

Abstract
We propose an unsupervised multi-omics integration pipeline, using deep-learning autoencoder algorithm, to predict the survival subtypes in bladder cancer (BC). We used TCGA dataset comprising mRNA, miRNA and methylation to infer two survival subtypes. We then constructed a supervised classification model to predict the survival subgroups of any new individual sample. Our training data gave two subgroups with significant survival differences (p-value=8e-4), where high-risk survival subgroup was enriched with KRT6/14 overexpression and PI3K-Akt pathways. We tested the robustness of model by randomly splitting the main dataset into multiple training and test folds, which gave overall significant p-values. Then, we successfully inferred the subtypes for a subset of samples kept as test dataset (p-value=0.03). We further applied our pipeline to predict the survival subgroups from another validation dataset with miRNA data (p-value=0.02). Conclusively, present pipeline is an effective approach to infer the survival subtype of a new sample, exemplified by BC.
Introduction
Multi-omics data integration approach is being preferred (as compared to be single omics) for addressing important biological questions1. While multi-omics methods have extensively been used for the subtype identification, these are naïve in the field of prognosis. State-of-the-art deep learning methods may be efficient in handling the multi-omics data for the development of efficient prognostic models. A plethora of bioinformatics studies used the deep learning algorithms to construct predictive models2. For instance, Cox-nnet is an artificial neural networks (ANN) based prognostic method that uses only single omics layer (transcriptomics)3. In contrast, DeepGene is a deep learning based method that classifies cancer types based on mutation profiles4. Deep learning algorithms present the advantage of handling high-dimensional data with numerous features and can exploit complex non-linear patterns, thanks to the activation functions. In particular, autoencoder is one of the DL algorithms which has gained popularity in recent times in biological domain. It has been used to find the unsupervised feature extraction in the breast cancer5.
Bladder cancer (BC) is the 9th most commonly diagnosed cancer worldwide6. In the US alone, ~80K new incidences and ~17K deaths are estimated in 2017. BC is also amongst the few highly sex-disparate cancers where males have 4-folds higher risk (both incidence and death) than women7. According to an estimate, bladder cancer will be responsible for 7% of cancer diagnoses and 4% of cancer deaths from all the cancers in USA7. Smoking is considered as the major risk factor associated with the bladder cancer6. Furthermore, this cancer is more common in aged people (>55 years). This disease is usually diagnosed at the later stage and the 5-year survival rate at advanced stage (IV) is around 15%8. Therefore, there is a pressing need for refining the diagnostic regimens to propose better risk-adapted therapies.
Two major subtypes are identified previously in bladder cancer-luminal and basal, where latter is more aggressive and characterized by TP53 and RB1 mutations. Further, two markers were found correlated with these two subtypes: GATA3 for luminal and KRT5/6 for basal9. Moreover, the study conducted by The Cancer Genome Atlas (TCGA), has identified four subtypes of urothelial bladder carcinoma based on the RNA-seq data10. In literature, a considerable work has been done to identify the survival related markers in BC from different individual omics layers. Studies show different genes, miRNA (miRs) and long non-coding RNAs (lncRNAs) for the survival prediction of bladder cancer11, 12. Though different studies identified several specific molecular subtypes of bladder cancer so far, but multi-omics integration along with utilization of survival information was not used while inferring the subtypes.
Here, we present a new extension of our original pipeline (namely DeepProg13), applied to the bladder cancer. DeepProg is a deep learning (autoencoder) based pipeline to infer the survival subgroups in unsupervised way followed by the prediction of survival subtype of the unknown sample. In the present work, we explored the use of autoencoder to transform multi-omics features followed by integration of survival information in order to identify subtypes linked with survival. To construct the model, we used the training dataset obtained from TCGA with three types of omics: mRNA, miRNA and methylation. For each omic, we constructed an individual autoencoder to produce new features linked to the survival. These features were then integrated together to infer the subtypes. We then developed a supervised classification model to identify the survival subtype of any new sample having common features with the training samples. This methodology has the advantages of being fast, robust and can be applied to various heterogeneous features potentially linked to survival. We anticipate that this pipeline will be a good starting point for data integration and survival-specific subtype identification.
