

Abstract
Rapid detection of nucleic acids is integral for clinical diagnostics and biotechnological applications. We recently developed a platform termed SHERLOCK (Specific High Sensitivity Enzymatic Reporter UnLOCKing) that combines isothermal pre-amplification with Cas13 to detect single molecules of RNA or DNA. Through characterization of CRISPR enzymology and application development, we report here four advances integrated into SHERLOCKv2: 1) 4-channel single reaction multiplexing using orthogonal CRISPR enzymes; 2) quantitative measurement of input down to 2 aM; 3) 3.5-fold increase in signal sensitivity by combining Cas13 with Csm6, an auxilary CRISPR-associated enzyme; and 4) lateral flow read-out. SHERLOCKv2 can detect Dengue or Zika virus ssRNA as well as mutations in patient liquid biopsy samples via lateral flow, highlighting its potential as a multiplexable, portable, rapid, and quantitative detection platform of nucleic acids.
Versatile, rapid, and portable sensing of nucleic acids is vital for applications in human health. The RNA-targeting CRISPR-associated enzyme Cas13(1, 2) has recently been adapted for such purpose. This detection platform, termed SHERLOCK (Specific High Sensitivity Enzymatic Reporter UnLOCKing) (3), can discriminate between inputs that differ by a single nucleotide at very low concentrations and can be lyophilized for portable deployment. However, this technology has several limitations, including the lack of quantitation and reliance on fluorescent detection equipment for readout. Here, we extend the SHERLOCK technology to address these limitations and further develop the utility of this platform.
Many applications require detection of more than one target molecule in a single reaction, and we therefore sought to create a multiplex platform that relies on unique cleavage preferences of Cas enzymes(2–5). To identify possible candidate enzymes compatible with multiplexing, we biochemically characterized three members of the CRISPR-Cas13a family and fourteen members of the CRISPR-Cas13b family(6, 7) (fig. S1, S2 and Table S1). We profiled cleavage preferences on homopolymer reporters, and found that most orthologs preferred either uridine, a combination of bases, or adenine (fig. S3 and Tables S2–5) and cleavage could be improved with buffer and crRNA design optimization (fig. S4–7, Supplementary Methods). Among the adenine cleaving enzymes, PsmCas13b was more sensitive than LbaCas13a (fig. S8). We refined the cleavage sequence preferences by evaluating collateral activity across di-nucleotide motifs (Fig. 1A), finding a large diversity of di-nucleotide cleavage motif preferences (figs. S9–10, Supplementary Methods). From these di-nucleotide cleavage screens, we found that the activities of LwaCas13a, CcaCas13b, LbaCas13a and PsmCas13b could all be independently measured with the four di-nucleotide reporters AU, UC, AC, and GA, respectively (Fig. 1B and fig. S11). Additionally, using a random in vitro RNA library motif cleavage screen, we identified numerous RNA 6-mers that allowed for further orthogonality between Cas13 enzymes (fig. S12–15 and Supplementary Methods).
Figure 1. Multiplexed SHERLOCK detection with orthogonal collateral activity of Class 2 enzymes.
