

Abstract
Lymphoma is characterized by heterogeneous biology, pathologic features, and clinical outcome. This has been proven by accumulating pathologic and molecular evidence attributed to underlying aberrant alterations at genetic, epigenetic, transcriptional, protein, microenvironmental levels, and dysregulated oncogenic signaling pathways. In the era of precision medicine, targeting oncogenic pathways to design drugs and to optimize treatment regimens for the lymphoma patients is feasible and clinically significant. As such, further understanding of the biology and the mechanisms behind lymphoma development and identification of oncogenic pathway activation and pathway-based biomarkers to better design precise therapies are challenging but hopeful. Furthermore, pathway-based targeted therapies in combination with traditional chemotherapy, single specific targeted antibody therapy, and immunotherapy might raise the hope for the patients with lymphoma, especially for relapsed and refractory lymphoma patients.
Keywords: biomarker, lymphoma, oncogenesis, signaling pathway, therapeutic target
I. INTRODUCTION
Lymphomas are a group of hematologic malignancies which originate in lymphocytes and are characterized with heterogeneous outcomes and variable responses to standard of care regimens. Behind the heterogeneity, there are complex molecular mechanisms, mainly including aberrant genetic alterations and dysregulated oncogenic signaling pathways.1,2 Advances in biotechnologies and understanding of tumor biology are paving the way to identify these dysregulations and oncogenic biomarkers that can be targeted and can predict the response to the targeted therapy.3
In the era of precision medicine, the theme is to stratify patients based on the omics and accordingly to provide an optimal intervention on the risk-specific grouped patients.3 Such strategy has attracted more attentions in the field. As such, targeted therapy and immunotherapy have become the hottest therapeutic fields in recent years. Identifying underlying mechanisms and potential therapeutic biomarkers are biologically important and urgent for designing efficient cancer treatment regimens in the era of precision medicine.3
In this review, we evaluate several major dysregulated signaling pathways that contribute to oncogenesis of malignant lymphoma and valuable oncogenic biomarkers that present in these oncogenic pathways and have potential to be therapeutically targeted.
II. ONCOGENIC SIGNALING PATHWAYS IN LYMPHOMAS AND THEIR THERAPEUTIC POTENTIAL
Growing evidence supports that many dysregulated oncogenic signaling pathways contribute to the oncogenesis of a wide variety of malignancies, including lymphomas. The most frequently aberrantly regulated signaling pathways in the oncogenesis of malignant lymphomas include the B-cell receptor (BCR) pathway, the nuclear factor-kappaB (NF-κB) pathway, the phosphoinositide-3-kinase/v-akt murine thymoma viral oncogene homolog1/mechanistic target of Rapamycin (PI3K/AKT/mTOR) pathway, the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, the apoptosis pathway, and the programmed death-1/programmed death-ligands (PD-1/PD-Ls) pathway (Fig. 1). Some given signaling pathways are more preferentially enriched in specific types of lymphomas.
FIG. 1.

Illustration of cross-communication network of BCR, PI3K, and NF-κB signaling pathways that harbor effective therapeutic targets in lymphoma. BCR signaling, PI3K/AKT/mTOR signaling, and NF-κB signaling are independent but interconnected and may form complex crosstalk network. One pathway may act as upstream or downstream of other pathways and some molecular targets function as key points and players involved in several pathways. These key molecules have been shown promise to be a good therapeutic target for effective treatment in lymphoid malignancies. (A) Tonic BCR signaling pathway and PI3K/AKT/mTOR signaling pathway; (B) chronic active BCR signaling; (C) canonical NF-κB signaling pathway; (D) noncanonical NF-κB signaling pathway. The arrows indicate direction of activating signaling steps; the light-colored bars indicate inhibitory signaling steps; the black bars show inhibition from therapeutic agents toward targets. BAFF-R, B-cell activating factor receptor; BCR, B-cell receptor; BTK, Bruton’s tyrosine kinase; CARD11, caspase recruitment domain family, member 11; IKK, IκB kinase; MALT1, mucosa-associated lymphoid tissue lymphoma translocation protein 1; NEMO, NF-κB essential modifier; NIK, NF-κB-inducing kinase; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3 kinase; TCR, T-cell receptor; TLRs, Toll-like receptors; TNFR, tumor necrosis factor receptor.
Targeting oncogenic signaling pathways that harbor oncogenic defects seem more feasible and are more applicable for the treatment of lymphoma patients than just based on the histological features. In fact, many pathway-based therapeutics have been demonstrated to have clinical activity across a broad variety types of lymphoma.
A. The BCR Signaling Pathway
The B-cell receptor is a transmembrane complex composed of a membrane-bound immunoglobulin that functions as an antigen-binding subunit and a linked heterodimer of CD79A/CD79B that acts to initiate and propagate signals. The signaling cascades and their corresponding targets control the precise function of normal B cells.4,5
There are three types of BCR signaling pathways based on their modes of initiation: chronic active BCR signaling, tonic BCR signaling, and autonomous BCR signaling.
In the chronic active BCR signaling pathway, BCR aggregation upon antigen binding extracellularly has been proposed as an initial driver. At the same time of signal initiating, the immune receptor tyrosine-based activation motifs (ITAM) domain of CD79A and CD79B, the cytoplasmic tail of BCR, are phosphorylated by SRC family members. Subsequently, the phosphorylated ITAM recruits and activates the spleen-associated tyrosine kinase (SYK). SYK thereby phosphorylates the Bruton tyrosine kinase (BTK) and the B-cell linker (BLNK), which act as key linker molecules to deliver multiple downstream signals, including the PI3K/AKT/mTOR signaling, the mitogen-activated protein kinase (MAPK) signaling, the NF-κB signaling, and the NF-AT signaling. During the signal delivering process, AKT and BTK utilizing phosphatidylinositol (3,4,5)-trisphosphate (PIP3) as a docking site phosphorylate and activate the PI3K/AKT/mTOR and the NF-κB pathway, respectively.2,6,7
This classical antigen-dependent BCR signaling and its downstream signaling result in a variety of cellular functions, including proliferation, survival, apoptosis, as well as differentiation. It is frequently and aberrantly activated in chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) with low level IgM expression, in a subset of active B-cell-like (ABC) diffuse large B-cell lymphoma (DLBCL) bearing CD79B and CARD11 mutations, in mucosa-associated lymphoid tissue lymphoma (MALT) with Hp infection, and in splenic marginal zone lymphoma with hepatitis C virus infection.2,8–10 With regard to the chronic stimulator, in addition to the driving aspects mentioned above, MALT1/API2 fusion and B-cell lymphoma 10 overexpression, PI3K mutation and phosphatase and tensin homolog (PTEN) gene loss of function are implicated in the oncogenesis of MALT and DLBCL, respectively.11,12
The second BCR pathway, called tonic BCR signaling, is characterized with signals initiating through temporary and stochastic interplays between the BCR and the Lck/Yes-related novel protein tyrosine kinase (LYN) and SYK, independent of antigen stimulating and BCR aggregation.7,13 The tonic BCR pathway exists in normal B-cell function and is commonly observed in Burkitt lymphoma (BL) that bears some distinctly different genetic abnormality compared to other lymphoma types, such as in transcription factor 3 (TCF3) and inhibitor of DNA binding 3(ID3) alterations.14
The third kind of BCR pathway called cell autonomous BCR signaling pathway has a specific feature in that BCR aggregates without external antigen but relies on an interaction between the two neighboring BCRs through CDR3 and FR2 crosslinking.15
Given that aberrantly activated BCR signaling has been increasingly implicated in oncogenesis of several types of B-cell hematologic malignancies, especially in CLL/SLL and ABC-DLBCL, the inhibitors targeting the BCR pathway render the patients with B-cell lymphomas an effective option for treatment.16 Several clinical trials targeting some oncogenic biomarkers in the BCR pathway and its downstream pathways have been showing promising efficacy to treat the malignant lymphomas with an aberrant signaling pathway activation (Tables 1 and 2).
TABLE 1.
Pathway-based therapeutically oncogenic biomarkers and their biological characteristics in lymphomas
| Oncogenic Biomarker |
Gene and Location |
Related Signaling Pathways |
Typically Upstream or Downstream Targets |
Protein Description |
Biological Functions | Oncogenic Modes | Mainly Involved Lymphoma Types |
|---|---|---|---|---|---|---|---|
| BCL2 | BCL2, 18q21.33 | Apoptosis pathway | p53, BAD, BAX, BAK, CDK1 | Anti-apoptotic protein | Suppresses apoptotic death | Translocation, amplification, overexpression | FL, DLBCL |
| Myc | MYC, 8q24.21 | Apoptosis pathway | CyclinD1, CyclinD2, CDK4, CDK6, BLIMP-1, MAX, p27KIP1, p21, p14ARF, SMAD3 | Proto-oncogene protein, pleiotropic transcription factor | Involves in cell cycle progression, apoptosis and cellular transformation | Translocation, overexpression, mutation | BL, DLBCL |
| p53 | TP53, 17p13.1 | Intrinsic apoptosis pathway | p14ARF, MDM2, ATM, ATR, p21, chk1, PUMA, NOXA, BAX | Nuclear transcription factor | Regulates transcription; Regulates cell cycle; induces growth arrest or apoptosis | Muatation, inactivation | MCL, CLL/SLL, BL, DLBCL, PMBL, HL |
| BTK | BTK, Xq22.1 | BCR, NF-kB, TLR, cytokine receptor signaling pathway | LYN, SYK, PLCγ2, PKCβ, MYD88, TLR8, TLR9 | Cytoplasmic tyrosine kinase | Modulates B-cell development, immune function, transcription, and apoptosis | Aberrant activation | CLL/SLL, ABC-DLBCL, FL, MCL, MZL |
| SYK | SYK, 9q22.2 | BCR, NF-kB, TCR, IL-2 signaling, PI3K pathway | BTK, SHP-1, BLNK, IKZF1, ROS, PLCγ1, STAT3 | Cytoplasmic non-receptor type tyrosine kinase | Regulates B-cell differentiation, T cells differentiation, adaptive immunity, cell adhesion, and vascular development | Aberrant activation | CLL/SLL, DLBCL, FL, MCL |
| PI3Kδ | PIK3CD, 1p36.22 | PI3K/AKT/mTOR, BCR, apoptosis, NF-kB pathway, JAK/STAT pathway | PIP3, AKT1, N-Ras, H-Ras, K-Ras, PI3K | Serine/threonine protein kinase | Involves in cell growth, proliferation, survival, and morphology | Mutation | DLBCL, FL, MCL |
| PI3Kγ | PIK3CG, 7q22.3 | PI3K/AKT/mTOR, BCR, TLR/MYD88 pathway | PIP3, AKT1, N-Ras, H-Ras | Serine/threonine protein kinase | Involves in B-cell, T-cell, and NK cell development, proliferation, migration, and cytokine production | Aberrant activation | B-cell lymphoma and T-cell lymphoma |
| AKT1 | AKT, 14q32.33 | PI3K/AKT/mTOR, BCR, apoptosis, JAK/STAT pathway, NF-KB pathway | PI3Kδ, MDM2, BAD, FOXO, Raf1 | Serine/threonine protein kinase | Regulates cell survival, growth, proliferation, and angiogenesis | Mutation, overexpression | DLBCL, MCL, PTCL |
| mTOR | MTOR, 1p36.22 | PI3K/AKT/mTOR, BCR, apoptosis, JAK/STAT, NF-KB pathway | AKT1, MAPKAP1, RPS6KB1, RPS6KB2, STAT1, STAT3 | Serine/threonine protein kinase | Regulates cellular metabolism, survival, growth; mTORC1 regulates mRNA translation, protein synthesis, and autophagy; mTORC2 regulates cell survival and cytoskeleton organization | Aberrant activation | CLL/SLL, DLBCL, FL, MCL, and other aggressive lymphoma |
| PTEN | PTEN, 10q23.31 | PI3K/AKT/mTOR, BCR, p53 pathway | miRNA17-92, AKT1, STAT5, PI3K | Tumor suppressor | Negatively modulates cell cycle progression, cell survival, proliferation, and migration | Mutation, inactivation | GCB-DLBCL, MCL, PTEN-deficient lymphomas |
| STAT3 | STAT3, 17q21.2 | JAK/STAT, EGFR, PI3K/AKT pathway, | EGFR, STAT1, Myc, mTOR, CyclinD1, JAK1, JAK2, TYK2, Src | Signal transducer and activator of transcription | Involves in cell growth and apoptosis | Mutation | NK/T cell lymphoma, cutaneous T-cell lymphoma, ALK+ALCL |
| STAT6 | STAT6, 12q13.3 | JAK/STAT pathway, IL-4 signaling pathway | JAK1, JAK2, MAPK8, SHP-1 | Signal transducer and activator of transcription | Activates transcription, involves in IL-4 signaling, induces antiapoptotic activitity | Mutation | PMBL, FL |
| JAK2 | JAK2, 9p24.1 | JAK/STAT pathway, IFNγ signaling pathway | STAT6, SOCS-1, SOCS-3, STAT1, STAT3, IL-6R, IFNγR1, EZH2, SHP-1 | Nonreceptor tyrosine kinase | Involves in cell growth, development, and differentiation; mediates adaptive and innate immunity | Amplification, overexpression, JAK2/SEC31A fusion, JAK2/PCM1 fusion, JAK2/FLT3 fusion | cHL, FL, DLBCL, PMBL, T-cell lymphoma |
| JAK3 | JAK3, 19p13.11 | JAK/STAT pathway, IL signaling pathway | CyclinD3, STAT3, STAT5, IL-2R, IL-6R, BCL2, IRF4, Myc, SHP-1 | Nonreceptor tyrosine kinase | Involves in cell growth, development, and differentiation; mediates adaptive and innate immunity | Mutation | NK/T cell lymphoma, ATLL |
| MYD88 | MYD88, 3p22.2 | IL-1 signaling, TLR, NF-kB pathway | TRAF6, TLR4, IL-1R1, NFKB2, IRAK2, IRAK4, CREBBP, EP300 | Cytosolic adapter protein | Involves in innate and adaptive immunity | MYD88 L256 mutation | LPL/WM, ABC-DLBCL, PCNSL |
| IL-6 | IL6, 7p15.3 | JAK/STAT, PI3K/AKT, and RAS/MAPK pathway | IL-6R, IL6ST | Pleiotropic cytokine | Functions in inflammation and B-cell maturation | Aberrant activation | B-NHL, Castleman’s disease |
| PD-1 | PDCD1, 2q37.3 | Immune system pathway, TCR pathway | PD-L1, PD-L2, SHP-2, | Cell surface membrane protein of the immunoglobulin superfamily receptor | Negatively regulates effector T-cell functions | Overexpression in infiltrating T-cells | FL, CLL/SLL |
| PD-L1 | CD274, 9p24.1 | Immune system pathway, TCR pathway | PD-1 | Immune inhibitory receptor ligand | Inhibits T-cell activation and cytokine production upon interaction with PD-1 | Amplification, overexpression | cHL, PMBL, ABC-DLBCL, ALK+ALCL |
| PD-L2 | PDCD1LG2, 9p24.1 | Immune system pathway, TCR pathway | PD-1 | Immune inhibitory receptor ligand | Inhibits T-cell activation and cytokine production upon interaction with PD-1 | Amplification, overexpression | cHL |
ABC, active B cell; AKT, v-akt murine thymoma viral oncogene homolog1; ALK, anaplastic lymphoma kinase; ATLL, adult T-cell leukemia/lymphoma; BCR, B-cell receptor; BL, Burkitt lymphoma; BTK, bruton tyrosine kinase; cHL, classical Hodgkin lymphoma; CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; GCB, germinal center B cell; HL, Hodgkin lymphoma; IL-6, interleukin 6; JAK, Janus kinase; LPL, Lymphoplasmacytic Lymphoma; MALT, mucosa associated lymphoid tissue lymphoma; MCL, mantle cell lymphoma; MDM2, mouse double-minute 2 protein; mTOR, mechanistic target of Rapamycin; mTORC, mTOR complex; MYC, v-Myc avian myelocytomatosis viral oncogene homolog; MYD88, myeloid differentiation primary response 88; MZL, marginal zone lymphoma; NF-κB, nuclear factor-kappaB; NHL, non-Hodgkin’s lymphoma; PCNSL, primary central nervous system lymphoma; PD-1, programmed death-1; PD-Ls, programmed death-ligands; PI3K, phosphoinositide-3-kinase; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PKCβ, protein kinase C β; PMBL, primary mediastinal B-cell lymphoma; PTEN, phosphatase and tensin homolog; SHP, Src homology region 2 domain-containing phosphatase; SLL, small lymphocytic lymphoma; STAT, signal transducer and activator of transcription; SOCS, suppressor of cytokine signaling; SYK, spleen-associated tyrosine kinase; TCR, T-cell receptors; TLR, Toll-like receptors; WM, Waldenstrom’s macroglobulinemia.