Methods
Dataset
We obtained the three omics dataset i.e. mRNA, miRNA and methylation of TCGA BC from the Genomics Data Commons web portal (https://portal.gdc.cancer.gov/), as of June 2017. We used TCGA-Assembler, an open source pipeline for downloading, assembling and processing of 402 BC samples14. For mRNA sequencing data, we used normalized RSEM (RNA-Seq by Expectation Maximization) quantification values15 from the Illumina HiSeq platform. For miRNA sequencing quantification data, we procured the reads per million miRNA mapped (RPM) data from the Illumina HiSeq platform for each sample. For methylation (Infinium HumanMethylation450 BeadChip platform), the average methylation value of all the CpG sites was calculated for each gene within the 1500 bp ahead of the transcription start site (TSS). The corresponding survival data for these 402 samples were obtained from the FireBrowse (http://firebrowse.org/) website hosted by the Broad Institute.
For the validation dataset, we mined the Gene Expression Omnibus (GEO) database and found miRNA sequencing data for muscle-invasive bladder cancer for 62 samples along with survival information (GSE84525)16. We processed the raw read count data and converted those to the RPM, the same normalization as that of the TCGA miRNA dataset.
Model building
Unsupervised inference of survival subtypes
We normalized each omic layer independently using the feature ranking and the per-sample correlation normalizations to produce three input matrices. Then, for each input matrix, we trained a denoising autoencoder to produce a new set of features specific to each omic. Finally, we searched for individual features linked to survival, stacked these features into a single matrix and clustered the samples (Figure 1).
Figure 1.
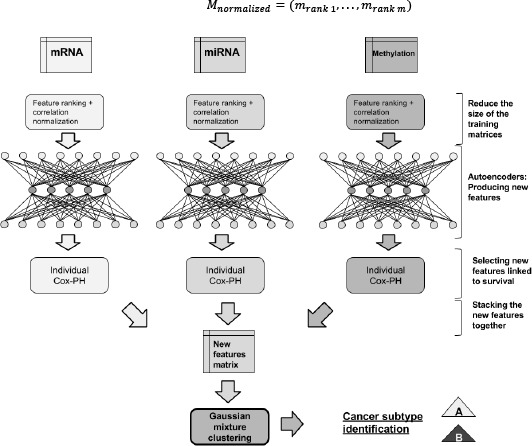
Unsupervised inference of the survival subtypes.
Normalization procedure
For each omic, we first normalized each sample independently using the rank normalization. For a given omic, we defined the input matrix M = (v1,…,vm) as the concatenation of m sample vectors v, having each n features. For a given sample vector v = (x1,…,xn), the function rank(xi) returns the rank of the feature xi in v (1 if x1 is the lowest value and n if it is the highest value). vrank is defined by:
Then, we normalized Mrank = (vrank 1,…, vrank m) by computing the Pearson correlation distance between each pair of samples:
Thus, Mcorr = {Mcoor(i,j) | i,j Em} is a square matrix of size m. Finally, for each sample vector of Mcorr = (m1,…, mm), we reapplied the rank normalization:
Autoencoder construction
For each input matrix, we trained a denoising autoencoder using one hidden layer. An autoencoder can be defined as a function f(v) = v’ where v is an input vector of size n and where size(v’) = size(v) = n. f aims to reconstruct v using a combination of nonlinear transformations of its features. In the case of an autoencoder with one hidden layer with h nodes and with a same activation function for both encoder and decoder f is defined by:
With W, and W’ represent the weight matrices with sizes h × m and m × h, and b and b’ are two biases vectors of sizes h and m, respectively. Finally, σ is an element-wise non-linear activation function such as the sigmoid or the relu functions. The training procedure of an autoencoder uses an optimization algorithm such as the Stochastic Gradient Descent to find iteratively the best W, W’, b and b’ that minimize a loss function loss(v, v ’), measuring the difference between v and v’. We trained denoising autoencoder by setting a specific percentage of the weight matrices coefficients randomly to 0 (dropout) at each training iteration. This technique is known to reduce overfitting. Once the autoencoder is trained, the transformation z of v is given by f(v) = σ(W.v+b). We used the python Keras framework to create and train autoencoders. For each autoencoder, we used h = 100 (hidden nodes). We used the logloss as loss function, the tanh as activation function and the adam optimizer to minimize the loss. Finally, we trained the autoencoders on 50 epochs and with 50% of dropout rate. We denote zOMIC as the transformed version of matrix
Identification of features linked to survival
We searched amongst the matrices produced by the autoencoder: zmRNA, zmiRNA and zMethylatlon, the features linked to survival. For each feature of these matrices, we built a univariate Cox-PH model to identify and retained those with log-rank pvalue < 0.01. We finally extracted these significant features and stacked them to form a new single matrix Z*. We used the functions of the R survival package to build the Cox-PH model and compute the p-values.
