

Abstract
Although several experimental studies showed cancer-preventive efficacy of supplemental dietary selenium, human clinical trials questioned this efficacy. Identifying its molecular targets and mechanism is important in understanding this discrepancy. Methylselenol, the active metabolite of selenium, reacts with lipid hydroperoxides bound to protein kinase C (PKC) and is oxidized to methylseleninic acid (MSA). This locally generated MSA selectively inactivates PKC by oxidizing its critical cysteine sulfhydryls. The peroxidatic redox cycle occurring in this process may explain how extremely low concentrations of selenium catalytically modify specific membrane-bound proteins compartmentally separated from glutathione and selectively induce cytotoxicity in promoting cells. Mammalian thioredoxin reductase (TR) is itself a selenoenzyme with a catalytic selenocysteine residue. Together with thioredoxin (Trx), it catalyzes reduction of selenite and selenocystine by NADPH generating selenide which in the presence of oxygen redox cycles producing reactive oxygen species. Trx binds with high affinity to PKC and reverses PKC inactivation. Therefore, established tumor cells overexpressing TR and Trx may escape the cancer-preventive actions of selenium. This suggests that in some cases, certain selenoproteins may counteract selenometabolite actions. Lower concentrations of selenium readily inactivate antiapoptotic PKC isoenzymes e and a which have a cluster of vicinal thiols, thereby inducing apoptosis. Higher concentrations of selenium also inactivate proapoptotic enzymes such as proteolytically activated PKCd fragment, holo-PKCz, caspase-3, and c-Jun N-terminal kinase, which all have a limited number of critical cysteine residues and make tumor cells resistant to selenium-induced apoptosis. This may explain the intriguing U-shaped curve that is seen with dietary selenium intake and the extent of cancer prevention.
Keywords: Cancer prevention, Fatty acid hydroperoxides, Protein kinase C, Protein thiol modification, Selenocompounds, Thioredoxin reductase
1. INTRODUCTION
Several epidemiologic studies showed an inverse relationship between the status of dietary selenium intake and the risk of cancer at various sites in humans [1]. However, a small number of epidemiologic studies did not find such an association [2, 3]. Numerous experimental studies of carcinogenesis in rodent models showed that supplementation using certain forms of selenium at doses (1 to 3 ppm) well above the nutritional requirement (0.1 ppm) decreased tumorigenesis in various organs [4–6]. Nevertheless, some studies did not find this effectiveness of selenium, and a limited number of studies even showed an increase in tumorigenesis with selenium supplementation [7, 8]. Early clinical trials in China showed that selenium was moderately effective in preventing both liver and esophageal cancers in humans [9]. Additionally, the Nutritional Prevention of Cancer (NPC) trial conducted by Clark et al in the United States suggested that supplemental selenium in the form of selenized yeast may reduce the incidence and mortality of cancers of the prostate, lung, and colon-rectum, but not cancers of the breast and skin [10]. However, the Selenium and Vitamin E Cancer Prevention trial (SELECT), a large-scale clinical trial employing selenomethionine, did not show any efficacy of selenium in preventing prostate cancer [11]. SELECT also showed a nonsignificant increase in diabetes with selenium supplementation [11]. Furthermore, additional clinical trials conducted with selenized yeast did not show efficacy of selenium in the prevention of cancer at various sites in the body [12]. Conceivably, the studies carried out over the past four to five decades support the notion that selenium supplementation may decrease the incidence of cancer in some cases, fail to do so in other cases, or may even cause unwanted effects and toxicity [12].
Although the form of selenium used in prevention studies may be an important factor in explaining some of the aforementioned discrepancies, understanding the cancer-preventive mechanism of selenium may shed light on why it works in some cases and fails in others. Selenium is a transition metal and its redox chemistry plays an important role in its actions in basic nutrition, cancer prevention, and toxicity. The ability of selenium to induce oxidation of protein thiols has been known for some time [13]. Protein thiol oxidation is emerging as an important mechanism in regulating key signaling enzymes such as protein kinase C (PKC), protein tyrosine phosphatases, and various transcriptional factors which influence tumorigenesis and cell death [14–16]. Therefore, this may be an important redox regulatory mechanism we can study to understand conflicting observations of the efficacy of selenium and to optimize its efficacy as a cancer-preventive agent.
The purpose of this review is to discuss the current state of knowledge regarding redox regulation of protein thiol homeostasis in order to illustrate how selenium selectively kills precancer cells, how tumor cells escape from it, and why a U-shaped curve is seen with dietary selenium intake and the extent of cancer prevention. We will use PKC isoenzymes and various other proteins as examples to illustrate selenium-induced protein thiol oxidations.
2. COMPLEXITY IN UNDERSTANDING THE CANCER-PREVENTIVE MECHANISM OF SELENIUM
2.1. Overview of Important Factors to Be Considered
In proposing a cancer-preventive mechanism for selenium, all the following important criteria must be taken into account: (1) the mechanism should consider the susceptibility of the promotional stage of carcinogenesis to selenium action as well as the efficacy of various forms of selenium for cancer prevention; (2) it should also clarify the relative role of selenoproteins and low-molecular weight selenometabolites, which are present at very low levels in tissues; (3) it should have specific and sensitive target(s) for selenium relevant to cell signaling in the events related to tumor promotion; (4) the proposed mechanism should address how key protein targets are oxidatively modified by selenium in the cell where millimolar concentrations of antioxidant thiol glutathione (GSH) are present; (5) It should explain how selenium induces selective toxicity in precancer cells but not in normal cells; (6) the mechanism should clarify how tumor cells can escape from selenium, leading to failure of chemoprevention; and finally, (7) the proposed mechanism should clarify why a U-shaped curve is seen when measuring the extent of cancer prevention with increasing selenium intake. In this review, we have addressed all these issues while discussing redox mechanisms that perturb protein thiol homeostasis in order to explain the success and failure of selenium as a cancer-preventive agent.
2.2. The Promotional Stage of Carcinogenesis Is Suitable for Chemoprevention
Selenium exerts its preventive effects at the initiation phase of tumorigenesis by inhibiting carcinogen binding to DNA [17]. In addition, selenium inhibits tumor promotion and angiogenesis as well as stimulates the immune system [18]. In cell culture experiments, a lower concentration of selenium is required for the inhibition of promotion of precancer cells than for the inhibition of growth of advanced malignant cells [19, 20]. Furthermore, there is limited evidence to support that dietary selenium at a level nontoxic to the host can prevent the growth of transplanted tumors in animals [21]. In vitro, higher doses of selenium induce apoptosis of advanced malignant cells [22]. These growth-inhibiting and apoptosis-inducing mechanisms may be important in the promotion stage of carcinogenesis, in which there is a clonal expansion of preneoplastic cells that escape death. The high incidence of prostate cancer and the decades of preneoplasia during its development make this cancer well-suited for chemoprevention trials. Although various stages of carcinogenesis are susceptible to the cancer-preventive actions of selenium, the promotion stage, with its longer duration, is most ideal.
2.3. Efficacy of Various Forms of Selenium for Cancer Prevention and Role of Thioredoxin System
The cancer-preventive efficacy of selenium varies depending on its chemical form [23, 24]. Naturally occurring organic forms of selenium, such as Se-methylselenocysteine, is efficacious in preventing cancer and is less toxic [25]. Although inorganic selenite has cancer-preventive potential, it is too toxic. The reason for its toxicity is the direct reaction with the thioredoxin system leading to redox cycling and production of reactive oxygen species (ROS) [26, 27]. Thioredoxin system comprising NADPH, thioredoxin reductase (TR) and thioredoxin (Trx) is present in all cells from bacteria to man and was originally discovered as the hydrogen donor for ribonucleotide reductase in E. coli [28, 29]. Each deoxyribonucleotide formed from the corresponding ribonucleotide gives rise to a disulfide in the enzyme which has to be reduced [30]. The thioredoxin system is now known to be a general disulfide reduction system and via peroxiredoxins control the level of hydrogen peroxide (H2O2) in cells [29]. Purification and characterization of the thioredoxin system from rat liver demonstrated that the TR enzyme was larger and had a broad substrate specificity compared to the smaller specific bacterial enzyme [31]. Substrates remarkably included selenite and selenodiglutathione which were shown to be direct substrates for the TR enzyme but also fast oxidants of reduced Trx [26, 27]. Of particular interest was that selenite in the presence of NADPH and TR resulted in a large non-stochiometric oxidation of NADPH in the presence of oxygen. This result showed two important facts. First, TR and Trx play a major role in generating the selenide for the cotranslational synthesis of the 25 selenoproteins in human genome. Second, it may help to explain why there is no free pool of selenocysteine since it will also redox cycle with NADPH and TR in the presence of oxygen [32]. Remarkably, these results were published before it was known that mammalian TR itself is a selenoprotein. This included the result showing that the human TR enzyme reduced lipid hydroperoxides directly by NADPH and free selenocystine strongly stimulated the reaction due to catalytically generated selenols [33]. Furthermore, it was discovered that dinitrochlorobenzene was an irreversible inhibitor of the enzyme where loss of thioredoxin reductase activity was accompanied by a large increase in NADPH oxidase activity (30-fold) [34]. The mechanism of this was later shown to involve modification of the selenocysteine in the active site [35] following the discovery that the enzyme is a selenoenzyme [36, 37] with a selenenylsulfide active site in the C-terminus and a selenolthiol being the catalytic active site. There are three genes for TR in mammalian cells with TR2 and Trx2 in mitochondria and TR1 and Trx1 in the cytosol/nucleus, respectively [30, 38]. Recent results have highlighted the role of the GSH/glutaredoxin and TR systems in carcinogenesis [39].