TABLE 2.
Pathway-based oncogenic biomarkers and their therapeutic applications in lymphomas
| Oncogenic Biomarker |
Therapeutics | Agent | Involved Lymphomas | Phase | Trial Registration or Reference |
|---|---|---|---|---|---|
| BTK | Btk inhibitor | Ibrutinib (PCI-32765) | Recurrent B-cell lymphoma, CLL/SLL, R/R MCL, ABC-DLBCL, FL, MZL, T-cell lymphoma | Phase 1/2 | NCT00849654; NCT01325701; NCT02169180; NCT01109069 |
| Btk inhibitor | BGB-3111 | R/R B-cell malignancies | Phase 1 | NCT02343120 | |
| Btk inhibitor | AVL-292 (cc-292) | R/R B-Cell NHL, CLL/SLL, WM | Phase 1 | NCT01351935 | |
| Btk inhibitor | M7583 | R/R B-cell malignancies, MCL, ABC-DLBCL | Phase 1/2 | NCT02825836 | |
| Btk inhibitor | Acalabrutinib (ACP-196) | R/R ABC-DLBCL, CLL/SLL, MCL | Phase 1/2 | NCT02112526; NCT02337829; NCT02029443; NCT02213926 | |
| SYK | Syk inhibitor | Fostamatinib (R788) (R406) | CLL/SLL, DLBCL, MCL, FL, T-cell lymphoma | Phase 1/2 | NCT00446095; NCT00798096 |
| Syk inhibitor | Cerdulatinib (PRT062070) | CLL/SLL, FL, NHL,T-cell lymphoma | Phase 1/2 | NCT01994382 | |
| Syk inhibitor | Entospletinib (GS-9973) | R/R CLL/SLL, MCL, DLBCL, FL; indolent NHL; MZL | Phase 2 | NCT01799889 | |
| PI3Ks | PI3Kδ inhibitor | Idelalisib (CAL-101, GS-1101) | R/R MCL, FL, SLL, LPL, MZL | Phase 1/2 | NCT00710528; NCT01282424 |
| PI3Kδ inhibitor | AMG 319 | Lymphoid malignancies | Phase 1 | NCT01300026 | |
| PI3Kδ/γ inhibitor | Duvelisib (IPI-145, INK1197) | R/R NHL, advanced hematologic malignancies | Phase 1/2 | NCT02598570; NCT01882803; NCT01476657 | |
| PI3Kδ inhibitor | Acalisib (GS-9820) (CAL-120) | R/R lymphoid malignancy | Phase 1 | NCT01705847 | |
| PI3Kγ inhibitor | Buparlisib (BKM120) | PCNSL, SCNSL, CLL/SLL | Phase 2 | NCT02301364; NCT02340780 | |
| PI3K inhibitor | SAR 245408 (XL147) | Lymphomas | Phase 1 | NCT00486135 | |
| AKT | Akt inhibitor | Perifosine (KRX-0401) | Hematologic malignancies, Lymphomas | Phase 1/2 | NCT00019656; NCT00389077 |
| Akt1/2/3 inhibitor | MK-2206 | Relapsed lymphoma, R/R DLBCL | Phase 2 | NCT01258998; NCT01481129 | |
| Akt inhibitor | GSK690693 | Lymphoma and solid tumor | Phase 1 | NCT00493818 | |
| mTOR | mTOR inhibitor | Ridaforolimus (AP23573, MK-8669) | Lymphoma, multiple myeloma, hematologic malignancies | Phase 2 | NCT00060632; NCT00060645; NCT00086125 |
| mTOR inhibitor | Temsirolimus (CCI-779) | R/R HL, R/R PCNSL, FL, CLL/SLL, R/R MCL | Phase 1/2/4 | NCT00838955; NCT00942747; NCT00033267; NCT01180049 | |
| mTOR inhibitor | Rapamycin (Sirolimus) | R/R Acute Lymphoblastic Leukemia/Lymphoma | Terminated | NCT01658007 | |
| mTORC1/mTORC2 inhibitor | Everolimus (RAD001) | R/R NHL, R/R MCL, R/R cutaneous T-cell lymphoma | Phase 1/2 | NCT00622258; NCT00516412; NCT01637090 | |
| mTORC1/mTORC2 inhibitor | AZD2014 | R/R non-GCB DLBCL | Phase 1 | NCT02780830 | |
| BCL2 | Bcl-2, Bcl-xl, Bcl-w inhibitor | Navitoclax (ABT263) | R/R lymphoid malignancy | Phase 2 | NCT00406809; NCT01557777 |
| BCL2 inhibitor | Venetoclax (ABT199) | NHL, CLL/SLL, MM, R/R NHL | Phase 1/2/3 | NCT01969695; NCT01328626; NCT02756611; NCT02966756 | |
| BCL2 inhibitor | Obatoclax Mesylate (GX15-070MS) | R/R HL | Phase 2 | NCT00359892 | |
| p53 | MDM2 inhibitor | APG-115 | Advanced solid tumors or lymphomas | Phase 1 | NCT02935907 |
| MDM2 inhibitor | DS-3032 | Advanced solid tumors or lymphomas | Phase 1 | NCT01877382 | |
| P53-MDM2 blockade | ALRN-6924 | Advanced solid tumors or lymphomas | Phase 1/2 | NCT02264613 | |
| MYC | BET inhibitor | CPI-0610 | Progressive lymphomas | Phase 1 | NCT01949883; NCT02158858 |
| BET/BRD4 inhibitor | AZD5153 | Hematologic malignancies | Preclinical | Reference 86 | |
| c-Myc-Max dimerization inhibitor | 10058-F4 | BL | Preclinical | Reference 81 | |
| Aurora A inhibitor | Alisertib | Myc-positive aggressive B-cell lymphomas | Phase 1 | NCT02700022 | |
| Small inhibitory RNA oligonucleotide targeting MYC | DCR-MYC | NHL, MM, solid tumors | Phase 1 | NCT02110563 | |
| BET inhibitor | JQ1 | Hematologic malignancies | Preclinical | Reference 82-84 | |
| JAKs | JAK3 inhibitor | Tofacitinib (CP-690550) | EBV-related NK/T-cell lymphoma | Preclinical | Reference 132 |
| JAK2 inhibitor | Pacritinib (SB1518) | R/R cHL, FL, MCL, and DLBCL | Phase 1/2 | NCT00741871; NCT01263899 | |
| JAK2 inhibitor | Ruxolitinib (INCB018424) | R/R T-cell or NK-cell lymphoma, R/R HL and PMBL | Phase 2 | NCT02974647; NCT01965119; NCT01877005 | |
| STAT3 | STAT3 inhibitor | Pyrimethamine | Relapsed CLL/SLL | Phase 1/2 | NCT01066663 |
| STAT3 inhibitor | IONIS-STAT3Rx (ISIS 481464) | DLBCL, lymphoma, advanced cancer | Phase 1/2 | NCT01563302 | |
| IL-6 | IL-6 inhibitor | Siltuximab (CNTO-328) | B-cell NHL, MM, or Castleman’s disease | Phase 1 | NCT00412321 |
| PD-1 | Anti-PD-1 antibody | MEDI0680 (AMP-514) | R/R aggressive B-cell lymphomas; advanced malignancies | Phase 1/2 | NCT02271945; NCT02013804 |
| Anti-PD-1 antibody | Pidilizumab (CT-011) | DLBCL and PMBL after ASCT; stage III–IV DLBCL; | Phase 2 | NCT00532259; NCT02530125 | |
| Anti-PD-1 antibody | Nivolumab (BMS-936558, MDX-1106, ONO-4538) | R/R DLBCL, PCNSL, PTL, FL, PTCL | Phase 2 | NCT02038933; NCT02857426; NCT02038946; NCT03075553 | |
| Anti-PD-1 antibody | Pembrolizumab (lambrolizumab, MK-3475) | R/R FL; HL, DLBCL and T-NHL after ASCT; T-cell or NK-cell lymphomas; R/R HL; recurrent PCNSL; R/R PMBL | Phase 2 | NCT02446457; NCT02362997; NCT03021057; NCT02453594; NCT02779101; NCT02576990 | |
| PD-L1 | Anti-PD-L1 antibody | Durvalumab (MEDI4736) | R/R lymphoma, solid tumor | Phase 1 | NCT02793466 |
| Anti-PD-L1 antibody | Avelumab (MSB0010718C) | Advanced cHL; R/R PTCL | Phase 1/2 | NCT02603419; NCT03046953 | |
| Anti-PD-L1 antibody | Atezolizumab (MPDL3280A) (RG7446) | Locally advanced or metastatic solid tumors or hematologic malignancies; R/R FL and DLBCL; R/R HL | Phase 1/2 | NCT01375842; NCT02220842; NCT02862275; NCT03120676 | |
| MYD88 | TLRs ligands inhibitor | IMO-8400 | R/R ABC-DLBCL with MYD88 L256 mutation | Phase 1/2 | NCT02252146 |
| IRAK4 inhibitor | ND-2158, ND-2110 | ABC-DLBCL, or other malignancies harboring aberrant MYD88 | Preclinical | Reference 138 | |
| IkBa | IkBa inhibitor | Bortezomib (Velcade, PS341) | MALT, R/R FL, DLBCL, R/R B-cell lymphomas | Phase 2 | NCT00210327; NCT00136591; NCT01226849; NCT00038571 |
| IkBa inhibitor | MLN4924 | ABC-DLBCL, MCL, hematologic malignancies | Phase 1 | NCT00722488 |
ABC, active B-cell; AKT, v-akt murine thymoma viral oncogene homolog1; ASCT, autologous stem cell transplantation; ATLL, Adult T-cell leukemia/lymphoma; BET, bromodomain and extra-terminal proteins; BRD4, bromodomain containing protein 4; BL, Burkitt lymphoma; BTK, bruton tyrosine kinase; cHL, classical Hodgkin lymphoma; CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; HL, Hodgkin lymphoma; IL-6, interleukin 6; JAK, Janus kinase; LPL, lymphoplasmacytic lymphoma; MALT, mucosa associated lymphoid tissue lymphoma; MCL, mantle cell lymphoma; MDM2, mouse double-minute 2 protein; MM, multiple myeloma; mTOR, mechanistic target of Rapamycin; mTORC, mTOR complex; MYC, v-Myc avian myelocytomatosis viral oncogene homolog; MYD88, myeloid differentiation primary response 88; MZL, marginal zone lymphoma; NHL, non-Hodgkin’s lymphoma; PCNSL, primary central nervous system lymphoma; PD-1, programmed death-1; PD-Ls, programmed death-ligands; PI3K, phosphoinositide-3-kinase; PMBL, primary mediastinal B-cell lymphoma; PTCL, peripheral T-cell lymphoma; PTL, primary testicular lymphoma; R/R, refractory and relapsed; SCNSL, secondary central nervous system lymphoma; SLL, small lymphocytic lymphoma; STAT, signal transducer and activator of transcription; SYK, spleen-associated tyrosine kinase; WM, Waldenstrom’s macroglobulinem.