Identification of the Survival subtypes
We used the Gaussian mixture algorithm to infer clusters from the matrix Z*. We used the gaussian mixture library from the Python 2.7 scikit-learn package with 1000 iterations, 100 initiations and a diagonal covariance type (meaning that each cluster has its diagonal covariance matrix). The best number of clusters K was estimated by computing the Silhouette and the Calinski-Harabasz scores.
Supervised inference of the survival subtype for a new sample
For each omic layer, we used the normalized matrices and the labels inferred by the clustering procedure to build supervised classification models. We first selected from these matrices the individual top features to most correlated to the cluster labels to create a Support Vector Machine (SVM) model for each individual omic but also for the three-omics stacked together. To classify a new sample, we first normalized it using the rank normalization and then by computing the correlation distance with each sample of the training set. Finally, according to the omic type of the sample, we used the corresponding classifier to infer the survival subtype.
Classifiers construction
For each normalized matrix: we used the Kruskal-Wallis test to select the top 10 features as the most discriminative according to the cluster labels. We then used these top features to construct an SVM model for each omic type. We also stacked the 30 features together to construct a “multi-omics” SVM model. Each SVM model was constructed using a grid-search procedure with a 5-fold cross-validation to find the best hyperparameters. As hyperparameter choices, we used two types of kernel: linear or Radial Basis Function (RBF) kernels. We also used an array of values for the penalty parameter C from 0.1 to 1000. We used the SVC and the model selection libraries from the scikit-learn package to construct the SVM models. In addition to the survival subtypes, we used the classifiers to infer the probability of a sample belonging to the subgroup with the lowest median survival (Figure 2).
Figure 2.
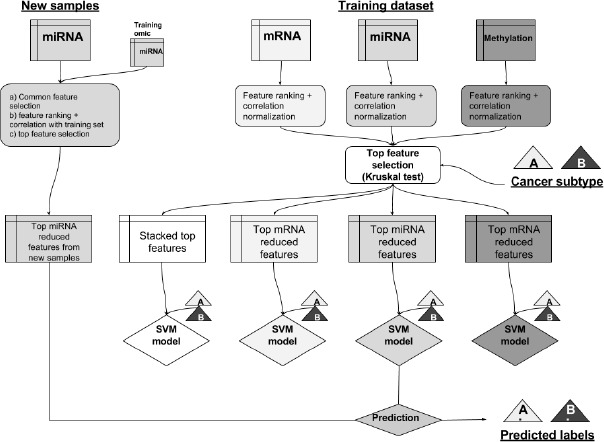
Supervised classification pipeline to infer the survival subtype, using the predicted labels, for a new sample
Normalization and classification of a new sample
A new sample can have features from one or multiple common omics with the training set. For each omic shared with the training samples, we selected the set of common features and applied the rank normalization on both, the new samples and the training set, using this subset of features. Then, we computed the Pearson correlation distance between the new sample and each sample of the training set. We used these values as new features which give a vector v. Finally, we applied again the rank normalization on v. Thus, the vector v and the matrices have the same features. We then selected from v the 10 top features identified during the classification construction procedure and classified v using the corresponding classifier (for example, if v was derived from miR features only we used the miR classifier).