Selenomethionine is the major form of selenium in typical diets [40]. However, it rapidly incorporates into proteins, whereas Se-methylselenocysteine does not. Some clinical trials such as NPC used selenized yeast, which contains high amounts of selenomethionine and significant amounts of several other forms of selenium [10]. Since there is a batch-to-batch variation in the composition of selenized yeast, selenomethionine, which is considered to be the major form of selenium in the selenized yeast, was chosen for SELECT [41]. Given that selenomethionine did not prevent tumorigenesis in various animal models, some speculated that selenomethionine is not an appropriate form to use for cancer prevention clinical trials [42, 43]. Selenomethionine can inhibit tumor cell growth and induce apoptosis in vitro, but requires high (micromolar) concentrations [44], which is likely due to the limited metabolism of this parent compound in culture. Synthetic methylseleninic acid (MSA), a second generation cancer-preventive selenocompound, is more effective than naturally occurring selenomethionine in both in vitro and in vivo models of cancer prevention [43, 45]. MSA is a stable oxidation product of volatile methylselenol, a presumed cancer-preventive metabolite of selenium [23]. Certain synthetic selenocompounds, such as 1,4-phenylene-bis(methylene)selenocyanate (p-XSC) were also shown to be more effective than selenomethionine in preventing carcinogenesis in rodent models [6, 42]. Why certain forms of selenium are more effective than others is not clearly understood.
2.4. Selenium Exerts Its Chemopreventive Actions as Selenoproteins—Antioxidants
At a nutritional level (0.1 ppm), selenium is essential to selenoprotein glutathione peroxidase (GPX), which protects cells from H2O2 and organic peroxides [46]. However, the amount of selenium required for cancer prevention (1 to 3 ppm) is well above levels required for optimal expression of GPX activity. This suggests involvement of additional mechanisms in the cancer-preventive actions of selenium.
Unlike other selenoproteins previously characterized, TR was observed to have a two-fold increase in activity upon dietary supplementation with selenite [47, 48]. However, this has not been observed with supplementation of other forms of selenium [49]. The expression of both Trx and TR was found to be increased several fold in some tumors compared to normal tissues [36, 50]. Overexpression of Trx inhibits apoptosis [51]. Given the importance of oxidative stress in tumor promotion, the antioxidant functions of GPX and TR are very relevant to the cancer-preventive actions of selenium. Nevertheless, their precise role is unknown.
2.5. Selenium Exerts Its Chemopreventive Actions as Selenometabolites—Prooxidants
Selenocompounds, such as selenite, selenomethionine, and Se-methylselenocysteine eventually generate selenide [23] which is ultimately incorporated into selenoproteins [46] (Figure 1). When selenide is generated in large amounts, it also reacts with oxygen to produce superoxide and, subsequently, H2O2, which results in toxicity [52]. Selenide is also sequentially methylated to methylselenol, dimethylselenide, and trimethylselenonium [53]. Both methylselenol and trimethylselenonium are excreted through the urine, while dimethylselenide is exhaled [23]. Although this methylation pathway was originally thought to be a detoxification pathway, later studies have suggested a role for methylselenol in mediating the anticarcinogenic actions of selenium [23]. Other important cancer-preventive selenocompounds such as p-XSC are also believed to mediate their cancer-preventive actions after their conversion to selenol metabolites [54]. Although methylselenol is a redox-active metabolite, whether or not its actions have any relation to oxidative stress associated with tumor promotion is not known. Furthermore, how volatile methylselenol is retained in the cell to mediate its action is not known.
FIGURE 1. Metabolism of dietary selenocompounds and excretion of selenometabolites.
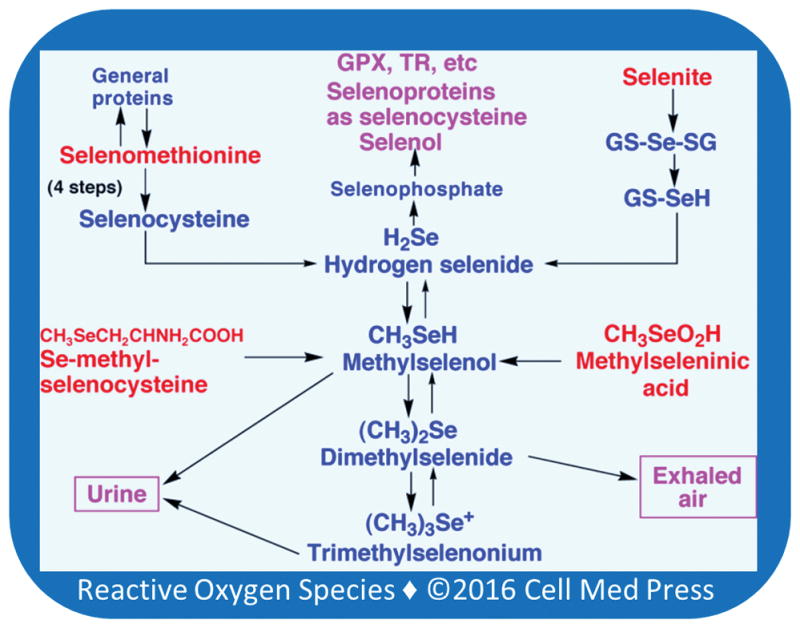
Various forms of selenocompounds (indicated in red) are administered for chemoprevention. They are eventually converted to hydrogen selenide which ultimately leads to the synthesis of selenocysteine residues in selenoproteins. Selenomethionine is also directly incorporated into various proteins in place of methionine. Excess selenide is sequentially methylated to methylselenol, dimethylselenide, and trimethylselenonium. Both methylselenol and trimethylselenonium are excreted through the urine, while dimethylselenide is exhaled.
2.6. Extremely Low Concentrations of Selenometabolites in Plasma and Tissues
At cancer-preventive doses, the total concentration of selenium is considered to be approximately 10 mM. Most (> 90%) selenium in the blood is bound to proteins or is present as selenoproteins [55], and only a limited amount (< 5%) is present as selenometabolites. This is important as it minimizes the occurrence of global toxicity of selenium. However, this raises an important challenge in understanding how such low tissue-available concentrations (nM) of selenometabolites can exert their cancer-preventive actions [56]. At the nutritionally adequate low dose (0.1 ppm), selenium’s actions are believed to be mediated primarily by selenoproteins, while at the toxic high dose (> 5 ppm), its actions are believed to be mediated by selenometabolites [1]. However, at the cancer-preventive intermediate doses (1 to 3 ppm), whether selenium exerts its actions exclusively as selenometabolites or through a combination of selenometabolites and selenoproteins is not known. Since selenium is a redox catalyst and protein thiols are very sensitive targets for redox modifications, proteins having clusters of cysteine residues are the most relevant targets for selenometabolites.
3. PKC ISOENZYMES AND THEIR RELEVANCE TO TUMOR PROMOTION
3.1. PKC—a Redox-Sensitive Cysteine-Rich Protein
PKC, a family of isoenzymes, is activated not only by lipid second messengers [57] but also by tumor promoters such as phorbol esters, oxidants, and fatty acid hydroperoxides [58–63]. A four-fold increase in the generation of diacylglycerol in prostate cancer tissue compared to benign tissue suggests a role for PKC in prostate tumorigenesis [64]. PKC has unique structural aspects that render it susceptible to activation by oxidants such as H2O2, periodate, and tobacco tumor promoters [14, 58–60].
There are two types of cysteine-rich regions present in PKC isoenzymes: one in the regulatory domain and the other in the catalytic domain (Figure 2). The cysteine-rich region present in the regulatory domain coordinates the binding of four zinc atoms [65]. Selective modification of these zinc-thiolates by tumor-promoting peroxides leads to collapse of the autoinhibitory region in the regulatory domain and cofactor-independent activation of PKC [14]. In contrast, modification of cysteine residues within the catalytic domain of the enzyme by alkylating agents and oxidants at high concentrations results in inactivation of PKC [66].
FIGURE 2. Cysteine-rich regions present in PKC isoenzymes.
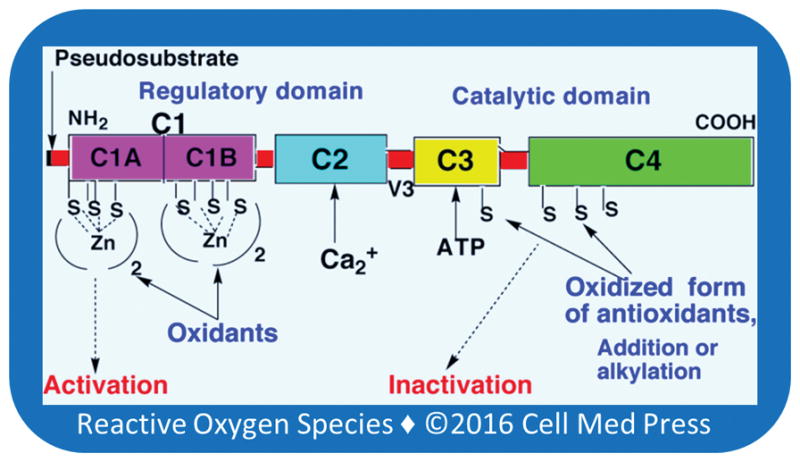
Unique structural aspects of PKC make it a receptor for not only tumor promoters but also anti-tumor promoters. C1: cysteine-rich constant region present in various PKC isoenzymes. C1A: first two zinc-thiolates in the C1 domain; C1B: second two zinc-thiolates in the C1 domain; C2: Ca2+-binding domain present in only conventional PKC isoenzymes (a, b, and g), but absent in novel PKC isoenzymes (d, e, h, and q) and atypical PKC isoenzymes (z, i, and l); C3: ATP-binding region in the catalytic domain; C4: protein substrate-binding region in the catalytic domain; V3: proteolysis susceptible variable region present in various PKC isoenzymes; pseudosubstrate: autoinhibitory region prevents the binding of protein substrate to the catalytic domain.
