100, are used to define equivalence of two dissolution profiles, which means an average difference ≤10% at each sampling time.
2.5.2. Dissolution efficiency
This concept has been presented by Khan and Rhodes in 1972 (Khan and Rhodes (1972)). For each sample, the percentage dissolution efficiency is calculated as the percentage ratio of the area under the dissolution curve up to time t to that of the area of the rectangle described by 100% dissolution at the same time point, and is defined as follows:
| (9) |
2.5.3. Mean dissolution time
The mean dissolution time is determined from the accumulative curves of dissolved API as function of time (Aguilar et al., 2008).
| (10) |
where ti is an intermediate time of the intervals of sampling time, ΔQi is the amount of API dissolved in every interval of t and Q∞ is the maximum of API dissolved.
Results of DE and MDT corresponding to the different meloxicam tablet brands were compared by one-way analysis of variance (ANOVA). The analysis was performed with the Infostat 2008 statistical package (Di Rienzo et al., 2008). After this analysis, a multiple-comparisons method (DGC) was applied (Di Rienzo et al., 2002).
3. Results and discussion
In vitro dissolution testing is a fundamental analytical methodology in the pharmaceutical industry. This methodology allows quality assurance of solid pharmaceutical forms for oral administration. The lot-to-lot quality assurance fosters the development of new formulations, guarantee quality homogeneity and ensure an acceptable API performance even after being modified. If all these requirements are met, the formulation optimization during the development phase is favoured, and stability studies and manufacturing process quality control can be carried out (Adams et al., 2001, Dressman et al., 1998, Ferraz et al., 2007).
Table 1 summarises the characteristics of the nine products. Brand I was taken as the reference product.
Table 1.
Formulation compositions.
| Brand | Other ingredients | Appearance |
|---|---|---|
| I | Sodium citrate, lactose, microcrystalline cellulose, povidone, colloidal silicon dioxide, insoluble povidone, magnesium stearate | Yellow, circular, with indented line in centre |
| II | Lactose, microcrystalline cellulose, povidone, insoluble povidone, colloidal silicon dioxide, sodium citrate, sunset yellow aluminum lake, magnesium stearate | Orange, circular, with indented line in centre |
| III | Lactose monohydrate, sodium citrate, povidone, magnesium stearate, colloidal silicon dioxide, polyethilene glycol 1500, microcrystalline cellulose, crospovidone | Yellow, circular, with indented line in centre |
| IV | Lactose monohydrate, microcrystalline cellulose, crospovidone, colloidal silicon dioxide, sodium citrate dehydrate, magnesium stearate, iron (III) oxide yellow | Orange, circular, with indented line in centre |
| V | Lactose monohydrate, microcrystalline cellulose, sodium citrate trihydrate, povidone K30, colloidal silicon dioxide, crospovidone, magnesium stearate | Yellow, circular, with indented line in centre |
| VI | Cellactose, crospovidone, povidone K30, sodium citrate, colloidal silicon dioxide, magnesium stearate | Yellow, circular, with indented line in centre |
| VII | Cellactose, sodium croscarmellose, colloidal silicon dioxide, sodium citrate dihydrate, magnesium stearate | Yellow, circular, with indented line in centre |
| VIII | Sodium citrate, lactose, microcrystalline cellulose, povidone, iron (III) oxide red, colloidal silicon dioxide, crospovidone, magnesium stearate | Orange, circular, with indented line in centre |
| IX | Sodium croscarmellose, sodium lauryl sulfate, cellactose 80, reticulated polyvinylpirrolidone, colloidal silicon dioxide, magnesium stearate, sodium citrate | Yellow, circular, with indented line in centre. |
The dissolution test for meloxicam tablets described in the USP (The United States Pharmacopeia, 2015b) indicates that no less than 70% (Q + 5%) of the API should be dissolved in 30 min. Table 2 summarises the mean percent dissolved at each time point, the relative standard deviation (RSD), and the upper and lower limits. As shown in Table 2, all Brands fulfilled the USP specifications.
Table 2.