B. The NF-κB Pathway
The NF-κB pathway, as one of the most common signaling pathways exists, and, functions transiently in normal lymphoid cells and several types of tissues. The constitutively activated NF-κB pathway has also been implicated by accumulating evidence in oncogenesis of many types of human malignancies including lymphomas.2,17
The NF-κB pathway, categorized by canonical and noncanonical pathways, includes p50, p65(RelA), RelB, p52, and c-Rel proteins. These subunits present in patterns of homodimers or heterodimers that transduce their functional signals.
The canonical NF-κB pathway is downstream of several signaling pathways, such as BCR, T-cell receptor (TCR), Toll-like receptor (TLR), and tumor necrosis factor receptor (TNFR) pathways. For example, in the downstream arm of the BCR pathway, the IκB kinase (IKK) complex is phosphorylated by the CARD11/MALT1/BCL10 complex and, thereby, directly activates IκBα. Activated IκBα subsequently activates the RelA/p50 heterodimer, leading to translocation of RelA/p50 into the nucleus and the execution of final functions, such as cell survival/antiapoptosis, proliferation, inflammation, and innate immunity.
In the noncanonical NF-κB pathway, the involved RelB subunit forms a heterodimer with its partner p52 or p50; the activators of the noncanonical signaling are preferentially BAFF-R and the IKK complex that contain two IKKα are activated by the NF-κB-inducing kinase (NIK). The target genes of the noncanonical NF-κB pathway function in lymphoid organogenesis, adaptive immunity, anti-inflammatory properties, and B-cell maturation.18–20
The constitutively activated canonical and non-canonical NF-κB signaling pathways can be observed more or less in almost all types of lymphoma. Particularly, ABC-DLBCL is the type affected by NF-κB through various mechanisms.21 These oncogenic events may partly result from a dysregulated upstream chronic active BCR signaling in which CARD11 and CD79B mutate or from the myeloid differentiation primary response 88 (MYD88)L265P mutation.8,9,22 Additionally, other oncogenic events, such as dysregulated upstream of the JAK/STAT pathway, NF-κB target genes alteration, chromosomal translocation, or EBV infection, may also contribute to the constitutive activation of the NF-κB pathway in ABC-DLBCL, MALT, BL, Hodgkin lymphoma (HL), or other types of lymphoma.2,18
The inhibition to the NF-κB pathway can be carried out by directly inhibiting NF-κB components or indirectly by inhibiting its upstream pathways. For example, the inhibition of p-IκBα using Bortezomib, a proteasome inhibitor, is a rational option for treating patients with constitutive NF-κB pathway activities and has shown good efficacy for DLBCL patients in combination with traditional chemotherapy.23 Another p-IκBα blockade, MLN 4924, was shown activity in mantle cell lymphoma (MCL) and DLBCL, regardless of the germinal center B-cell-like or the ABC subtype.24–26
C. The PI3K/AKT/mTOR Signaling Pathway
The PI3K/AKT/mTOR signaling pathway is activated following the BCR pathway, the TNFR pathway, or other upstream pathways, and functions as a regulatory part in cell growth, survival, apoptosis, proliferation, metabolism, and motility processes.27,28 The PI3K family consists of four major isoforms on the basis of different structures and functions. Among the PI3 kinases, PI3Kδ and PI3Kγ are preferentially critical to B- and T-cell development and immune function.28–30 In the PI3K/AKT signaling, PI3K delivers an active signal to AKT through PIP3 which acts as a docking site for AKT; this process can be negatively regulated by PTEN.31 Then, AKT as a principal oncogenic effector of this pathway targets many molecules and executes its functions.32 These include the indirect activation of the mTOR complex 1 (mTORC1) and the mTOR complex 2 (mTORC2), which increase mRNA translation, protein synthesis, and cellular proliferation or negatively regulate this pathway with a negative feedback.28,33
The aberrant activation of the PI3K/AKT/mTOR pathway can confer oncogenicity in many types of cancer. This oncogenic activation is commonly related to PIK3CD mutation in DLBCL, loss of function PTEN in GCB-DLBCL, and PIK3CA copy number increasing in MCL.34–36 Moreover, p-AKT has been shown to be overexpressed and acts as an inferior outcome indicator in DLBCL, MCL, and peripheral T-cell lymphoma.37–39 Considering the critical status of the PI3K/AKT/mTOR pathway in lymphoma, PI3K, AKT, and mTOR are all potential targets to design drugs for the treatment of lymphoma patients (Tables 1 and 2).
D. The JAK/STAT Signaling Pathway
The JAK/STAT pathway is an essential signaling pathway as a key mediator of cytokine signaling, in which a network of molecules is involved and functions aberrantly. Cytokines, cytokine receptors, JAKs and STATs participate in the positive regulation of the JAK/STAT pathway, while tyrosine phosphatases, inhibitors of STATs, suppressor of cytokine signaling (SOCS) proteins, and cytokine-inducible SH2-containing (CIS) protein are the negative regulators.40–42 Upon binding of cytokines, basally inactive JAKs engaged to cytokine receptors are phosphorylated and, thereby, phosphorylate STATs. After dimerization, phosphorylated STATs translocate from the cytoplasm to the nucleus and then bind targeted genes, causing transcriptional activation and functioning in apoptosis, survival, proliferation, angiogenesis, and metastasis. This pathway, in turn, is negatively regulated through SOCS and CIS.
The underlying molecular mechanisms in the dysregulation of the JAK/STAT pathway in lymphoma still remain unclear. But major aberrations like JAK2 aberration are companied by Jumonji domain-containing protein 2C (JMJD2C) abnormality, loss-of-function mutation of SOCS1, and protein tyrosine phosphatase non-receptor type 2 (PTPN2) deletion may all be contributors to the oncogenic JAK/STAT signaling in primary mediastinal B-cell lymphoma (PMBL), classical Hodgkin lymphoma (cHL), and ABC-DLBCL.40,41,43–45 Particularly, aberrations of STAT3, STAT5, and JAK2 have been shown to be the most common signature in these malignancies.40,46–48
The increasing evidence described above has paved the way for targeting the JAK/STAT pathway to treat hematologic patients with a dysregulated JAK/STAT pathway. The inhibitors targeting JAKs, STATs, or ILs, and other related biomarkers have been developed or are under investigation. They are being expected to show efficacy in lymphoma via many rational ways such as inducing growth arrest and inhibiting angiogenesis (Fig. 2, Tables 1 and 2).47,49,50
FIG. 2.
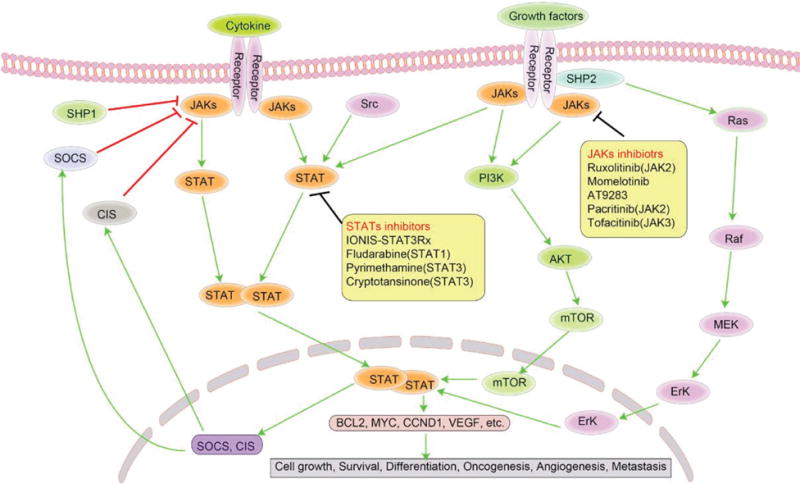
Illustration of JAK/STAT signaling pathway that inspires effective therapeutic targets in lymphoma. JAK/STAT pathway is a key mediator of cytokine signaling in both physiological and pathological conditions. Upon binding cytokines, basally inactive JAKs engaged to cytokine receptors are phosphorylated and thereby phosphorylate STATs. After being dimerization, phosphorylated STATs translocate from cytoplasm to nucleus and then bind to specific targeted genes. In turn, this pathway is negatively regulated by SOCS and CIS. The inhibitors targeting JAKs, STATs, or ILs are being expected to show expected treatment efficacy in some lymphomas. JAK, Janus kinase; STAT, signal transducer and activator of transcription; SHP, Src homology region 2 domain-containing phosphatase; SOCS, suppressor of cytokine signaling; CIS, cytokine-inducible SH2-containing protein; PI3K, phosphoinositide-3-kinase; AKT, v-akt murine thymoma viral oncogene homolog1; mTOR, mechanistic target of Rapamycin; ERK, extracellular signal-regulated kinase; CCND1, cyclin D1; VEGF, vascular endothelial growth factor.
E. The Apoptosis Signaling Pathway
Apoptosis is a process of programmed cell death that occurs naturally and is critical for the normal organism development, biological evolution, internal environmental stability, and abnormal cells clearance. This process, being a complex network, is controlled by a series of highly conserve molecules, like the BCL2 family, the caspase family, and the tumor suppressor protein p53.51
The apoptosis pathway can function via at least two distinct but interconnected pathways: the extrinsic and the intrinsic apoptosis pathways. One of the extrinsic apoptotic pathways is triggered by some death receptors outside a cell upon ligands binding, rapidly activating death caspases and subsequently causing apoptotic signaling cascades. Among the death receptors, Fas and TNFR-1 are the well-known and best-characterized death receptors, which bind their correspondent ligands. Fas and its cofactor, fas-associated via death domain (FADD), and caspase 8 form a death-inducing signaling complex that commits the cell to apoptosis through direct activation of downstream caspases such as caspase 3 and caspase 7, or through the mitochondrial-dependent pathway, which involves the release of cytochrome C, formation of apoptosome, activation of caspase cascades, and regulation by the BCL2 family.52,53 Similarly, after being activated, TNFR1 forms complexes with many death domain-containing proteins like TNFRSF1A associated via death domain (TRADD), TNF receptor associated factor 2 (TRAF2), thereby committing the cell to apoptosis through ways that Fas mediates, or recruiting IKK and finally activating NF-κB function.54,55
The intrinsic apoptosis pathway occurs mainly due to intracellular stresses, including oncogenic stress, DNA damage, as well as hypoxia. This intrinsic apoptosis pathway related with the mitochondria is mediated by p53 which interacts with other upstream or downstream DNA checkpoint proteins and BCL2 family members. Similar to the extrinsic pathway, the final effectors of the intrinsic pathway are caspase 3 and caspase 7, which cause proteolysis and apoptosis.52,56,57
In the pathologic process, the normal apoptosis signaling pathway is interrupted by oncogenic defects and cells grow unchecked, leading to the oncogenesis of a wild variety of cancer, including hematologic malignancies. Interfering pathologic apoptosis pathway and restoring its normal function seem to have therapeutic potential. Among the therapeutic targets, BCL2, MYC, and p53 might be the suitable target candidate (Fig. 3, Tables 1 and 2).
FIG. 3.
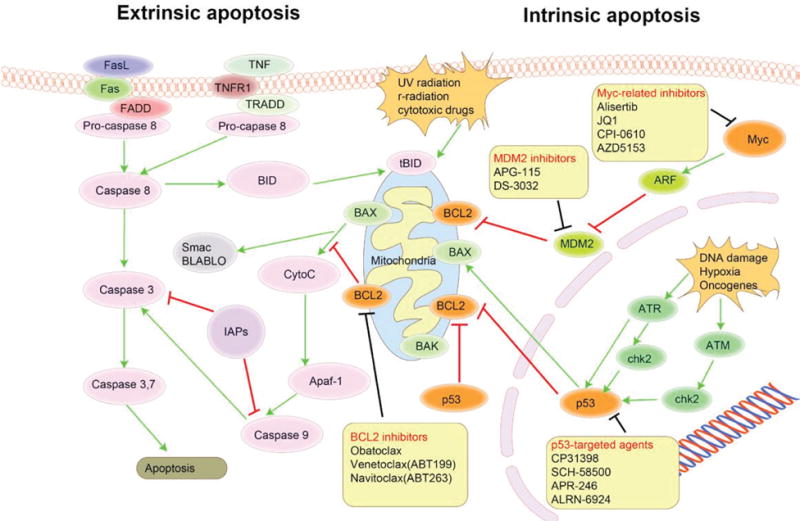
Overview of apoptosis signaling pathway that contains effective therapeutic targets in lymphoma. The naturally occurring apoptosis pathway functions through extrinsic and intrinsic signaling and is controlled by a series of highly conserve molecules. Interfering pathologic apoptosis pathway and restoring its normal function seem to have therapeutic potential in many types of cancer. Among these therapeutic targets, BCL2, MYC, and p53 might be the suitable target candidates. FasL, Fas ligand; TNF, tumor necrosis factor; TNFR1, tumor necrosis factor receptor 1; FADD, fas associated via death domain; TRADD, TNFRSF1A associated via death domain; BID, BH3 interacting domain death agonist; tBID, truncated BID; BAX, BCL2 associated X; BCL2, B-cell lymphoma 2; BAK, BCL2 antagonist/killer; Myc, v-Myc avian myelocytomatosis viral oncogene homolog; ARF, alternate open reading frame; MDM2, mouse double-minute 2 protein; ATR, Ataxia telangiectasia and Rads-related protein; ATM, ataxia telangiectasia mutated; chk2, checkpoint kinase 2.
F. The PD-1/PD-Ls Signaling Pathway
PD-1 and PD-Ls are harnessed by the immune system to balance its immune function. Upon binding to its ligands, PD-1 delivers co-inhibitory signals, negatively regulating the functions of T and B cells to maintain the immune balance.58 However, in the cases of tumor, overexpression of PD-Ls in tumor cells and infiltrating cells, owing to genetic alteration, virus infection, or other intrinsic and extrinsic factors, can activate PD-1 signaling and lead to exhaustion of effector T cells after the interaction of PD-1 and PD-Ls.59,60 Such an immune escaping signaling has been demonstrated to be a major mechanism contributing to oncogenesis, tumor aggression and metastasis.59
Apart from the PD-1/PD-Ls signaling, many other immune checkpoint pathways and molecules function between tumor cells and their surrounding microenvironment. Many costimulatory signals and coinhibitory signals participate in this pathway, which modulates a strict immune function, such as cytotoxic T-lymphocyte associated protein 4 (CTLA-40/CD86/CD80, B- and T-lymphocyte–associated/Herpes virus entry mediator A (BTLA/HVEM), lymphocyte activating 3/major histocompatibility complex II (LAG-3/MHCII), and T-cell immunoglobulin mucin receptor 3 (TIM-3)/Galectin9 (Fig. 4).61
FIG. 4.
