

Abstract
The overlap of signs and symptoms between Parkinson disease and the atypical parkinsonian syndromes, such as progressive supranuclear palsy (PSP), multiple system atrophy (MSA), corticobasal degeneration (CBD), and dementia with Lewy bodies (DLB), can render clinical diagnoses challenging. The continued evolution of diagnostic criteria to reflect the increasingly recognized heterogeneous presentations of these diseases further complicates timely recognition and diagnoses. This review provides a diagnostic approach to the classic atypical parkinsonian syndromes (PSP, CBD, MSA, and DLB) with emphasis on the key clinical and pathological features of each and the recognition of “red flags” in the setting of recent advances in diagnosis and treatment.
Keywords: Parkinson disease, atypical parkinsonism, proteinopathy, neurodegeneration, multidisciplinary care
INTRODUCTION
Parkinsonism refers to a constellation of symptoms including rest tremor, muscle rigidity, akinesia and/or bradykinesia, and postural instability.[1] While the most common neurodegenerative cause of Parkinsonism is Parkinson disease (PD), the causes of parkinsonism can be many, including secondary causes, hereditary neurodegenerative disorders, and the atypical parkinsonian or “Parkinson-plus” syndromes, all of which can have overlapping signs and symptoms (Figure 1). The atypical Parkinson disorders commonly include progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), multiple system atrophy (MSA), and dementia with Lewy body (DLB). Although less common than PD, it is important to distinguish the atypical parkinsonian syndromes, as disease progression and functional decline is typically more rapid and standard therapies for PD often provide only partial or waning benefit with more side effects. Moreover, patients suffering from these disorders have complex care needs that benefit from a multidisciplinary approach. This review describes the diagnostic approach to the atypical parkinsonian syndromes in order to help the practitioner recognize key clinical and pathological features and to highlight recent advances in diagnostics and treatment for these disorders.
Figure 1.

Atypical Parkinsonisms overlap and share common features with the other parkinsonian syndromes. (NBIAs, neuronal brain iron accumulation disorders; NPH, normal pressure hydrocephalus)
DISTINGUISHING ATYPICAL PARKINSONISM FROM PARKINSON DISEASE
The major challenge in distinguishing atypical parkinsonian syndromes from PD is the considerable overlap in symptoms and features between them, particularly early on in the disease course. Further complicating the issue are many primary neurodegenerative disorders that have features that mimic PD (see Table 1). Although brain imaging can be helpful, to date there is no single reliable test or specific biomarker used to diagnose PD and to distinguish it from atypical parkinsonism; thus, diagnosis remains primarily based on clinical evaluation. Expert practitioners are reasonably good at diagnosing PD based on key clinical features—indeed 75 to 95% of PD diagnoses are confirmed at autopsy[2–5]—and accuracy as expected increases with disease duration.[6] However, the sensitivity of diagnosis for atypical parkinsonian syndromes is much lower, leading to potential delays and errors in diagnosis. Increasingly, neuropathological and clinical features are being combined in a clinicopathologic approach to better delineate diagnoses and shed light on disease pathophysiology.
Table 1.
Causes of Atypical Parkinsonism
| Multisystem diseases |
| Progressive supranuclear palsy (PSP) |
| Corticobasal syndrome (CBS) |
| Multiple system atrophy (MSA) |
| Shy-Drager, Striatonigral degeneration (SND/MSA-P) |
| Olivopontocerebellar degeneration (OPCA/MSA-C) |
| Dementia with Lewy bodies (DLB) |
| Parkinsonism-dementia-ALS complex |
|
|
| Hereditary neurodegenerative diseases |
| Huntington disease |
| Spinocerebellar ataxias (esp. 2, 3, 17) |
| Wilson disease |
| Hereditary ceruloplasmin deficiency |
| Frontotemporal lobar dementia w/ Parkinsonism (FTLD-17) |
| Neuronal brain iron accumulation disorders (e.g., pantothenate kinase-associated degeneration) |
| Hereditary dystonia-parkinsonisms (e.g., X-linked Lubag syndrome) |
| Gerstmann-Strausler-Scheinker |
| Neuroacanthocytosis |
| Neuronal ceroid lipofuscinoses |
| Mitochondrial cytopathies |
| Hereditary hemochromatosis |
| Familial basal ganglia calcification (Fahr syndrome) |
| Familial progressive subcortical gliosis |
| Disinhibition-dementia-parkinsonism-amyotrophy complex |
(Adapted with permission from Jankovic J, Lang AE, Saunders. 2008 [Saunders, an imprint of Elsevier])
A common pathological feature of neurodegenerative diseases is the presence of abnormal proteinaceous deposits in brain, and thus the term “neuroproteinopathy” has been coined to describe these disorders. More specific terms such as “tauopathy” for PSP, CBD, and frontotemporal dementia (FTD) and “synucleinopathy” for PD, DLB, and MSA have been liberally applied. Various proteins accumulate abnormally both intracellularly (i.e., tau tangles, Lewy bodies) and extracellularly (i.e., plaques) and are linked to neurodegenerative diseases. Within a disorder there may be overlap of protein pathology, such as the co-occurrence of amyloid-beta plaques and Lewy bodies in DLB (Figure 2).[7]
Figure 2.

Clinicopathologic overlap exists among neurodegenerative proteinopathies including alpha-synuclein (synucleinopathy), tau (tauopathy), amyloid and TDP-43 pathology. (AD, Alzheimer disease; ALS, amyotrophic lateral sclerosis; DLB, dementia with Lewy bodies; CBD, corticobasal degeneration; FTD, frontotemporal dementia; LPA, logopenic primary progressive aphasia; MND, motor neuron disease; MSA, multiple system atrophy; PSP, progressive supranuclear palsy)
To date, no reliable biomarkers have been established as diagnostic for any of the atypical parkinsonisms; however, ancillary brain imaging (as described in subsequent sections) is increasingly used as adjunctive diagnostic tools in select cases. While many features of the atypical parkinsonian syndromes overlap with those of PD, certain key features and “red flags” have been identified that can be helpful in distinguishing the atypical parkinsonisms (see Table 2, diagnostic criteria for PD from the Movement Disorders Society).[8] As in PD, core parkinsonian motor features must be present in atypical parkinsonisms, including bradykinesia in combination with either rest tremor, rigidity, or both. Although the absence or paucity of tremor is sometimes reported as a “red flag” for atypical parkinsonian syndromes, this finding is non-specific and may also be observed in certain PD subtypes. Critically, the progression of symptoms in atypical parkinsonian syndromes differs from PD, with a relatively rapid functional decline with early gait impairment and falls (often requiring an assist device or wheelchair within 5 years of disease onset) and often with bulbar dysfunction with severe dysarthria, dysphonia, and dysphagia. While a therapeutic response to dopaminergic therapy is typical in PD, in atypical parkinsonisms the response may be poor or absent and adverse effects can be more significant. Motor fluctuations and levodopa-induced dyskinesias are uncommon in atypical parkinsonisms and support the diagnosis of PD when present. Additional “plus” features may include oculomotor abnormalities, early prominent dysautonomia, pyramidal tract or cerebellar signs (ataxia), dystonia (especially antecollis or dystonia leading to limb contractures), laryngeal stridor, myoclonus, alien-limb phenomenon, apraxia, and early dementia. Non-motor features such as REM-sleep behavior disorder (RBD), restless leg syndrome (RLS), and periodic limb movements of sleep are not common in tau-disorders such as PSP and CBD whereas they are common in synucleinopathies such as PD.
Table 2.
MDS Clinical Diagnostic Criteria for PD
| Supportive | Red Flags | Exclusionary |
|---|---|---|
|
| ||
| Cardinal features: rest tremor, rigidity, bradykinesia (documented) Clear response to dopaminergic therapy (subjective or >30% change in UPDRS III with treatment) Marked on/off fluctuations, end-dose wearing-off Presence of levodopa-induced dyskinesias Olfactory loss Cardiac sympathetic denervation on MIBG scintigraphy |
Rapid progression of gait impairment, requiring a wheelchair within 5 yrs of onset Absence of progression of motor symptoms or signs over 5 yrs or more (absent treatment) Early bulbar dysfunction (< 5 yrs from onset) Inspiratory respiratory dysfunction (stridor, frequent sighs) Severe autonomic failure (< 5 yrs from onset) Recurrent (>1/yr) falls from imbalance within 3 yrs of onset Anterocollis or limb contractures within 10 yrs of onset Absence of common non-motor features (sleep dysfunction, RBD, RLS/PLMS, daytime somnolence, autonomic dysfunction, hyposmia, mood disturbance) Otherwise unexplained pyramidal tract signs Bilateral symmetric parkinsonism |
Unequivocal cerebellar abnormalities (e.g., cerebellar gait, limb ataxia, oculomotor abnormalities) Downward vertical supranuclear gaze palsy or selective slowing of vertical saccades Probable behavioral variant FTD or PPA within <5 yrs onset Restriction of parkinsonism to lower extremities >3 yrs Drug-induced parkinsonism (documented prior use and link to drug, i.e., antipsychotic, etc.) Absence of response to high-dose levodopa Cortical sensory loss (evidence of graphesthesia, astereognosis) ideomotor apraxia, or primary aphasia Normal dopamine transporter imaging (presynaptic) Documentation of an alternative condition |
(Adapted with permission from Postuma RB et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 2015; 30, 1591–1601, doi:10.1002/mds.26424) [8].