Dissolution data and descriptive statistics of nine meloxicam tablet brands.
| Time (min) | Brand | mean% | RSD | Lower limit | Upper limit |
|---|---|---|---|---|---|
| I | 32.3 | 30.4 | 14.0 | 48.4 | |
| II | 24.2 | 8.6 | 20.8 | 27.8 | |
| III | 60.3 | 9.8 | 49.4 | 67.7 | |
| IV | 59.9 | 9.8 | 51.7 | 66.8 | |
| 5 | V | 28.7 | 8.3 | 25.2 | 32.0 |
| VI | 69.8 | 13.4 | 49.7 | 82.7 | |
| VII | 66.7 | 10.1 | 56.5 | 78.3 | |
| VIII | 78.8 | 1.9 | 76.0 | 81.2 | |
| IX | 59.2 | 2.6 | 56.7 | 62.8 | |
| I | 66.9 | 7.5 | 58.6 | 74.0 | |
| II | 64.2 | 8.6 | 55.0 | 74.2 | |
| III | 88.7 | 6.5 | 77.0 | 96.6 | |
| IV | 83.0 | 5.4 | 77.2 | 89.2 | |
| 15 | V | 74.0 | 2.5 | 71.4 | 77.2 |
| VI | 86.9 | 7.0 | 79.3 | 94.8 | |
| VII | 89.8 | 8.7 | 84.9 | 113.3 | |
| VIII | 97.8 | 2.5 | 95.3 | 102.5 | |
| IX | 85.0 | 4.6 | 78.1 | 92.9 | |
| I | 83.2 | 5.7 | 74.4 | 88.7 | |
| II | 79.5 | 8.3 | 68.8 | 89.0 | |
| III | 98.5 | 7.0 | 83.9 | 109.7 | |
| IV | 93.4 | 2.8 | 88.9 | 98.2 | |
| 30 | V | 92.2 | 2.9 | 88.6 | 96.7 |
| VI | 91.9 | 4.3 | 87.0 | 97.8 | |
| VII | 99.7 | 3.6 | 95.8 | 107.4 | |
| VIII | 102.2 | 1.7 | 99.7 | 105.4 | |
| IX | 99.9 | 3.0 | 92.4 | 105.6 | |
| I | 89.6 | 5.7 | 80.8 | 97.0 | |
| II | 95.0 | 4.7 | 85.8 | 102.8 | |
| III | 100.1 | 6.9 | 88.0 | 112.4 | |
| IV | 97.9 | 2.4 | 93.3 | 102.4 | |
| 45 | V | 93.9 | 1.4 | 91.6 | 96.1 |
| VI | 93.2 | 5.5 | 85.3 | 102.0 | |
| VII | 98.0 | 3.6 | 94.4 | 105.6 | |
| VIII | 102.2 | 2.7 | 99.4 | 108.1 | |
| IX | 105.0 | 4.0 | 98.7 | 111.0 | |
| I | 97.0 | 3.1 | 90.0 | 102.0 | |
| II | 98.8 | 5.2 | 87.1 | 107.0 | |
| III | 101.5 | 6.2 | 89.7 | 114.1 | |
| IV | 98.5 | 4.3 | 89.6 | 107.1 | |
| 60 | V | 95.3 | 2.0 | 92.9 | 98.8 |
| VI | 96.0 | 5.9 | 89.2 | 109.5 | |
| VII | 98.4 | 5.8 | 91.4 | 106.4 | |
| VIII | 103.0 | 0.7 | 101.8 | 104.4 | |
| IX | 105.8 | 2.6 | 100.0 | 110.4 | |
Dissolution curves indicated that the analysed products presented different dissolution profiles (Fig. 1). As meloxicam is a class 2 BCS drug, the dissolution test may be formulation-dependent, and the decision related to generics must be made based on the in vivo bioequivalence studies.
Fig. 1.

Dissolution profiles of meloxicam tablets.
