

Abstract
Protein kinases generate nearly a thousand different protein products and regulate the majority of cellular pathways and signal transduction. It is therefore not surprising that the deregulation of kinases has been implicated in many disease states. In fact, kinase inhibitors are the largest class of new cancer therapies. Understanding polypharmacology within the full kinome, how drugs interact with many different kinases, would allow for the development of safer and more efficacious cancer therapies. A full understanding of these interactions is not experimentally feasible making highly accurate computational predictions extremely useful and important. This work aims at making a machine learning model useful for investigating the full kinome. We evaluate many feature sets for our model and get better performance over molecular docking with all of them. We demonstrate that you can achieve a nearly 60% increase in success rate at identifying binding compounds using our model over molecular docking scores.
1. Introduction
Protein kinases represent a large number of proteins in our body with essential functions. Because of this, any disruption in normal kinase activity may lead to a disease state. Additionally, due to high sequence and structural identity, selectively inhibiting a kinase is difficult. This means a drug intended to target one kinase will likely also target multiple other kinases. If these other kinases are normally expressed and not implicated in the given disease it could lead to toxic off-target effects. Pharmaceutical companies test drug interactions with many different kinases in the beginning of the drug discovery process. They do this as early as possible before lots of time and money has gone into drug development [1]. Drugs failing late in the pharmaceutical pipeline can be very costly, driving up the cost of drugs that do make it to market when they have to recuperate the cost for the failed drugs. It can also be fatal when they fail during clinical trial, because animal testing does not always give a good indication of serious side-effects [2]. Therefore, our interest in accurate computational models to study kinases is to develop better and safer cancer therapies, using efficient computational predictions that reduce the time and cost of bringing a drug to market.
We propose to use machine learning techniques to increase the accuracy of computational drug discovery in order to make better predictions as early as possible. We have seen in our own work that a small number of calculated features similar to ones used in this study can identify active compounds for a given protein with greater than 99% accuracy. These same drug features have been used in machine learning models in combination with docking scores to rescore interactions with one candidate drug to multiple proteins [3]. The individual components of a molecular docking scoring function can be used as features in a machine learning model to greatly improve the accuracy of identifying active compounds in models specific for one protein [4]. From a different perspective, protein features have been used in machine learning models to predict the druggability of a protein [5]. The goal of this work is to combine all these components in one model that would vastly improve the accuracy of predicting the effects of new proteins and classes of drugs. The specific goal of this paper is to present machine learning models that can accurately predict the drug interaction for a class of functionally related proteins (kinases), an important class of proteins for drug discovery as already stated.
2. Methods
Our goal is to estimate the probability that a kinase-drug pair is active (binding) or decoy (not binding), a binary classification task. We propose to use a random forest classification method to address this task. A key focus of our effort is in investigating which features are most informative for this task. To support this effort, we created a large dataset of kinase-drug pairs and computed a wide variety of different features.
The data used in this study comes from the kinase subset of the Directory of Useful Decoys - enhanced (DUD-e) [6]. It is important to note that the ratio of active to decoy compounds in DUD-e is approximately 1:50.
2.1. Data Collection
Protein Descriptors The human canonical sequences were collected for each protein from UniProt [7]. The sequences were submitted to three different webservers to collect features: ExPasy [8], Porter, PaleAle 4.0 [9], and PROFEAT Protein Feature Server[10]. These three tools were used to ensure we collect all features used in the DrugMiner [5] project. Additional features that these tools calculate are also collected. ExPasy calculates many features, such as the length, weight, half-life, isoelectric point, extinction coefficient assuming all pairs of Cys residues form cysteines, extinction coefficient assuming all Cys residues are reduced, instability index, aliphatic index, Grand average of hydropathicity (GRAVY), and the frequency of single amino acids, amino acid types (tiny, small, aliphatic, nonpolar, aromatic, polar, charged, basic, acidic, hydrophobic, hydrophilic, positive, and negative), and atom types. Porter calculates the predicted secondary structure based on the amino acid sequence and classifies each amino acid as helical, beta strand, or coil. PaleAle calculates the predicted relative solvent accessibility based on the amino acid sequence and classifies each amino acid as completely buried (0-4% exposed), partly buried (4-25% exposed), partly exposed (25-50% exposed), or completely exposed (50+% exposed). PROFEAT calculates features using many different tools including features based on the dipeptide composition of the protein sequence.
Pocket Descriptors Inner point features are collected using PRANK [11], software used to predict and rank binding sites. PRANK first calculates feature vectors for heavy solvent exposed atoms (AFVs), including residue and atomic level features. Then feature vectors are calculated for inner pocket points (IFVs) by summing all AFVs within an 8 Å radius using a distance weight function and then appending features specific to the inner pocket point, such as the number of H-bond donors and acceptors in its local neighborhood. The IFV from the inner pocket point with the closest distance to the center of the docking box calculated for molecular docking is used.
Drug Descriptors Drug features are calculated using the Dragon Software [12]. Dragon can calculate over 5 thousand molecular descriptors, including the simplest atom types, functional groups and fragment counts, topological and geometrical descriptors, and three-dimensional descriptors. It also includes several property estimations like logP and drug-like alerts like Lipinski’s alert. In this study 3-dimensional descriptors are left out because the input structures for Dragon are the predocking structures and not those predicted by molecular docking.