3.2. Relevance of PKC Isoenzymes to Apoptosis
A variety of agents that induce PKC inactivation induce apoptosis [67, 68] (Figure 3). The inactivation of PKC activates sphingomyelinase to increase the generation of ceramide [69–71]. Although apoptosis can be prevented by Bcl-2 [72], its anti-apoptotic function is compromised when Bcl-2 is not phosphorylated by PKCa and downstream ERK [73]. The ceramide-induced changes in mitochondria trigger the release of cytochrome c into cytosol where it induces the activation of caspase-3, a key protease involved in the initiation of apoptotic events [74, 75].
FIGURE 3. Selenium-induced inactivation of PKC induces apoptosis—opposing roles of PKC isoenzymes.
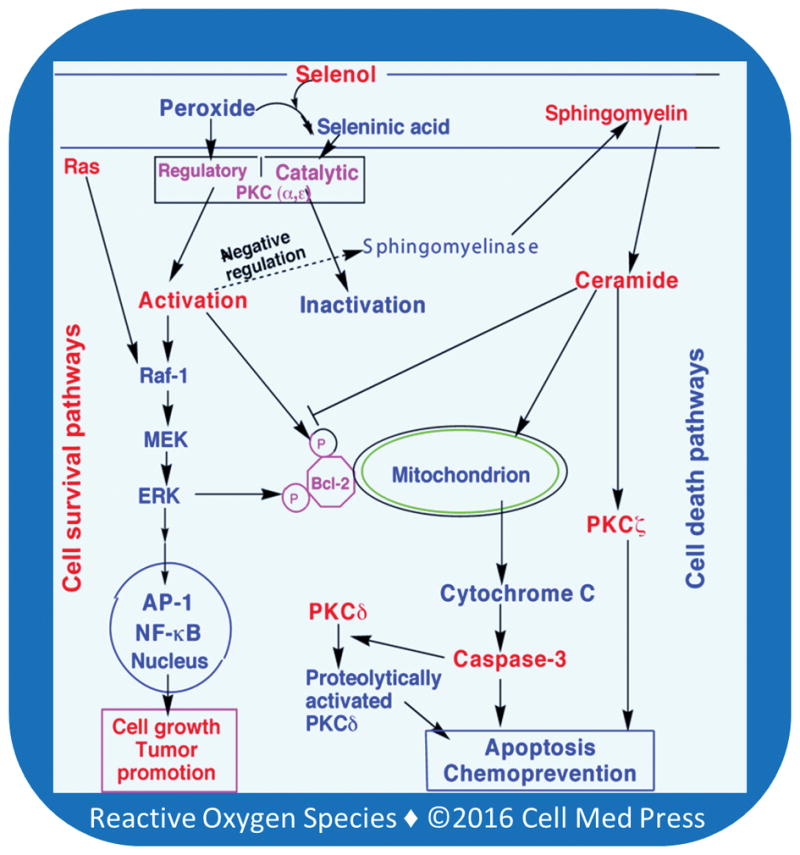
Activation of PKC a and e isoenzymes leads to activation of cell survival pathways. It also induces a negative regulation of sphingomyelinase and decreases the generation of cell death messenger ceramide. Selenium-induced inactivation of PKC a and e removes the negative regulation of sphingomyelinase. As a result, the sphingomyelinase activity increases and in turn enhances the generation of ceramide. Ceramide induces cell death pathways by inhibiting phosphorylation of Bcl-2, affecting mitochondria to release cytochrome C, which subsequently induces activation of caspase-3. Caspase-3 induces proteolytic activation of PKCd, which plays a key role in executing the apoptotic process. Ceramide also activates PKCz, which plays an important role in apoptosis.
Various PKC isoenzymes respond differently to stimuli that cause tumor promotion, cell growth, and cell death [76]. The role of PKCa in cell growth and cell death varies depending on cell type [77]. PKCe has oncogenic potential and is a promitogenic and prosurvival enzyme [76]. PKCe expression significantly increases in prostate cancer in a manner correlating with the aggressiveness of the disease [78]. Unlike PKCe, PKCd is a proapoptotic and antitumorigenic isoenzyme. A limited proteolysis of PKCd at its hinge region by caspase-3 separates its autoinhibitory regulatory domain from the catalytic domain [76]. This cofactor-independent, constitutively active catalytic fragment of PKCd mediates an important role in executing apoptosis. PKCz that is activated by ceramide also plays a role in inducing apoptosis [79]. Therefore, it is important to know whether selenium differentially inactivates antiapoptotic and proapoptotic PKC isoenzymes causing either tumor cell death or survival.
4. HYDROPEROXIDE-MEDIATED SELENIUM-INDUCED INACTIVATION OF PKC
4.1. Tumor Promoter Lipid Hydroperoxide-Mediated Redox Cycling of Selenium Inactivates PKC
Fatty acid hydroperoxides reversibly activate PKC isoenzymes [63]. Direct reactivity of the lipid hydroperoxides with sulfhydryls is very low; therefore, it will not directly oxidize cysteine residues. However, methylselenol reacts with the lipid hydroperoxides that are bound to PKC and reduces them, and in turn, methylselenol is oxidized to MSA (Figure 4). This locally generated MSA specifically oxidizes PKC cysteine sulfhydryl residues present within the vicinity [80]. Given that a cluster of cysteine residues present within the PKC catalytic domain does not coordinate binding of metal ions, MSA readily oxidizes these cysteine residues and inactivates the enzyme. During this process, at least two disulfide bonds are formed in the calcium-dependent classic PKC isoenzymes (a, b, and g). Although the PKC regulatory domain also has a cluster of 12 cysteine residues, they coordinate binding of four zinc ions. This makes them less susceptible to oxidation induced by MSA. However, if MSA is formed in high amounts, it can also oxidize the zinc-thiolates present within the regulatory domain [80]. In these oxidative reactions, MSA is reduced back to methylselenol, which repeats the reduction of fatty acid hydroperoxides. The methylselenol-MSA redox cycle allows selenium to catalytically oxidize many sulfhydryls in PKC even at low nanomolar concentrations. It is intriguing that what is a receptor for various tumor promoters is also a critical target for cancer prevention. As a result, selenium may effectively halt tumor promotion.
FIGURE 4. Hydroperoxide-mediated methylselenol-MSA redox cycle inactivates PKC.
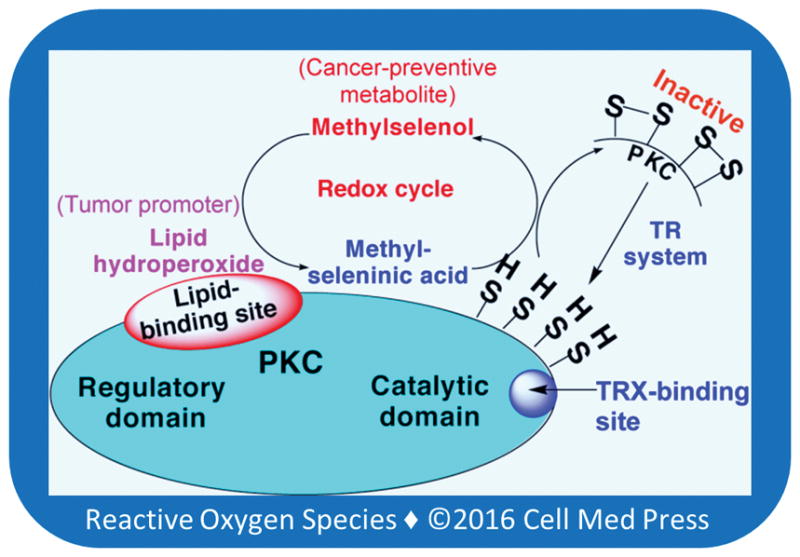
Initially, methylselenol generated by the TR system reduces lipid hydroperoxides bound to PKC and it, in turn, is oxidized to MSA. This locally generated MSA oxidizes PKC cysteine sulfhydryls present within the vicinity and inactivates the kinase. This reaction regenerates methylselenol, which reacts with another molecule of lipid hydroperoxide. The methylselenol-MSA redox cycle makes low concentrations of selenium to catalytically oxidize protein sulfhydryls. TRX binds with a high affinity to the PKC catalytic domain. Therefore, the TR system efficiently reduces disulfides formed within the PKC catalytic domain and regenerates its lost kinase activity.
4.2. PKC is a Specific Target to Locally Generated Methylseleninic Acid
A complete reduction of MSA to methylselenol requires four thiols. It involves an addition reaction and intermediate formation of selenosulfide: CH3SeO2H + 4 RSH → CH3SeH + 2 RSSR + 2 H2O. Thus, two pairs of juxtaposed sulfhydryls may be more effectively oxidized than one cysteine residue or two vicinal cysteine residues present in proteins. It is important to note that if MSA is formed distant from PKC in the cell its reactivity with PKC will be lower and less specific. Therefore, a local generation of MSA and redox cycle of methylselenol-MSA within the vicinity of PKC occurring with the membrane lipid hydroperoxides makes PKC a sensitive and specific target for selenium in the cell membrane compartment.