The dissolution profiles corresponding to the nine products were evaluated by fitting experimental data to the zero and first-order, the Hixson-Crowell, the Higuchi, and the Weibull models. In Table 3, the following parameters are presented: dissolution constants (k) and the determination constant (r2), for zero and first order, Hixson-Crowell and Higuchi models, and the shape parameter β, the determination constant (r2) and Td which is the time interval necessary to dissolve or release 63.2% of the drug present in the pharmaceutical dosage form, for the Weibull model. No significant variations were found among all β values (β < 1). The Weibull model provided the best adjustment curve for the nine brands, with the higher determination coefficients (r2) and smallest AIC values for all the brands tested (bold print indicating the best fits).
Table 3.
Parameters of the mathematical models and descriptive statistics for the dissolution data.
| Brands |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Model | Statistics | I | II | III | IV | V | VI | VII | VIII | IX |
| Zero Order | r2 | 0.8257 | 0.8414 | 0.6774 | 0.7601 | 0.6720 | 0.7272 | 0.5990 | 0.5931 | 0.7807 |
| k | 0.1385 | 0.1644 | 0.0842 | 0.0834 | 0.1374 | 0.0529 | 0.0637 | 0.0468 | 0.1028 | |
| AIC | 12.38 | 13.53 | 11.47 | 9.32 | 16.49 | 5.63 | 10.37 | 7.43 | 10.83 | |
| First Order | r2 | 0.9553 | 0.9667 | 0.8080 | 0.8689 | 0.7903 | 0.8205 | 0.6411 | 0.6815 | 0.9182 |
| k | 0.0268 | 0.0319 | 0.0244 | 0.0220 | 0.0268 | 0.0143 | 0.0179 | 0.0180 | 0.0350 | |
| AIC | −9.59 | −9.35 | −2.40 | −5.69 | −1.28 | −8.17 | −1.18 | −2.03 | −3.68 | |
| Hixson-Crowell | r2 | 0.9195 | 0.9361 | 0.7647 | 0.8352 | 0.7523 | 0.7896 | 0.6304 | 0.6507 | 0.8791 |
| k | 0.0151 | 0.0179 | 0.0121 | 0.0113 | 0.0147 | 0.0073 | 0.0090 | 0.0082 | 0.0162 | |
| AIC | −14.16 | −13.71 | −10.15 | −13.03 | −7.82 | −15.87 | −9.83 | −11.27 | −11.21 | |
| Higuchi | r2 | 0.9243 | 0.9342 | 0.8102 | 0.8788 | 0.8069 | 0.8464 | 0.7440 | 0.7338 | 0.8951 |
| k | 1.4778 | 1.7464 | 0.9283 | 0.9039 | 1.5180 | 0.5751 | 0.7153 | 0.5250 | 1.1102 | |
| AIC | 8.22 | 9.13 | 8.82 | 5.91 | 13.85 | 2.76 | 8.14 | 5.31 | 7.14 | |
| Weibull | r2 | 0.9985 | 0.9955 | 0.9896 | 0.9961 | 0.9745 | 0.9934 | 0.9470 | 0.9635 | 0.9866 |
| Td | 13.25 | 14.93 | 2.53 | 3.22 | 11.60 | 1.30 | 1.50 | 0.32 | 2.85 | |
| β | 0.4265 | 0.5320 | 0.2763 | 0.2541 | 0.4749 | 0.1632 | 0.2167 | 0.1761 | 0.3306 | |
| AIC | −25.20 | −17.60 | −19.95 | −25.68 | −9.97 | −27.51 | −14.00 | −18.03 | −16.86 | |
The determination of DE and MDT values are useful methods to reduce each curve to a single number, which may be referred to the dissolution rate constant (Fig. 2, Fig. 3). Statistical differences were found among the brands analysed (Tables 4a,4b., and 5a.,5b.). The DGC test indicated that brands III, IV, VI and IX were similar, whereas brand VII was similar to brand VIII. MDT values for brands III, IV and VI were found to be similar to each other, whereas brand VII was similar to brand VIII. In both cases, brand I was not found to be similar to any other.
Fig. 2.

Statistical comparison of the Dissolution Efficiency (DE) among the nine commercial products. The average is indicated near the boxes. Bars indicate standard deviation.
Fig. 3.