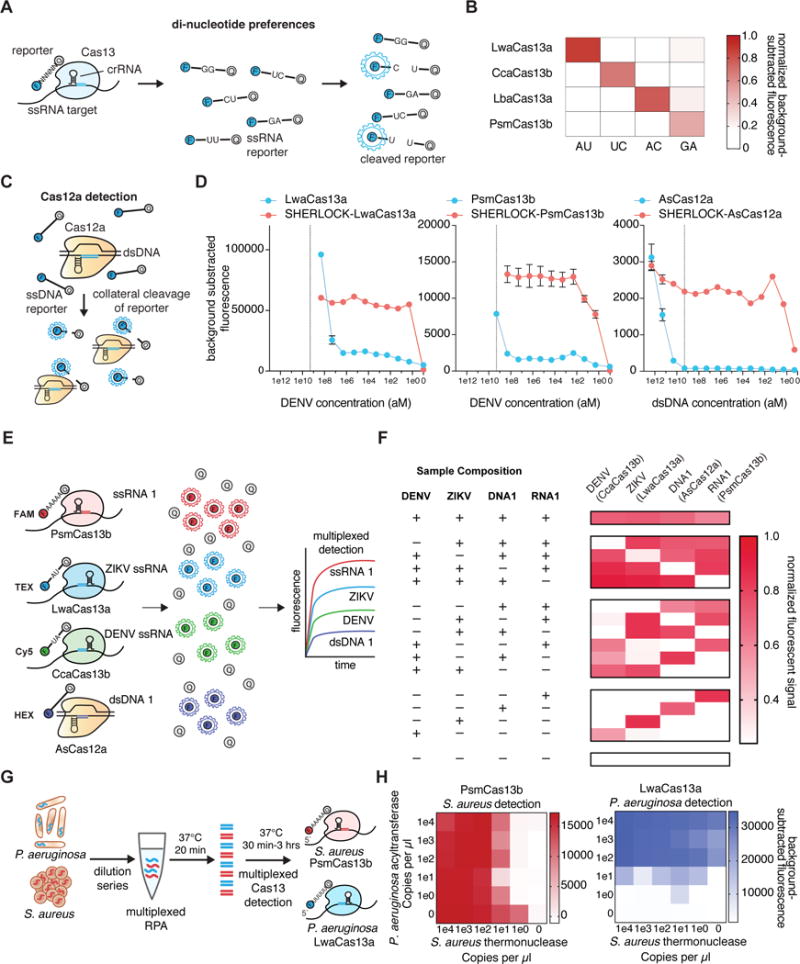
A) Schematic of assay for determining di-nucleotide preferences of Cas13a/b enzymes.
B) Collateral activity of LwaCas13a, CcaCas13b, LbaCas13a, and PsmCas13b on orthogonal di-nucleotide reporters.
C) Schematic of collateral activity of Cas12a activated by dsDNA.
D) Comparison of collateral activity and pre-amplification enhanced collateral activity (SHERLOCK) of LwaCas13a, PsmCas13b, and AsCas12a. The dotted line denotes 2e9 (aM), the limit of AsCas12a sensitivity without preamplification. Values represent mean +/− S.E.M.
E) Schematic of in-sample 4 channel multiplexing using orthogonal Cas13 and Cas12a enzymes.
F) In-sample multiplexed detection of ZIKV ssRNA, ssRNA 1, DENV ssRNA, and dsDNA 1 with LwaCas13a, PsmCas13b, CcaCas13b, and AsCas12a.
G) Schematic of in-sample multiplexed detection of S. aureus thermonuclease and P. aeruoginosa acyltransferase synthetic targets with LwaCas13a and PsmCas13b.
H) In-sample multiplexed RPA and collateral detection at decreasing concentrations of S. aureus thermonuclease and P. aeruoginosa acyltransferase synthetic targets with LwaCas13a and PsmCas13b.
Using these unique cleavage preferences, we were able to detect synthetic Zika virus (ZIKV) ssRNA in the HEX channel and synthetic Dengue virus (DENV) ssRNA in the FAM channel in the same reaction (fig. S16). To expand the in-sample multiplexing capabilities of SHERLOCK, we engineered a detection system based on Cas12a, which also exhibits collateral activity(8) (Fig. 1C). Although AsCas12a collateral activity did not produce a detectable signal at input concentrations below 100nM, preamplification with recombinase polymerase amplification (RPA) enabled single-molecule detection at 2aM (Fig. 1D, S17) (unless otherwise noted, all SHERLOCK reactions that involve a pre-amplification are performed in two steps with the RPA reaction being directly added into the Cas13 assay without any purification step). For triplex detection, we designed a LwaCas13a uridine reporter in the Cy5 channel, a PsmCas13b adenine reporter in the FAM channel, and an AsCas12a ssDNA reporter in the HEX channel (fig S18A). We were able to detect three targets (a synthetic ssDNA target, ZIKV ssRNA, and DENV ssRNA) in a single reaction (fig. S18B). We further extended detection to four targets by leveraging orthogonal di-nucleotide motifs, with reporters for LwaCas13a, PsmCas13b, CcaCas13b, and AsCas12a in FAM, TEX, Cy5, and HEX channels, respectively (Fig. 1E), and were able to distinguish all combinations of targets (Fig. 1F). When combined with RPA, we detected two DNA targets (the P. aeruginosa acyltransferase gene and the S. aureus thermonuclease gene) (Fig. 1G) down to the attomolar range (Fig. 1H). Similarly, multiplexed SHERLOCK with PsmCas13b and LwaCas13a achieved attomolar multiplexed detection of ZIKV and DENV RNA dilutions as well as allele-specific genotyping of human saliva samples (fig. S19). These advances in in-sample multiplexing via orthogonal base preferences allow for many targets to be detected at scale and for cheaper cost.