Model performance assessment
We split our dataset into training and test datasets. We then inferred the survival subtypes from the training set and built a classification model to infer the labels from the test set. We used the labels and label probabilities inferred for the test, training and the full datasets to fit Cox-PH models and used the log-rank p-values as performance criteria. Moreover, we reported the geometric mean of the p-values obtained for both the training and the test sets.
Validation
Survival
We used the survival package17 in R to analyze the survival difference between two subtypes in the main TCGA dataset and validation dataset. We used the survdiff function to compute the survival differences between two subtypes and plot the Kaplan-Meier curves with the log-rank P-values.
Functional analysis post-hoc analysis
After getting the cluster labels for the main TCGA cohort for the two subtypes, we performed the post-hoc analysis to find out the differentially expressed features in the individual omic layers. We used the edgeR R package18 to perform the differential gene expression and filtered the upregulated and downregulated genes and miRs with a filter of log fold change >2 and false discovery rate (FDR) <0.05. For the differentially expressed genes, we mined the enriched pathways in the two subtypes using the enrichr tool19 and selected the final pathways satisfying the adjusted p-value <0.05. For the differentially methylated genes, we first converted the beta values to the M-values using lumi R package20 and then used limma R package21 for the differential methylation gene identification using logFC >1 and adjusted p-value <0.05.
Results
The inferred survival subtypes present significant and robust survival differences
Using the silhouette score (top silhouette score: 0.39 for K=2) and the Calinski-Harabasz criterion, our model identified the optimal number of 2 subtypes amongst the BC population. Finally, on 20 successive and random splits of the TCGA 3-omics dataset in training (66%) and test (33%) sets, we obtained an overall p-value of 0.0008 and 0.04, using the cluster labels as predictor, for training and test set respectively (Table 1).
Table 1.
Log-rank p-value of Cox-PH models for the training, test and validation datasets. The Cox-PH models were constructed using either the cluster label or the probability to belong the cluster with the lowest survival as single variable.
| Dataset | p-value label | p-value label probability |
|---|---|---|
| Training folds (20 iteration) | 8e-4 (geo. mean) | 7e-4 (geo. mean) |
| Test folds (20 iteration) | 0.04 (geo. mean) | 0.03 (geo. mean) |
| Final training dataset | 0.002 | 0.001 |
| Final test dataset | 0.03 | 0.02 |
| Validation dataset | 0.02 | 0.02 |
These results proved that the model used is robust towards the random process inherent to the autoencoder construction and the choice of the training sample. Moreover, the two subtypes in the full TCGA cohort presented a K-M estimates with clear segregation between the curves with log-rank p-value 1.8e-05 (Figure 3A). Validation cohort also showed the significant differences with p-value=0.014 (Figure 3B).
Figure 3.
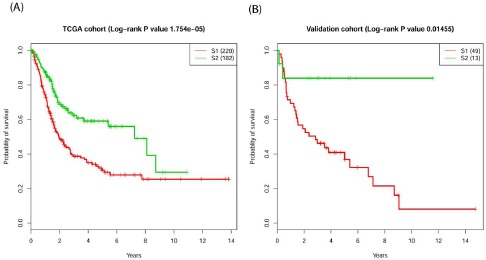
Survival profiles of the two survival subtypes for the (A) TCGA full cohort and (B) the validation cohort.
Specific molecular signatures are linked to bladder cancer survival subtypes
Differential gene expression analysis revealed 801 upregulated and 228 downregulated genes in the S1 cluster as compared to S2 subtype (Figure 4A). We found KRT6 and KRT14 upregulated in the aggressive subtype (S1), which are in corroboration with the previous finding where these genes were reported to be the markers of basal (aggressive) subtype of bladder cancer9. Among the top upregulated genes in S1 subtype were DLK1, TRH, DEFB103B, MYBPH and KRT38, in contrast among top downregulated genes were AMELX, MUC17, CYP1A2, SI and GC.
Figure 4.