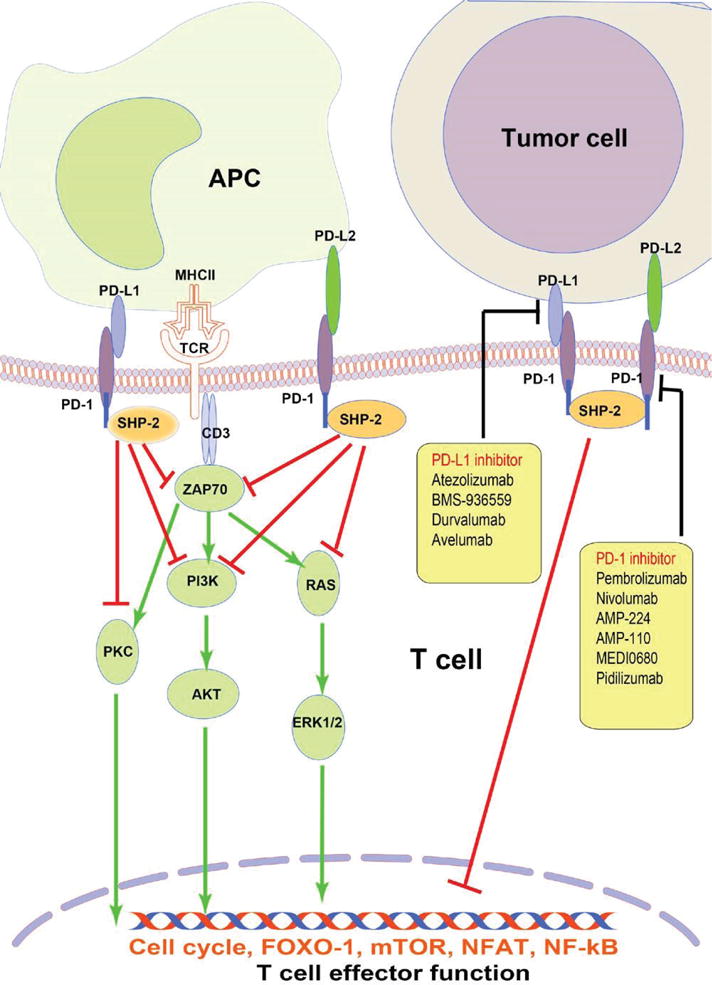
Overview of PD-1/PD-Ls signaling pathway that acts as effective therapeutic targets in lymphoma. Upon binding to its ligands PD-Ls, PD-1 delivers co-inhibitory signals, negatively regulating T-cell function to maintain immune balance. In the cases of tumor, overexpressed PD-Ls activate PD-1 signaling, leading to exhaustion of effector T cell after interaction of PD-1 and PD-Ls. Targeting PD-1 and/or PD-L1 are the current focus of immunotherapy and clinical management in several solid tumor and hematologic malignancies. APC, antigen presenting cell; PD-1, programmed death-1; PD-L, programmed death-ligand; MHCII, major histocompatibility complex, class II; TCR, T-cell receptor; SHP, Src homology region 2 domain-containing phosphatase; ZAP70, zeta chain of T cell receptor associated protein kinase 70; PI3K, phosphoinositide-3-kinase; AKT, v-akt murine thymoma viral oncogene homolog1; ERK, extracellular signal-regulated kinase; FOXO-1, forkhead box 1; mTOR, mechanistic target of Rapamycin; NFAT, nuclear factor of activated T cells; NF-kB, nuclear factor-kappa B.
Agents blocking these immune checkpoint path ways bring a promise to treat a broad spectrum of malignancies. For relapsed/refractory cHL, DLBCL, and follicular lymphoma (FL), immune checkpoint blockades showed encouraging efficacy. Among these blockades, antibodies targeting PD-1 or PD-L1 are the current focus. More and more of the checkpoint inhibitors have been approved and translated into clinical immunotherapy management (Tables 1 and 2).59,61
G. The Complex Interplay Among Different Oncogenic Pathways in Lymphomas
Detailed genomic and biologic studies have revealed the complexity of different oncogenic pathway networks in lymphoma. Many oncogenic pathways are independent but interconnected in a specific type of lymphoma. One type of lymphoma attributes to several dysregulated pathways that interact with each other; one activator can activate several pathways; one pathway may act as upstream or downstream of other pathways; and some molecules function as key points involved in several pathways. The complex crosstalk among the different oncogenic pathways may contribute to oncogenesis and heterogeneity of lymphoma, raising questions about how to design efficient therapeutic drugs targeting these complex oncogenic pathways and how to prevent targeted drugs from resistance and toxicity.
III. THERAPEUTIC BIOMARKERS RELATED TO ONCOGENIC PATHWAYS IN LYMPHOMAS
It is well known that targeted therapies are established toward specific biomarkers that might be key components of oncogenic pathways or other related molecules in cases of mutation, deletion, translocation, and overexpression. These biomarkers can be tested by means of DNA, RNA, or protein using reliable and reproducible technique platforms. In addition to being targeted for rational drugs design, some of these biomarkers might be able to predict treatment outcome and to be used for the selection of patients who may benefit from the pathway-based therapy management.
A. BCL2
BCL2 is encoded by the BCL2 gene located on chromosome 18q21.3. As a member of the BCL2 family and an apoptosis regulator, BCL2 functions to suppress apoptotic death by interacting with caspases and other BCL2 family members. The BCL2 family includes antiapoptotic and proapoptotic members, which programmatically control cell death. Among them, BCL2 is the key molecule at the crossing point in the apoptosis pathway that is involved in cell survival, apoptosis, and proliferation.62,63
t(14;18)/IgH/BCL2, involving BCL2 juxtaposed with IgH, is a molecular hallmark of FL and can be found in GCB-DLBCL.64,65 But interestingly, BCL2 is essentially untranslocated but is frequently amplified in the ABC-DLBCL subtype.66,67 Both translocation and overexpression were identified to be associated with some types of lymphoma with aggressive clinical features and adverse outcome.
All the BCL2 family members have a BCL2 homology domain (BH). Particularly, the BH3 domain is the region that can be frequently mimicked to develop inhibitors through binding BCL2 or BCL-XL.68
BCL2 appears to be an ideal druggable target to develop its inhibitor. Obatoclax is such a BCL2 inhibitor that can restore apoptosis. Obatoclax can be used as a single agent to treat relapsed/refractory HL and to treat previously untreated FL while in combination with Rituximab.69,70 Venetoclax (ABT199), another BCL2 inhibitor, offers promise to patients with relapsed/refractory lymphoid malignancies.71
B. p53
p53, a nuclear transcription factor, regulates the expression of a broad variety of target genes involved in physiological cellular functions. As a tumor suppressor protein, p53 functions as an important mediator in the apoptosis pathways under oncogenic stress, DNA damage, and hypoxia through transcriptional activation of pro-apoptotic BCL2 family members and repression of antiapoptotic BCL2 proteins, inducing cell apoptosis and growth arrest.72,73
p53 inactivation and the dysregulation of p53 signaling pathway contribute to oncogenesis of cancers at various frequencies. Abnormality of components in the p53 pathway, like MDM2 or p14ARF, are frequent oncogenic events in cancers, including in lymphomas.72,74
Therapeutically, the p53 pathway-targeting appears rational to treat p53-related malignancies. Mechanistically, such therapeutic approaches may include to restore the tumor suppressor function of p53, to block MDM2-p53 interaction, and to reactivate p53-mediated apoptosis pathways, using p53-delivering adenovirus, MDM2 inhibitors, and vaccine.72,75–77 APG-115, an MDM2 inhibitor, has shown antitumor activities in multiple human cancer xenografts and is being expected to show efficacy and safety in advanced solid tumors and lymphomas (Clinical Trials Regisrtration: NCT02935907).
C. Myc
The proto-oncogene Myc encodes the Myc protein, which is a typical pleiotropic transcription factor and is involved in almost every process of cell biology and oncology by regulating thousands of target genes.78,79 In many aggressive cancers, including aggressive lymphomas, Myc functions as a driver along with other cofactors, involving in oncogenesis, malignant transformation, and aggressive clinical features.
The chromosomal translocation of Myc often presents in some aggressive lymphomas, typically in BL and a subset of DLBCL. Myc on chromosome 8q24 is juxtaposed with one of the heavy- or light-chain immunoglobulin gene loci on the derivative chromosome 14 or other, which is defined as t(8;14) (q24;q32), t(2;8)(p12;q24), or t(8;22)(q24;q11).78,80 The small subset of DLBCL in which the Myc translocation occurs along with BCL2 or BCL6 is generally described as double-hit lymphoma or triple-hit lymphoma, which confers adverse outcomes. Similar to the double-hit lymphoma, the double-expression lymphoma, which is characterized with Myc and BCL2 co-overexpression, also exhibits inferior prognosis.
Unlike other oncogenic biomarkers, Myc itself was not considered druggable because of its unique structure and function. Alternatively, targeting options include Myc activation by reducing MycC/Myc associated factor X (MAX) heterodimerization, and DNA binding, targeting Myc-associated chromatin modification, targeting cell-cycle kinases, and interfering with its downstream target genes.81 For example, the bromodomain containing protein 4 (BRD4) now acts as an important and promising target whose inhibitors, JQ1 and RVX2135, are small molecules to downregulate Myc function and expression. JQ1 also shows efficacy in multiple myeloma and other Myc-driven malignancies.82–84 Another indirect Myc inhibitor is Alisertib, a selective Aurora A kinase inhibitor, which has been shown to induce response in relapsed/refractory aggressive B- and T-cell lymphomas. Aurora A is essential in Myc-driven lymphoma.85 AZD5153, an available BET/BRD4 inhibitor, has a rational potential for the treatment of hematologic malignancies owing to its antitumor activity by transcriptionally affecting Myc, E2F, and mTOR.86
D. BTK
BTK, a member of the Tec family, is a cytoplasmic tyrosine kinase and is mainly expressed in B cells and myeloid cells but not in plasma cells and T cells. Its normal function is limited in B cells. In addition to its critical function in B lymphocyte development and differentiation, BTK is an indispensable component for BCR signaling, responsible for being activated by SYK and subsequently phosphorylating and activating phospholipase C γ2 (PLCγ2) and protein kinase Cβ (PKCβ). Phosphorylated PLCγ2 and PKCβ finally trigger the downstream NF-κB pathway, which can induce multiple genes with antiapoptotic reaction and cell survival.16,87,88 In addition, BTK plays key roles in the TLR pathway and cytokine receptor signaling pathway, that are involved in immune function, transcription regulation, and apoptosis modulation.89
BTK as a therapeutic biomarker can be targeted by its inhibitors to interrupt BCR signaling and NF-kB signaling. Treatment with ibrutinib (PCI-32765) has been shown to induce considerable efficacy to patients with untreated and relapsed/refractory CLL, ABC-DLBCL, FL, MCL, marginal zone lymphoma, and lymphoplasmacytic lymphoma.90–93 However, ibrutinib may have certain limitations. First, some lymphomas do not just depend on the BCR and NF-κB pathways but may depend on other BTK-independent pathways or genetic mutations; therefore, these lymphomas may show constitutive resistance to ibrutinib. Second, BTK has shown to take part in other pathways apart from the BCR pathway. Ibrutinib in combination with bendamustine and rituximab is also being tested for treatment of relapsed/refractory aggressive non-Hodgkin lymphomas, including PMBL, double-hit lymphoma, transformed indolent lymphoma, high-grade B-cell lymphoma, and CLL/SLL.94,95 Other BTK inhibitors, such as acalabrutinib (ACP196), ONO/GS-4059, BGB3111, and M7583, are being evaluated in various clinical trials.96,97
E. SYK
SYK, another cytoplasmic nonreceptor-type tyrosine kinase, is extensively expressed in B cells, T cells, and myeloid cells and regulates a diverse range of cell functions, including adaptive immunity, cell adhesion, and vascular development.98 SYK is indispensable for the maturation of B cells, especially at the differentiation stage from pro-B to pre-B, and functions downstream of the BCR signaling pathway.16,99 SYK directly binds the phosphorylated ITAMs of CD79A/B or is phosphorylated by activated LYN, and thereby phosphorylates and activates BLNK or PKCβ, ultimately generating downstream signaling effects.16,100 SYK is also involved in T cells differentiation and T-cell receptor signaling.16
One of inhibitors of SYK, Fostamatinib (R406, R788), has been evaluated in several types of lymphoma and showed effects on some lymphomas. Among them, the highest response rate is 55% in CLL patients. The response rate in DLBCL, MCL, and FL was 22%, 11%, and 10%, respectively.16,101
F. PI3Ks
The PI3K family comprises three classes of kinases with different structures and functions. Class I is most commonly involved in cancer and contains four isoforms: p110α, p110β, p110δ, and p110γ.28 p110δ, encoded by PIK3CD, is generally expressed in leukocytes and the thymus, and it acts to activate signaling cascades to influence cell growth, proliferation, survival, and morphology through the PIP3 function. p110γ, encoded by PIK3CG, is involved in immune function. PI3Ks, as important kinases, are involved in the PI3K signaling, particularly p110δ and p110γ, and play essential roles in B-cell, T-cell, and NK-cell development, proliferation, migration, as well as cytokine production.28,102,103
The representative inhibitor of the p110δ iso-form is idelalisib, which has been shown clinical activity in relapsed CLL, MCL, FL, and DLBCL. The efficacy of idelalisb in lymphoma may result from the direct effect of inhibiting p110δ or the indirect effect of controlling the microenvironmental signals such as cytokines and chemokines secretion.103–105 Other pan-class I PI3K inhibitors include buparlisib (BKM120), SAR245408, and BAY 80-6946;28 duvelisib (IPI-145) is an inhibitor against p110δ/p110γ isoform, which shows efficacy in both T- and B-cell lymphomas.106,107
G. AKT
AKT belongs to the serine/threonine kinase and acts as a downstream mediator of the PI3K pathway, targeting numerous target genes and regulating biological processes related to cell survival, growth, proliferation, and angiogenesis.