MIBG, 123I-metaiodobenzylguanidine; RBD, REM-sleep behavior disorder; RLS, restless leg syndrome; PLMS, periodic limb movements of sleep; FTD, frontotemporal dementia; PPA, primary progressive aphasia; UPDRS, Unified Parkinson Disease Rating Scale.
PROGRESSIVE SUPRANUCLEAR PALSY
Progressive Supranuclear Palsy (PSP) was first described by Drs Steele, Richardson, and Olszewski in 1963.[9] Approximately 5–6% of those presenting with parkinsonism are diagnosed with PSP, making it one of the most common forms of atypical parkinsonism. The estimated prevalence is about 5 per 100,000 individuals,[10–12] and the reported annual incidence is 5.3 per 100,000 in men and women between the ages of 50 to 99.[13] However, these estimates are likely low due to under-recognition and misdiagnosis. Unlike PD, diagnosis of PSP by general and movement disorders neurologists is correct in approximately 75% of cases.[14] The most common misdiagnoses include PD and vascular parkinsonism, as well as the atypical parkinsonisms MSA and CBS.[15] Further complicating diagnosis is the recent recognition of the heterogeneous presentation of PSP and the identification of PSP variants.
Classic PSP, or Richardson syndrome, is characterized by early gait and postural instability, unexplained falls, supranuclear gaze palsy, axial rigidity, bradykinesia, dysarthric speech, and progressive dementia. The onset of symptoms in PSP typically occurs in the 60’s with an average age of onset between 63 and 66 years old and a mean survival of about 5 to 9 years from disease onset.
A key feature of PSP that distinguishes it from other parkinsonian syndromes is the presence of early gait dysfunction and postural instability. Gait in PSP is often described as clumsy and stiff, and is characterized by step asymmetry and large lateral deviations sometimes described as “like a drunken sailor.” PSP patients also pivot somewhat recklessly rather than turning en bloc as in PD, resulting in more instability and falls. Falls occur within the first year or two and often lead to significant injury including bruises and fractures.[16] Falls can occur due to due to multiple factors including axial rigidity, bradykinesia, freezing, reduced/absent postural reflex, visual-vestibular deficits, and decreased insight. Indeed, patients with PSP who have lost insight into their postural instability can appear to “rocket” out of their chair recklessly instead of waiting for assistance, often prompting a fall.
In addition to gait impairment, visual symptoms are also a hallmark of PSP. These include blurred vision, photosensitivity, diplopia, and difficulty reading due to both the inability to look down and due to convergence insufficiency. Vertical gaze impairment in general is often associated with PSP, but this is nonspecific and can be seen in other neurodegenerative disorders and normal aging.[17] By contrast, downgaze impairment is the most sensitive predictor for PSP, though both upgaze and horizontal ophthalmoparesis can progressively develop as well. Supranuclear gaze palsy can be overcome by changes in head position or oculocephalic maneuvers, but testing is often limited by axial rigidity. Slowed vertical saccades and reduction of optokinetic nystagmus are increasingly recognized as early signs and predictors of supranuclear gaze palsy in PSP. Square-wave jerks are also common and characterized by mini-saccadic intrusions during fixation at a distance. In addition, patients may suffer from blepharospasm and apraxia of eye opening. This apraxia can in select cases result in functional blindness.
Additional symptoms of PSP can include a change in personality, emotional withdrawal, and a characteristic facial appearance in which bradykinesia and dystonia combine with reduced eye blinking to produce a “worried” or “astonished” expression. Eyebrow furrowing (procerus contraction) and vertical wrinkling of the forehead are common and referred to as the “Procerus sign.” [18] Bulbar symptoms include progressive dysarthria characterized by spastic, hypernasal, hypokinetic, and monotonous speech. Stuttering, echolalia, and occasional involuntary vocalizations may also be present, as well as an apraxia of phonation. In addition, progressive dysphagia occurs early and increases the risk of aspiration, pneumonia, and early death.
Mood disturbance and cognitive decline are also frequent in PSP. Mood disturbance is primarily characterized by apathy more than depression, but may also include disinhibition, dysphoria, anxiety, irritability, and agitation.[19] Pseudobulbar affect (laughing or crying incongruous with mood) can also occur with emotional lability or incontinence and is more common in PSP relative to other parkinsonian syndromes, and can be a cause of significant distress for both the patient and caregivers.[20]
PSP is also associated with dementia. Similar to other parkinsonian syndromes, progressive frontal, subcortical dementia is typical and includes slowed processing, reduced verbal fluency, and executive dysfunction. Perseveration of automatic behaviors (such as that seen with the three-clap test or “applause sign”) and grasping, imitative behaviors may also be present. Ideomotor apraxia occurs in both PSP and CBS leading to confusion in diagnosis, but is more prominent in CBS.[21]
Diagnosis: Distinguishing Novel PSP Variants
The NINDS-SPSP (National Institute of Neurological Disorders and Stroke and the Society for PSP International Workshop) established diagnostic criteria for possible and probable PSP. They include progressive symptoms with onset after the age 40, postural instability, frequent falls, and slowed vertical saccades or vertical gaze palsy.[22] Definite PSP additionally required pathologic evidence. Supportive findings included symmetric rigidity, diminished response to levodopa, and early cognitive impairment, whereas exclusionary factors included encephalitis, focal brain lesion, hallucinations, prominent dysautonomia, and alien limb phenomenon. Although previously thought to be exclusionary, cerebellar features have recently been described in variant PSP.[23]
The diagnosis of classic PSP, or Richardson syndrome, is relatively straightforward. However, the clinical presentation of PSP is increasingly recognized as heterogeneous and includes several clinical variants or phenotypes.[24] Table 3 describes at least five phenotypic variants have been described: PSP-Parkinsonism (PSP-P), PSP-Pure Akinesia Gait Freezing (PSP-PAGF), PSP-Corticobasal Syndrome (PSP-CBS), PSP-behavioral variant of FTD (PSP-bvFTD), and two other possible PSP variants with features that overlap with either primary lateral sclerosis (PSP-PLS) or cerebellar ataxia (PSP-C). While the presentation of each PSP variant is different, they all progress to develop more typical PSP features including postural instability, falls, and supranuclear gaze palsy, as well as classic tau pathology.
Table 3.
PSP Syndromes
| Variant PSP | Clinical Features | Regional Pathology* |
|---|---|---|
| PSP-RS (Richardson Syndrome) | Early gait instability, falls, supranuclear gaze palsy, axial rigidity, dysarthria, dysphagia, progressive dementia | Dentate, globus pallidus, striatum, midbrain, and superior cerebellar (CBL) peduncle |
| PSP-Parkinsonism | Tremor, rigid-bradykinesia, Levodopa responsive** Late cognitive decline Longer life expectancy (9+ yrs) |
Substantia nigra, subthalamic nucleus |
| PSP-PAGF (Pure akinesia-gait failure) | Early gait difficulty, freezing of gait/motor block, micrographia, speech impairment, hypophonia Disease duration (11–15 yr) |
Motor cortex, pons, cerebellum |
| PSP-CBS | Dystonia, dyspraxia, cortical sensory loss, apraxia speech | Frontal and parietal cortex |
| PSP-bvFTD (behavioral variant FTD) | Predominant cognitive, personality change, late Parkinsonism | Frontotemporal cortex |
| PSP-PLS (primary lateral sclerosis) | Bulbar, limb weakness, upper motor neuron signs/spasticity | Frontal predominant, corticospinal tract |
| PSP-cerebellar | Cerebellar ataxia | Deep cerebellar nuclei |
Levodopa response for PSP-Parkinsonism diminishes later in disease.
(Reprinted with permission from McFarland NR. Diagnostic Approach to Atypical Parkinsonian Syndromes. Continuum 2016;22(4):1123. doi: 10.1212/CON.0000000000000348)
PSP-Parkinsonism
The most common variant of PSP is PSP-Parkinsonism with features of tremor, asymmetric bradykinesia and rigidity that mimic PD.[25] Ocular motility may be relatively normal early in disease; however, slowed vertical saccades and reduced optokinetic nystagmus typically develop over time. Parkinsonism is frequently partially levodopa-responsive early on, with waning of response and the development of more typical PSP symptoms as the disease progresses. Mean survival for patients with PSP-Parkinsonism is usually longer than in Richardson syndrome, averaging nine or more years from disease onset.
PSP-Pure Akinesia with Gait Freezing (PSP-PAGF)
PSP-PAGF was first described in 1974 and is characterized by progressive freezing of gait or speech, usually with initiation, frequent falls, micrographia, and facial hypomimia. [26] [27] Tremor is typically absent and more classic signs of PSP such rigidity, supranuclear palsy, and dementia develop much later in the disease course. Evidence of significant leukoaraiosis on brain imaging is exclusionary and response to levodopa is typically poor or transient.
PSP-Corticobasal Syndrome (PSP-CBS)
PSP and CBD overlap both clinically and pathologically. Indeed, PSP pathology is reported in up to 40% of clinically diagnosed CBD cases.[28] A relative rare variant of PSP, PSP-CBS presents initially like CBD with asymmetric rigidity, bradykinesia, dyspraxia, alien limb phenomenon, and dystonia.[29] Slowed saccades, particularly toward the more affected side, are common as opposed to vertical gaze palsy. More classic features of PSP such as gait and postural instability develop relatively later.