4.3. Differential Sensitivities of PKC Isoenzymes to Redox-Active Selenium
Among PKC isoenzymes, the conventional isoenzymes a, b, and g are more sensitive to both MSA and selenite than the novel isoenzymes e and d, whereas atypical isoenzyme z is the least sensitive among PKC isoezymes tested [80, 81]. This is consistent with the lower number of conserved cysteine residues present in the catalytic domain of PKCz compared to that of other PKC isoenzymes. Our previous studies suggested that the PKCa inhibition alone is not sufficient for MSA-induced apoptosis and that inactivation of PKCe is required as well [80]. Inactivation of PKCe, an oncogenic, promitogenic, and prosurvival enzyme, by selenium decreases cell proliferation and induces apoptosis. This hypothesis is further supported by the fact that the prostate cancer cells or premalignant cells overexpressing PKCe become less sensitive to MSA. Conversely, knockout of PKCe expression makes these cells more sensitive to MSA. PKCd is also inactivated by MSA at a rate equal to that of PKCe. Since methylselenol-MSA redox cycles occur with the hydroperoxide bound to the regulatory domain, it is more likely that the caspase-3-mediated proteolytic separation of the PKCd regulatory domain from the catalytic domain makes the latter domain less susceptible to inactivation by the methylselenol-MSA redox cycle. Thus, it is possible that at low concentrations, this redox cycle selectively inactivates cell survival isoenzymes such as PKCe and spares the activity of proteolytically activated catalytic fragment of PKCd and holo-PKCz, which are needed for executing apoptosis.
4.4. Significance of Hydroperoxide-Methylselenol Reaction in Promoting Precancer Cells
Fatty acid hydroperoxides have been implicated in tumor promotion in various sites including the prostate [14]. Such hydroperoxides may be generated by the overexpression of lipoxygenases and nonenzymatic oxidation of unsaturated fatty acids by oxidative stress and inflammation associated with tumor promotion [82, 83]. In promoting precancer cells where fatty acid hydroperoxides or other oxidants are in high concentrations, volatile methylselenol is retained after conversion to nonvolatile MSA, causing selective toxicity to these cells. It is intriguing that fatty acid hydroperoxides and other oxidants that induce tumor promotion may also help in mediating the chemopreventive actions of selenium. In contrast, in the normal cells where lipid hydroperoxides or oxidants are very low, volatile methylselenol is not oxidized to nonvolatile MSA and thus leaves normal cells or gets converted to excretory metabolites. Thus, methylselenol is less toxic to normal cells.
4.5. Selenium “Restores” Cell Death during Tumor Promotion
Oxidant tumor promoters are capable of inducing cell death in normal cells. However, some premalignant or malignant cells escape oxidant-induced cell death and accumulate mutations induced by oxidants, which ultimately lead to tumor promotion and progression [84]. Low concentrations of selenol, retained and amplified by peroxides, block tumor cells from escaping death and “restore” apoptosis. The concept of selenium “restoring” cell death is different from selenium per se inducing cell death. Selenium per se induces cell death only at high concentrations by a global toxic mechanism. With a direct selenium cytotoxicity approach (global toxicity), it is difficult to explain selenium’s selective toxicity to precancer cells.
4.6. Protein Thiol Oxidation Is Compartmentally Separated from GSH
Although several cases of protein thiol oxidation by selenium or oxidants have been demonstrated, it is often not addressed how protein thiol oxidation occurs in the reducing environment of the cell where there are millimolar concentrations of GSH, a thiol antioxidant present in the cytoplasm. PKC translocates from the cytoplasm to the cell membrane in cells treated with various tumor promoters [85]. A cellular depletion of GSH did not enhance inactivation of PKC by selenocompounds [20]. Since fatty acid hydroperoxides are abundant in the cell membrane, which is subjected to oxidative stress, the peroxidatic redox cycle of methylselenol-MSA oxidizes thiol-rich regions in the membrane-bound proteins compartmentally separated from GSH.
5. OTHER PROTEIN THIOLS OXIDIZED BY SELENIUM
5.1. Proteins with Zinc-Thiolates
In metallothioneins, each zinc atom is bound tetrahedrally to four cysteine residues to form extensive zinc-thiolate cluster networks between 7 zinc atoms and 20 cysteine thiolates [86]. Redox-active selenocompounds oxidize these zinc-thiolate clusters with low redox potential and release the bound zinc from metallothioneins [86]. Enzymes involved in DNA repair, such as formmidopyrimidine-DNA glycosylase and xeroderma pigmentosum group A protein, have zinc thiolates and as such, various reducible selenocompounds oxidize these thiolates and release zinc from the enzymes, causing the collapse of their DNA-binding sites [87]. Estrogen receptor-beta also has two zinc-thiolate clusters in the DNA-binding domain. However, MSA treatment of prostate cancer cells did not affect the receptor binding to estrogen response element or the estrogen-regulated gene expression [88]. As mentioned before, the zinc-thiolates in PKC are oxidized to a lesser extent than the free thiolates present within the catalytic domain which are not coordinating zinc [80, 81]. Overall, the rate of the selenium-induced oxidation of thiolates depends on their redox potential, which is in turn influenced by adjacent amino acid residues in the protein. Therefore, zinc-thiolates in different proteins may have different degree of susceptibility to selenium-induced oxidation. Since sulfhydryl oxidation by redox-active selenocompounds involves initial addition reaction and formation of selenosulfide, it is possible that the binding of redox-inert zinc to thiolates makes them oxidized at a lower rate than free thiolates. However, further studies are needed to address this issue.
5.2. Transcriptional Factors
A decrease in transcriptional factor AP-1 binding to DNA was observed in selenite and selenodiglutathione-treated cells [89]. The authors have suggested that cysteine-modification in TRX contributes to a decrease in Ref-1 activity, which is required for activation of Jun and Fos proteins. In addition, MSA has been shown to inhibit the binding of transcriptional factor NF-kB to DNA and the expression of antiapoptotic genes in the prostate cancer cells [90]. Furthermore, MSA significantly downregulated Keap1, thereby inducing the activation of Nrf2 and antioxidant response element (ARE) promoter activity in esophageal squamous cell carcinoma cells [91]. The authors suggested that the downregulation of Keap1 occurred by MSA-induced miR-200a expression. However, no experiments were carried out in this study to determine whether MSA indeed induced a redox modification of two vicinal cysteines in Keap1, which are highly susceptible to oxidation and addition reactions [92]. Another organic selenocompound, ebselen, has been shown to induce redox modification of cysteine 151 in Keap1 and liberation and activation of Nrf2 [93]. Both selenocompounds selenite and p-XSC decreased the binding of transcription factors Sp1 and Sp3 to their consensus site in DNA [94]. In addition, some authors have shown activation of p53, a tumor suppressor protein, by selenomethionine in lung cancer cells [95]. In this process, a reduction of its two cysteine residues that require Ref-1 was observed. The authors suggested the significance of this selenium-dependent activation of p53 on DNA repair in cancer prevention.
5.3. Apoptosis-Related Proteins
Selenite can activate apoptotic machinery through redox-dependent activation of Bax, a Bcl-2 family protein, by modifying two conserved cysteine residues present in this protein [96]. On the contrary, selenite inactivates proapoptotic protease caspase-3 in HEK293 cells [97]. Indeed, selenite directly inactivates recombinant caspase-3 by inducing reversible thiol redox modification of critical cysteine residue present within the active site. In cells, selenite also inactivates enzymes such as c-Jun N-terminal kinase and p38, which are involved in inducing cell death in response to external stresses [98]. Selenite directly inactivates c-Jun N-terminal kinase by a direct modification of a single cysteine residue. However, the inactivation of p38 is caused by the effect of selenite on target(s) upstream to p38 rather than by the direct modification of the enzyme [98].
6. RELATIONSHIP BETWEEN SELENOPROTEINS AND SELENOMETABOLITES
6.1. Selenoproteins Antagonize Selenometabolite Actions—Resistance to Selenium
In studies carried out with intact tumor cells or purified PKC isoenzymes, the TR system reduced the selenium-induced disulfides in PKC regenerating its lost kinase activity [80]. It is intriguing that selenometabolite action on PKC is nullified by selenoprotein TR. It is also interesting to note that TRX or related proteins bind with high affinity to the PKC catalytic domain [99]. Inhibition of TR activity by its specific inhibitor auranofin was shown to increase MSA-induced inactivation of PKC in cells [80]. Therefore, in cells treated with methylselenol or MSA, TR is a limiting factor for the reduction of oxidized sulfhydryls in PKC to regenerate its kinase activity.
An opposing relationship may exist between the actions of selenometabolites and selenoproteins. It is important to elucidate this relationship in order to clarify the complexity of selenium as a pro-oxidant and an antioxidant. Selenometabolites, as pro-oxidants, generate free radicals and cause lipid peroxidation and DNA damage [100]. Selenoproteins, as antioxidants, negate the actions of selenometabolites. For example, selenometabolites such as selenite produce H2O2 and result in the formation of lipid hydroperoxides [101], which are removed by selenoprotein GPX. Similarly, selenometabolite-induced protein thiol oxidation is reversed by the TR system as seen in the PKC model discussed above. Furthermore, TR converts protein sulfhydryl-oxidizing MSA into volatile methylselenol, which cannot directly oxidize protein thiols. In addition, removal of fatty acid hydroperoxides by GPX-4 could take away the hydroperoxides necessary for the conversion of methylselenol to MSA.
The opposing relationship between selenometabolites and selenoproteins may provide an interesting scenario to explain why cancer preventive concentrations of selenium are safe to normal cells while being selectively cytotoxic to precancer cells but not to advanced malignant cells (Figure 5). Since volatile selenometabolites are retained to a lesser extent in normal cells due to presence of low amounts of lipid hydroperoxides, selenometabolite toxicity is low and selenoproteins can effectively safeguard against this toxicity. On the contrary, in precancer promoting cells, due to presence of high amounts of lipid peroxides, selenometabolites are retained to a greater extent and cause greater toxicity. This overwhelms selenoproteins, leading to selective selenium toxicity to precancer cells. An increased expression of TR was observed in many tumors compared to their normal counterparts [36, 50]. Various skin tumor promoters have been shown to increase the expression of TR and TRX [102]. This can cause escape of tumor cells from selenium-induced cell death, leading to failure of chemoprevention by selenium.