We next focused on tuning the output of the SHERLOCK signal to make it more quantitative, sensitive, and robust to broaden the utility of the technology. SHERLOCK relies on an exponential pre-amplification, which saturates quickly and hinders accurate quantitation, but we observed that more dilute primer concentrations increased both raw signal and quantitative accuracy, indicating that at lower primer concentrations, the reaction does not saturate (Fig. 2A,B and fig. S20A–E). We tested a range of primer concentrations and found that 240nM exhibited the greatest correlation between signal and input (fig. S20F), and quantification was sustainable across a large range of sample concentrations down to the attomolar range (Fig. 2C and fig. S20G). Many applications of nucleic acid detection, such as HIV detection(9, 10), require single molecule/mL sensitivity, and we therefore tested if the detection limit could be pushed beyond 2aM, allowing for more dilute sample inputs into SHERLOCK. By scaling up the pre-amplification RPA step, we found that LwaCas13a could give detection signal for 200, 80, and 8zM input samples and allow for single-molecule volume inputs of 250μL and 540μL (fig. S21A–B), and PsmCas13b could detect 200zM input samples in 250μL reactions (fig. 21C).
Figure 2. Single molecule quantitation and enhanced signal with SHERLOCK and Csm6.
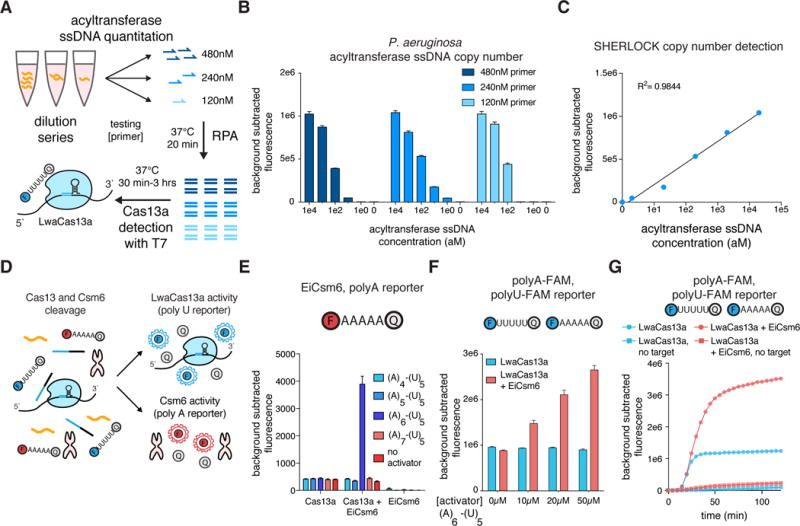
A) Schematic of DNA reaction scheme for quantitation of P. aeroginosa synthetic DNA
B) Quantitation of P. aeroginosa synthetic DNA at various RPA primer concentrations. Values represent mean +/− S.E.M.
C) Correlation of P. aeroginosa synthetic DNA concentration with detected fluorescence. Values represent mean +/− S.E.M.
D) Schematic of independent readout of LwaCas13a and Csm6 cleavage activity with orthogonal reporters.
E) Activation of EiCsm6 by LwaCas13a cleavage of adenine-uridine activators with different length adenine tracts. LwaCas13a is targeting synthetic DENV ssRNA. Values represent mean +/− S.E.M.
F) Combined LwaCas13a and EiCsm6 signal for increasing concentrations of (A)6-(U)5 activator detecting 20nM of DENV ssRNA. Values represent mean +/− S.E.M.
G) Kinetics of EiCsm6-enhanced LwaCas13a SHERLOCK detection of P. aeruoginosa acyltransferase synthetic target.