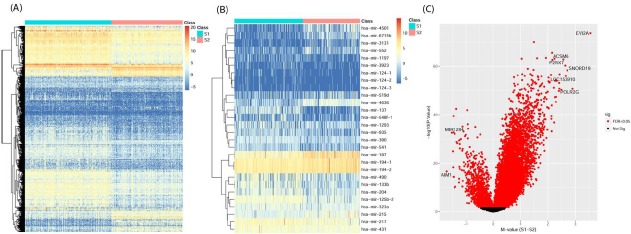
Molecular signatures of the two survival subtypes. (A) mRNA differential expression (B) miRNA differential expression and (C) Volcano plot of differential methylated genes.
18 upregulated and 10 downregulated miRs were identified in the differentially expressed miRs analysis in S1 as compared to S2 subtype (Figure 4B). mir-194 was found downregulated in S1 aggressive subtype down in bladder cancer and miR-194 was reported to be acting as a tumor repressor and found downregulated in BC22. Upregulated mir-133b in S1 (aggressive) subtype was reported earlier to be associated with high risk and was also reported as a prognostic marker in progression free survival23.
For genes with differential methylation status, 918 hyper- and 46 hypo-methylated genes were obtained (Figure 4C). Out of the hypermethylated genes, 6 were having mean difference greater than 2.5 and 2 hypomethylated genes were with mean difference >1.5. KRT13 was hypermethylated in S1 and it has been reported to be associated with high-grade disease in non-invasive bladder cancer24. FHIT and LAMC2 which were found hypermethylated in S1 are also in corroboration with earlier work, where the methylated region in these two gene was reported to be associated with poor survival in patients 25.
Enriched pathway analysis revealed 31 activated pathways in S1 and 15 depleted pathways in S1 as compared to S2. Among the top activated pathways, cytokine-cytokine receptor interaction pathway (adj. p-value=1.97e-13), PI3K-Akt signaling pathway (adj. p-value=3.16e-06), and ECM-receptor interaction pathway (adj. p-value=5.13e-06) were enriched in S1 subtype (Table 2). PI3K-Akt signaling pathway was targeted earlier to inhibit the BC cell growth26. Cytokine-cytokine receptor interaction has been reported to be associated with BC progression27. Surprisingly, Staphylococcus aureus infection pathway has popped up as a top significant pathway. In the past this bacterium has shown the association with the schistosome infection which is known to be one of the risk factors in bladder cancer patients6. The association of parasites, bladder cancer and bacterium, may explain the significance of this pathway. In the enriched pathways for downregulated genes in S1, metabolism and drug and glucose metabolism apart from PPAR signaling pathways were present (Table 2).
Table 2.
Pathway enrichment analysis for the two subtypes.
| Pathway upregulated in SI | Adjusted lvalue |
|---|---|
| Cytokine-cytokine receptor interaction | 1.97E-13 |
| Staphylococcus aureus infection | 3.91E-07 |
| PI3K-Akt signaling pathway | 3.16E-06 |
| Amoebiasis | 3.77E-06 |
| Complement and coagulation cascades | 3.77E-06 |
| ECM-receptor interaction | 5.13E-06 |
| Hematopoietic cell lineage | 6.61E-05 |
| Focal adhesion | 1.94E-04 |
| Tuberculosis | 2.47E-04 |
| Chemokine signaling pathway | 4.32E-04 |
| Rheumatoid arthritis | 3.03E-04 |
| Jak-STAT signaling pathway | 1.Q8E-03 |
| Cell adhesion molecules (CAMs) | 9.81E-04 |
| Pertussis | 7.78E-04 |
| Osteoclast differentiation | 3.84E-03 |
| Neuroactive ligand-receptor interaction | 4.43E-03 |
| Transcriptional misregulation in cancer | 4.43E-03 |
| Protein digestion and absorption | 3.77E-03 |
| Hypertrophic cardiomyopathy (HCM) | 5.41E-03 |
| Phagosome | 5.41E-03 |
| Leishmaniasis | 7.02E-03 |
| D i lated ca idiom yopat hy | 9.32E-03 |
| Malaria | 7.08E-03 |
| Toll-like receptor signaling pathway | 2.98E-02 |
| Systemic lupus erythematosus | 2.70E-02 |
| Calcium signaling pathway | 4.48E-02 |
| Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 2.53E-02 |
| Inflammatory bowel disease (IBD) | 3.56E-02 |
| Prion diseases | 2.35E-02 |