FIGURE 5. Relationship between selenoproteins and selenometabolites in normal, precancer, and advanced malignant cells.

In normal cells, selenometabolites are low and selenoproteins effectively safeguard against cytotoxcity induced by selenometabolites. In contrast, in precancer cells, high selenometabolite-induced cytotoxicity overwhelms the selenoprotein safeguards, resulting in cell death. However, in advanced malignant cells, both selenometabolite effects and selenoprotein safeguards are high, thereby resulting in resistance to selenium-induced cytotoxicity.
6.2. Selenoproteins Support Selenometabolite-Induced Cell Death
In some conditions, selenoproteins may support selenometabolite actions. For example, high amounts of TR may increase selenite conversion to selenide, which then undergoes the redox cycle, induces oxidative stress, inhibits lipoxygenases, and induces apoptosis [103, 104]. There are also suggestions that in some cases, TR can function as a cell death-inducing enzyme instead of a cell survival enzyme [105]. Paradoxical roles of TR and GPX in cellular survival [106] further complicate understanding their role in selenium-induced cancer prevention. Conceivably, knowledge of this complex relationship between selenometabolites and selenoproteins may help further understanding of why certain cancers are prevented by selenium and others are not.
7. U-SHAPED CURVE IN SELENIUM-INDUCED CANCER PREVENTION
An intriguing U-shaped dose-response relationship was observed between dietary selenium intake and the extent of DNA damage in the canine prostate [107]. In the NPC trial, the initial selenium status of the people was found to be an important determining factor for the efficacy of cancer prevention by selenium [10]. Selenium supplementation was found to decrease cancer incidence in men with baseline plasma selenium levels that fell in the lowest two tertiles (< 122 ng/mL). In contrast, those in the highest tertile showed an increase in cancer incidence. Similarly, in the Southwest Oncology Group S9917 high-grade prostatic intraepithelial neoplasia trial, selenomethionine supplementation (200 mg/day) reduced prostate cancer risk in men with the baseline plasma selenium level in the lowest quartile (<106 ng/mL) [108]. In our studies, we found a biphasic effect of MSA on growth inhibition and induction of cell death in a human prostate cancer cell line (WPE1-NB26); lower concentrations of MSA induced cell growth inhibition and cell death whereas higher concentrations of MSA did not (unpublished results).
The observed biphasic effect of MSA may be due to a variation in sensitivity of proteins to redox-active selenium and their differences in subcellular localization (Figure 6). More sensitive protein targets having a cluster of vicinal thiols and the ability to bind membrane hydroperoxides are inactivated by low concentrations of selenium that undergoing catalytic redox cycle compartmentally separated from GSH [20, 80]. These targets include enzymes such as PKC (a and e isoenzymes), whose inactivation may induce apoptosis [80]. On the other hand, low affinity targets that have limited cysteine residues or are located in the cytoplasmic compartment, such as caspase-3, PKCz, and proteolytically activated PKCd, are not affected by low concentrations of selenium. Therefore, the apoptotic process is executed in the cell by low concentrations of MSA [80]. With an increase in MSA concentration, it is possible that these low affinity proapoptotic targets are also inactivated by MSA, and as such, it fails to induce apoptosis. Similarly, modification of one or two cysteine residues in Keap1 may be expected to occur only at higher concentrations of MSA, leading to the activation of Nrf2. The activation of Nrf2 lsubsequently results in induction of antioxidant/cytoprotective enzymes, including the selenoproteins TR and GPX [92]. This may protect cells from MSA-induced cell death. Although various studies identified protein thiol targets for different redox-active selenocompounds in different cell systems, studies assessing the relative sensitivity of various targets to a given selenocompound in the same cell type are needed to understand further this biphasic effect of selenium in cancer prevention.
FIGURE 6. Possible mechanism for biphasic effects of selenium in inducing apoptosis in tumor cells.
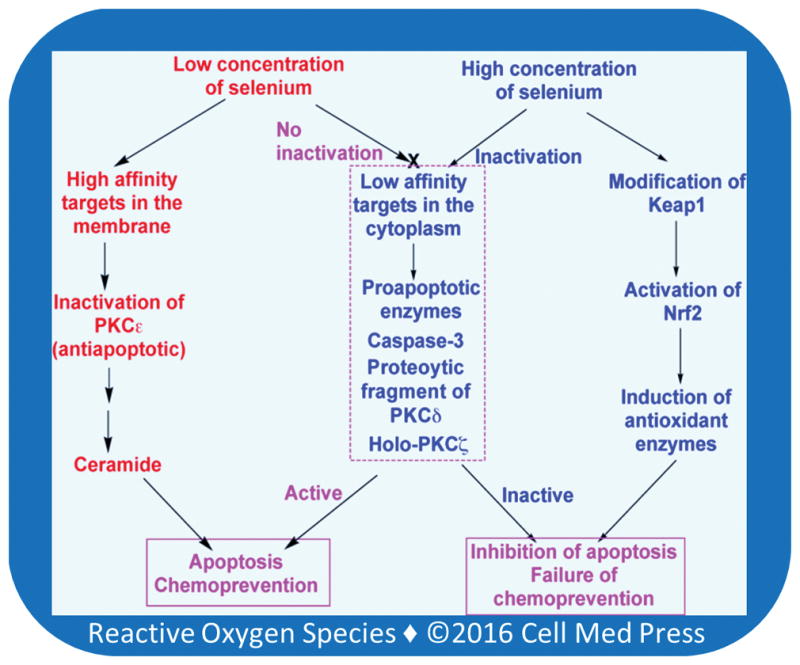
At low concentrations, selenium inactivates high affinity membrane targets such as antiapoptotic enzymes PKCa and e. Nevertheless, such low concentrations of selenium do not inactivate low affinity cytoplasmic targets such as proapototic enzymes caspase-3, proteolytically activated PKCd and holo-PKCz. Therefore, these enzymes execute the apoptotic processes. At higher concentrations, besides inactivating antiapoptotic enzymes, selenium inactivates proapoptotic enzymes as well. Thus, apoptosis is not executed. Furthermore, selenium activates Nrf2, which induces antioxidant enzymes that protect cells from death.
8. CONCLUSIONS AND PERSPECTIVES
Given the ability of redox-active selenocompounds to induce protein thiol oxidation, understanding redox regulation of critical cellular proteins which influence cell death and survival is important to elucidate why selenium prevents cancer in some cases only. PKC isoenzymes are well-qualified targets for protein thiol regulation in cancer-preventive actions of selenium. Several other proteins with critical cysteine residues are also oxidatively modified by selenium. However, additional critical studies are needed to address the significance of such modifications in cancer prevention.
In this review, we considered various criteria necessary to propose a cancer-preventive mechanism for selenium. First, we address how the promotional stage of carcinogenesis, with its long duration, is well suited for chemoprevention. PKC isoenzymes are targets for both endogenous and exogenous tumor promoters. Therefore, as a receptor for tumor promoters, PKC is a well-qualified target for preventing tumor promotion. At the same time, it has unique structural features that render it susceptible to selenium-induced thiol-redox regulation. The intriguing aspect is that selenium uses the same receptor to which tumor promoters bind and thus effectively block the tumor promotion process. Second, methylselenol and other selenol metabolites of synthetic compounds are implicated in mediating the cancer-preventive actions of selenium. The mechanism proposed involves a reaction of this form of selenium with tumor-promoting lipid hydroperoxides bound to PKC; the resulting locally generated MSA inactivates PKC by oxidizing critical thiols present within the catalytic domain. The peroxidatic methylselenol-MSA redox cycle makes low nanomolar concentrations of selenium to catalytically oxidize sulfhydryl clusters in the proteins. While this occurs with the membrane-bound lipid hydroperoxides and proteins, this reaction occurs compartmentally separated from cytoplasmic GSH. It is astonishing that antitumor promoter selenium utilizes tumor promoting lipid hydroperoxides, restores cell death, and thereby prevents tumor promotion. The abundance of hydroperoxides in precancer cells makes them more susceptible to selenium-induced cytotoxicity than normal cells, where lipid hydroperoxides are less abundant. Third, we examine the reversal of selenometabolite-induced thiol oxidation of PKC isoenzymes by selenoprotein TR-containing system. This reversal mechanism is strongly supported by the presence of high affinity site for TRX on the catalytic domain of PKC. Another selenoprotein GPX may inhibit methylselenol-MSA redox cycle by removing hydroperoxides. Conceivably, in some cases selenoproteins can counteract the cytotoxicity induced by selenometabolites. This is especially important given that TR is overexpressed in advanced malignant cells, which may make them escape cancer-preventive actions of selenium. Finally, the relative susceptibility of antiapoptotic and proapoptotic enzymes to selenium-induced inactivation may explain the intriguing U-shaped curve that is seen in the dietary selenium intake and extent of cancer-prevention. Selenium at lower concentrations oxidatively inactivates antiapoptotic PKC isoenzymes (e and a) having both a lipid hydroperoxide-binding site and a cluster of critical thiols in the catalytic domain. However, at low concentrations selenium cannot inactivate low affinity targets such as proapoptotic enzymes (PKC d catalytic fragment, PKCz, caspase-3, and c-Jun N-terminal kinase) having a limited number of critical thiols. These spared proapoptotic enzymes induce apoptosis at lower concentrations of selenium. On the other hand, tumor cells will become resistant to apoptosis when these proapoptotic enzymes are also inactivated by higher concentrations of selenium.