In order to amplify the detection signal, we leveraged the CRISPR type-III effector nuclease Csm6(11–17), which is activated by cyclic adenylate molecules or linear adenine homopolymers terminated with a 2′,3′-cyclic phosphate(18, 19). LwaCas13a and PsmCas13b collateral activity generates cleavage products with hydroxylated 5′ ends and 2′,3′-cyclic phosphate ends (fig. S22), suggesting that Cas13 collateral activity could generate Csm6 activating species, which would allow for amplified signal detection in the SHERLOCK assay. By testing RNA adenylate molecules of different lengths and 3′ end modifications (Fig. S23 and S24A; Table S6), we found that Csm6 from Enterococcus italicus (EiCsm6) and Csm6 from Lactobacillus salivarius (LsCsm6) were efficiently activated by hexadenylates containing 2′,3′-cyclic phosphate ends (Fig. S24B,C). Moreover, EiCsm6, LsCsm6, and Csm6 from Thermus thermophilus (TtCsm6) demonstrated a strong cleavage preference for A- and C-rich sensors based on sensor screening, enabling independent measurements of LwaCas13a and Csm6 cleavage activity in separate channels (Fig. 2D and fig. S24B–D, S25, S26A–E).
To couple the activity of Cas13 with Csm6 activation, we designed protected RNA activators that contained a poly-A stretch followed by a protecting poly-U stretch that could be cleaved by a uracil preferring Cas13 enzyme, with the rationale that LwaCas13a could degrade all the uridines down to the homopolymeric A stretch since it had robust activity on UU and AU two-base motifs (fig. S9). We found that, upon addition of target and LwaCas13a-crRNA complex, EiCsm6 and LsCsm6 were activated by the (A)6-(U)5 activator, consistent with the finding that the A6 activator is optimal for Csm6 activation and confirmed by mass spectrometry (Fig. 2E and fig. S26F, S27–S28). We combined the reporters for both Csm6 and Cas13 in the same reaction within the same fluorescence channel, and found that increasing the activator concentration increased the synergistic activation of Csm6 by Cas13 for DENV ssRNA detection (Fig. 2F), and that increasing the Csm6-specific polyA reporter also increased the Csm6 signal, leading to a larger increase in signal upon activator addition (fig. S29A,B). After optimization (fig S30), we found that Csm6-enhanced LwaCas13a increased the overall signal and kinetics of synthetic acyltransferase gene detection by SHERLOCK (Fig. 2G).
Another goal of SHERLOCKv2 was engineering a visual readout of activity requiring no additional instrumentation. We first tested a colorimetric RNase reporter based upon gold nanoparticle cluster disaggregation(20, 21), but this readout required a level of RNase activity beyond what Cas13 collateral activity could achieve (fig. S31). We then designed a lateral-flow readout that was based on the destruction of a FAM-biotin reporter, allowing for detection on commercial lateral flow strips. Abundant reporter accumulates anti-FAM antibody-gold nanoparticle conjugates at the first line on the strip, preventing binding of the antibody-gold conjugates to protein A on the second line; cleavage of reporter would reduce accumulation at the first line and result in signal on the second line (Fig. 3A). We tested this design for instrument-free detection of ZIKV or DENV ssRNA, and found that detection was possible in under 90 minutes with sensitivities down to the 2 aM condition (Fig. 3B,C and fig. S32). Moreover, we found that we could do rapid genomic DNA extraction from human saliva (<10min) and input this directly into SHERLOCK without purification for rapid genotyping in under 23 minutes by fluorescence and 2 hours by lateral flow (fig. S33). This exemplifies a closed-tube assay format with the entire SHERLOCK reaction being performed in a one-pot assay without any sample purification.
Figure 3. Adapting SHERLOCK for lateral flow detection.