PSP-behavioral variant Frontal Temporal Dementia (PSP-bvFTD)
Also rare, PSP-bvFTD often presents with behavioral and cognitive dysfunction. Typical PSP symptoms follow later including gait difficulty, falls, supranuclear palsy and levodopa unresponsive parkinsonism.[30] Until other parkinsonian features develop, a misdiagnosis of frontal temporal dementia or other primary dementia is common.[31]
Progressive Supranuclear Palsy-Primary Lateral Sclerosis (PSP-PLS)
PSP and primary lateral sclerosis (PLS) share common features including bulbar symptoms and spastic gait. Although supranuclear gaze palsy is not usually present, ocular dysmotility is sometimes apparent in PLS. Recent reports suggest a possible PSP-PLS overlap syndrome[32,33] with tau pathology found in areas linked to the corticospinal tract.[34]
Cerebellar variant of PSP (PSP-C)
Increasing reports suggest a novel variant of PSP with features of cerebellar ataxia and tau pathology in the deep cerebellar nuclei.[23,35] Although still rare and under-recognized, based on recent reports PSP-C may be more prevalent in Asian populations.
The definition and variants of PSP continue to undergo revision and new criteria are often being established. Respondek and colleagues (2014) recently proposed an approach to describing the phenotypic spectrum of PSP based on the predominant clinical features seen within the first 2 years of presentation.[24] “Predominance types” were identified including Richardson syndrome, postural instability versus oculomotor predominant, Parkinsonism, corticobasal syndrome, FTD, and unclassified, and validation. These predominance types, as well as “atypical” PSP variants, are still being validated.
Pathology
PSP is pathologically characterized by abnormal tau deposition and associated pathology (Figure 3). The hallmark of PSP is the tufted astrocyte, but the pathology includes also neurofibrillary tangles, pretangles and coiled bodies.[36] Tau pathology is typically present in the basal ganglia, pons, oculomotor nuclei, and usually spares the cortex. There is associated gliosis and degeneration marked by loss of pigmented cells in the substantia nigra and progressive atrophy of the midbrain, subthalamic nucleus, superior cerebellar and middle cerebellar peduncles, dentate, and frontal cortex.
Figure 3. Typical neuropathological findings in progressive supranuclear palsy.

The photomicrographs show classic pathological findings including (A) neurofibrillary tangles (arrows), (B) neuropil threads (arrowheads), and a (E) tufted astrocyte stained with PHF1 antibody to human tau.
Abnormal deposition of hyperphosphorylated tau is characteristic of the “tauopathies” including PSP, CBD, AD, and FTD. The tau protein is encoded by MAPT (17q21) and has six isoforms that include three or four repeat (R) domains that bind microtubules. In PSP and CBD the ratio of 4R:3R tau in brain is increased compared to unaffected individuals, whereas in frontotemporal dementia 3R tau is predominant. Although MAPT variants are closely linked to parkinsonisms including PSP, specific MAPT mutations are rare in PSP. In addition to MAPT, recent genome-wide association studies (GWAS) identified novel variants and putative genes associated with PSP including STX6 (encodes syntaxin 6), EIF2AK3 (PERK), and MOBP (myelin-associated oligodendrocytic basic protein).[37] Efforts are underway to validate these genes and their potential role in PSP (and other tauopathies).
Diagnostic Modalities and Biomarkers
Brain imaging has been the most useful adjunctive diagnostic tool for PSP to date. The midbrain and superior cerebellar peduncles are typically atrophied in PSP, which can help distinguish it from other parkinsonian disorders.[38–43] Several methodologies have been proposed to measure midbrain atrophy on MRI, including measurement of the suprapontine midline anteroposterior (AP) diameter versus the midsagittal area of the midbrain tegmentum and the ratio of these measurements to the pons size. The midbrain:pons ratio is significantly reduced compared to that in PD, MSA-parkinsonism, and healthy controls.[44] The midsagittal view of the brainstem in PSP consequently takes on a beaked appearance like a “hummingbird” or “penguin body” (Figure 4).[39,44] Superior cerebellar atrophy can also be assessed and compared to the size of the middle cerebral peduncle.[45] In addition, both gray and white matter reductions, in particular in the anterior and medial thalamic nuclei, can be detected by diffusion tensor imaging, or DTI.[46] Positron emission tomography (PET) is also increasingly being used and shows hypometabolism in the frontal cortex, cingulate, caudate, thalamus, and midbrain.[47] Although still controversial, specific PET ligands such as 18F-T807/8 and 18F-THK523 are likewise being developed and show promise for detecting tau pathology in PSP (and other tau disorders).[48]
Figure 4.
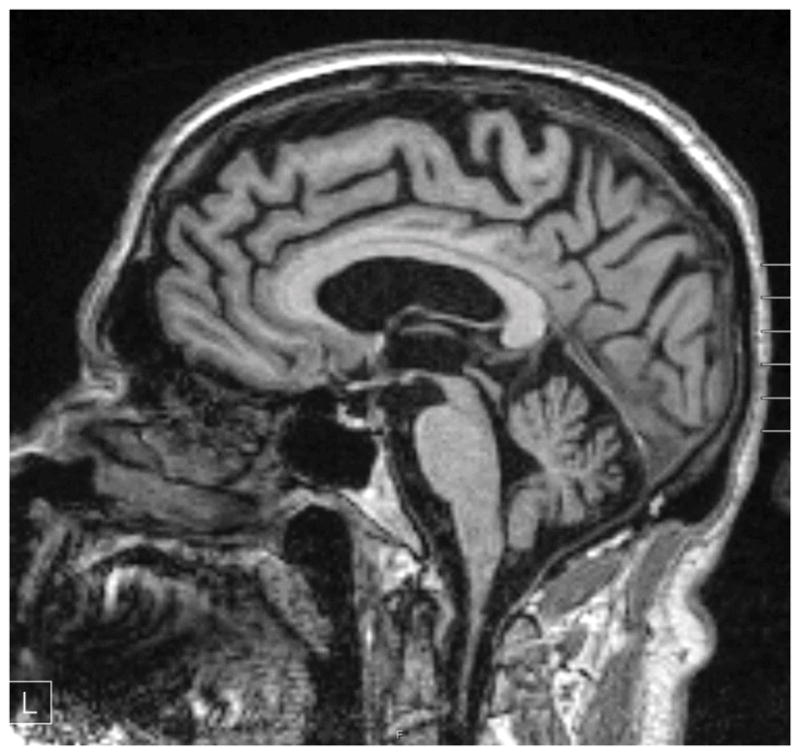
Sagittal T1-weighted MRI image of a patient with PSP-Richardson syndrome. Marked midbrain atrophy is present suggesting the appearance of a “humming bird” or “penguin” sign. Note also cortical atrophy that is frontal-predominant and ex-vacuo ventricular enlargement.
Therapeutic Strategies
Treatment of PSP remains primarily symptomatic and should utilize a multidisciplinary team approach including physical and occupational therapists, speech pathology, neuropsychology, psychiatry, social work, and palliative care. Physical and occupational therapy are critical to prevent falls, maintain mobility, and help with activities of daily living. Speech therapy early on focuses on dysarthria and may include cognitive therapy for speech apraxia. As speech becomes more dysarthric or anarthric, alternative means of communication may be needed. Dysphagia develops early in PSP relative to PD, increasing aspiration risk, morbidity and mortality. Early and regular swallowing evaluations are critical to assess aspiration risk and to intervene with swallow therapy or modify diet. In advanced disease, the question of a feeding tube may arise. The decision to place a feeding tube should be discussed early with the patient, family/caregiver, and speech pathologist. Importantly, although a percutaneous gastrostomy tube can help with nutrition, hydration, and medication administration, it does not reduce risk of aspiration or change overall disease prognosis. Ethical, cultural, and personal considerations regarding quality of life are important to discuss when deciding whether gastrostomy is appropriate. Palliative care options and frank discussion of advanced care directives should be discussed. A social worker or case manager is often helpful to discuss care planning and needs with patients, family and caregivers.
Drug therapy for PSP, while limited, focuses on symptoms (see Table 4 for summary). Parkinsonism may respond to carbidopa-levodopa but often doses up to 300 mg three to four times daily for a few months are required to determine responsiveness. PSP-Parkinsonism in particular can respond to levodopa initially, whereas Richardson syndrome and other variants are more often refractory. Dopamine agonists typically provide minimal benefit in PSP. Limited evidence suggests that amantadine may help gait and dysphagia.[49] Alternative therapies include the supplement coenzyme Q10, which has been shown in a small randomized clinical trial to provide modest benefit in PSP, including slight improvement in cognitive function.[50] More recently, a larger trial treatment with coenzyme Q10 2400 mg daily showed no benefit compared to placebo,[51] though this trial was limited by a high drop-out rate of nearly 40%. Painful dystonic posturing of the neck and limbs, blepharospasm, and eye-opening apraxia can be treated with botulinum toxin. For cognitive decline and dementia, cholinesterase inhibitors such as rivastigmine provide only a modest benefit. Mood disturbance, when identified, may include depression, anxiety, and irritability and should be treated with antidepressants. Some selective serotonin reuptake inhibitors (SSRI’s) may worsen apathy; instead, practitioners may consider more activating antidepressants such as bupropion, venlafaxine, sertraline or fluoxetine. For pseudobulbar affect dextromethorphan-quinidine may be helpful if refractory to antidepressant therapy.
Table 4.