Considering that ROS are generated in the action of a variety of cancer-preventive agents, understanding the cellular protein thiol redox status may help optimize the cancer-preventive efficacy of not only selenium but also other chemopreventive agents. Similarly, the imbalance in protein thiol oxidations may contribute to selenium-induced clinical complications such as diabetes. Finally, the protein thiol regulations may contribute to cell death in degenerative diseases and thus this subject has broader implications beyond the realm of cancer prevention.
Acknowledgments
The authors’ work was supported by grants from the National Cancer Institute/National Institutes of Health (CA099216) and the Swedish Cancer Society (961).
ABBREVIATIONS
- GPX
glutathione peroxidase
- GSH
glutathione
- MSA
methylseleninic acid
- PKC
protein kinase C
- p-XSC
1,4-phenylene-bis(methylene)selenocyanate
- ROS
reactive oxygen species
- TR
thioredoxin reductase
- Trx
thioredoxin
References
- 1.Combs GF, Jr, Gray WP. Chemopreventive agents: selenium. Pharmacol Ther. 1998;79(3):179–92. doi: 10.1016/s0163-7258(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 2.Coates RJ, Weiss NS, Daling JR, Morris JS, Labbe RF. Serum levels of selenium and retinol and the subsequent risk of cancer. Am J Epidemiol. 1988;128(3):515–23. doi: 10.1093/oxfordjournals.aje.a114999. [DOI] [PubMed] [Google Scholar]
- 3.Glattre E, Thomassen Y, Thoresen SO, Haldorsen T, Lund-Larsen PG, Theodorsen L, et al. Prediagnostic serum selenium in a case-control study of thyroid cancer. Int J Epidemiol. 1989;18(1):45–9. doi: 10.1093/ije/18.1.45. [DOI] [PubMed] [Google Scholar]
- 4.Milner JA. Effect of selenium on virally induced and transplantable tumor models. Fed Proc. 1985;44(9):2568–72. [PubMed] [Google Scholar]
- 5.Ip C. Prophylaxis of mammary neoplasia by selenium supplementation in the initiation and promotion phases of chemical carcinogenesis. Cancer Res. 1981;41(11 Pt 1):4386–90. [PubMed] [Google Scholar]
- 6.el-Bayoumy K, Rao CV, Reddy BS. Multiorgan sensitivity to anticarcinogenesis by the organoselenium 1,4-phenylenebis(methylene)selenocyanate. Nutr Cancer. 2001;40(1):18–27. doi: 10.1207/S15327914NC401_6. [DOI] [PubMed] [Google Scholar]
- 7.Dorado RD, Porta EA, Aquino TM. Effects of dietary selenium on hepatic and renal tumorigenesis induced in rats by diethylnitrosamine. Hepatology. 1985;5(6):1201–8. doi: 10.1002/hep.1840050623. [DOI] [PubMed] [Google Scholar]
- 8.Perchellet JP, Abney NL, Thomas RM, Guislain YL, Perchellet EM. Effects of combined treatments with selenium, glutathione, and vitamin E on glutathione peroxidase activity, ornithine decarboxylase induction, and complete and multistage carcinogenesis in mouse skin. Cancer Res. 1987;47(2):477–85. [PubMed] [Google Scholar]
- 9.Li JY, Taylor PR, Li B, Dawsey S, Wang GQ, Ershow AG, et al. Nutrition intervention trials in Linxian, China: multiple vitamin/mineral supplementation, cancer incidence, and disease-specific mortality among adults with esophageal dysplasia. J Natl Cancer Inst. 1993;85(18):1492–8. doi: 10.1093/jnci/85.18.1492. [DOI] [PubMed] [Google Scholar]
- 10.Clark LC, Combs GF, Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin: a randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276(24):1957–63. [PubMed] [Google Scholar]
- 11.Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301(1):39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinbrenner H, Speckmann B, Sies H. Toward understanding success and failures in the use of selenium for cancer prevention. Antioxid Redox Signal. 2013;19(2):181–91. doi: 10.1089/ars.2013.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganther HE, Corcoran C. Selenotrisulfides. II. Cross-linking of reduced pancreatic ribonuclease with selenium. Biochemistry. 1969;8(6):2557–63. doi: 10.1021/bi00834a044. [DOI] [PubMed] [Google Scholar]
- 14.Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28(9):1349–61. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 15.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312(5782):1882–3. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 16.Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10(8):1343–74. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milner JA, Pigott MA, Dipple A. Selective effects of selenium selenite on 7,12-dimethylbenz(a)anthracene-DNA binding in fetal mouse cell cultures. Cancer Res. 1985;45(12 Pt 1):6347–54. [PubMed] [Google Scholar]
- 18.Medina D. Mechanisms of selenium inhibition of tumorigenesis. Adv Exp Med Biol. 1986;206:465–72. doi: 10.1007/978-1-4613-1835-4_33. [DOI] [PubMed] [Google Scholar]
- 19.Sharma S, Stutzman JD, Kelloff GJ, Steele VE. Screening of potential chemopreventive agents using biochemical markers of carcinogenesis. Cancer Res. 1994;54(22):5848–55. [PubMed] [Google Scholar]
- 20.Gopalakrishna R, Chen ZH, Gundimeda U. Selenocompounds induce a redox modulation of protein kinase C in the cell, compartmentally independent from cytosolic glutathione: its role in inhibition of tumor promotion. Arch Biochem Biophys. 1997;348(1):37–48. doi: 10.1006/abbi.1997.0335. [DOI] [PubMed] [Google Scholar]
- 21.Ip C, Ip MM, Kim U. Dietary selenium intake and growth of the MT-W9B transplantable rat mammary tumor. Cancer Lett. 1981;14(1):101–7. doi: 10.1016/0304-3835(81)90015-x. [DOI] [PubMed] [Google Scholar]
- 22.Lu J, Kaeck M, Jiang C, Wilson AC, Thompson HJ. Selenite induction of DNA strand breaks and apoptosis in mouse leukemic L1210 cells. Biochem Pharmacol. 1994;47(9):1531–5. doi: 10.1016/0006-2952(94)90528-2. [DOI] [PubMed] [Google Scholar]
- 23.Ip C, Ganther HE. Activity of methylated forms of selenium in cancer prevention. Cancer Res. 1990;50(4):1206–11. [PubMed] [Google Scholar]
- 24.Weekley CM, Harris HH. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem Soc Rev. 2013;42(23):8870–94. doi: 10.1039/c3cs60272a. [DOI] [PubMed] [Google Scholar]
- 25.Medina D, Thompson H, Ganther H, Ip C. Se-methylselenocysteine: a new compound for chemoprevention of breast cancer. Nutr Cancer. 2001;40(1):12–7. doi: 10.1207/S15327914NC401_5. [DOI] [PubMed] [Google Scholar]
- 26.Kumar S, Bjornstedt M, Holmgren A. Selenite is a substrate for calf thymus thioredoxin reductase and thioredoxin and elicits a large non-stoichiometric oxidation of NADPH in the presence of oxygen. Eur J Biochem. 1992;207(2):435–39. doi: 10.1111/j.1432-1033.1992.tb17068.x. [DOI] [PubMed] [Google Scholar]
- 27.Bjornstedt M, Kumar S, Holmgren A. Selenodiglutathione is a highly efficient oxidant of reduced thioredoxin and a substrate for mammalian thioredoxin reductase. J Biol Chem. 1992;267(12):8030–4. [PubMed] [Google Scholar]
- 28.Holmgren A. Thioredoxin. Annu Rev Biochem. 1985;54:237–71. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 29.Holmgren A, Lu J. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochem Biophys Res Commun. 2010;396(1):120–4. doi: 10.1016/j.bbrc.2010.03.083. [DOI] [PubMed] [Google Scholar]
- 30.Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267(20):6102–9. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 31.Luthman M, Holmgren A. Rat liver thioredoxin and thioredoxin reductase: purification and characterization. Biochemistry. 1982;21(26):6628–33. doi: 10.1021/bi00269a003. [DOI] [PubMed] [Google Scholar]
- 32.Lu J, Berndt C, Holmgren A. Metabolism of selenium compounds catalyzed by the mammalian selenoprotein thioredoxin reductase. Biochim Biophys Acta. 2009;1790(11):1513–9. doi: 10.1016/j.bbagen.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 33.Bjornstedt M, Hamberg M, Kumar S, Xue J, Holmgren A. Human thioredoxin reductase directly reduces lipid hydroperoxides by NADPH and selenocystine strongly stimulates the reaction via catalytically generated selenols. J Biol Chem. 1995;270(20):11761–4. doi: 10.1074/jbc.270.20.11761. [DOI] [PubMed] [Google Scholar]
- 34.Arner ES, Bjornstedt M, Holmgren A. 1-Chloro-2,4-dinitrobenzene is an irreversible inhibitor of human thioredoxin reductase: loss of thioredoxin disulfide reductase activity is accompanied by a large increase in NADPH oxidase activity. J Biol Chem. 1995;270(8):3479–82. doi: 10.1074/jbc.270.8.3479. [DOI] [PubMed] [Google Scholar]
- 35.Nordberg J, Zhong L, Holmgren A, Arner ES. Mammalian thioredoxin reductase is irreversibly inhibited by dinitrohalobenzenes by alkylation of both the redox active selenocysteine and its neighboring cysteine residue. J Biol Chem. 1998;273(18):10835–42. doi: 10.1074/jbc.273.18.10835. [DOI] [PubMed] [Google Scholar]