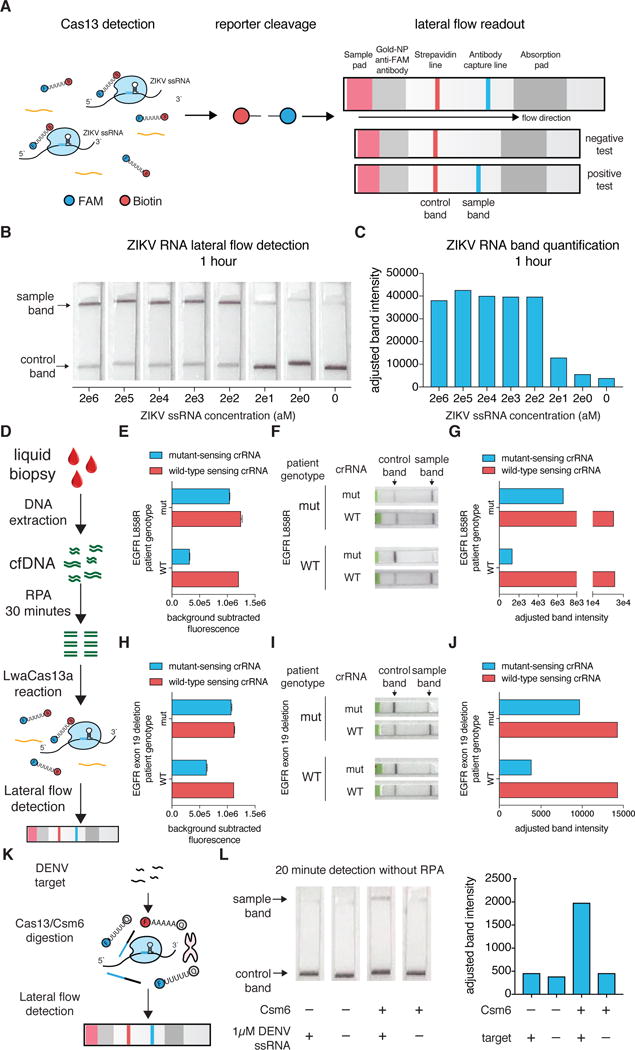
A) Schematic of lateral flow detection with SHERLOCK
B) Detection of synthetic ZIKV ssRNA using lateral flow SHERLOCK with 1 hour of LwaCas13a reaction
C) Quantitation of band intensity from detection in (B)
D) Schematic of lateral flow detection of therapeutically relevant EGFR mutations from patient liquid biopsy samples.
E) Detection of EGFR L858R mutation in patient-derived cell-free DNA samples with either L858R or WT cancer mutations. Values represent mean +/− S.E.M.
F) Lateral-flow detection of EGFR L858R mutation in patient-derived cell-free DNA samples with either L858R or WT alleles.
G) Quantitation of band intensity from detection in (E).
H) Detection of EGFR exon 19 deletion mutation in patient-derived cell-free DNA samples with either exon 19 deletion or WT alleles. Values represent mean +/− S.E.M.
I) Lateral-flow detection of EGFR exon 19 deletion mutation in patient-derived cell-free DNA samples with either exon 19 deletion or WT alleles.
J) Quantitation of band intensity from detection in (H).
K) Schematic of lateral flow readout of EiCsm6-enhanced LwaCas13a detection of DENV ssRNA
L) EiCsm6-enhanced lateral flow detection of synthetic DENV RNA in combination with LwaCas13a without preamplification by RPA. Band intensity quantitation is shown to the right.
We also applied the system to create a rapid and portable paper test for mutation detection in liquid biopsies of non-small cell lung cancer (NSCLC) patients. We designed SHERLOCK assays to detect either the EGFR L858R mutation or the exon 19 deletion (5 amino acids) and isolated cfDNA from patients with or without these mutations (Fig. 3D), as verified by targeted sequencing (Table S7). SHERLOCK successfully detected these mutations, both with fluorescence based readout (Fig. 3E,H) and lateral flow-based readout (Fig. 3F,G,I,J fig. S34A–D). Fluorescence-based SHERLOCK was also able to detect a different common EGFR mutation, T790M, in synthetic and patient cfDNA liquid biopsy samples (fig. S34E,F).
To improve the robustness of the detection and reduce the likelihood of false positive readout, we combined Csm6 with Cas13 detection on lateral flow (Fig. 3K). We tested lateral flow reporters of various sequence and length in the presence of Csm6 and activator, and found that a long A-C reporter demonstrated strong cleavage signal (fig. S35A,B). We used this reporter in combination with the Cas13 lateral flow reporter for rapid detection of DENV ssRNA relying solely on Csm6 for amplification (i.e., in the absence of RPA) (Fig. 3L). We subsequently combined RPA, Cas13/Csm6, and lateral flow readout to detect an acyltransferase target, and found that the increase in signal conferred by Csm6 allowed for more rapid detection by lateral flow (fig. S35C–D) with reduced background.