AKT as a therapeutic target can be inhibited by inhibitory agents. Perifosine, a first-generation inhibitor against AKT, has not been put into clinical practice owing to its moderate toxicity.108,109 MK-2206 is a second-generation AKT inhibitor used for trials in patients with relapsed/refractory lymphoma.110 Notably, the phosphorylated-Akt protein expression by immunohistochemistry from pre- to post treatments can be used as a prognostic or predictive biomarker to assess the outcome or treatment response.38,39
H. mTOR
mTOR, a serine/threonine-protein kinase, regulates cellular metabolism, survival, and growth, by regulating the phosphorylation of thousands of target proteins. Both of mTORC1 and mTORC2 use mTOR as a functional and structural unit and the phosphorylated activation comes from upstream AKT. Activated mTORC1 positively regulates mRNA translation and protein synthesis and negatively regulates autophagy. mTORC2 regulates cellular processes including cell survival and cytoskeleton organization.
Concerning mTOR inhibitors, most of the present agents inhibit mTORC1. Temsirolimus alone or combined with rituximab showed 38% and 59% of objective response rates in relapsed MCL, respectively.111 Temsirolimus has also been evaluated in FL, CLL/SLL, and other aggressive lymphomas.112 Another mTORC1 inhibitor, Everolimus, has also shown preclinical or clinical efficacy in a variety of relapsed aggressive lymphomas, including DLBCL, HL, and even T-cell lymphoma.113–115 A new inhibitor targeting both mTORC1 and mTORC2 is also being developed and tested in lymphoma and is being looked to for its greater efficacy with minimized toxicity.
I. PTEN
PTEN, known as a tumor suppressor gene, normally functions through suppression of the PI3K/AKT/mTOR pathway.116 Loss-of-function of the PTEN protein, resulted from genetic mutation and deletion, abnormal epigenetic regulation, as well as dysregulated protein–protein interactions, may lead to elevated AKT and mTOR activities which promote tumor growth.117,118 The PTEN-deficient lymphomas mainly include GCB-DLBCL (~11%) and MCL, suggesting the exclusive role of the functional deficiency of PTEN in oncogenesis of these lymphomas.38,66
As for the therapeutic implication of PTEN, it mainly relies on the inhibition of some components of the PI3K/AKT/mTOR pathway. This is due to the fact that inactivation of PTEN is significantly associated with activation of the PI3K/AKT/mTOR pathway.117–119 Such inhibitory agents include the pan-PI3K inhibitor like buparlisib, the p110δ-specific PI3K inhibitor like idelalisib, and the mTOR inhibitor like everolimus,105,113,120 which have the potential to intervene in both advanced solid tumors with PTEN deficiency and PTEN-deficient lymphomas.
J. STATs
The STATs family includes seven members: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. These DNA-binding proteins play a critical role in survival, proliferation, and immune modulation.121 Significantly elevated expression of STAT3, IL6, and IL10 can be seen in ABC-DLBCL, which commonly shows constitutive NF-kB pathway activation.48,122 STAT3 and STAT5 activation in cutaneous T-cell lymphoma are also unique features.123 In anaplastic large T-cell lymphoma, STAT3 is upregulated significantly, partly owing to the nucleophosmin-1/anaplastic lymphoma receptor tyrosine kinase (NPM-1/ALK) fusion.124 In contrast, STAT6 mutation occurs in 36% of PMBL and 11% of FL.125 Given the therapeutic potential of STATs, pyrimethamine, a STAT3 inhibitor for the treatment of relapsed CLL/SLL, is in phase I/II study sponsored by the Dana-Farber Cancer Institute (Clinical Trials Registration: NCT01066663); SD1008 is a STAT3/JAK2 inhibitor, which could induce apoptosis.126
K. JAKs
JAKs are JAK family members composed of four cytoplasmic tyrosine kinases: JAK1, JAK2, JAK3, and tyrosine kinase 2. JAK1 and JAK2 mainly function in hematopoiesis and host defense, while JAK3 and TYK2 basically participate in the immune response. The JAKs aberrations in lymphomas have been demonstrated and implicated in oncogenesis. Aberrant JAK2 and JMJD2C amplification have been shown as a significant signature in cHL and PMBL, frequently accompanying with loss of function SOCS1 and PTPN2, which are negative regulators of the JAK/STAT pathway.40,41,45,50 The JAK2/SEC31A fusion involving t(4;9)(q21;p24) has been commonly seen in cHL;127 the JAK2/PCM1 fusion deriving from t(8;9)(p22;p24) in patients with T-cell lymphoma has also been reported.128 The JAK2/FLT3 fusion has been reported, and its inhibitor pacritinib (SB1518) was designed to treat relapsed/refractory cHL, FL, and DLBCL and ademonstrates clinical safety and efficacy.50,129 JAK3 mutations can be found in NK T-cell lymphoma and adult T-cell leukemia/lymphoma, and may be involved in cytokine-independent JAK/STAT activation.130,131 Tofacitinib, a JAK3 inhibitor, has therapeutic potential to treat EBV-associated NK- and T-cell lymphomas by decreasing STAT5 phosphorylation, suppressing proliferation, and reducing EBV latency.132
L. Interleukin 6 (IL-6)
IL-6, as a pleiotropic cytokine, functions in inflammation and B-cell maturation and acts as an initiator of the JAK/STAT pathway, the PI3K/AKT pathway, and the RAS/MAPK pathway.133 IL-6 also acts to promote tumor, including IL-6-driven B-cell lymphoma transformation.134
Targeting IL-6 or IL-6 receptor using monoclonal antibodies or inhibiting IL-6–related pathway to treat IL-6-driven malignancies is becoming feasible and promising.135 Siltuximab (CNTO-328), an IL-6 ligand-inhibiting monoclonal antibody, has been evaluated in B-cell non-Hodgkin lymphoma, multiple myeloma, and Castelman disease.96,134,136
M. MYD88
MYD88 is an activator of both NF-κB and JAK/STAT signaling pathways by stimulating TLR/IL-1 receptor signaling. The TLR/IL-1/MYD88 signaling pathway has also been implicated in oncogenesis of a broad range of malignancies and has therapeutically targeted potential.22 MYD88L256 mutation is identified as a hallmark of LPL/WM and can also be found in a portion of ABC-DLBCL (~30%), primary central nervous system lymphoma (PCNSL), and marginal zone B-cell lymphoma (MZL).22 One study has made an attempt to develop drugs to inhibit MYD88 homodimerization on the basis of its structural and functional domains to block MYD88-dependent pathways like the TLR and IL-1 pathways.137 IMO-8400, an oligonucleotide, used to inhibit ligands activation of TLRs7, 8, and 9, has completed its phase I/II clinical trial for the treatment of relapsed/refractory ABC-DLBCL with MYD88L265P mutation (Clinical Trials Registration: NCT02252146). Another therapeutic strategy to treat ABC-DLBCL or other malignancies bearing aberrant MYD88 is to selectively inhibit interleukin-1 receptor–associated kinase 4 (IRAK4), which is responsible for all the biological function of MYD88.138
N. PD-1 and PD-Ls
PD-1, encoded by programmed cell death 1 (PDCD1) at locus 2q37.3, is mainly expressed on activated T-cells. PD-1 functions as an inhibitory cell surface receptor to negatively regulate effector T-cell functions via phosphatases Src homology region 2 domain-containing phosphatase (SHP)-1/2-mediated inhibition toward TCR/CD3/ZAP70 signaling. PD-1 can also affect cell differentiation and survival by affecting cell survival factor Bcl-xL and effector function-associated transcription factors.58,139
PD-L1, also called B7-H1, is encoded by CD274 and in normally expressed on antigen-presenting cells while PD-L2, is encoded by programmed cell death 1 ligand 2 (PDCD1LG2), is mainly expressed on activated dendritic cells and some macrophages. PD-L1 overexpression in lymphoma cells and stromal cells may result from many different mechanisms. Apart from common amplification or translocation at 9p24.1, activation by other oncogenic pathways like the JAK/STAT or AP1 pathways, viral infection, and chemokines could mediate the constitutive expression of PD-L1. In the microenvironment, as overexpressed PD-L1 engaging to PD-1, effector T-cells are gradually exhausted, leading to immune function impairment and malignant cells escape from immune clearance.58,59,139
In many lymphomas, PD-1 and PD-Ls are aberrantly expressed with variable degrees and different patterns attributed to distinct histone types and different molecular mechanisms. CD274 amplification on chromosome 9q24.1 and PD-L1 overexpression are typical features in nearly 40% of cHL, 70% of PMBL, a small part of ABC-DLBCL and ALK+ an-aplastic large T-cell lymphoma.60,140,141 In contrast, in FL and CLL/SLL, PD-1-positive infiltrating T cells are observed to be increased in lymph node follicles or CLL tumor cells;142,143 in virus-associated lymphomas, not only PD-L1 overexpression is especially significant but PD-1 expression on nonmalignant infiltrating T-cells is also increased.140,144,145 Meaningfully, the aberrant expression of PD-1 and PD-L1 has been associated with patient outcome and treatment response. Therefore, the immunohistochemical expression of PD-1 and PD-L1 can be used as predictive biomarkers.142,146,147 The study about PD-L2 and CTLA-4 expressions or other checkpoint molecules in lymphomas remains rare or absent.
Targeting by PD-1/PD-L1 pathway has been demonstrated to be a very encouraging strategy in a variety of tumors, including solid tumors and hematologic malignancies.148,149 Monoclonal antibodies targeting PD-1, pidilizumab, nivolumab and pembrolizumab, all have been approved to be used and have shown encouraging efficacy in hematologic malignancies, including cHL, PMBL, FL, CLL/SLL, particularly in relapsed/refractory patients or in combination with autologous hematopoietic stem cell transplantation.59,148,150–152 Some other PD1/PD-L1 monoclonal antibodies against PD-L1 are also under clinical evaluation in patients with solid tumors and hematologic malignancies. Durvalumab, atezolizumab, and avelumab are the representative anti-PD-L1 agents which have been approved by the FDA for the treatment of some solid tumors.153–155
IV. PERSPECTIVES AND CHALLENGES
With the advance of biotechnology, especially NGS, and further understanding of the mechanisms of disease, identifying responsible oncogenic signaling pathways and biomarkers became manageable. While more and more investigations are focusing on immunotherapy, the combination of targeting genetic defects and modulating immune functions through immunotherapy holds promise in treating tumors in the era of precision medicine.
However, numerous critical challenges stand in front of precision medicine for lymphoma treatment. First, although pathway-based oncogenic biomarkers are rationally of good values to be as targets rather than targets without oncogenesis involvement, the present targeted drugs did not show ideal and adequate effects in some lymphoma patients; this might be due to tumor heterogeneity and the complex interactivity among oncogenic pathways in lymphoma. Therefore, it is imperative to select ‘driver’ oncogenic biomarkers to develop drugs or to guide the selection of patients who may likely benefit from targeted drugs. Nonetheless, identifying such optimal “driver” oncogenic biomarkers will be a major challenge. It requires further understanding of the pathobiology and the mechanisms. Fortunately, numerous investigators are on their way to elucidate the biology and mechanisms at the genetic, epigenetic, transcriptional, protein, and microenvironmental levels.
Additionally, patient stratification based on oncogenic pathways will be another outstanding problem. Reliable and reproducible biomarkers to accurately convey activated oncogenic pathways in clinical patients will be needed. For example, PD-L1 expressed on the tumor cells can be used as a predictive biomarker to evaluate the response to the PD-L1 blockade. But in practice, the robustness of PD-L1 may depend on the specificity and sensitivity of the antibody, the platform or method to test, the status of intra-tumor heterogeneity, and other factors.
Like other tumors, many subtypes of lymphoma may be characterized by several and different oncogenic pathways. For this reason, it is challenging to determine whether the combination of multiple pathway inhibitors will be more effective than a single targeted agent, and how the best targeted combination can be chosen. Combination regimens should be established using predictive biomarkers from individual patients, and long-time validation should be carried in large cohort. Moreover, pathway-based targeted agents combined with traditional chemotherapy or immune checkpoint therapies might be optimal options for treating lymphoma patients.
As noted, the heterogeneity behind the biology, pathologic feature, molecular and clinical outcomes in lymphoma still stands in the way of precise medicine. Same as in practice, more attention should be given not only to further detailed insights of disease heterogeneity and underlying mechanisms but also to many issues related to heterogeneity, such as drug resistance, biomarker screening, biomarker testing, disease diagnosis, and patient selection. Particularly, effectively overcoming the resistance of pathway-based therapeutics is the major problem that researchers and clinicians must face.
Until now, precision medicine seems to be a paradox. Precision oncology pairing patients with malignancies with drugs for mutations or “pathway” mutations has been questioned. Some people debate whether the termed precision medicine or precision oncology will truly bring the greatest medical breakthrough at the price of significant cost and too high expectation. For this matter, the objective understanding of the advantages and disadvantages of precision medicine will be rational.
V. CONCLUSION
Oncogenic signaling pathways and their harboring oncogenic alterations have been well recognized to play important roles in the development of lymphoma. In the era of precision medicine, identifying the activation of these oncogenic pathways and oncogenic biomarkers to pair patient treatment strategies with pathway-based targeted therapy is imperative and valuable. It is challenging, but it still holds promise. Lymphoma is complex and heterogeneous in diagnosis and treatment, which require more effort by investigators and physicians, and there is long way to go. Pathway-based targeted therapy in combination with traditional chemotherapy, single, specific, targeted antibody therapy, and immunotherapy raise hope for the patients with lymphomas, especially for relapsed and refractory lymphoma patients.