Symptomatic Treatment Options for PSP/CBS
| Symptom | Treatment Options |
|---|---|
| Parkinsonism | Levodopa trial (200–300 mg TID to QID as tolerated) to determine responsiveness of motor symptoms (especially PSP-Parkinsonism).[9] Dopamine agonists – minimal benefit Amantadine – possible benefit[49] |
| Gait imbalance, falls | Physical therapy and assistive devices |
| Dystonic posturing, spasticity, blepharospasm | Botulinum toxin, muscle relaxants (e.g., baclofen, cyclobenzaprine); avoid anticholinergics due to potential for confusion |
| Dysarthria | Speech therapy, adaptive assistive communication device |
| Dysphagia | Swallow evaluation (with fluoroscopy) and therapy; modified diet, aspiration precautions |
| Sialorrhea | Anticholinergics (glycopyrrolate, scopolamine) may worsen cognition; botulinum toxin in parotid and submandibular glands (can worsen dysphagia); sublingual atropine 1% ophthalmic drops (palliative) |
| Ocular symptoms | Zolpidem – short term improvement in voluntary saccades[95,96] Balance and eye movement therapy, better control downward saccades Dry eyes – ophthalmic lubricants Photosensitivity – sunglasses Diplopia – ophthalmology referral, prism glasses Eye opening apraxia – eyelid crutches or myomectomy (not recommended) |
| Urinary frequency | Antispasmodics (e.g., troposium, solifenacin—newer agents are preferred over oxybutynin as less likely to cross blood-brain barrier and cause delirium) versus tricyclic antidepressants (e.g., amitriptyline, desipramine). Consider desmopressin for frequent nocturia. |
| Dementia | Cholinesterase inhibitors (rivastigmine, donepezil) |
| Depression/Anxiety | Tricyclic antidepressants, SSRI’s, SNRI’s |
| Pseudobulbar affect | Dextromethorphan plus quinidine |
(Reprinted with permission from McFarland NR. Diagnostic Approach to Atypical Parkinsonian Syndromes. Continuum 2016;22(4):1129. doi: 10.1212/CON.0000000000000348)
CORTICOBASAL SYNDROME
Corticobasal (or cortico-basal ganglionic) degeneration (CBD) classically describes an atypical parkinsonian syndrome that presents with asymmetric parkinsonism, dystonia, and ideomotor apraxia. The term corticobasal syndrome (CBS) is increasingly used during life as the clinical presentation thought initially to be specific to CBD has been shown to occur due to other neuropathologies, including Alzheimer disease and even PSP. Most commonly however, patients present with marked asymmetric hand dysfunction followed by bradykinesia, tremor, rigidity, and a frontal syndrome.[52] Additional features include a coarse rest/action tremor; limb dystonia resulting in contractures; alien limb phenomenon; hand, limb, gait, or speech apraxia; myoclonus; cortical sensory loss; language deficits; frontal/cortical dementia; supranuclear gaze palsy; dysarthria/dysphagia; gait and postural instability; and hyperreflexia with extensor plantar response. Onset of disease generally occurs in the mid 60’s and prognosis is often poor with a mean survival of approximately 7 years from diagnosis.
Features somewhat unique to CBS include ideomotor apraxia, myoclonus, and alien limb phenomenon.[53] Inability to perform a skilled motor task with intact language, motor, and sensory function defines ideomotor apraxia. Examples include inability to imitate gestures or mime a certain task (e.g., cut with a pair of scissors or show a Victory sign). As symptoms progress, limb-kinetic apraxia, which is independent of modality (imitation versus miming) may become prominent and difficult to distinguish from ideomotor apraxia. Alien limb typically manifests as spontaneous levitation of an arm or leg, but can also include abnormal grasping, posturing, or pursuit versus avoidance of a tactile stimulus in the contralateral limb. Dementia in CBS is a later feature and sematic memory is often preserved.[54] Neuropsychiatric testing demonstrates deficits in attention, concentration, verbal fluency, language, praxis, executive and visuospatial function.[55] Other cortical findings such as aphasia, limb apraxia, and graphesthesia depend on the predominant hemisphere affected.
Clinical criteria for the diagnosis of CBS have evolved with the recognition of variants and diverse pathology. In 2013, Armstrong and colleagues proposed new diagnostic criteria for CBS including four phenotypes: classic CBS, frontal behavioral-spatial syndrome, nonfluent/agrammatic primary progressive aphasia (naPPA), and a PSP syndrome.[28] Probable and possible criteria were described for CBS and include insidious onset, gradual progression of symptoms for more than 1 year, and age of onset 50 years or more, with or without a similar family history or known tau mutations.[28] The frontal behavioral variant CBS involves executive dysfunction, behavioral or personality changes. Progressive nonfluent/agrammatic PPA is characterized by effortful, agrammatic speech with impaired grammar/sentence comprehension and groping, or distorted speech production (i.e., apraxia of speech). The PSP-CBS as expected includes axial or symmetric limb rigidity/akinesia, postural instability, falls, urinary incontinence, behavioral changes, and supranuclear vertical gaze palsy.
Pathology
CBS (as CBD) has classically been considered a distinct clinicopathological entity, but more recently it has been shown to have different pathologies. Indeed, classic CBD pathology is found in only 25% to 56% of suspected CBS cases.[28] CBD pathology is characterized by widespread neurodegeneration, and despite the typical asymmetrical clinical presentation, involves both cortical hemispheres including the basal ganglia, thalamus, substantia nigra, subthalamic nucleus, and red nucleus. As in PSP, widespread 4R-predominant tau pathology is present in neurons and glia. While tufted astrocytes are typically absent, “coiled bodies” are frequent in oligodendroglia, as well as in astrocytic plaques and corticobasal inclusions.[56] Intranuclear inclusions in subcortical nuclei such as the substantia nigra are characteristic of globose neurofibrillary tangles, whereas those in the cortex are granular, crescent-shaped, or globular.[57]
Diagnostic and Therapeutic Strategies
To date there are no specific diagnostic biomarkers for CBS. Neuroimaging findings such asymmetric frontoparietal atrophy seen on CT/MRI, however, may be helpful in differentiating CBS from other parkinsonian syndromes.[58] Functional imaging such as [F18]fluorodeoxyglucose positron emission tomography (FDG-PET) may also reveal asymmetric cortical metabolism. Similarly, striatal uptake on [18F]dopa–positron emission tomography (DOPA-PET) and [123I]β-CIT (2beta-carbomethoxy-3beta-(4-iodophenyl)tropane) binding (SPECT imaging, similar to DaT-Scan™) are reduced and asymmetric, consistent with dopaminergic denervation—although nonspecific to CBS. In addition, both tau and beta-amyloid ligands are being explored to diagnose and differentiate CBS from other disorders.[59]
Treatment of CBS remains primarily supportive. Similar to PSP, a multidisciplinary approach involving rehabilitation services and palliative care is critical to maintain function and comfort despite continued disease progression. Regular exercise and activity early in the disease helps with independent mobility and may delay disability. Although dopaminergic therapy rarely provides improvement in classic CBS, a levodopa trial up to 250–300 mg administered three to four times daily should in most cases be attempted as it sometimes provides partial benefit for rigidity. Muscle relaxants (e.g., baclofen) and botulinum toxin can help painful rigidity and dystonic (contractures) that may limit function. Myoclonus in CBS is typically cortical and may respond to levetiracetam or clonazepam, but should only be treated if bothersome.
MULTIPLE SYSTEM ATROPHY
MSA is an adult-onset, sporadic, progressive, multisystem, neurodegenerative disease characterized by a variable expression of parkinsonism, cerebellar and pyramidal signs, and autonomic dysfunction. First described in the 1960s, MSA has been called by a variety in different names including striatonigral degeneration (SND), Shy Drager syndrome, and olivopontocerebellar atrophy (OPCA), depending on the predominant presenting symptoms.[60] The discovery of pathologic oligodendroglial cytoplasmic inclusions (GCI) and multisystem neurodegeneration regardless of the clinical phenotype, however, suggests a common pathology and this the current nomenclature which can be confusing to even experts. The current consensus statement on MSA now recognizes two clinical phenotypes: MSA-P which is distinguished by predominant parkinsonism and MSA-C which has dominant cerebellar features.[61] The prevalence of MSA is similar to PSP and estimated at 3.4 to 4.9 cases per 100,000 individuals.[62] The median age of onset for MSA is earlier at 58 years of age and cutoff for early diagnosis 40 years-old. Disease progression is generally more rapid than in PD and the mean survival is 6 to 10 years,[63,64] consistent with more widespread neurodegeneration.
Clinical Features
Although MSA-P presents with predominant parkinsonism, there are several features that distinguish it from PD. In contrast to the typical pill-rolling type of tremor typically seen in PD, tremor in MSA-P is often higher in frequency and lower in amplitude with a jerky, stimulus-sensitive myoclonic postural/action component.[65] Parkinsonism in MSA is further differentiated from PD by its more symmetrical appearance, dystonia (axial and appendicular), early dysarthria/dysphonia, gait and postural instability (though later compared to that in PSP), rapid progression, and autonomic dysfunction. Speech is often characterized by a mixed spastic, hypokinetic dysarthria with a vocal flutter or dysphonia. Dysphagia also occurs earlier and is more severe than in PD. Some patients may also develop respiratory or laryngeal stridor requiring CPAP. Other features include hyperreflexia, disproportionate anterocollis, and early striatal deformities. Parkinsonism in MSA may initially responds to dopaminergic therapy, but often doses are limited by side effects including exacerbation of orthostatic symptoms and dyskinesia (in particular orofacial dyskinesia and dystonia).