Finally, we applied SHERLOCKv2 in a simulated approach that involves Cas13 serving as both a companion diagnostic and the therapy itself, as Cas13 has been developed for a variety of applications in mammalian cells including RNA knockdown, imaging, and editing (22, 23)(Fig. 4A and Table S8). We recently harnessed Cas13b from Prevotella sp. P5-125 (PspCas13b) to correct mutations in genetic diseases using a system called RNA Editing for Programmable A-to-I Replacement (REPAIR)(23). To direct and monitor the outcome of a treatment, we tested if SHERLOCK could be used both for genotyping to guide the REPAIR treatment and as a readout of the edited RNA to track the efficiency of the therapy. We used a mutation in APC (APC:c.1262G>A) implicated in Familial adenomatous polyposis 1 (Fig. 4B,C) (24), and transfected synthetic healthy and mutant cDNAs of the fragment surrounding the mutation into HEK293FT cells. We harvested DNA from these cells and successfully genotyped the correct samples using single-sample multiplexed SHERLOCK with LwaCas13a and PsmCas13b (Fig. 4D). Concurrently, we designed and cloned guide RNAs for the REPAIR system and transfected cells that had the diseased genotype with the guide RNA and dPspCas13b-ADAR2dd(E488Q) REPAIR system. We confirmed editing by next-generation sequencing (NGS) analysis, finding that 43% editing was achieved with the REPAIR system (Fig. 4E), and we were able to detect this editing with SHERLOCK (Fig. 4F and fig. S36).
Figure 4. Combined therapeutics and diagnostics with Cas13 enzymes.
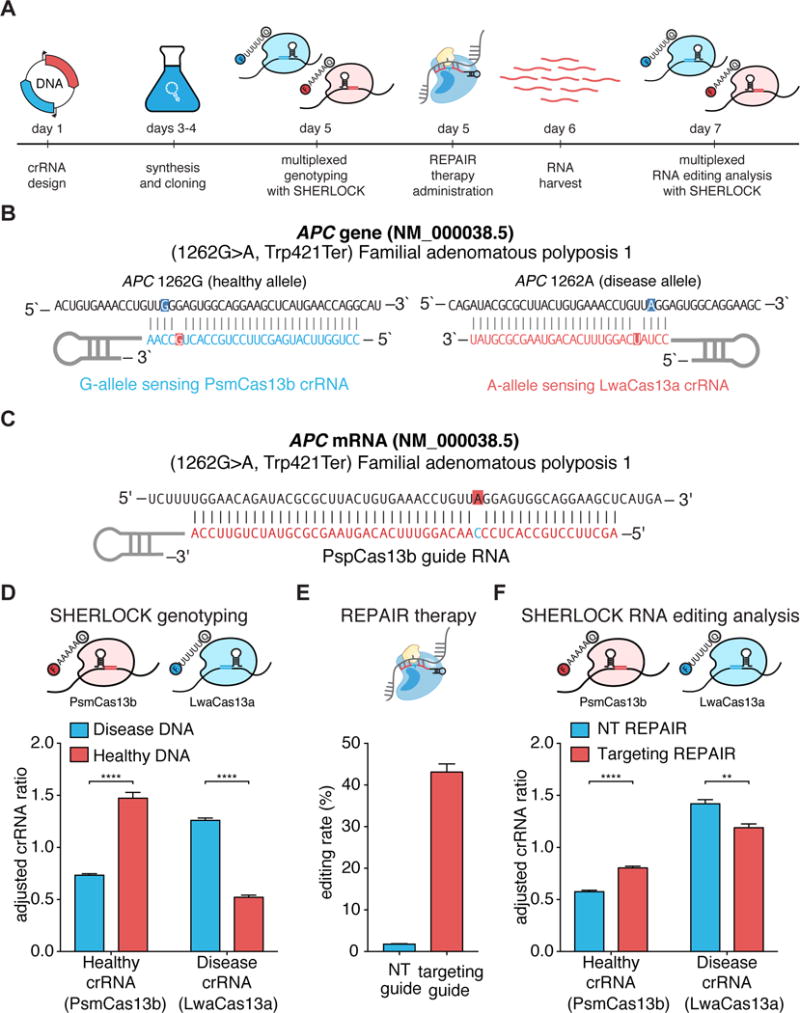
A) Schematic of timeline for detection of disease alleles, correction with REPAIR, and assessment of REPAIR correction.