- 36.Gladyshev VN, Jeang KT, Stadtman TC. Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc Natl Acad Sci USA. 1996;93(12):6146–51. doi: 10.1073/pnas.93.12.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong L, Arner ES, Ljung J, Aslund F, Holmgren A. Rat and calf thioredoxin reductase are homologous to glutathione reductase with a carboxyl-terminal elongation containing a conserved catalytically active penultimate selenocysteine residue. J Biol Chem. 1998;273(15):8581–91. doi: 10.1074/jbc.273.15.8581. [DOI] [PubMed] [Google Scholar]
- 38.Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol. 2006;16(6):420–6. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 39.Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27(2):211–22. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 40.Schrauzer GN. Selenomethionine: a review of its nutritional significance, metabolism and toxicity. J Nutr. 2000;130(7):1653–6. doi: 10.1093/jn/130.7.1653. [DOI] [PubMed] [Google Scholar]
- 41.Klein EA, Thompson IM, Lippman SM, Goodman PJ, Albanes D, Taylor PR, et al. SELECT: the next prostate cancer prevention trial. Selenum and Vitamin E Cancer Prevention Trial. J Urol. 2001;166(4):1311–5. doi: 10.1016/s0022-5347(05)65759-x. [DOI] [PubMed] [Google Scholar]
- 42.Reddy BS, Hirose Y, Lubet RA, Steele VE, Kelloff GJ, Rao CV. Lack of chemopreventive efficacy of DL-selenomethionine in colon carcinogenesis. Int J Mol Med. 2000;5(4):327–30. doi: 10.3892/ijmm.5.4.327. [DOI] [PubMed] [Google Scholar]
- 43.Lu J, Zhang J, Jiang C, Deng Y, Ozten N, Bosland MC. Cancer chemoprevention research with selenium in the post-SELECT era: Promises and challenges. Nutr Cancer. 2016;68(1):1–17. doi: 10.1080/01635581.2016.1105267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Redman C, Scott JA, Baines AT, Basye JL, Clark LC, Calley C, et al. Inhibitory effect of selenomethionine on the growth of three selected human tumor cell lines. Cancer Lett. 1998;125(1–2):103–10. doi: 10.1016/s0304-3835(97)00497-7. [DOI] [PubMed] [Google Scholar]
- 45.Lu J, Jiang C. Selenium and cancer chemoprevention: hypotheses integrating the actions of selenoproteins and selenium metabolites in epithelial and non-epithelial target cells. Antioxid Redox Signal. 2005;7(11–12):1715–27. doi: 10.1089/ars.2005.7.1715. [DOI] [PubMed] [Google Scholar]
- 46.Burk RF, Hill KE. Regulation of selenoproteins. Annu Rev Nutr. 1993;13:65–81. doi: 10.1146/annurev.nu.13.070193.000433. [DOI] [PubMed] [Google Scholar]
- 47.Gallegos A, Berggren M, Gasdaska JR, Powis G. Mechanisms of the regulation of thioredoxin reductase activity in cancer cells by the chemopreventive agent selenium. Cancer Res. 1997;57(21):4965–70. [PubMed] [Google Scholar]
- 48.Hill KE, McCollum GW, Boeglin ME, Burk RF. Thioredoxin reductase activity is decreased by selenium deficiency. Biochem Biophys Res Commun. 1997;234(2):293–5. doi: 10.1006/bbrc.1997.6618. [DOI] [PubMed] [Google Scholar]
- 49.Ganther H, Ip C. Thioredoxin reductase activity in rat liver is not affected by supranutritional levels of monomethylated selenium in vivo and is inhibited only by high levels of selenium in vitro. J Nutr. 2001;131(2):301–4. doi: 10.1093/jn/131.2.301. [DOI] [PubMed] [Google Scholar]
- 50.Berggren M, Gallegos A, Gasdaska JR, Gasdaska PY, Warneke J, Powis G. Thioredoxin and thioredoxin reductase gene expression in human tumors and cell lines, and the effects of serum stimulation and hypoxia. Anticancer Res. 1996;16(6B):3459–66. [PubMed] [Google Scholar]
- 51.Baker A, Payne CM, Briehl MM, Powis G. Thioredoxin, a gene found overexpressed in human cancer, inhibits apoptosis in vitro and in vivo. Cancer Res. 1997;57(22):5162–7. [PubMed] [Google Scholar]
- 52.Spallholz JE. On the nature of selenium toxicity and carcinostatic activity. Free Radic Biol Med. 1994;17(1):45–64. doi: 10.1016/0891-5849(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 53.Ganther HE. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: complexities with thioredoxin reductase. Carcinogenesis. 1999;20(9):1657–66. doi: 10.1093/carcin/20.9.1657. [DOI] [PubMed] [Google Scholar]
- 54.El-Bayoumy K, Upadhyaya P, Sohn OS, Rosa JG, Fiala ES. Synthesis and excretion profile of 1,4-[14C]phenylenebis(methylene)selenocyanate in the rat. Carcinogenesis. 1998;19(9):1603–7. doi: 10.1093/carcin/19.9.1603. [DOI] [PubMed] [Google Scholar]
- 55.Deagen JT, Butler JA, Zachara BA, Whanger PD. Determination of the distribution of selenium between glutathione peroxidase, selenoprotein P, and albumin in plasma. Anal Biochem. 1993;208(1):176–81. doi: 10.1006/abio.1993.1025. [DOI] [PubMed] [Google Scholar]
- 56.Combs GF., Jr Status of selenium in prostate cancer prevention. Br J Cancer. 2004;91(2):195–9. doi: 10.1038/sj.bjc.6601974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258(5082):607–14. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 58.Gopalakrishna R, Anderson WB. Ca2+– and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc Natl Acad Sci USA. 1989;86(17):6758–62. doi: 10.1073/pnas.86.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gopalakrishna R, Anderson WB. Reversible oxidative activation and inactivation of protein kinase C by the mitogen/tumor promoter periodate. Arch Biochem Biophys. 1991;285(2):382–7. doi: 10.1016/0003-9861(91)90377-u. [DOI] [PubMed] [Google Scholar]
- 60.Gopalakrishna R, Chen ZH, Gundimeda U. Tobacco smoke tumor promoters, catechol and hydroquinone, induce oxidative regulation of protein kinase C and influence invasion and metastasis of lung carcinoma cells. Proc Natl Acad Sci USA. 1994;91(25):12233–7. doi: 10.1073/pnas.91.25.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kass GE, Duddy SK, Orrenius S. Activation of hepatocyte protein kinase C by redox-cycling quinones. Biochem J. 1989;260(2):499–507. doi: 10.1042/bj2600499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larsson R, Cerutti P. Translocation and enhancement of phosphotransferase activity of protein kinase C following exposure in mouse epidermal cells to oxidants. Cancer Res. 1989;49(20):5627–32. [PubMed] [Google Scholar]
- 63.O’Brian CA, Ward NE, Weinstein IB, Bull AW, Marnett LJ. Activation of rat brain protein kinase C by lipid oxidation products. Biochem Biophys Res Commun. 1988;155(3):1374–80. doi: 10.1016/s0006-291x(88)81293-2. [DOI] [PubMed] [Google Scholar]
- 64.Gavrielides MV, Frijhoff AF, Conti CJ, Kazanietz MG. Protein kinase C and prostate carcinogenesis: targeting the cell cycle and apoptotic mechanisms. Curr Drug Targets. 2004;5(5):431–43. doi: 10.2174/1389450043345380#sthash.v5cjScgj.dpuf. [DOI] [PubMed] [Google Scholar]
- 65.Kazanietz MG, Wang S, Milne GW, Lewin NE, Liu HL, Blumberg PM. Residues in the second cysteine-rich region of protein kinase C delta relevant to phorbol ester binding as revealed by site-directed mutagenesis. J Biol Chem. 1995;270(37):21852–9. doi: 10.1074/jbc.270.37.21852. [DOI] [PubMed] [Google Scholar]
- 66.Gopalakrishna R, Gundimeda U. Antioxidant regulation of protein kinase C in cancer prevention. J Nutr. 2002;132(12):3819S–23S. doi: 10.1093/jn/132.12.3819S. [DOI] [PubMed] [Google Scholar]
- 67.Jarvis WD, Turner AJ, Povirk LF, Traylor RS, Grant S. Induction of apoptotic DNA fragmentation and cell death in HL-60 human promyelocytic leukemia cells by pharmacological inhibitors of protein kinase C. Cancer Res. 1994;54(7):1707–14. [PubMed] [Google Scholar]
- 68.Couldwell WT, Gopalakrishna R, Hinton DR, He S, Weiss MH, Law RE, et al. Hypericin: a potential antiglioma therapy. Neurosurgery. 1994;35(4):705–9. doi: 10.1227/00006123-199410000-00017. discussion 9–10. [DOI] [PubMed] [Google Scholar]
- 69.Hannun YA. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274(5294):1855–9. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 70.Chmura SJ, Nodzenski E, Weichselbaum RR, Quintans J. Protein kinase C inhibition induces apoptosis and ceramide production through activation of a neutral sphingomyelinase. Cancer Res. 1996;56(12):2711–4. [PubMed] [Google Scholar]
- 71.Haimovitz-Friedman A, Kolesnick RN, Fuks Z. Ceramide signaling in apoptosis. Br Med Bull. 1997;53(3):539–53. doi: 10.1093/oxfordjournals.bmb.a011629. [DOI] [PubMed] [Google Scholar]
- 72.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75(2):241–51. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 73.Ruvolo PP, Deng X, Carr BK, May WS. A functional role for mitochondrial protein kinase Calpha in Bcl2 phosphorylation and suppression of apoptosis. J Biol Chem. 1998;273(39):25436–42. doi: 10.1074/jbc.273.39.25436. [DOI] [PubMed] [Google Scholar]