Acknowledgments
This study was supported by the National Institutes of Health/National Cancer Institute grants R01CA138688 and 1RC1CA146299 to KHY. RS is the recipient of scholarship award. KHY is supported by the University of Texas MD Anderson Cancer Center Institutional Research and Development Fund, an Institutional Research Grant Award, an MD Anderson Cancer Center Lymphoma Specialized Programs on Research Excellence (SPORE) Research Development Program Award, an MD Anderson Cancer Center Myeloma SPORE Research Development Program Award, and an MD Anderson Myeloma SPORE Research Developmental Program Award and the University Cancer Foundation via the Sister institution network Fund at the University of Texas MD Anderson Cancer Cancer. The study is also partially supported by P50CA136411 and P50CA142509 and the MD Anderson Cancer Center Support Grant CA016672
ABBREVIATIONS
- ABC
active B-cell
- AKT
V-AKT murine thymoma viral oncogene homolog1
- BCR
B-cell receptor
- BL
Burkitt lymphoma
- BTK
Bruton tyrosine kinase
- cHL
classical Hodgkin lymphoma
- CLL
chronic lymphocytic leukemia
- DLBCL
diffuse large B-cell lymphoma
- FL
follicular lymphoma
- GCB
germinal center B-cell
- HL
Hodgkin lymphoma
- IKK
IkB kinase
- IL-6
interleukin 6
- ITAM
immune receptor tyrosine-based activation motifs
- JAK
Janus kinase
- MALT
mucosa-associated lymphoid tissue lymphoma
- MCL
mantle cell lymphoma
- MDM2
double-minute 2 protein
- mTOR
mechanistic target of rapamycin
- mTORC
mTOR complex
- MYC
v-Myc avian myelocytomatosis viral oncogene homolog
- MYD88
myeloid differentiation primary response 88
- NF-κB
nuclear factor-kappaB
- PD-1
programmed death-1
- PD-Ls
programmed death-ligands
- PI3K
phosphoinositide-3-kinase
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- PKCβ
protein kinase C β
- PMBL
primary mediastinal B-cell lymphoma
- PTEN
phosphatase and tensin homolog
- SLL
small lymphocytic lymphoma
- STAT
signal transducer and activator of transcription
- SOCS
suppressor of cytokine signaling
- SYK
spleen-associated tyrosine kinase
- TLR
Toll-like receptors
- TNFR
tumor necrosis factor receptor
References
- 1.Swerdlow SH, Campo E, Harris NL. World Health Organization classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: Lark; 2008. [Google Scholar]
- 2.Shaffer AL, 3rd, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol. 2012;30:565–610. doi: 10.1146/annurev-immunol-020711-075027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Younes A. Beyond chemotherapy: new agents for targeted treatment of lymphoma. Nat Rev Clin Oncol. 2011;8(2):85–96. doi: 10.1038/nrclinonc.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570–80. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2011;118(16):4313–20. doi: 10.1182/blood-2011-06-338855. [DOI] [PubMed] [Google Scholar]
- 6.Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Immunol. 2004;41(6–7):599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 7.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12(3):229–43. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, Xu W, Shaffer AL, Wright G, Xiao W, Powell J, Jiang JK, Thomas CJ, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Johnson NA, Rimsza LM, Campo E, Jaffe ES, Wilson WH, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Pierce SK, Staudt LM. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu WH, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Chan WC, Staudt LM. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319(5870):1676–9. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 10.Peveling-Oberhag J, Arcaini L, Hansmann ML, Zeuzem S. Hepatitis C–associated B-cell non-Hodgkin lymphomas. Epidemiology, molecular signature and clinical management. J Hepatol. 2013;59(1):169–77. doi: 10.1016/j.jhep.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 11.Hosokawa Y. Anti-apoptotic action of API2-MALT1 fusion protein involved in t(11;18)(q21;q21) MALT lymphoma. Apoptosis. 2005;10(1):25–34. doi: 10.1007/s10495-005-6059-6. [DOI] [PubMed] [Google Scholar]
- 12.Wotherspoon AC, Doglioni C, Diss TC, Pan L, Moschini A, de Boni M, Isaacson PG. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993;342(8871):575–7. doi: 10.1016/0140-6736(93)91409-f. [DOI] [PubMed] [Google Scholar]
- 13.Lam KP, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90(6):1073–83. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- 14.Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, Liu X, Powell J, Yang Y, Xu W, Zhao H, Kohlhammer H, Rosenwald A, Kluin P, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Ogwang MD, Reynolds SJ, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Pittaluga S, Wilson W, Waldmann TA, Rowe M, Mbulaiteye SM, Rickinson AB, Staudt LM. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490(7418):116–20. doi: 10.1038/nature11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55–65. [PubMed] [Google Scholar]
- 16.Niemann CU, Wiestner A. B-cell receptor signaling as a driver of lymphoma development and evolution. Semin Cancer Biol. 2013;23(6):410–21. doi: 10.1016/j.semcancer.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2(6):a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu L, Li L, Medeiros LJ, Young KH. NF-kappaB signaling pathway and its potential as a target for therapy in lymphoid neoplasms. Blood Rev. 2017;31(2):77–92. doi: 10.1016/j.blre.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagel D, Vincendeau M, Eitelhuber AC, Krappmann D. Mechanisms and consequences of constitutive NF-kappaB activation in B-cell lymphoid malignancies. Oncogene. 2014;33(50):5655–65. doi: 10.1038/onc.2013.565. [DOI] [PubMed] [Google Scholar]
- 20.Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, Korneluk RG, Cheng G. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9(12):1371–8. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappa B activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194(12):1861–74. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Staudt LM. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–9. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruan J, Martin P, Furman RR, Lee SM, Cheung K, Vose JM, Lacasce A, Morrison J, Elstrom R, Ely S, Chadburn A, Cesarman E, Coleman M, Leonard JP. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29(6):690–7. doi: 10.1200/JCO.2010.31.1142. [DOI] [PubMed] [Google Scholar]
- 24.Shah JJ, Jakubowiak AJ, O’Connor OA, Orlowski RZ, Harvey RD, Smith MR, Lebovic D, Diefenbach C, Kelly K, Hua Z, Berger AJ, Mulligan G, Faessel HM, Tirrell S, Dezube BJ, Lonial S. Phase I study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res. 2016;22(1):34–43. doi: 10.1158/1078-0432.CCR-15-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, Dick LR, Garnsey JJ, Koenig E, Langston SP, Manfredi M, Narayanan U, Rolfe M, Staudt LM, Soucy TA, Yu J, Zhang J, Bolen JB, Smith PG. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-kB-dependent lymphoma. Blood. 2010;116(9):1515–23. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- 26.Czuczman NM, Barth MJ, Gu J, Neppalli V, Mavis C, Frys SE, Hu Q, Liu S, Klener P, Vockova P, Czuczman MS, Hernandez-Ilizaliturri FJ. Pevonedistat, a NEDD8-activating enzyme inhibitor, is active in mantle cell lymphoma and enhances rituximab activity in vivo. Blood. 2016;127(9):1128–37. doi: 10.1182/blood-2015-04-640920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dutton A, Reynolds GM, Dawson CW, Young LS, Murray PG. Constitutive activation of phosphatidyl-inositide 3 kinase contributes to the survival of Hodgkin’s lymphoma cells through a mechanism involving Akt kinase and mTOR. J Pathol. 2005;205(4):498–506. doi: 10.1002/path.1725. [DOI] [PubMed] [Google Scholar]
- 28.Westin JR. Status of PI3K/Akt/mTOR pathway inhibitors in lymphoma. Clin Lymphoma Myeloma Leuk. 2014;14(5):335–42. doi: 10.1016/j.clml.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 30.Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318(5857):1744–8. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 31.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6(3):184–92. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 32.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 34.Abubaker J, Bavi PP, Al-Harbi S, Siraj AK, Al-Dayel F, Uddin S, Al-Kuraya K. PIK3CA mutations are mutually exclusive with PTEN loss in diffuse large B-cell lymphoma. Leukemia. 2007;21(11):2368–70. doi: 10.1038/sj.leu.2404873. [DOI] [PubMed] [Google Scholar]
- 35.Baohua Y, Xiaoyan Z, Tiecheng Z, Tao Q, Daren S. Mutations of the PIK3CA gene in diffuse large B cell lymphoma. DiagN Mol Pathol. 2008;17(3):159–65. doi: 10.1097/PDM.0b013e31815d0588. [DOI] [PubMed] [Google Scholar]
- 36.Psyrri A, Papageorgiou S, Liakata E, Scorilas A, Rontogianni D, Kontos CK, Argyriou P, Pectasides D, Harhalakis N, Pappa V, Kolialexi A, Economopoulou C, Kontsioti F, Maratou E, Dimitriadis G, Economopoulou P, Economopoulos T. Phosphatidylinositol 3’-kinase catalytic subunit alpha gene amplification contributes to the pathogenesis of mantle cell lymphoma. Clin Cancer Res. 2009;15(18):5724–32. doi: 10.1158/1078-0432.CCR-08-3215. [DOI] [PubMed] [Google Scholar]
- 37.Uddin S, Hussain AR, Siraj AK, Manogaran PS, Al-Jomah NA, Moorji A, Atizado V, Al-Dayel F, Belgaumi A, El-Solh H, Ezzat A, Bavi P, Al-Kuraya KS. Role of phosphatidylinositol 3’-kinase/AKT pathway in diffuse large B-cell lymphoma survival. Blood. 2006;108(13):4178–86. doi: 10.1182/blood-2006-04-016907. [DOI] [PubMed] [Google Scholar]
- 38.Rudelius M, Pittaluga S, Nishizuka S, Pham TH, Fend F, Jaffe ES, Quintanilla-Martinez L, Raffeld M. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood. 2006;108(5):1668–76. doi: 10.1182/blood-2006-04-015586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai Q, Deng H, Xie D, Lin T, Lin T. Phosphorylated AKT protein is overexpressed in human peripheral T-cell lymphomas and predicts decreased patient survival. Clin Lymph Myelom Leuk. 2012;12(2):106–12. doi: 10.1016/j.clml.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 40.Rui L, Emre NC, Kruhlak MJ, Chung HJ, Steidl C, Slack G, Wright GW, Lenz G, Ngo VN, Shaffer AL, Xu W, Zhao H, Yang Y, Lamy L, Davis RE, Xiao W, Powell J, Maloney D, Thomas CJ, Moller P, Rosenwald A, Ott G, Muller-Hermelink HK, Savage K, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Weisenburger DD, Chan WC, Gascoyne RD, Levens D, Staudt LM. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell. 2010;18(6):590–605. doi: 10.1016/j.ccr.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi S, Calhoun HC, Xia F, Li J, Le L, Li WX. JAK signaling globally counteracts heterochromatic gene silencing. Nat Genet. 2006;38(9):1071–6. doi: 10.1038/ng1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valentino L, Pierre J. JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol. 2006;71(6):713–21. doi: 10.1016/j.bcp.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 43.Ilangumaran S, Rottapel R. Regulation of cytokine receptor signaling by SOCS1. Immunol Rev. 2003;192:196–211. doi: 10.1034/j.1600-065x.2003.00020.x. [DOI] [PubMed] [Google Scholar]
- 44.Iqbal J, Naushad H, Bi C, Yu J, Bouska A, Rohr J, Chao W, Fu K, Chan WC, Vose JM. Genomic signatures in B-cell lymphoma: How can these improve precision in diagnosis and inform prognosis? Blood Rev. 2016;30(2):73–88. doi: 10.1016/j.blre.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 45.Kleppe M, Tousseyn T, Geissinger E, Kalender Atak Z, Aerts S, Rosenwald A, Wlodarska I, Cools J. Mutation analysis of the tyrosine phosphatase PTPN2 in Hodgkin’s lymphoma and T-cell non-Hodgkin lymphoma. Haematol. 2011;96(11):1723–7. doi: 10.3324/haematol.2011.041921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joliot V, Cormier F, Medyouf H, Alcalde H, Ghysdael J. Constitutive STAT5 activation specifically cooperates with the loss of p53 function in B-cell lymphomagenesis. Oncogene. 2006;25(33):4573–84. doi: 10.1038/sj.onc.1209480. [DOI] [PubMed] [Google Scholar]
- 47.Holtick U, Vockerodt M, Pinkert D, Schoof N, Sturzen-hofecker B, Kussebi N, Lauber K, Wesselborg S, Loffler D, Horn F, Trumper L, Kube D. STAT3 is essential for Hodgkin lymphoma cell proliferation and is a target of tyrphostin AG17 which confers sensitization for apoptosis. Leukemia. 2005;19(6):936–44. doi: 10.1038/sj.leu.2403750. [DOI] [PubMed] [Google Scholar]
- 48.Ding BB, Yu JJ, Yu RY, Mendez LM, Shaknovich R, Zhang Y, Cattoretti G, Ye BH. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood. 2008;111(3):1515–23. doi: 10.1182/blood-2007-04-087734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol. 2012;30(9):1005–14. doi: 10.1200/JCO.2010.31.8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Derenzini E, Younes A. Targeting the JAK-STAT pathway in lymphoma: a focus on pacritinib. Expert Opin Investig Drugs. 2013;22(6):775–85. doi: 10.1517/13543784.2013.775244. [DOI] [PubMed] [Google Scholar]
- 51.Pirnia F, Schneider E, Betticher DC, Borner MM. Mitomycin C induces apoptosis and caspase-8 and -9 processing through a caspase-3 and Fas-independent pathway. Cell Death Differ. 2002;9(9):905–14. doi: 10.1038/sj.cdd.4401062. [DOI] [PubMed] [Google Scholar]