B) Sequences of targets and crRNA designs used for detection of APC alleles.
C) Sequences of target and REPAIR guide design used for correction of APC alleles.
D) In-sample multiplexed detection of APC alleles from healthy- and disease-simulating samples with LwaCas13a and PsmCas13b. Adjusted crRNA ratio allows for comparisons between different crRNAs that will have different overall signal levels (see supplementary methods for more details). Values represent mean +/− S.E.M.
E) Quantitation of REPAIR editing efficiency at the targeted APC mutation. Values represent mean +/− S.E.M.
F) In-sample multiplexed detection of APC alleles from REPAIR targeting and non-targeting samples with LwaCas13a and PsmCas13b. Values represent mean +/− S.E.M.
The additional refinements presented here for Cas13-based detection allow for quantitative, visual, more sensitive, and multiplexed readouts, enabling additional applications for nucleic acid detection, especially in settings where portable and instrument-free analysis are necessary (Table S9). SHERLOCKv2 can be used for multiplexed genotyping to inform pharmacogenomic therapeutic development and application, detecting genetically modified organisms in the field, or determining the presence of co-occurring pathogens. Moreover, the rapid, isothermal readout of SHERLOCKv2, enabled by lateral flow and Csm6, provides an opportunity for detection in settings where power or portable readers are unavailable, even for rare species like circulating DNA. In the future, it might be possible to make solution-based colorimetric readouts and multiplex lateral flow assays containing multiple test strips for different targets. Improved CRISPR-dx nucleic acid tests make it easier to detect the presence of nucleic acids in a range of applications across biotechnology and health and are now field-ready for rapid and portable deployment.
Supplementary Material
Acknowledgments
We would like to thank S. Trauger and the Harvard Small Molecule Mass Spectrometry facility for mass spectrometry assistance, J. Strecker and I. M. Slaymaker for protein purification assistance, D.B.T.C. for assistance with Cas13b gene synthesis, B. Franklin, V. Verdine, and A.H. Le for additional experimental assistance, L.M. Sholl and J.A. Golden for providing cell-free DNA samples, L. Hao and S. Bhatia for assistance with gold nanoparticle experiments, C.A. Freije, C. Myrhvold, and P.C. Sabeti for assistance with manuscript preparation, and R. Macrae, R. Belliveau, E. Blackwell, and the entire Zhang lab for discussions and support.
Funding: O.O.A. is supported by a Paul and Daisy Soros Fellowship and a NIH F30 NRSA 1F30-CA210382. J.J.C. is supported by the Defense Threat Reduction Agency grant HDTRA1-14-1-0006, the Paul G. Allen Frontiers Group, and the Wyss Institute. F.Z. is a New York Stem Cell Foundation–Robertson Investigator. F.Z. is supported by NIH grants (1R01-HG009761, 1R01-MH110049, and 1DP1-HL141201); the Howard Hughes Medical Institute; the New York Stem Cell, Simons, Paul G. Allen Family, and Vallee Foundations; the Poitras Center for Affective Disorders Research at MIT; the Hock E. Tan and K. Lisa Yang Center for Autism Research at MIT; the Skolkovo Institute of Science and Technology; and J. and P. Poitras, R. Metcalfe, and D. Cheng.
Footnotes
Author contributions: O.O.A, J.S.G, and F.Z. conceived and designed the study. O.O.A, J.S.G, and M.J.K participated in the design and execution of all experiments. J.J. designed and performed the RNA motif screens. O.O.A, J.S.G, M.J.K, J.J.C. and F.Z. wrote the paper with contributions from all authors.
Competing interests: J.S.G., O.O.A., J.J.C. and F.Z are co-inventors on patent applications filed by the Broad Institute relating to work in this manuscript.
Data and materials availability: Sequencing data are available at Sequence Read Archive under BioProject accession number PRJNA433191. The authors plan to make the reagents widely available to the academic community through Addgene and to provide software tools via the Zhang lab website (www.genome-engineering.org) and GitHub (github.com/fengzhanglab).