MSA-C is one of the most common causes of sporadic, adult-onset ataxia, occurring in the setting of parkinsonism, autonomic dysfunction, and a relatively rapid progression compared to PD.[65] In MSA-C, cerebellar dysfunction manifests as gait and balance impairment, limb ataxia, dysarthria, and oculomotor dysmetria (hypometric/hypermetric saccades, jerky pursuits, and nystagmus). Features common to both subtypes of MSA include sleep disturbances (i.e., sleep apnea, RBD and respiratory and autonomic dysfunction, which can precede motor signs by several months to years.[66] Orthostatic hypotension is common and frequently exacerbated by treatment. Urogenital dysfunction is manifest by incomplete bladder emptying or urinary frequency/urgency/incontinence, particularly in women, and erectile dysfunction in men. Additional features can include Pisa syndrome, dystonic lateral flexion of the spine, abnormal forward truncal flexion (camptocormia), respiratory insufficiency, and pseudobulbar affect.[67] These “red flags” may help to distinguish MSA from PD (Table 5). Cognitive dysfunction and dementia are also increasingly recognized in MSA.[68]
Table 5.
Differentiating features of MSA-P and MSA-C from PD (“Red flags”)
| MSA-P | MSA-C |
|---|---|
| Symmetrical onset | Cerebellar limb, gait ataxia |
| Rapid symptom progression | Early gait instability, falls |
| Jerky, myoclonic, postural/action tremor | Dysarthria (scanning, ataxic) |
| Contractures of hands and feet | Dysphagia |
| Anterocollis, axial dystonia (camptocormia ± lateral flexion, or Pisa syndrome) | Gaze impairment (hypo/hyperkinetic saccades) |
| Early gait difficulty, falls | Lower and upper motor neuron signs |
| Severe dysphonia, dysarthria | Emotionality, depression, anxiety |
| New/increased snoring, sleep apnea | Progressive dementia |
| Respiratory/laryngeal stridor | No family history of ataxia or parkinsonsism |
| Hyperreflexia, Babinski’s | |
| Pseudobulbar affect (emotional lability) | |
| Cold hands/feet | |
| Dysautonomia (69% vs 5% in PD) | |
| Poor/unsustained levodopa response (~30%) | |
| Orofacial dyskinesia/dystonia |
(Reprinted with permission from McFarland NR. Diagnostic Approach to Atypical Parkinsonian Syndromes. Continuum 2016;22(4):1134. doi: 10.1212/CON.0000000000000348)
Pathology
MSA is characterized by oligodendroglial cytoplasmic inclusions (GCIs) that are enriched with α-synuclein and correlate with the severity of neuronal loss and disease duration.[69] Neurodegeneration and gliosis are widespread and involve the putamen, substantia nigra, pons, inferior olivary nucleus, cerebellum, and brainstem.[70] The degree of abnormalities found in the nigrostriatal system, pons, and cerebellum correlate with the subtype of MSA. Pathology of the dorsal motor nucleus of the vagus, locus coeruleus, ventrolateral medulla, and intermediolateral cell columns of the spinal cord has been for example associated with autonomic dysfunction.
Diagnostic and Therapeutic Strategies
Adjunctive diagnostic testing may aide in the distinction of MSA from PD and other parkinsonian syndromes. While serum biomarkers such as α-synuclein—total, phosphorylated, and oligomeric forms—remain controversial, neuroimaging can be helpful along with careful clinical assessment.[71] Brain MRI in MSA-P may demonstrate T2 hypointensity in the posterolateral putamen, representing neurodegeneration and iron deposition, and slit hyperintensity at the lateral margin of the putamen.[72,73] In MSA-C olivoponotocerebellar atrophy is predominant, and pontine atrophy and gliosis may manifest as a “hot cross bun”-like pattern on T2 imaging (Figure 5). PET and SPECT imaging have also been shown to help with MSA diagnosis and show decreased metabolism and reduced binding of 125I-ioflupane in the striatum.[74] Voxel-based morphometry and functional MRI demonstrate reduced volumes in the basal ganglia, cerebellum, and pons in MSA compared to PD.[75] In addition to imaging, autonomic testing such as tilt-table testing and 24-hour ambulatory blood pressure and heart rate monitoring can help assess baroreceptor sensitivity and risk for orthostatic hypotension. While not readily available in most centers, 123I-metaiodobenzylguanidine (MIBG) scintography can be useful to differentiate MSA from PD and shows reduced cardiac uptake in PD but not in MSA.[76] Other tests to consider include a gastric emptying study for gastroparesis (though not specific for MSA or PD), urodynamics, and sudomotor testing. A formal sleep study can also help identify sleep apnea, stridor, periodic limb movements, or RBD.
Figure 5.
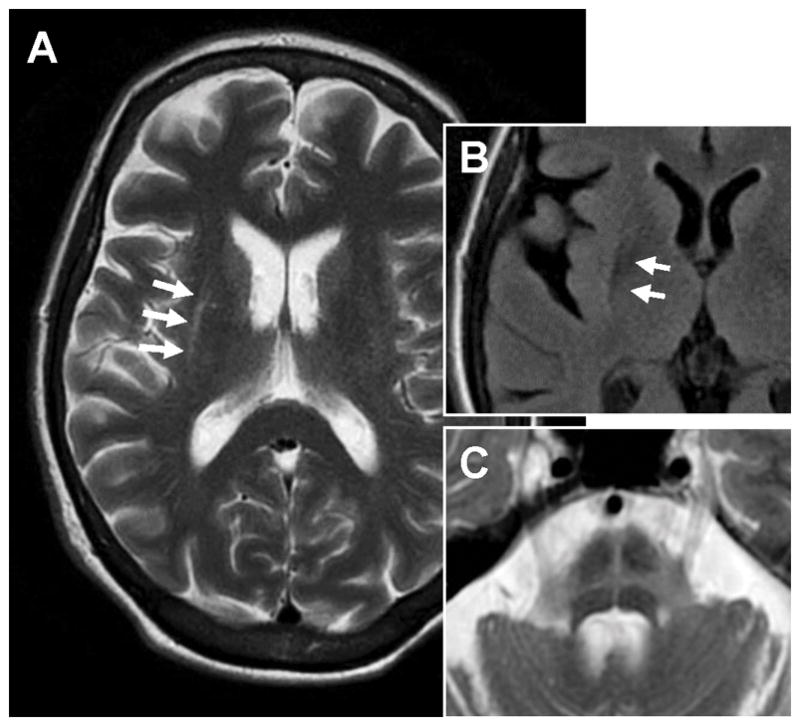
Axial T2-weighted MRI images of a MSA-parkinsonism patient demonstrating (A) slit-like hyperintensity (arrows) at the rim of the right putamen, (B) hypointensity of posterior putamen (better seen on FLAIR), and (C) typical “hot cross bun” sign representing atrophy and gliosis of the pons. (Reprinted with permission from McFarland NR. Diagnostic Approach to Atypical Parkinsonian Syndromes. Continuum 2016;22(4):1137. doi: 10.1212/CON.0000000000000348)
Treatment of MSA requires a multisystem team approach by allied health care providers that involves symptomatic therapy, rehabilitation services, and supportive care. To date, there are no effective medications that treat the cerebellar ataxia or pyramidal symptoms of MSA. However, Parkinsonism in MSA initially responds to dopaminergic therapy in about a third of patients, but response frequently wanes over time in contrast to what is seen in PD. A trial of up to 200 to 300 mg levodopa three to four times daily (1000–1200 mg/d) as tolerated for a period of three months is recommended to assess response, [62,77] though both levodopa-induced dyskinesias (orofacial) and other side effects may limit therapy. Dopamine agonists can be used as a second-line treatment, but are generally inferior to levodopa for MSA and have not been carefully studied. Recently, rasagiline has been studied in MSA-P but failed to show a benefit as assessed by the Unified MSA Rating Scale.[78] Amantadine is another option for treatment of Parkinsonism in MSA, but lacks strong evidence and the potential adverse effects may be relatively high.[79] Deep brain stimulation is generally not recommended in MSA due to poor response despite occasionally reducing tremor or dyskinesia.
Treatment of autonomic dysfunction such as orthostatic hypotension should include conservative measures such as oral hydration, increased salt intake, and compression stockings/abdominal binders. Pharmacological options include fludrocortisone (0.1 mg 1–3/day) which increases blood volume, but may be contraindicated in heart failure patients. Midodrine (2.5–10 mg) and droxidopa (100–300 mg) given two to three times daily can also be used to treat neurogenic orthostatic hypotension, but have the potential to cause supine hypertension. Pyridostigmine is an alternative that increases blood pressure without supine hypertension, but may have a modest effect and require high doses that result in intolerance. Octreotide can be used for postprandial hypotension. For neurogenic bladder, antispasmodics (e.g., oxybutynin) or botulinum toxin injections can be helpful. Intermittent self-catheterization or placement of a suprapubic catheter may be required in some patients. Sleep apnea and inspiratory stridor benefit from ventilation by continuous positive airway pressure (CPAP).
DEMENTIA WITH LEWY BODIES
Dementia with Lewy bodies (DLB) is the second most common form of neurodegenerative dementia, after AD, and has similar features to other dementias including PD dementia (PDD). Indeed, DLB is of the PD disease spectrum and is characterized by is early-onset, rapidly progressive dementia. The clinical criteria for DLB include 1) parkinsonism that is coincident with or follows the onset of dementia, 2) mental status fluctuations, and 3) recurrent visual hallucinations.[80] Additional features include gait instability, falls, orthostatic hypotension (sometimes causing confusion with MSA), RBD, depression, delusions/paranoia (psychosis) and neuroleptic sensitivity.