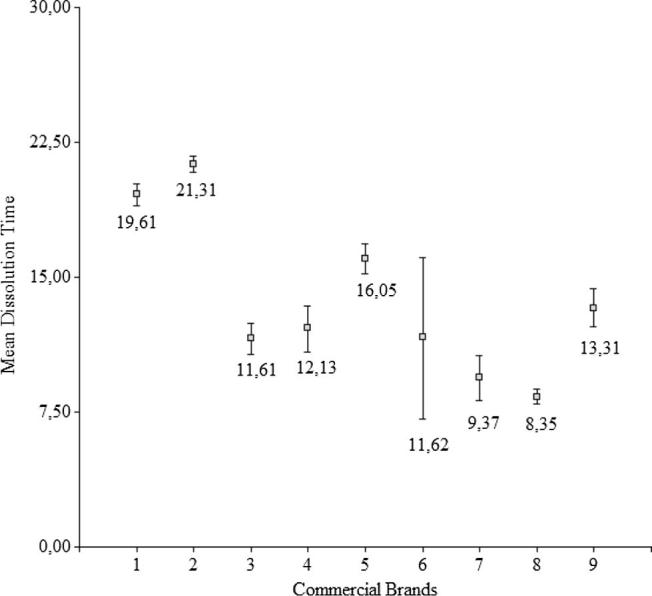
Statistical comparison of the Mean Dissolution Time (MDT) among the nine commercial products. The average is indicated near the boxes. Bars indicate the standard deviation.
Table 4a.
Variance analysis (ANOVA) of DE Values for the nine pharmaceutical products.
| Variation source | Square sum | Degrees of freedom | Medium square | Fa |
|---|---|---|---|---|
| Between treatments | 3733.05 | 8 | 466.63 | 72.67 |
| Within treatments | 635.68 | 99 | 6.42 | |
| Total | 4368.73 | 107 |
Fcritical = 1.890.
Significant for P > 0.05.
Table 4b.
DGC test of DE values for the nine pharmaceutical products.
| Test:DGC Alfa = 0.05 PCALT = 2.1382 | ||||||||
|---|---|---|---|---|---|---|---|---|
|
Error: 6.4210 df: 99 | ||||||||
| Brands | Average | n | E.E. | |||||
| II | 73.25 | 12 | 0.73 | A | ||||
| I | 75.56 | 12 | 0.73 | B | ||||
| V | 81.26 | 12 | 0.73 | C | ||||
| IX | 84.64 | 12 | 0.73 | D | ||||
| IV | 86.24 | 12 | 0.73 | D | ||||
| VI | 86.77 | 12 | 0.73 | D | ||||
| III | 87.03 | 12 | 0.73 | D | ||||
| VII | 90.25 | 12 | 0.73 | E | ||||
| VIII | 91.45 | 12 | 0.73 | E | ||||
Table 5a.
Variance analysis (ANOVA) of MDT values for the nine Pharmaceutical products.
| Variation source | Square sum | Degrees of freedom | Medium square | Fa |
|---|---|---|---|---|
| Between treatments | 1885.17 | 8 | 235.65 | 77.90 |
| Within treatments | 299.49 | 99 | 3.03 | |
| Total | 2184.66 | 107 |
Fcritical = 2.033.
Significant for P > 0.05.
Table 5b.
DGC test of MDT values for the nine Pharmaceutical products.
| Test:DGC Alfa = 0.05 PCALT = 1.4676 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
Error: 3.0251 df: 99 | |||||||||
| Brands | Average | n | E.E. | ||||||
| VIII | 8.35 | 12 | 0.50 | A | |||||
| VII | 9.37 | 12 | 0.50 | A | |||||
| III | 11.61 | 12 | 0.50 | B | |||||
| VI | 11.62 | 12 | 0.50 | B | |||||
| IV | 12.13 | 12 | 0.50 | B | |||||
| IX | 13.31 | 12 | 0.50 | C | |||||
| V | 16.05 | 12 | 0.50 | D | |||||
| I | 19.61 | 12 | 0.50 | E | |||||
| II | 21.31 | 12 | 0.50 | F | |||||
Fit factors are important quantitative parameters recommended by the FDA to compare dissolution profiles (Food and Drug Administration, 1997). The results obtained with each brand using brand I as reference are shown in Table 6. The similarity factor f2 is more sensitive in finding dissimilarities between dissolution curves than the difference factor f1, and the fit factor values are dependent on the number of sampling time point selected. According to the FDA, f1 values less that 15 and f2 values greater than 50 should ensure equivalence between the dissolution curves, indicating an average difference of no more than 10% at the sample time points. According to this guideline, the dissolution curves corresponding to brand II and brand V would be similar to that obtained with the reference formulation.