- 52.Munoz-Pinedo C. Signaling pathways that regulate life and cell death: evolution of apoptosis in the context of self-defense. Adv Exp Med Biol. 2012;738:124–43. doi: 10.1007/978-1-4614-1680-7_8. [DOI] [PubMed] [Google Scholar]
- 53.Yin XM. Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res. 2000;10(3):161–7. doi: 10.1038/sj.cr.7290045. [DOI] [PubMed] [Google Scholar]
- 54.Luschen S, Falk M, Scherer G, Ussat S, Paulsen M, Adam-Klages S. The Fas-associated death domain protein/caspase-8/c-FLIP signaling pathway is involved in TNF-induced activation of ERK. Exp Eell Res. 2005;310(1):33–42. doi: 10.1016/j.yexcr.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 55.Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11(9):372–7. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 56.Tian E, Gazitt Y. The role of p53, bcl-2 and bax network in dexamethasone induced apoptosis in multiple myeloma cell lines. Int J Oncol. 1996;8(4):719–26. doi: 10.3892/ijo.8.4.719. [DOI] [PubMed] [Google Scholar]
- 57.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22(56):9030–40. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 58.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Ann Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xia Y, Jeffrey Medeiros L, Young KH. Signaling pathway and dysregulation of PD1 and its ligands in lymphoid malignancies. Biochim Biophys Acta. 2016;1865(1):58–71. doi: 10.1016/j.bbcan.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, Chapuy B, Takeyama K, Neuberg D, Golub TR, Kutok JL, Shipp MA. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116(17):3268–77. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–14. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13(15):1899–911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 63.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev cell. 2011;21(1):92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Freedman A. Follicular lymphoma: 2014 update on diagnosis and management. Am J Hematol. 2014;89(4):429–36. doi: 10.1002/ajh.23674. [DOI] [PubMed] [Google Scholar]
- 65.Iqbal J, Sanger WG, Horsman DE, Rosenwald A, Pickering DL, Dave B, Dave S, Xiao L, Cao K, Zhu Q, Sherman S, Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Ott G, Muller-Hermelink HK, Delabie J, Braziel RM, Jaffe ES, Campo E, Lynch JC, Connors JM, Vose JM, Armitage JO, Grogan TM, Staudt LM, Chan WC. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am J Pathol. 2004;165(1):159–66. doi: 10.1016/s0002-9440(10)63284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, Powell J, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Campo E, Jaffe ES, Delabie J, Smeland EB, Rimsza LM, Fisher RI, Weisenburger DD, Chan WC, Staudt LM. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105(36):13520–5. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kendrick SL, Redd L, Muranyi A, Henricksen LA, Stanislaw S, Smith LM, Perry AM, Fu K, Weisenburger DD, Rosenwald A, Ott G, Gascoyne RD, Jaffe ES, Campo E, Delabie J, Braziel RM, Cook JR, Tubbs RR, Staudt LM, Chan WC, Steidl C, Grogan TM, Rimsza LM. BCL2 antibodies targeted at different epitopes detect varying levels of protein expression and correlate with frequent gene amplification in diffuse large B-cell lymphoma. Hum Pathol. 2014;45(10):2144–53. doi: 10.1016/j.humpath.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5(11):876–85. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- 69.Oki Y, Copeland A, Hagemeister F, Fayad LE, Fanale M, Romaguera J, Younes A. Experience with obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist in patients with relapsed or refractory classical Hodgkin lymphoma. Blood. 2012;119(9):2171–2. doi: 10.1182/blood-2011-11-391037. [DOI] [PubMed] [Google Scholar]
- 70.Goy A, Berger M, Ford P, Feldman T, Mato A, Bejot C, Fung HC. Sequential single-agent obatoclax mesylate (GX15-070MS) followed by combination with rituximab in patients with previously untreated follicular lymphoma. Leuk Lymph. 2014;55(12):2932–4. doi: 10.3109/10428194.2014.900760. [DOI] [PubMed] [Google Scholar]
- 71.King AC, Peterson TJ, Horvat TZ, Rodriguez M, Tang LA. Venetoclax: A first-in-class oral BCL-2 inhibitor for the management of lymphoid malignancies. Ann Pharmacother. 2017;5(15):410–6. doi: 10.1177/1060028016685803. [DOI] [PubMed] [Google Scholar]
- 72.Xu-Monette ZY, Medeiros LJ, Li Y, Orlowski RZ, Andreeff M, Bueso-Ramos CE, Greiner TC, McDonnell TJ, Young KH. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood. 2012;119(16):3668–83. doi: 10.1182/blood-2011-11-366062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boehme KA, Blattner C. Regulation of p53—insights into a complex process. Crit Rev Biochem Mol Biol. 2009;44(6):367–92. doi: 10.3109/10409230903401507. [DOI] [PubMed] [Google Scholar]
- 74.Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16(11):528–36. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cillessen SA, Meijer CJ, Notoya M, Ossenkoppele GJ, Oudejans JJ. Molecular targeted therapies for diffuse large B-cell lymphoma based on apoptosis profiles. J Pathol. 2010;220(5):509–20. doi: 10.1002/path.2670. [DOI] [PubMed] [Google Scholar]
- 76.Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4(5):371–81. doi: 10.1016/s1535-6108(03)00272-1. [DOI] [PubMed] [Google Scholar]
- 77.Prabhu VV, Allen JE, Hong B, Zhang S, Cheng H, El-Deiry WS. Therapeutic targeting of the p53 pathway in cancer stem cells. Expert Opin Ther Targets. 2012;16(12):1161–74. doi: 10.1517/14728222.2012.726985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meyer N, Penn LZ. MYC—Timeline Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8(12):976–90. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 79.Dang CV. MYC on the path to Cancer. Cell. 2012;149(1):22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hao S, Sanger W, Onciu M, Lai R, Schlette EJ, Medeiros LJ. Mantle cell lymphoma with 8q24 chromosomal abnormalities: a report of 5 cases with blastoid features. Mod Pathol. 2002;15(12):1266–72. doi: 10.1097/01.MP.0000037310.82136.99. [DOI] [PubMed] [Google Scholar]
- 81.Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22(40):6151–9. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- 82.Trabucco SE, Gerstein RM, Evens AM, Bradner JE, Shultz LD, Greiner DL, Zhang H. Inhibition of bromodomain proteins for the treatment of human diffuse large B-cell lymphoma. Clin Cancer Res. 2015;21(1):113–22. doi: 10.1158/1078-0432.CCR-13-3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bhadury J, Nilsson LM, Muralidharan SV, Green LC, Li Z, Gesner EM, Hansen HC, Keller UB, McLure KG, Nilsson JA. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc Natl Acad Sci U S A. 2014;111(26):E2721–30. doi: 10.1073/pnas.1406722111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, Vakoc CR. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–8. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Friedberg JW, Mahadevan D, Cebula E, Persky D, Lossos I, Agarwal AB, Jung J, Burack R, Zhou X, Leonard EJ, Fingert H, Danaee H, Bernstein SH. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol. 2014;32(1):44–50. doi: 10.1200/JCO.2012.46.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rhyasen GW, Hattersley MM, Yao Y, Dulak A, Wang W, Petteruti P, Dale IL, Boiko S, Cheung T, Zhang J, Wen S, Castriotta L, Lawson D, Collins M, Bao L, Ahdesmaki MJ, Walker G, O’Connor G, Yeh TC, Rabow AA, Dry JR, Reimer C, Lyne P, Mills GB, Fawell SE, Waring MJ, Zinda M, Clark E, Chen H. AZD5153: a novel bivalent BET bromodomain inhibitor highly active against hematologic malignancies. Mol Cancer Ther. 2016;15(11):2563–74. doi: 10.1158/1535-7163.MCT-16-0141. [DOI] [PubMed] [Google Scholar]
- 87.Humphries LA, Dangelmaier C, Sommer K, Kipp K, Kato RM, Griffith N, Bakman I, Turk CW, Daniel JL, Rawlings DJ. Tec kinases mediate sustained calcium influx via site-specific tyrosine phosphorylation of the phospholipase Cgamma Src homology 2-Src homology 3 linker. J Biol Chem. 2004;279(36):37651–61. doi: 10.1074/jbc.M311985200. [DOI] [PubMed] [Google Scholar]
- 88.Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol. 2012;31(2):119–32. doi: 10.3109/08830185.2012.664797. [DOI] [PubMed] [Google Scholar]
- 89.Horwood NJ, Page TH, McDaid JP, Palmer CD, Campbell J, Mahon T, Brennan FM, Webster D, Foxwell BM. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol. 2006;176(6):3635–41. doi: 10.4049/jimmunol.176.6.3635. [DOI] [PubMed] [Google Scholar]
- 90.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, Kolibaba KS, Furman RR, Rodriguez S, Chang BY, Sukbuntherng J, Izumi R, Hamdy A, Hedrick E, Fowler NH. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, Johnson P, Spurgeon SE, Li L, Zhang L, Newberry K, Ou Z, Cheng N, Fang B, McGreivy J, Clow F, Buggy JJ, Chang BY, Beaupre DM, Kunkel LA, Blum KA. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–16. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, O’Brien S. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Noy A, de Vos S, Thieblemont C, Martin P, Flowers CR, Morschhauser F, Collins GP, Ma S, Coleman M, Peles S, Smith S, Barrientos JC, Smith A, Munneke B, Dimery I, Beaupre DM, Chen R. Targeting BTK with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood. 2017 doi: 10.1182/blood-2016-10-747345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maddocks K, Christian B, Jaglowski S, Flynn J, Jones JA, Porcu P, Wei L, Jenkins C, Lozanski G, Byrd JC, Blum KA. A phase 1/1b study of rituximab, bendamustine, and ibrutinib in patients with untreated and relapsed/refractory non-Hodgkin lymphoma. Blood. 2015;125(2):242–8. doi: 10.1182/blood-2014-08-597914. [DOI] [PubMed] [Google Scholar]
- 95.Hallek M, Kay NE, Osterborg A, Chanan-Khan AA, Mahler M, Salman M, Wan Y, Sun S, Zhuang SH, Howes A. The HELIOS trial protocol: a phase III study of ibrutinib in combination with bendamustine and rituximab in relapsed/refractory chronic lymphocytic leukemia. Future Oncol. 2015;11(1):51–9. doi: 10.2217/fon.14.119. [DOI] [PubMed] [Google Scholar]
- 96.Kurzrock R, Voorhees PM, Casper C, Furman RR, Fayad L, Lonial S, Borghaei H, Jagannath S, Sokol L, Usmani SZ, van de Velde H, Qin X, Puchalski TA, Hall B, Reddy M, Qi M, van Rhee F. A phase I, open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma, multiple myeloma, or Castleman disease. Clin Cancer Res. 2013;19(13):3659–70. doi: 10.1158/1078-0432.CCR-12-3349. [DOI] [PubMed] [Google Scholar]
- 97.Walter HS, Rule SA, Dyer MJ, Karlin L, Jones C, Cazin B, Quittet P, Shah N, Hutchinson CV, Honda H, Duffy K, Birkett J, Jamieson V, Courtenay-Luck N, Yoshizawa T, Sharpe J, Ohno T, Abe S, Nishimura A, Cartron G, Morschhauser F, Fegan C, Salles G. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood. 2016;127(4):411–9. doi: 10.1182/blood-2015-08-664086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10(6):387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature. 1995;378(6554):303–6. doi: 10.1038/378303a0. [DOI] [PubMed] [Google Scholar]
- 100.Rowley RB, Burkhardt AL, Chao HG, Matsueda GR, Bolen JB. Syk protein-tyrosine kinase is regulated by tyrosine-phosphorylated Ig alpha/Ig beta immunoreceptor tyrosine activation motif binding and autophosphorylation. J Biol Chem. 1995;270(19):11590–4. doi: 10.1074/jbc.270.19.11590. [DOI] [PubMed] [Google Scholar]
- 101.Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, Schaefer-Cutillo J, De Vos S, Sinha R, Leonard JP, Cripe LD, Gregory SA, Sterba MP, Lowe AM, Levy R, Shipp MA. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578–85. doi: 10.1182/blood-2009-08-236471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Swat W, Montgrain V, Doggett TA, Douangpanya J, Puri K, Vermi W, Diacovo TG. Essential role of PI3Kdelta and PI3Kgamma in thymocyte survival. Blood. 2006;107(6):2415–22. doi: 10.1182/blood-2005-08-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim N, Saudemont A, Webb L, Camps M, Ruckle T, Hirsch E, Turner M, Colucci F. The p110delta catalytic isoform of PI3K is a key player in NK-cell development and cytokine secretion. Blood. 2007;110(9):3202–8. doi: 10.1182/blood-2007-02-075366. [DOI] [PubMed] [Google Scholar]
- 104.Cheah CY, Fowler NH. Idelalisib in the management of lymphoma. Blood. 2016;128(3):331–6. doi: 10.1182/blood-2016-02-702761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gilbert JA. Idelalisib: targeting PI3Kdelta in B-cell malignancies. Lancet Oncol. 2014;15(3):e108. doi: 10.1016/s1470-2045(14)70052-x. [DOI] [PubMed] [Google Scholar]
- 106.Vangapandu HV, Jain N, Gandhi V. Duvelisib: A phosphoinositide-3 kinase delta/gamma inhibitor for chronic lymphocytic leukemia. Expert Opin Investig Drugs. 2017 doi: 10.1080/13543784.2017.1312338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Balakrishnan K, Peluso M, Fu M, Rosin NY, Burger JA, Wierda WG, Keating MJ, Faia K, O’Brien S, Kutok JL, Gandhi V. The phosphoinositide-3-kinase (PI3K)-delta and gamma inhibitor, IPI-145 (Duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia. 2015;29(9):1811–22. doi: 10.1038/leu.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chiarini F, Del Sole M, Mongiorgi S, Gaboardi GC, Cappellini A, Mantovani I, Follo MY, McCubrey JA, Martelli AM. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia. 2008;22(6):1106–16. doi: 10.1038/leu.2008.79. [DOI] [PubMed] [Google Scholar]