Pathology
The pathological hallmark of DLB (and similarly in PDD) is diffuse Lewy bodies, characterized by intracellular inclusions enriched with α-synuclein (Figure 6). Incidental Lewy body pathology is found at autopsy in 10 to 12% healthy individuals without signs of parkinsonism and may indicate preclinical disease.[81–83] The degree of cortical Lewy body pathology in DLB correlates well with dementia severity.[84] Spread of Lewy body pathology, or synucleinopathy, from brainstem to neocortex as described by Braak and colleagues is thought to contribute to progressive cognitive impairment.[85] Most cases of DLB (and PDD) have concurrent Alzheimer-type pathology that likely also contributes to dementia.[86]
Figure 6.
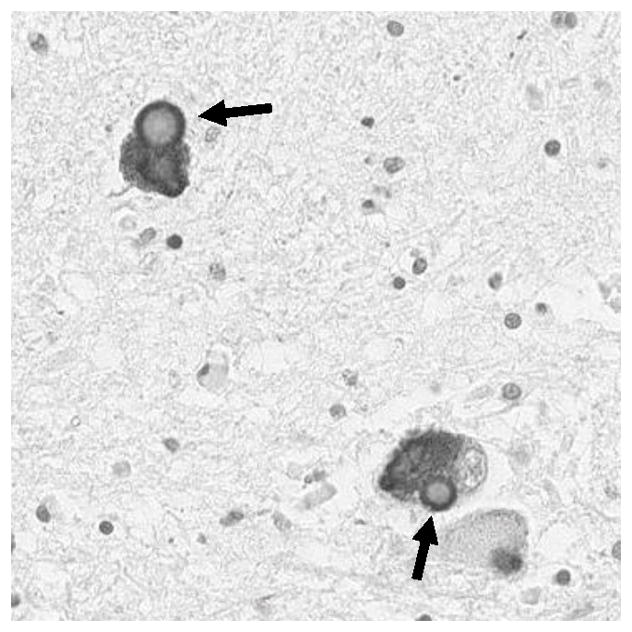
Typical Lewy bodies in are found in the pigmented cells of the substantial nigra in both Parkinson’s disease and Lewy body dementia. The photo shows a Lewy body is stained with an antibody specific to the phosphorylated form (serine-129) of α-synuclein found enriched in Lewy bodies. Note the characteristic round appearance with lighter core and dark halo.
Diagnostic and Therapeutic Strategies
Developing neuroimaging biomarkers currently hold the most promise for distinguishing DLB from AD and other Parkinsonian syndromes.[87] Brain MRI typically demonstrates diffuse cerebral atrophy with relative preservation of occipital and mesiotemporal lobes compared to that seen in AD.[88] Functional imaging such as SPECT and PET may also help discriminate DLB from AD and show occipital hypoperfusion. In addition, preserved metabolism of the posterior cingulate gyrus relative to the precuneus on FDG-PET has recently been shown to correlate with DLB and is coined the “cingulate island” sign.[89] Dopaminergic imaging (i.e., DAT SPECT) while nonspecific may be abnormal in early DLB and demonstrate nigrostriatal denervation. Likewise, amyloid burden can be assessed with PiB (Pittsburg compound B) scanning and associates more with PDD than DLB.[90] In addition to imaging, CSF analysis of tau and α-synuclein levels may be useful in differentiating DLB from AD.[91]
As with other atypical parkinsonian syndromes, treatment of DLB is primarily symptomatic and requires a multidisciplinary team approach. Parkinsonism in DLB typically responds to levodopa, however treatment must be balanced against the potential for exacerbating psychosis and hallucinations. Dopaminergic agonists are not preferred, and non-dopaminergic agents such as monoamine oxidase inhibitors (MAOI’s), amantadine, and anticholinergics are often avoided due to their potential to cause confusion and worsen psychosis. First-line treatment for hallucinations and psychosis is reduction in dopaminergic medications, but if necessary atypical antipsychotics are added. Frequently, quetiapine is used and can be helpful to reduce agitation associated with sundowning, but studies do not support its efficacy for hallucinations. Clozapine has been shown to be proven effective but requires routine blood monitoring for agranulocytosis and may exacerbate orthostatic hypotension and depression.[92] Pimavanserin, a selective 5-HT2 inverse agonist, has recently been (FDA) approved for treatment of psychosis in PD but not DLB.[93] Cognitive impairment and fluctuations in DLB benefit from treatment with cholinesterase inhibitors such as rivastigmine and donepezil.[94] Comorbid depression, apathy, and anxiety should also be treated if identified.
CONCLUSIONS
Although there is considerable overlap of features between the atypical parkinsonian syndromes and PD, attention to historical details (such as mode of presentation and the rate of disease progression) and specific clinical features or “red flags” may aide in the differentiation of these overlapping entities. Frequent revision of clinical criteria to better encompass the clinical heterogeneity and the varied phenotypes of these syndromes may also help with diagnosis and will become increasingly important with the emergence of novel therapies. Understanding of the clinicopathological correlates and the development of novel diagnostic biomarkers will help further efforts toward disease-modifying therapeutics for both PD and the atypical parkinsonian syndromes.
Acknowledgments
Dr. McFarland has received support from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (K08-NS067024), the Michael J. Fox Foundation, and the Huntington Disease Society of America. Dr. Hess receives grant support from the University of Florida Clinical and Translational Research Institute, which is supported in part by NIH award KL2 TR001429. He has served as a research committee member for the Michael J. Fox Foundation and as a speaker for the National Parkinson Foundation, the Parkinson’s Disease Foundation, and the Davis Phinney Foundation. Dr. Hess has participated in CME and educational activities on movement disorders sponsored by Allergan, Ipsen, Mertz Pharmaceuticals, Peerview Online, and QuantiaMD. This center’s work was supported by a Center Excellence grant from the Parkinson’s Foundation. Drs. McFarland and Hess report no relevant conflicts of interests for this publication.
Abbreviations
- AD
Alzheimer disease
- CBD
Corticobasal degeneration
- CBS
Corticobasal syndrome
- CSF
Cerebrospinal fluid
- DAT-SPECT
Dopamine transporter single-photon emission computed tomography
- DLB
Dementia with Lewy bodies
- DTI
Diffusion-tensor imaging
- FDG-PET
18F-fluorodeoxyglucose positron emission tomography
- FTD
Frontotemporal dementia
- MRI
Magnetic resonance imaging
- MSA
Multiple system atrophy
- MSA-C
Multiple system atrophy–cerebellar type
- MSA-P
Multiple system atrophy–parkinsonian type
- PD
Parkinson disease
- PiB
Pittsburg compound B
- PET
Positron emission tomography
- PLS
Primary lateral sclerosis
- PSP
Progressive supranuclear palsy
- PSP-bvFTD
Progressive supranuclear palsy behavioral variant of FTD
- PSP-C
Progressive supranuclear palsy-cerebellar ataxia
- PSP-CBS
Progressive supranuclear palsy-Corticobasal Syndrome
- PSP-P
Progressive supranuclear palsy-Parkinsonism
- PSP-PAGF
Progressive supranuclear palsy-Pure Akinesia Gait Freezing
- PSP-PLS
Progressive supranuclear palsy- primary lateral sclerosis
- REM
Rapid Eye Movement
- SNRI
Serotonin norepinephrine reuptake inhibitor
- SPECT
Single-photon emission computed tomography
- SSRI
Selective serotonin reuptake inhibitor
REFERENCES (Thieme style)
- 1.Daniel SE, Lees AJ. Parkinson’s Disease Society Brain Bank, London: overview and research. J Neural Transm Suppl. 1993;39:165–172. [PubMed] [Google Scholar]
- 2.Litvan I, MacIntyre A, Goetz CG, et al. Accuracy of the clinical diagnoses of Lewy body disease, Parkinson disease, and dementia with Lewy bodies: a clinicopathologic study. Archives of neurology. 1998;55:969–978. doi: 10.1001/archneur.55.7.969. [DOI] [PubMed] [Google Scholar]
- 3.Hughes AJ, Daniel SE, Kilford L, et al. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson’s disease. Neurology. 2001;57:1497–1499. doi: 10.1212/wnl.57.8.1497. [DOI] [PubMed] [Google Scholar]
- 5.Rajput AH, Rozdilsky B, Rajput A. Accuracy of Clinical-Diagnosis in Parkinsonism - a Prospective-Study. Can J Neurol Sci. 1991;18:275–278. doi: 10.1017/s0317167100031814. [DOI] [PubMed] [Google Scholar]
- 6.Adler CH, Beach TG, Hentz JG, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology. 2014;83:406–412. doi: 10.1212/WNL.0000000000000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mark MH, Dickson DW, Sage JI, et al. The clinicopathologic spectrum of Lewy body disease. Adv Neurol. 1996;69:315–318. [PubMed] [Google Scholar]
- 8.Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30:1591–1601. doi: 10.1002/mds.26424. [DOI] [PubMed] [Google Scholar]
- 9.Richardson JC, Steele J, Olszewski J. Supranuclear ophthalmoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight cases of "heterogenous system degeneration". Trans Am Neurol Assoc. 1963;88:25–29. [PubMed] [Google Scholar]
- 10.Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet. 1999;354:1771–1775. doi: 10.1016/s0140-6736(99)04137-9. [DOI] [PubMed] [Google Scholar]
- 11.Golbe LI, Davis PH, Schoenberg BS, et al. Prevalence and natural history of progressive supranuclear palsy. Neurology. 1988;38:1031–1034. doi: 10.1212/wnl.38.7.1031. [DOI] [PubMed] [Google Scholar]