Table 6.
Fit factors for the nine brands of meloxicam tablets based on the average of twelve tablets.
| Fit factor |
||
|---|---|---|
| Brand | f1 | f2 |
| I/II | 6 | 65 |
| I/III | 22 | 37 |
| I/IV | 17 | 40 |
| I/V | 7 | 62 |
| I/VI | 19 | 36 |
| I/VII | 23 | 35 |
| I/VIII | 31 | 28 |
| I/IX | 23 | 37 |
4. Conclusions
This study found variations in the dissolution profiles of meloxicam tablets commercially available in Argentina. The dissolution profiles obtained with the analysed products were found to be quite different from each other, thus demonstrating that the results obtained with the dissolution test are formulation-dependent. All brands fulfilled the specifications of the dissolution test established by the USP 38.
The Weibull model kinetic curve rendered the best adjustment. The best fittings were obtained with brands I, II, IV and VI, with maximum determination coefficients and smallest AIC values.
The ANOVA and DGC tests applied to DE and MDT values demonstrated that brand I was not similar to any other.
If fit factors are considered, only brands I, II and V were similar.
In conclusion, significant differences were observed between the in vitro dissolution profiles of meloxicam tablets of different commercial preparations. As meloxicam is a class II BCS drug, interchangeability between commercial products must be demonstrated with in vivo bioequivalence studies, which remain to be implemented in our country.
Acknowledgment
This work was supported by grant 20020130100342BA to A. I. Segall from UBA and PIP No: 11420110100380 from CONICET.
Footnotes
Peer review under responsibility of King Saud University.
References
- Adams E., Coomans D., Smeyers-Verbeke J., Massart D.L. Application of linear mixed effects models to the evaluation of dissolution profiles. Int. J. Pharm. 2001;226:107–125. doi: 10.1016/s0378-5173(01)00775-x. [DOI] [PubMed] [Google Scholar]
- Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT), 1999. Disposición No. 3185, Buenos Aires, Argentina.
- Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT), 2009. Disposición No. 5568, Buenos Aires, Argentina.
- Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT), 2009. Disposición No. 5568, Buenos Aires, Argentina.
- Aguilar Ros, A., Caamaño Somoza, M., Martín Martín, F. R., Montejo Rubio, M.C., 2008. Parámetros amodelísticos: comparación de perfiles. In Biofarmacia y Farmacocinética, ejercicios y problemas resueltos, first ed. Elsevier España S. L., Barcelona, España, pp. 3–4.
- Al Ameri M.N., Nayuni N., Kumar K.G.A., Johnston A. The differences between the branded and generic medicines using solid dosage forms: in vitro dissolution testing. Results Pharma. Sci. 2012;2:1–8. doi: 10.1016/j.rinphs.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa P., Souza Lobo J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001;13:123–133. doi: 10.1016/s0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- Costa F.O., Sousa J.J.S., Pais A.A.C.C., Formosinho S.J. Comparison of dissolution profiles of Ibuprofen Pellets. J. Control Release. 2003;89:199–212. doi: 10.1016/s0168-3659(03)00033-6. [DOI] [PubMed] [Google Scholar]
- Di Rienzo, J.A., Casanoves, F., Balzarini, M.G., Gonzalez, L., Tablada, M., Robledo, C.W., 2008. InfoStat, 2008 version, Grupo InfoStat, FCA, Universidad Nacional de Córdoba, Argentina. <http://www.infostat.com.ar>.