- 109.Friedman DR, Lanasa MC, Davis PH, Allgood SD, Matta KM, Brander DM, Chen Y, Davis ED, Volkheimer AD, Moore JO, Gockerman JP, Sportelli P, Weinberg JB. Perifosine treatment in chronic lymphocytic leukemia: results of a phase II clinical trial and in vitro studies. Leuk Lymph. 2014;55(5):1067–75. doi: 10.3109/10428194.2013.824080. [DOI] [PubMed] [Google Scholar]
- 110.Oki Y, Fanale M, Romaguera J, Fayad L, Fowler N, Copeland A, Samaniego F, Kwak LW, Neelapu S, Wang M, Feng L, Younes A. Phase II study of an AKT inhibitor MK2206 in patients with relapsed or refractory lymphoma. Br J Haematol. 2015;171(4):463–70. doi: 10.1111/bjh.13603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Witzig TE, Geyer SM, Ghobrial I, Inwards DJ, Fonseca R, Kurtin P, Ansell SM, Luyun R, Flynn PJ, Morton RF, Dakhil SR, Gross H, Kaufmann SH. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23(23):5347–56. doi: 10.1200/JCO.2005.13.466. [DOI] [PubMed] [Google Scholar]
- 112.Smith SM, van Besien K, Karrison T, Dancey J, McLaughlin P, Younes A, Smith S, Stiff P, Lester E, Modi S, Doyle LA, Vokes EE, Pro B. Temsirolimus has activity in non-mantle cell non-Hodgkin’s lymphoma subtypes: The University of Chicago phase II consortium. J Clin Oncol. 2010;28(31):4740–6. doi: 10.1200/JCO.2010.29.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Witzig TE, Reeder CB, LaPlant BR, Gupta M, Johnston PB, Micallef IN, Porrata LF, Ansell SM, Colgan JP, Jacobsen ED, Ghobrial IM, Habermann TM. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2011;25(2):341–7. doi: 10.1038/leu.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Johnston PB, Inwards DJ, Colgan JP, Laplant BR, Kabat BF, Habermann TM, Micallef IN, Porrata LF, Ansell SM, Reeder CB, Roy V, Witzig TE. A Phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol. 2010;85(5):320–4. doi: 10.1002/ajh.21664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Witzig TE, Reeder C, Han JJ, LaPlant B, Stenson M, Tun HW, Macon W, Ansell SM, Habermann TM, Inwards DJ, Micallef IN, Johnston PB, Porrata LF, Colgan JP, Markovic S, Nowakowski GS, Gupta M. The mTORC1 inhibitor everolimus has antitumor activity in vitro and produces tumor responses in patients with relapsed T-cell lymphoma. Blood. 2015;126(3):328–35. doi: 10.1182/blood-2015-02-629543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 117.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22(14):2954–63. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 118.Wang X, Huang H, Young KH. The PTEN tumor suppressor gene and its role in lymphoma pathogenesis. Aging. 2015;7(12):1032–49. doi: 10.18632/aging.100855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xu G, Zhang W, Bertram P, Zheng XF, McLeod H. Pharmacogenomic profiling of the PI3K/PTEN-AKT-mTOR pathway in common human tumors. Int J Oncol. 2004;24(4):893–900. [PubMed] [Google Scholar]
- 120.Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann S, Fritsch C, Dorsch M, Chene P, Shoemaker K, De Pover A, Menezes D, Martiny-Baron G, Fabbro D, Wilson CJ, Schlegel R, Hofmann F, Garcia-Echeverria C, Sellers WR, Voliva CF. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–28. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- 121.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nature reviews. Mol Cell Biol. 2002;3(9):651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 122.Mughal TI, Girnius S, Rosen ST, Kumar S, Wiestner A, Abdel-Wahab O, Kiladjian JJ, Wilson WH, Van Etten RA. Emerging therapeutic paradigms to target the dysregulated Janus kinase/signal transducer and activator of transcription pathway in hematological malignancies. Leuk Lymph. 2014;55(9):1968–79. doi: 10.3109/10428194.2013.863307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang Q, Nowak I, Vonderheid EC, Rook AH, Kadin ME, Nowell PC, Shaw LM, Wasik MA. Activation of Jak/STAT proteins involved in signal transduction pathway mediated by receptor for interleukin 2 in malignant T lymphocytes derived from cutaneous anaplastic large T-cell lymphoma and Sezary syndrome. Proc Natl Acad Sci U S A. 1996;93(17):9148–53. doi: 10.1073/pnas.93.17.9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Han Y, Amin HM, Franko B, Frantz C, Shi X, Lai R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood. 2006;108(8):2796–803. doi: 10.1182/blood-2006-04-017434. [DOI] [PubMed] [Google Scholar]
- 125.Springuel L, Renauld JC, Knoops L. JAK kinase targeting in hematologic malignancies: a sinuous pathway from identification of genetic alterations towards clinical indications. Haematol. 2015;100(10):1240–53. doi: 10.3324/haematol.2015.132142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Duan Z, Bradner J, Greenberg E, Mazitschek R, Foster R, Mahoney J, Seiden MV. 8-benzyl-4-oxo-8-azabicyclo[3.2.1]oct-2-ene-6,7-dicarboxylic acid (SD-1008), a novel Janus kinase 2 inhibitor, increases chemotherapy sensitivity in human ovarian cancer cells. Mol Pharmacol. 2007;72(5):1137–45. doi: 10.1124/mol.107.038117. [DOI] [PubMed] [Google Scholar]
- 127.Van Roosbroeck K, Cox L, Tousseyn T, Lahortiga I, Gielen O, Cauwelier B, De Paepe P, Verhoef G, Marynen P, Vandenberghe P, De Wolf-Peeters C, Cools J, Wlodarska I. JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood. 2011;117(15):4056–64. doi: 10.1182/blood-2010-06-291310. [DOI] [PubMed] [Google Scholar]
- 128.Adelaide J, Perot C, Gelsi-Boyer V, Pautas C, Murati A, Copie-Bergman C, Imbert M, Chaffanet M, Birnbaum D, Mozziconacci MJ. A t(8;9) translocation with PCM1-JAK2 fusion in a patient with T-cell lymphoma. Leukemia. 2006;20(3):536–7. doi: 10.1038/sj.leu.2404104. [DOI] [PubMed] [Google Scholar]
- 129.Hart S, Goh KC, Novotny-Diermayr V, Hu CY, Hentze H, Tan YC, Madan B, Amalini C, Loh YK, Ong LC, William AD, Lee A, Poulsen A, Jayaraman R, Ong KH, Ethirajulu K, Dymock BW, Wood JW. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia. 2011;25(11):1751–9. doi: 10.1038/leu.2011.148. [DOI] [PubMed] [Google Scholar]
- 130.Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L, Chong SC, Ong WS, Tay K, Tao M, Quek R, Loong S, Yeoh KW, Yap SP, Lee KA, Lim LC, Tan D, Goh C, Cutcutache I, Yu W, Ng CC, Rajasegaran V, Heng HL, Gan A, Ong CK, Rozen S, Tan P, Teh BT, Lim ST. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012;2(7):591–7. doi: 10.1158/2159-8290.CD-12-0028. [DOI] [PubMed] [Google Scholar]
- 131.Elliott NE, Cleveland SM, Grann V, Janik J, Waldmann TA, Dave UP. FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood. 2011;118(14):3911–21. doi: 10.1182/blood-2010-12-319467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ando S, Kawada JI, Watanabe T, Suzuki M, Sato Y, Torii Y, Asai M, Goshima F, Murata T, Shimizu N, Ito Y, Kimura H. Tofacitinib induces G1 cell-cycle arrest and inhibits tumor growth in Epstein-Barr virus-associated T and natural killer cell lymphoma cells. Oncotarget. 2016;7(47):76793–805. doi: 10.18632/oncotarget.12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nat Clin Prac Rheumatol. 2006;2(11):619–26. doi: 10.1038/ncprheum0338. [DOI] [PubMed] [Google Scholar]
- 134.Ferrario A, Merli M, Basilico C, Maffioli M, Passamonti F. Siltuximab and hematologic malignancies. A focus in non Hodgkin lymphoma. Expert Opin Investig Drugs. 2017;26(3):367–73. doi: 10.1080/13543784.2017.1288213. [DOI] [PubMed] [Google Scholar]
- 135.Nishimoto N. Interleukin-6 as a therapeutic target in candidate inflammatory diseases. Clin Pharmacol Ther. 2010;87(4):483–7. doi: 10.1038/clpt.2009.313. [DOI] [PubMed] [Google Scholar]
- 136.Barquero N. Siltuximab: a new option for the management of Castleman’s disease. Drugs Today (Barc) 2015;51(1):21–8. doi: 10.1358/dot.2015.51.1.2234002. [DOI] [PubMed] [Google Scholar]
- 137.Loiarro M, Ruggiero V, Sette C. Targeting the Toll-like receptor/interleukin 1 receptor pathway in human diseases: rational design of MyD88 inhibitors. Clin Lymph Myelom Leuk. 2013;13(2):222–6. doi: 10.1016/j.clml.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 138.Kelly PN, Romero DL, Yang Y, Shaffer AL, 3rd, Chaudhary D, Robinson S, Miao W, Rui L, Westlin WF, Kapeller R, Staudt LM. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J Exp Med. 2015;212(13):2189–201. doi: 10.1084/jem.20151074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, Yu H, Fletcher CD, Freeman GJ, Shipp MA, Rodig SJ. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. 2013;19(13):3462–73. doi: 10.1158/1078-0432.CCR-13-0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yamamoto R, Nishikori M, Tashima M, Sakai T, Ichinohe T, Takaori-Kondo A, Ohmori K, Uchiyama T. B7-H1 expression is regulated by MEK/ERK signaling pathway in anaplastic large cell lymphoma and Hodgkin lymphoma. Cancer Sci. 2009;100(11):2093–100. doi: 10.1111/j.1349-7006.2009.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Carreras J, Lopez-Guillermo A, Roncador G, Villamor N, Colomo L, Martinez A, Hamoudi R, Howat WJ, Montserrat E, Campo E. High numbers of tumor-infiltrating programmed cell death 1-positive regulatory lymphocytes are associated with improved overall survival in follicular lymphoma. J Clin Oncol. 2009;27(9):1470–6. doi: 10.1200/JCO.2008.18.0513. [DOI] [PubMed] [Google Scholar]
- 143.Brusa D, Serra S, Coscia M, Rossi D, D’Arena G, Laurenti L, Jaksic O, Fedele G, Inghirami G, Gaidano G, Malavasi F, Deaglio S. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematol. 2013;98(6):953–63. doi: 10.3324/haematol.2012.077537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Paydas S, Bagir E, Seydaoglu G, Ercolak V, Ergin M. Programmed death-1 (PD-1), programmed death-ligand 1 (PD-L1), and EBV-encoded RNA (EBER) expression in Hodgkin lymphoma. Ann Hematol. 2015;94(9):1545–52. doi: 10.1007/s00277-015-2403-2. [DOI] [PubMed] [Google Scholar]
- 145.Ni L, Ma CJ, Zhang Y, Nandakumar S, Zhang CL, Wu XY, Borthwick T, Hamati A, Chen XY, Kumaraguru U, Moorman JP, Yao ZQ. PD-1 modulates regulatory T cells and suppresses T-cell responses in HCV-associated lymphoma. Immunol Cell Biol. 2011;89(4):535–9. doi: 10.1038/icb.2010.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Muenst S, Hoeller S, Dirnhofer S, Tzankov A. Increased programmed death-1+tumor-infiltrating lymphocytes in classical Hodgkin lymphoma substantiate reduced overall survival. Hum Pathol. 2009;40(12):1715–22. doi: 10.1016/j.humpath.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 147.Kiyasu J, Miyoshi H, Hirata A, Arakawa F, Ichikawa A, Niino D, Sugita Y, Yufu Y, Choi I, Abe Y, Uike N, Nagafuji K, Okamura T, Akashi K, Takayanagi R, Shiratsuchi M, Oshima K. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood. 2015 doi: 10.1182/blood-2015-02-629600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, Zhu L, Grosso JF, Kim SY, Timmerman JM, Shipp MA, Armand P. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–9. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, Kaminski MS, Holland HK, Winter JN, Mason JR, Fay JW, Rizzieri DA, Hosing CM, Ball ED, Uberti JP, Lazarus HM, Mapara MY, Gregory SA, Timmerman JM, Andorsky D, Or R, Waller EK, Rotem-Yehudar R, Gordon LI. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31(33):4199–206. doi: 10.1200/JCO.2012.48.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, Millenson MM, Cohen AD, Schuster SJ, Lebovic D, Dhodapkar M, Avigan D, Chapuy B, Ligon AH, Freeman GJ, Rodig SJ, Cattry D, Zhu L, Grosso JF, Bradley Garelik MB, Shipp MA, Borrello I, Timmerman J. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J Clin Oncol. 2016;34(23):2698–704. doi: 10.1200/JCO.2015.65.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Armand P, Shipp MA, Ribrag V, Michot JM, Zinzani PL, Kuruvilla J, Snyder ES, Ricart AD, Balakumaran A, Rose S, Moskowitz CH. Programmed death-1 blockade with pembrolizumab in patients with classical hodgkin lymphoma after brentuximab vedotin failure. J Clin Oncol. 2016 doi: 10.1200/JCO.2016.67.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Guibert N, Mazieres J. Nivolumab for treating non-small cell lung cancer. Expert Opin Biol Ther. 2015;15(12):1789–97. doi: 10.1517/14712598.2015.1114097. [DOI] [PubMed] [Google Scholar]
- 154.Ning YM, Suzman D, Maher VE, Zhang L, Tang S, Ricks T, Palmby T, Fu W, Liu Q, Goldberg KB, Kim G, Pazdur R. FDA approval summary: atezolizumab for the treatment of patients with progressive advanced urothelial carcinoma after platinum-containing chemotherapy. Oncol. 2017;22(6):743–749. doi: 10.1634/theoncologist.2017-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kotsakis A, Georgoulias V. Avelumab, an anti-PD-L1 monoclonal antibody, shows activity in various tumour types. Lancet Oncol. 2017;18(5):556–557. doi: 10.1016/S1470-2045(17)30227-9. [DOI] [PubMed] [Google Scholar]