- 12.Nath U, Ben-Shlomo Y, Thomson RG, et al. The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK. Brain. 2001;124:1438–1449. doi: 10.1093/brain/124.7.1438. [DOI] [PubMed] [Google Scholar]
- 13.Bower J, Maraganore D, McDonnell K, et al. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology. 1997;49:1284–1288. doi: 10.1212/wnl.49.5.1284. [DOI] [PubMed] [Google Scholar]
- 14.Josephs KA, Dickson DW. Diagnostic accuracy of progressive supranuclear palsy in the Society for Progressive Supranuclear Palsy brain bank. Mov Disord. 2003;18:1018–1026. doi: 10.1002/mds.10488. [DOI] [PubMed] [Google Scholar]
- 15.Pastor P, Tolosa E. Progressive supranuclear palsy: clinical and genetic aspects. Curr Opin Neurol. 2002;15:429–437. doi: 10.1097/00019052-200208000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Williams DR, Watt HC, Lees AJ. Predictors of falls and fractures in bradykinetic rigid syndromes: a retrospective study. J Neurol Neurosurg Psychiatry. 2006;77:468–473. doi: 10.1136/jnnp.2005.074070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brett FM, Henson C, Staunton H. Familial diffuse Lewy body disease, eye movement abnormalities, and distribution of pathology. Archives of neurology. 2002;59:464–467. doi: 10.1001/archneur.59.3.464. [DOI] [PubMed] [Google Scholar]
- 18.Batla A, Nehru R, Vijay T. Vertical wrinkling of the forehead or Procerus sign in Progressive Supranuclear Palsy. Journal of the neurological sciences. 2010;298:148–149. doi: 10.1016/j.jns.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Litvan I, Mega MS, Cummings JL, et al. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology. 1996;47:1184–1189. doi: 10.1212/wnl.47.5.1184. [DOI] [PubMed] [Google Scholar]
- 20.Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257:1382–1387. doi: 10.1007/s00415-010-5550-3. [DOI] [PubMed] [Google Scholar]
- 21.Pharr V, Uttl B, Stark M, et al. Comparison of apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology. 2001;56:957–963. doi: 10.1212/wnl.56.7.957. [DOI] [PubMed] [Google Scholar]
- 22.Litvan I, Hauw JJ, Bartko JJ, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol. 1996;55:97–105. doi: 10.1097/00005072-199601000-00010. [DOI] [PubMed] [Google Scholar]
- 23.Kanazawa M, Shimohata T, Toyoshima Y, et al. Cerebellar involvement in progressive supranuclear palsy: A clinicopathological study. Movement disorders : official journal of the Movement Disorder Society. 2009;24:1312–1318. doi: 10.1002/mds.22583. [DOI] [PubMed] [Google Scholar]
- 24.Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. 2014;29:1758–1766. doi: 10.1002/mds.26054. [DOI] [PubMed] [Google Scholar]
- 25.Williams DR, de Silva R, Paviour DC, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain. 2005;128:1247–1258. doi: 10.1093/brain/awh488. [DOI] [PubMed] [Google Scholar]
- 26.Imai H, Narabayashi H. Akinesia-concerning two cases of pure akinesia. 1974;18:787–794. [Google Scholar]
- 27.Williams DR, Holton JL, Strand K, et al. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord. 2007;22:2235–2241. doi: 10.1002/mds.21698. [DOI] [PubMed] [Google Scholar]
- 28.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80:496–503. doi: 10.1212/WNL.0b013e31827f0fd1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8:270–279. doi: 10.1016/S1474-4422(09)70042-0. [DOI] [PubMed] [Google Scholar]
- 30.Hassan A, Parisi JE, Josephs KA. Autopsy-proven progressive supranuclear palsy presenting as behavioral variant frontotemporal dementia. Neurocase. 2011 doi: 10.1080/13554794.2011.627345. [DOI] [PubMed] [Google Scholar]
- 31.Kaat L, Boon A, Azmani A, et al. Familial aggregation of parkinsonism in progressive supranuclear palsy. Neurology. 2009;73:98–105. doi: 10.1212/WNL.0b013e3181a92bcc. [DOI] [PubMed] [Google Scholar]
- 32.King A, Curran O, Al-Sarraj S. Atypical progressive supranuclear palsy presenting as primary lateral sclerosis. J Neurol Sci. 2013;329:69. doi: 10.1016/j.jns.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 33.Nagao S, Yokota O, Nanba R, et al. Progressive supranuclear palsy presenting as primary lateral sclerosis but lacking parkinsonism, gaze palsy, aphasia, or dementia. J Neurol Sci. 2012;323:147–153. doi: 10.1016/j.jns.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Dickson DW, Ahmed Z, Algom AA, et al. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. 2010;23:394–400. doi: 10.1097/WCO.0b013e32833be924. [DOI] [PubMed] [Google Scholar]
- 35.Jellinger K. Cerebellar involvement in progressive supranuclear palsy. Movement disorders : official journal of the Movement Disorder Society. 2010;25:1104–1105. doi: 10.1002/mds.23045. [DOI] [PubMed] [Google Scholar]
- 36.Stamelou M, de Silva R, Arias-Carrion O, et al. Rational therapeutic approaches to progressive supranuclear palsy. Brain. 2010;133:1578–1590. doi: 10.1093/brain/awq115. [DOI] [PubMed] [Google Scholar]
- 37.Höglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oba H, Yagishita A, Terada H, et al. New and reliable MRI diagnosis for progressive supranuclear palsy. Neurology. 2005;64:2050–2055. doi: 10.1212/01.WNL.0000165960.04422.D0. [DOI] [PubMed] [Google Scholar]
- 39.Groschel K, Kastrup A, Litvan I, et al. Penguins and hummingbirds: midbrain atrophy in progressive supranuclear palsy. Neurology. 2006;66:949–950. doi: 10.1212/01.wnl.0000203342.77115.bf. [DOI] [PubMed] [Google Scholar]
- 40.Paviour DC, Price SL, Jahanshahi M, et al. Longitudinal MRI in progressive supranuclear palsy and multiple system atrophy: rates and regions of atrophy. Brain. 2006;129:1040–1049. doi: 10.1093/brain/awl021. [DOI] [PubMed] [Google Scholar]
- 41.Paviour DC, Thornton JS, Lees AJ, et al. Diffusion-weighted magnetic resonance imaging differentiates Parkinsonian variant of multiple-system atrophy from progressive supranuclear palsy. Mov Disord. 2007;22:68–74. doi: 10.1002/mds.21204. [DOI] [PubMed] [Google Scholar]
- 42.Slowinski J, Imamura A, Uitti RJ, et al. MR imaging of brainstem atrophy in progressive supranuclear palsy. J Neurol. 2008;255:37–44. doi: 10.1007/s00415-007-0656-y. [DOI] [PubMed] [Google Scholar]
- 43.Tsuboi Y, Slowinski J, Josephs KA, et al. Atrophy of superior cerebellar peduncle in progressive supranuclear palsy. Neurology. 2003;60:1766–1769. doi: 10.1212/01.wnl.0000068011.21396.f4. [DOI] [PubMed] [Google Scholar]
- 44.Oba H, Yagishita A, Terada H, et al. New and reliable MRI diagnosis for progressive supranuclear palsy. Neurology. 2005;64:2050–2055. doi: 10.1212/01.WNL.0000165960.04422.D0. [DOI] [PubMed] [Google Scholar]
- 45.Longoni G, Agosta F, Kostic VS, et al. MRI measurements of brainstem structures in patients with Richardson’s syndrome, progressive supranuclear palsy-parkinsonism, and Parkinson’s disease. Mov Disord. 2011;26:247–255. doi: 10.1002/mds.23293. [DOI] [PubMed] [Google Scholar]
- 46.Knake S, Belke M, Menzler K, et al. In vivo demonstration of microstructural brain pathology in progressive supranuclear palsy: a DTI study using TBSS. Mov Disord. 2010;25:1232–1238. doi: 10.1002/mds.23054. [DOI] [PubMed] [Google Scholar]
- 47.Renard D, Collombier L, Castelnovo G, et al. Teaching NeuroImages: FDG-PET in progressive supranuclear palsy. Neurology. 2010;74:e60. doi: 10.1212/WNL.0b013e3181d7d871. [DOI] [PubMed] [Google Scholar]
- 48.Ariza M, Kolb HC, Moechars D, et al. Tau Positron Emission Tomography (PET) Imaging: Past, Present, and Future. J Med Chem. 2015;58:4365–4382. doi: 10.1021/jm5017544. [DOI] [PubMed] [Google Scholar]
- 49.Norris FH, Panitch HS, Denys EH, et al. The treatment of subacute sclerosing panencephalitis with interferon: a case report. J Neurol. 1986;233:102–107. doi: 10.1007/BF00313855. [DOI] [PubMed] [Google Scholar]
- 50.Stamelou M, Reuss A, Pilatus U, et al. Short-term effects of coenzyme Q10 in progressive supranuclear palsy: a randomized, placebo-controlled trial. Mov Disord. 2008;23:942–949. doi: 10.1002/mds.22023. [DOI] [PubMed] [Google Scholar]
- 51.Apetauerova D, Scala SA, Hamill RW, et al. CoQ10 in progressive supranuclear palsy: A randomized, placebo-controlled, double-blind trial. Neurol Neuroimmunol Neuroinflamm. 2016;3:e266. doi: 10.1212/NXI.0000000000000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wenning GK, Litvan I, Jankovic J, et al. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry. 1998;64:184–189. doi: 10.1136/jnnp.64.2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Litvan I, Agid Y, Goetz C, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology. 1997;48:119–125. doi: 10.1212/wnl.48.1.119. [DOI] [PubMed] [Google Scholar]