- 74.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275(5303):1132–6. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 75.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 76.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7(4):281–94. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 77.Gonzalez-Guerrico AM, Meshki J, Xiao L, Benavides F, Conti CJ, Kazanietz MG. Molecular mechanisms of protein kinase C-induced apoptosis in prostate cancer cells. J Biochem Mol Biol. 2005;38(6):639–45. doi: 10.5483/bmbrep.2005.38.6.639. [DOI] [PubMed] [Google Scholar]
- 78.Wu D, Foreman TL, Gregory CW, McJilton MA, Wescott GG, Ford OH, et al. Protein kinase cepsilon has the potential to advance the recurrence of human prostate cancer. Cancer Res. 2002;62(8):2423–9. [PubMed] [Google Scholar]
- 79.Wang G, Silva J, Krishnamurthy K, Tran E, Condie BG, Bieberich E. Direct binding to ceramide activates protein kinase Czeta before the formation of a pro-apoptotic complex with PAR-4 in differentiating stem cells. J Biol Chem. 2005;280(28):26415–24. doi: 10.1074/jbc.M501492200. [DOI] [PubMed] [Google Scholar]
- 80.Gundimeda U, Schiffman JE, Chhabra D, Wong J, Wu A, Gopalakrishna R. Locally generated methylseleninic acid induces specific inactivation of protein kinase C isoenzymes: relevance to selenium-induced apoptosis in prostate cancer cells. J Biol Chem. 2008;283(50):34519–31. doi: 10.1074/jbc.M807007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gopalakrishna R, Gundimeda U, Chen ZH. Cancer-preventive selenocompounds induce a specific redox modification of cysteine-rich regions in Ca2+-dependent isoenzymes of protein kinase C. Arch Biochem Biophys. 1997;348(1):25–36. doi: 10.1006/abbi.1997.0334. [DOI] [PubMed] [Google Scholar]
- 82.Ghosh J, Myers CE. Arachidonic acid stimulates prostate cancer cell growth: critical role of 5-lipoxygenase. Biochem Biophys Res Commun. 1997;235(2):418–23. doi: 10.1006/bbrc.1997.6799. [DOI] [PubMed] [Google Scholar]
- 83.Nie D, Hillman GG, Geddes T, Tang K, Pierson C, Grignon DJ, et al. Platelet-type 12-lipoxygenase in a human prostate carcinoma stimulates angiogenesis and tumor growth. Cancer Res. 1998;58(18):4047–51. [PubMed] [Google Scholar]
- 84.Marsman DS, Barrett JC. Apoptosis and chemical carcinogenesis. Risk Anal. 1994;14(3):321–6. doi: 10.1111/j.1539-6924.1994.tb00247.x. [DOI] [PubMed] [Google Scholar]
- 85.Gopalakrishna R, Barsky SH, Thomas TP, Anderson WB. Factors influencing chelator-stable, detergent-extractable, phorbol diester-induced membrane association of protein kinase C. Differences between Ca2+-induced and phorbol ester-stabilized membrane bindings of protein kinase C. J Biol Chem. 1986;261(35):16438–45. [PubMed] [Google Scholar]
- 86.Jacob C, Maret W, Vallee BL. Selenium redox biochemistry of zinc-sulfur coordination sites in proteins and enzymes. Proc Natl Acad Sci USA. 1999;96(5):1910–4. doi: 10.1073/pnas.96.5.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blessing H, Kraus S, Heindl P, Bal W, Hartwig A. Interaction of selenium compounds with zinc finger proteins involved in DNA repair. Eur J Biochem. 2004;271(15):3190–9. doi: 10.1111/j.1432-1033.2004.04251.x. [DOI] [PubMed] [Google Scholar]
- 88.Parker TL, Eggett DL, Christensen MJ. Estrogen receptor activation and estrogen-regulated gene expression are unaffected by methylseleninic acid in LNCaP prostate cancer cells. J Nutr Biochem. 2007;18(11):746–52. doi: 10.1016/j.jnutbio.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 89.Spyrou G, Bjornstedt M, Kumar S, Holmgren A. AP-1 DNA-binding activity is inhibited by selenite and selenodiglutathione. FEBS Lett. 1995;368(1):59–63. doi: 10.1016/0014-5793(95)00599-5. [DOI] [PubMed] [Google Scholar]
- 90.Christensen MJ, Nartey ET, Hada AL, Legg RL, Barzee BR. High selenium reduces NF-kappaB-regulated gene expression in uninduced human prostate cancer cells. Nutr Cancer. 2007;58(2):197–204. doi: 10.1080/01635580701328701. [DOI] [PubMed] [Google Scholar]
- 91.Liu M, Hu C, Xu Q, Chen L, Ma K, Xu N, et al. Methylseleninic acid activates Keap1/Nrf2 pathway via up-regulating miR-200a in human oesophageal squamous cell carcinoma cells. Biosci Rep. 2015;35(5) doi: 10.1042/BSR20150092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 93.Sakurai T, Kanayama M, Shibata T, Itoh K, Kobayashi A, Yamamoto M, et al. Ebselen, a seleno-organic antioxidant, as an electrophile. Chem Res Toxicol. 2006;19(9):1196–204. doi: 10.1021/tx0601105. [DOI] [PubMed] [Google Scholar]
- 94.Youn BW, Fiala ES, Sohn OS. Mechanisms of organoselenium compounds in chemoprevention: effects on transcription factor-DNA binding. Nutr Cancer. 2001;40(1):28–33. doi: 10.1207/S15327914NC401_7. [DOI] [PubMed] [Google Scholar]
- 95.Seo YR, Kelley MR, Smith ML. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc Natl Acad Sci USA. 2002;99(22):14548–53. doi: 10.1073/pnas.212319799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huang F, Nie C, Yang Y, Yue W, Ren Y, Shang Y, et al. Selenite induces redox-dependent Bax activation and apoptosis in colorectal cancer cells. Free Radic Biol Med. 2009;46(8):1186–96. doi: 10.1016/j.freeradbiomed.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 97.Park HS, Huh SH, Kim Y, Shim J, Lee SH, Park IS, et al. Selenite negatively regulates caspase-3 through a redox mechanism. J Biol Chem. 2000;275(12):8487–91. doi: 10.1074/jbc.275.12.8487. [DOI] [PubMed] [Google Scholar]
- 98.Park HS, Park E, Kim MS, Ahn K, Kim IY, Choi EJ. Selenite inhibits the c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) through a thiol redox mechanism. J Biol Chem. 2000;275(4):2527–31. doi: 10.1074/jbc.275.4.2527. [DOI] [PubMed] [Google Scholar]
- 99.Watson JA, Rumsby MG, Wolowacz RG. Phage display identifies thioredoxin and superoxide dismutase as novel protein kinase C-interacting proteins: thioredoxin inhibits protein kinase C-mediated phosphorylation of histone. Biochem J. 1999;343(Pt 2):301–5. [PMC free article] [PubMed] [Google Scholar]
- 100.Wycherly BJ, Moak MA, Christensen MJ. High dietary intake of sodium selenite induces oxidative DNA damage in rat liver. Nutr Cancer. 2004;48(1):78–83. doi: 10.1207/s15327914nc4801_11. [DOI] [PubMed] [Google Scholar]
- 101.Spallholz JE. Free radical generation by selenium compounds and their prooxidant toxicity. Biomed Environ Sci. 1997;10(2–3):260–70. [PubMed] [Google Scholar]
- 102.Kumar S, Holmgren A. Induction of thioredoxin, thioredoxin reductase and glutaredoxin activity in mouse skin by TPA, a calcium ionophore and other tumor promoters. Carcinogenesis. 1999;20(9):1761–7. doi: 10.1093/carcin/20.9.1761. [DOI] [PubMed] [Google Scholar]
- 103.Holmgren A. Selenite in cancer therapy: a commentary on “Selenite induces apoptosis in sarcomatoid malignant mesothelioma cells through oxidative stress”. Free Radic Biol Med. 2006;41(6):862–5. doi: 10.1016/j.freeradbiomed.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 104.Bjornstedt M, Odlander B, Kuprin S, Claesson HE, Holmgren A. Selenite incubated with NADPH and mammalian thioredoxin reductase yields selenide, which inhibits lipoxygenase and changes the electron spin resonance spectrum of the active site iron. Biochemistry. 1996;35(26):8511–6. doi: 10.1021/bi9528762. [DOI] [PubMed] [Google Scholar]
- 105.Hofmann ER, Boyanapalli M, Lindner DJ, Weihua X, Hassel BA, Jagus R, et al. Thioredoxin reductase mediates cell death effects of the combination of beta interferon and retinoic acid. Mol Cell Biol. 1998;18(11):6493–504. doi: 10.1128/mcb.18.11.6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lei XG, Zhu JH, Cheng WH, Bao Y, Ho YS, Reddi AR, et al. Paradoxical Roles of Antioxidant Enzymes: Basic Mechanisms and Health Implications. Physiol Rev. 2016;96(1):307–64. doi: 10.1152/physrev.00010.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Waters DJ, Shen S, Glickman LT, Cooley DM, Bostwick DG, Qian J, et al. Prostate cancer risk and DNA damage: translational significance of selenium supplementation in a canine model. Carcinogenesis. 2005;26(7):1256–62. doi: 10.1093/carcin/bgi077. [DOI] [PubMed] [Google Scholar]
- 108.Marshall JR, Tangen CM, Sakr WA, Wood DP, Jr, Berry DL, Klein EA, et al. Phase III trial of selenium to prevent prostate cancer in men with high-grade prostatic intraepithelial neoplasia: SWOG S9917. Cancer Prev Res (Phila) 2011;4(11):1761–9. doi: 10.1158/1940-6207.CAPR-10-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]