References
- 1.Shmakov S, et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abudayyeh OO, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gootenberg JS, et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–442. doi: 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.East-Seletsky A, et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016;538:270–273. doi: 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.East-Seletsky A, O’Connell MR, Burstein D, Knott GJ, Doudna JA. RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes. Mol Cell. 2017;66:373–383 e373. doi: 10.1016/j.molcel.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shmakov S, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017;15:169–182. doi: 10.1038/nrmicro.2016.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smargon AA, et al. Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol Cell. 2017;65:618–630 e617. doi: 10.1016/j.molcel.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen JS, Ma E, Harrington LB, Tian X, Doudna JA. CRISPR-Cas12a target binding unleashes single-stranded DNase activity. bioRxiv. 2017 doi: 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.W. H. Organization. Guidelines for Using HIV Testing Technologies in Surveillance: Selection, Evaluation and Implementation: 2009 Update. Geneva: 2009. [PubMed] [Google Scholar]
- 10.Barletta JM, Edelman DC, Constantine NT. Lowering the detection limits of HIV-1 viral load using real-time immuno-PCR for HIV-1 p24 antigen. Am J Clin Pathol. 2004;122:20–27. doi: 10.1309/529T-2WDN-EB6X-8VUN. [DOI] [PubMed] [Google Scholar]
- 11.Deng L, Garrett RA, Shah SA, Peng X, She Q. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol Microbiol. 2013;87:1088–1099. doi: 10.1111/mmi.12152. [DOI] [PubMed] [Google Scholar]
- 12.Goldberg GW, Jiang W, Bikard D, Marraffini LA. Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature. 2014;514:633–637. doi: 10.1038/nature13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang W, Samai P, Marraffini LA. Degradation of Phage Transcripts by CRISPR-Associated RNases Enables Type III CRISPR-Cas Immunity. Cell. 2016;164:710–721. doi: 10.1016/j.cell.2015.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niewoehner O, Jinek M. Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein Csm6. RNA. 2016;22:318–329. doi: 10.1261/rna.054098.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samai P, et al. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell. 2015;161:1164–1174. doi: 10.1016/j.cell.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staals RH, et al. RNA targeting by the type III-A CRISPR-Cas Csm complex of Thermus thermophilus. Mol Cell. 2014;56:518–530. doi: 10.1016/j.molcel.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamulaitis G, et al. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol Cell. 2014;56:506–517. doi: 10.1016/j.molcel.2014.09.027. [DOI] [PubMed] [Google Scholar]
- 18.Kazlauskiene M, Kostiuk G, Venclovas C, Tamulaitis G, Siksnys V. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017;357:605–609. doi: 10.1126/science.aao0100. [DOI] [PubMed] [Google Scholar]
- 19.Niewoehner O, et al. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017;548:543–548. doi: 10.1038/nature23467. [DOI] [PubMed] [Google Scholar]
- 20.Zhao W, Ali MM, Aguirre SD, Brook MA, Li Y. Paper-based bioassays using gold nanoparticle colorimetric probes. Anal Chem. 2008;80:8431–8437. doi: 10.1021/ac801008q. [DOI] [PubMed] [Google Scholar]
- 21.Zhao W, Lam JC, Chiuman W, Brook MA, Li Y. Enzymatic cleavage of nucleic acids on gold nanoparticles: a generic platform for facile colorimetric biosensors. Small. 2008;4:810–816. doi: 10.1002/smll.200700757. [DOI] [PubMed] [Google Scholar]
- 22.Abudayyeh OO, et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284. doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cox DBT, et al. RNA editing with CRISPR-Cas13. Science. 2017;358:1019–1027. doi: 10.1126/science.aaq0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cottrell S, Bicknell D, Kaklamanis L, Bodmer WF. Molecular analysis of APC mutations in familial adenomatous polyposis and sporadic colon carcinomas. Lancet. 1992;340:626–630. doi: 10.1016/0140-6736(92)92169-g. [DOI] [PubMed] [Google Scholar]
- 25.Chen CY. DNA polymerases drive DNA sequencing-by-synthesis technologies: both past and present. Front Microbiol. 2014;5:305. doi: 10.3389/fmicb.2014.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome research. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye J, et al. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.