| Graft-versus-host disease | 4.25 E-02 |
| Nicotinate and nicotinamide metabolism | 4.25E-02 |
| Metabolism of xenobiotics by cytochrome P450 | 2.37E-09 |
| Chemical carcinogenesis | 1.15E-09 |
| Retinol metabolism | 7.61E-09 |
| Drug metabolism - cytochrome P450 | 1.12E-08 |
| Steroid hormone biosynthesis | 4.81E-07 |
| PPAR signaling pathway | 2.97E-04 |
| Arachidonic acid metabolism | 1.40E-03 |
| Bile secretion | 2.62E-03 |
| Starch and sucrose metabolism | 5.26E-03 |
| Ascorbateand aldarate metabolism | 3.45 E-03 |
| Metabolic pathways | 1.74E-02 |
| Linoleic acid metabolism | 4.12E-03 |
| Pentose and glucuronate inteiconversions | 8.01E-03 |
| Porphyrin and chlorophyll metabolism | 1.33E-02 |
| Drug metabolism - other enzymes | 1.74E-02 |
mRNA transcriptomic features are the most informative features for survival prediction
We computed the importance of each omic layer to predict the survival subtype, by estimating the average number of new features linked to survival for each omic layer. RNA had the most important contribution with the average 14 features (out of 100) linked to survival followed by miRNA (6 features) and Methylation (2 features). Furthermore, this trend was reflected from the average p-value obtained with models using individual omic layer i.e. mRNA (0.001), miR (0.002) and methylation (0.004) with 20 iterations.
Discussion
This is the cardinal study to use deep learning to integrate multi-omics data in order to find the survival risk stratification for bladder cancer samples. This new version of our DeepProg pipeline provides a flexible normalization procedure and a modularized architecture that can incorporate any new omic layer. Using samples from TCGA with 3-omics layers as the training set, we inferred two subtypes with significant and robust survival difference. Our pipeline achieved a significant stratification of the validation dataset, which contains only a single omic layer. Also, any new sample with a subset of features common with the training samples can be processed by DeepProg. The only preprocessing required is the distances computation between the new sample and the training samples. Thus, this strategy is more practical in the real-life situation where multi-omics for the external dataset are difficult to find. Finally, the modularity of the pipeline, i.e. an autoencoder is built for each omic layer, can be used to integrate any additional omic layer or survival related features, such as clinical image data28.
Several individual omic layer features are in agreement with the previously found markers including KRT14 overexpression and enriched PI3K-Akt pathway in aggressive subtype. Thus, we postulate that our strategy can be useful in the future to create analytical pipelines with different degree of clinical interests. Furthermore, an improvement of the overall performance is attributable to the data quality (tumor purity) on which models are made in addition to the confounders viz. tumor heterogeneity, risk factors. It has been reported in a recent study that bladder cancer samples from TCGA belong to one of the least pure cancer type29, which makes survival analysis a challenging task. We anticipate that big cohort studies in the future with good quality of samples will be instrumental in improving the performance of our pipeline.
Acknowledgments
This research was supported by grants K01ES025434 awarded by NIEHS through funds provided by the trans-NIH Big Data to Knowledge (BD2K) initiative (www.bd2k.nih.gov), P20 COBRE GM103457 awarded by NIH/NIGMS, R01 LM012373 awarded by NLM, R01 HD084633 awarded by NICHD and Hawaii Community Foundation Medical Research Grant 14ADVC-64566 to L. X. Garmire.