- Di Rienzo J.A., Guzmán A.W., Casanoves F. Multiple Comparisons method based on the distribution of the root node distance of a binary tree. J. Agric. Biol. Environ. S. 2002;7(2):1–14. [Google Scholar]
- Dressman J.B., Amidon G.L., Reppas L., Shah V.P. Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms. Phar Res. 1998;15(1):11–22. doi: 10.1023/a:1011984216775. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency . Committee for Medicinal Products for Human Use (CHMP); London UK: 2010. Guideline on the investigation of bioequivalence. [Google Scholar]
- Ferraz H.G., Carpentieri L.N., Pereira Watanabe S. Dissolution profile evaluation of solid pharmaceutical forms containing chloramphenicol marketed in Brazil. Braz. Arch. Biol. Technol. 2007;50(1):57–65. [Google Scholar]
- Food and Drug Administration, 1997. Guidance for Industry, Testing of Immediate Release Solid Oral Dosage Forms. U.S. Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), U.S. Government Printing Office: Washington, DC.
- Intra-Agency Agreement Between the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the U.S. Food and Drug Administration (FDA) Oral Formulations Platform—Report 1. <https://bpca.nichd.nih.gov/collaborativeefforts/initiatives/Documents/Formulations_Platform_Report1.pdf> (accessed June 5, 2017).
- Jafar M., Mhg D., Shareef A. Enhancement of dissolution and antiinflammatory effect of meloxicam using solid dispersions. Int. J. Appl. Pharm. 2010;2(1):22–27. [Google Scholar]
- Khan F., Li M., Schlindwein W. Comparison of in vitro dissolution tests for commercially available aspirin tablets. Diss. Techol. 2013;20:48–58. [Google Scholar]
- Khan K.A., Rhodes C.T. Effect of compaction pressure on the dissolution efficiency of some direct compression systems. Pharm. Acta Helv. 1972;47:594–607. [PubMed] [Google Scholar]
- LeBlond D., Altan S., Novick S., Peterson J., Shen Y., Yang H. In vitro dissolution curve comparisons: a critique of current practice. Diss. Technol. 2016;23(1):14–23. [Google Scholar]
- Moore J.W., Flanner H.H. Mathematical comparison of dissolution profiles. Pharm. Technol. 1996;20:64–74. [Google Scholar]
- Noolkar S.B., Jadhav N.R., Bhende S.A., Killedar S.G. Solid-state characterization and dissolution properties of meloxicam-moringa coagulant–PVP ternary solid dispersions. AAPS PharmSciTech. 2013;14(2):569–577. doi: 10.1208/s12249-013-9941-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappich M.G., Martinez M.N. Applying biopharmaceutical classification system (BCS) criteria to predict oral absorption of drugs in dogs: challenges and pitfalls. AAPS J. 2015;17(4):948–964. doi: 10.1208/s12248-015-9743-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polli J.E., Rekhi G.S., Ausburger L.L., Shah V.P. Methods to compare dissolution profiles and a rationale for wide dissolution specifications for metoprolol tartrate tablets. J. Pharm. Sci. 1997;86:690–700. doi: 10.1021/js960473x. [DOI] [PubMed] [Google Scholar]
- Sugihara M., Takeuchi S., Sugita M., Higaki K., Kataoka M., Yamashita S. Analysis of intra- and intersubject variability in oral drug absorption in human bioequivalence studies of 113 generic products. Mol. Pharmaceutics. 2015;12:4405–4413. doi: 10.1021/acs.molpharmaceut.5b00602. [DOI] [PubMed] [Google Scholar]
- The United States Pharmacopeia, 2015a. 38th Ed. (Spanish version) U.S. Pharmacopeial Convention, Rockville, MD; pp. 520–530.
- The United States Pharmacopeia, 2015b. 38th Ed. (Spanish version) U.S. Pharmacopeial Convention, Rockville, MD; pp. 4640–4642.
- World Health Organization, 2006. Technical Report Series No. 937, Annex 7, Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish Interchangeability.
- Yuksel N., Kanik A.E., Baykara T. Comparison of in vitro dissolution profiles by ANOVA-based, model-dependent and -independent methods. Int. J. Pharm. 2000;209:57–67. doi: 10.1016/s0378-5173(00)00554-8. [DOI] [PubMed] [Google Scholar]