- 54.Graham NL, Bak TH, Hodges JR. Corticobasal degeneration as a cognitive disorder. Mov Disord. 2003;18:1224–1232. doi: 10.1002/mds.10536. [DOI] [PubMed] [Google Scholar]
- 55.Pillon B, Gouider-Khouja N, Deweer B, et al. Neuropsychological pattern of striatonigral degeneration: comparison with Parkinson’s disease and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 1995;58:174–179. doi: 10.1136/jnnp.58.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dickson DW, Bergeron C, Chin SS, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61:935–946. doi: 10.1093/jnen/61.11.935. [DOI] [PubMed] [Google Scholar]
- 57.Kouri N, Whitwell JL, Josephs KA, et al. Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol. 2011;7:263–272. doi: 10.1038/nrneurol.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whitwell JL, Jack CR, Jr, Boeve BF, et al. Imaging correlates of pathology in corticobasal syndrome. Neurology. 2010;75:1879–1887. doi: 10.1212/WNL.0b013e3181feb2e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murray ME, Kouri N, Lin WL, et al. Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias. Alzheimers Res Ther. 2014;6:1. doi: 10.1186/alzrt231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stefanova N, Bucke P, Duerr S, et al. Multiple system atrophy: an update. Lancet Neurol. 2009;8:1172–1178. doi: 10.1016/S1474-4422(09)70288-1. [DOI] [PubMed] [Google Scholar]
- 61.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–676. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fanciulli A, Wenning GK. Multiple-system atrophy. The New England journal of medicine. 2015;372:249–263. doi: 10.1056/NEJMra1311488. [DOI] [PubMed] [Google Scholar]
- 63.Wenning GK, Colosimo C, Geser F, et al. Multiple system atrophy. Lancet Neurol. 2004;3:93–103. doi: 10.1016/s1474-4422(03)00662-8. [DOI] [PubMed] [Google Scholar]
- 64.Wenning GK, Geser F, Krismer F, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol. 2013;12:264–274. doi: 10.1016/S1474-4422(12)70327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kollensperger M, Geser F, Seppi K, et al. Red flags for multiple system atrophy. Mov Disord. 2008;23:1093–1099. doi: 10.1002/mds.21992. [DOI] [PubMed] [Google Scholar]
- 66.Jecmenica-Lukic M, Poewe W, Tolosa E, et al. Premotor signs and symptoms of multiple system atrophy. Lancet Neurol. 2012;11:361–368. doi: 10.1016/S1474-4422(12)70022-4. [DOI] [PubMed] [Google Scholar]
- 67.Kollensperger M, Geser F, Ndayisaba JP, et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord. 2010;25:2604–2612. doi: 10.1002/mds.23192. [DOI] [PubMed] [Google Scholar]
- 68.Brown RG, Lacomblez L, Landwehrmeyer BG, et al. Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain. 2010;133:2382–2393. doi: 10.1093/brain/awq158. [DOI] [PubMed] [Google Scholar]
- 69.Wenning GK, Jellinger KA. The role of alpha-synuclein in the pathogenesis of multiple system atrophy. Acta Neuropathol. 2005;109:129–140. doi: 10.1007/s00401-004-0935-y. [DOI] [PubMed] [Google Scholar]
- 70.Ahmed Z, Asi YT, Sailer A, et al. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol. 2012;38:4–24. doi: 10.1111/j.1365-2990.2011.01234.x. [DOI] [PubMed] [Google Scholar]
- 71.Brooks DJ, Seppi K Neuroimaging Working Group on MSA. Proposed neuroimaging criteria for the diagnosis of multiple system atrophy. Mov Disord. 2009;24:949–964. doi: 10.1002/mds.22413. [DOI] [PubMed] [Google Scholar]
- 72.Tha KK, Terae S, Tsukahara A, et al. Hyperintense putaminal rim at 1. 5 T: prevalence in normal subjects and distinguishing features from multiple system atrophy. BMC Neurol. 2012;12:39. doi: 10.1186/1471-2377-12-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bhattacharya K, Saadia D, Eisenkraft B, et al. Brain magnetic resonance imaging in multiple-system atrophy and Parkinson disease: a diagnostic algorithm. Archives of neurology. 2002;59:835–842. doi: 10.1001/archneur.59.5.835. [DOI] [PubMed] [Google Scholar]
- 74.Perju-Dumbrava LD, Kovacs GG, Pirker S, et al. Dopamine transporter imaging in autopsy-confirmed Parkinson’s disease and multiple system atrophy. Mov Disord. 2012;27:65–71. doi: 10.1002/mds.24000. [DOI] [PubMed] [Google Scholar]
- 75.Planetta PJ, Kurani AS, Shukla P, et al. Distinct functional and macrostructural brain changes in Parkinson’s disease and multiple system atrophy. Human brain mapping. 2015;36:1165–1179. doi: 10.1002/hbm.22694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chung EJ, Lee WY, Yoon WT, et al. MIBG scintigraphy for differentiating Parkinson’s disease with autonomic dysfunction from Parkinsonism-predominant multiple system atrophy. Mov Disord. 2009;24:1650–1655. doi: 10.1002/mds.22649. [DOI] [PubMed] [Google Scholar]
- 77.Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci. 1999;163:94–98. doi: 10.1016/s0022-510x(98)00304-9. [DOI] [PubMed] [Google Scholar]
- 78.Poewe W, Seppi K, Fitzer-Attas CJ, et al. Efficacy of rasagiline in patients with the parkinsonian variant of multiple system atrophy: a randomised, placebo-controlled trial. The Lancet Neurology. 2015;14:145–152. doi: 10.1016/S1474-4422(14)70288-1. [DOI] [PubMed] [Google Scholar]
- 79.Rajrut AH, Uitti RJ, Fenton ME, et al. Amantadine effectiveness in multiple system atrophy and progressive supranuclear palsy. Parkinsonism Relat Disord. 1997;3:211–214. doi: 10.1016/s1353-8020(97)00022-9. [DOI] [PubMed] [Google Scholar]
- 80.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 81.DelleDonne A, Klos KJ, Fujishiro H, et al. Incidental Lewy body disease and preclinical Parkinson disease. Archives of neurology. 2008;65:1074–1080. doi: 10.1001/archneur.65.8.1074. [DOI] [PubMed] [Google Scholar]
- 82.Gibb WR. Idiopathic Parkinson’s disease and the Lewy body disorders. Neuropathol Appl Neurobiol. 1986;12:223–234. doi: 10.1111/j.1365-2990.1986.tb00136.x. [DOI] [PubMed] [Google Scholar]
- 83.Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51:745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology. 2000;54:1916–1921. doi: 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- 85.Braak H, Rub U, Jansen Steur EN, et al. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology. 2005;64:1404–1410. doi: 10.1212/01.WNL.0000158422.41380.82. [DOI] [PubMed] [Google Scholar]
- 86.Tsuboi Y, Uchikado H, Dickson DW. Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat Disord. 2007;13(Suppl 3):S221–224. doi: 10.1016/S1353-8020(08)70005-1. [DOI] [PubMed] [Google Scholar]
- 87.Sinha N, Firbank M, O’Brien JT. Biomarkers in dementia with Lewy bodies: a review. Int J Geriatr Psychiatry. 2012;27:443–453. doi: 10.1002/gps.2749. [DOI] [PubMed] [Google Scholar]
- 88.Whitwell JL, Weigand SD, Shiung MM, et al. Focal atrophy in dementia with Lewy bodies on MRI: a distinct pattern from Alzheimer’s disease. Brain. 2007;130:708–719. doi: 10.1093/brain/awl388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Graff-Radford J, Murray ME, Lowe VJ, et al. Dementia with Lewy bodies: basis of cingulate island sign. Neurology. 2014;83:801–809. doi: 10.1212/WNL.0000000000000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology. 2008;71:903–910. doi: 10.1212/01.wnl.0000326146.60732.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mollenhauer B, Bibl M, Trenkwalder C, et al. Follow-up investigations in cerebrospinal fluid of patients with dementia with Lewy bodies and Alzheimer’s disease. J Neural Transm. 2005;112:933–948. doi: 10.1007/s00702-004-0235-7. [DOI] [PubMed] [Google Scholar]
- 92.Hack N, Fayad SM, Monari EH, et al. An eight-year clinic experience with clozapine use in a Parkinson’s disease clinic setting. PloS one. 2014;9:e91545. doi: 10.1371/journal.pone.0091545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet. 2014;383:533–540. doi: 10.1016/S0140-6736(13)62106-6. [DOI] [PubMed] [Google Scholar]
- 94.Rolinski M, Fox C, Maidment I, et al. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst Rev. 2012;3:CD006504. doi: 10.1002/14651858.CD006504.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Daniele A, Moro E, Bentivoglio AR. Zolpidem in progressive supranuclear palsy. N Engl J Med. 1999;341:543–544. doi: 10.1056/NEJM199908123410721. [DOI] [PubMed] [Google Scholar]
- 96.Cotter C, Armytage T, Crimmins D. The use of zolpidem in the treatment of progressive supranuclear palsy. J Clin Neurosci. 2010;17:385–386. doi: 10.1016/j.jocn.2009.05.038. [DOI] [PubMed] [Google Scholar]