References
- 1.Huang S, Chaudhary K, Garmire LX. More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front Genet. 2017;8:84. doi: 10.3389/fgene.2017.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform. 2016 doi: 10.1093/bib/bbw068. [DOI] [PubMed] [Google Scholar]
- 3.Ching T, Zhu X, Garmire L. Cox-nnet: an artificial neural network Cox regression for prognosis prediction. bioRxiv. 2016:093021. doi: 10.1371/journal.pcbi.1006076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan Y, et al. DeepGene: an advanced cancer type classifier based on deep learning and somatic point mutations. BMC Bioinformatics. 2016;17(Suppl 17):476. doi: 10.1186/s12859-016-1334-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan J, Ung M, Cheng C, Greene CS. Unsupervised feature construction and knowledge extraction from genome-wide assays of breast cancer with denoising autoencoders. Pac Symp Biocomput. 2015:132–43. [PMC free article] [PubMed] [Google Scholar]
- 6.Antoni S, et al. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur Urol. 2017;71(1):96–108. doi: 10.1016/j.eururo.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 7.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 8.Society AC. Bladder Cancer. 2017.
- 9.Dadhania V, et al. Meta-Analysis of the Luminal and Basal Subtypes of Bladder Cancer and the Identification of Signature Immunohistochemical Markers for Clinical Use. EBioMedicine. 2016;12:105–17. doi: 10.1016/j.ebiom.2016.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–22. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bao Z, Zhang W, Dong D. A potential prognostic lncRNA signature for predicting survival in patients with bladder urothelial carcinoma. Oncotarget. 2017;8(6):10485–97. doi: 10.18632/oncotarget.14441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Q, Diao R, Feng G, Mu X, Li A. Risk score based on three mRNA expression predicts the survival of bladder cancer. Oncotarget. 2017 doi: 10.18632/oncotarget.18642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaudhary K, Poirion OB, Lu L, Garmire L. Deep Learning based multi-omics integration robustly predicts survival in liver cancer. bioRxiv. 2017:114892. doi: 10.1158/1078-0432.CCR-17-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu Y, Qiu P, Ji Y. TCGA-assembler: open-source software for retrieving and processing TCGA data. Nat Methods. 2014;11(6):599–600. doi: 10.1038/nmeth.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochoa AE, et al. Specific micro-RNA expression patterns distinguish the basal and luminal subtypes of muscle-invasive bladder cancer. Oncotarget. 2016;7(49):80164–74. doi: 10.18632/oncotarget.13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.T T. A Package for Survival Analysis in S. version 2.38. 2015.
- 18.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–7. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24(13):1547–8. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 21.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang M, Zhuang Q, Cui L. MiR-194 inhibits cell proliferation and invasion via repression of RAP2B in bladder cancer. Biomed Pharmacother. 2016;80:268–75. doi: 10.1016/j.biopha.2016.03.026. [DOI] [PubMed] [Google Scholar]
- 23.Dyrskjot L, et al. Genomic profiling of microRNAs in bladder cancer: miR-129 is associated with poor outcome and promotes cell death in vitro. Cancer Res. 2009;69(11):4851–60. doi: 10.1158/0008-5472.CAN-08-4043. [DOI] [PubMed] [Google Scholar]
- 24.Marsit CJ, et al. Identification of methylated genes associated with aggressive bladder cancer. PLoS One. 2010;5(8):e12334. doi: 10.1371/journal.pone.0012334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kandimalla R, van Tilborg AA, Zwarthoff EC. DNA methylation-based biomarkers in bladder cancer. Nat Rev Urol. 2013;10(6):327–35. doi: 10.1038/nrurol.2013.89. [DOI] [PubMed] [Google Scholar]
- 26.Yang Y, Guo JX, Shao ZQ, Gao JP. Matrine inhibits bladder cancer cell growth and invasion in vitro through PI3K/AKT signaling pathway: An experimental study. Asian Pac J Trop Med. 2017;10(5):515–19. doi: 10.1016/j.apjtm.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 27.Fang ZQ, et al. Gene expression profile and enrichment pathways in different stages of bladder cancer. Genet Mol Res. 2013;12(2):1479–89. doi: 10.4238/2013.May.6.1. [DOI] [PubMed] [Google Scholar]
- 28.Clark K, et al. The Cancer Imaging Archive (TCIA): maintaining and operating a public information repository. J Digit Imaging. 2013;26(6):1045–57. doi: 10.1007/s10278-013-9622-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun 2015. 6:8971. doi: 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]