Binding Descriptors The receptor files from DUD-e that were optimized for docking are used in this study. The dimension and center of the docking boxes are calculated using a VMD [13] tcl script to draw a box around the co-crystallized ligand included in the DUD-e dataset and it is extended by 5 Å in each direction. Compounds are prepared for docking using modified ADT scripts and a wrapper script for automation. Docking was performed using VinaMPI [14], which allows the distribution of a large number of Autodock Vina [15] docking jobs on MPI-enabled high-performance computers. The results of the docking jobs were submitted to Autodock Vina using the “–score-only” option to collect the individual terms calculated in the scoring function. This includes terms for gauss1, gauss2, repulsion, hydrophobic, and hydrogen interactions. The values for the first model and averages of each term for all models are kept.
2.2. Feature Selection & Classification
The only form of preprocessing we performed was eliminating features with too many missing values. Specifically, we eliminated 21 features computed by the Dragon software package that had more than 5% missing values. The eliminated features had between 23.1%-99.9% missingness. There were 167 additional Dragon features that had less than 5% missing values and we imputed these values by using the column average. The final full dataset contains 5,410 features and 361,786 examples. After initial preprocessing, we train a classifier for various subsets of features and perform feature selection.
Random forests [16] are known to produce robust classifiers that are less prone to overfitting than ordinary decision trees. For a brief review, random forests contain a number of decision trees, a parameter that is chosen prior to training, each of which take random samples from the training data and random subsets of features to grow decision trees that are often limited in depth to create “weak” learners that underfit the testing data. By combining the “weak” learners that specialize in different regions of the feature space, random forests are able to learn complex functions that are robust to label imbalance or overfitting, two properties that are of great importance in our classification problem. The decision trees that make up a random forest compute orthogonal splits in feature space that attempt to maximize separation between the positive and negative classes minimize what is known as the GINI Impurity. From training a random forest, one can compute feature importances by measuring the average after-split impurity of the feature across all trees in the forest.
The feature selection method we use is similar to those used by Menze et al. [17] and Anaissi et al. [18] who also apply iterative feature selection method for classification to learn important sets of features. In our feature selection method, we input an initial set of features F for which we use to train a random forest classifier. The input data, after preprocessing, is partitioned into training and testing sets using an 80/20 stratified split, with the test set containing the same proportion of positives to negatives as the training set. We fix several parameters of the random forest classifier by using an out-of-bag score to protect against overfitting, balanced class weighting when computing impurities for the forests which inversely adjust the weights according to class frequency to get measures of the F1-score that better reflect the random forest’s performance in correctly predicting the active class, bootstrap sampling which allows training examples to be used in the building of more than one tree, and the GINI impurity criterion for which to compute the split quality when building the tree. We then perform model selection by sampling from distributions of hyperparameters, shown to be as effective as exhaustive parameter grid searching by Louppe et al. [19], for the random forest including the number of trees to include (30-100 trees) in the forest, the minimum number of samples required to create a leaf node (1-100 samples), and the maximum number of features f ∈ F to sample from F for each decision tree Given the distributions over hyperparameter values, we sample 100 possible settings of hyperparameters each iteration, evaluating the performance of each candidate model using k-fold cross validation, with k = 3. We define the best model trained on the feature set to be the one which maximizes the weighted F1-score on the testing data. We then compute the mean importance, more specifically the mean decrease in impurity , for the set of features F, and retain all features that have above mean importance. This strategy is employed in order to remove features with near 0 importance that contribute negligible information to the classification model and do not have a significant affect on performance. After computing the set of features to keep, F becomes the set of features identified as relevant, reducing the dimensionality of the input data. The iteration process continues until either a maximum number of iterations have completed or if there are no remaining features.
Algorithm 1 Feature Selection
1: procedure SELECTION FOREST(F
2: while F ≠ 0 and step < max-steps do
3: X,y = load_data(features = features_to_keep)
4: Xtrain, Xtest, ytrain, ytest = train_test_split(X,y)
5: best_forest = RandomizedGridSearch(RandomForest, Xtrain, ytrain). best_estimator
6: feature_importances = best_forest.importances
7:
8: F = features_to_keep
We use principal component analysis (PCA) to visualize the various feature representations. For our purpose of visualization, we reduce the dimensionality to 2 principal components.
2.3. Test set
We used a fixed seed when creating the test set in order to make sure all models are tested with the same data. The test and training sets are stratified by kinase, keeping the same proportion of active and decoy compounds for each.
Table 1 gives the representation of each kinase in the test set. The whole dataset ‘total’ column gives the total number of active and decoy compounds for the given kinase in the whole dataset. The whole dataset ‘ratio 0:1’ column gives the ratio of negative to positive class for the entire given kinase’s dataset. The remaining columns are particular to the test set. The ‘0’ column gives the number of decoys (negative class) in the test set. The ‘1’ column gives the number of actives (positive class) in the test set. The ‘percent 0’ column gives the percentage of the given kinase’s dataset that is in the negative class test set. The ‘percent 1’ column gives the percentage of the given kinase’s dataset that is in the positive class test set. The ‘total %’ column gives the percentage of the given kinase’s dataset that is in the test set. The ‘ratio 0:1’ gives the ratio of negative to positive test cases for the given kinase in the test set.
Table 1:
Representation of each kinase in the test set
| whole dataset | test dataset | |||||||
|---|---|---|---|---|---|---|---|---|
| kinase | total | ratio 0:1 | 0 | 1 | percent 0 | percent 1 | total % | ratio 0:1 |
| abl1 | 11,180 | 37 | 2,105 | 60 | 0.188 | 0.005 | 0.194 | 35 |
| akt1 | 16,999 | 39 | 3,298 | 73 | 0.194 | 0.004 | 0.198 | 45 |
| akt2 | 7,142 | 37 | 1,462 | 34 | 0.205 | 0.005 | 0.209 | 43 |
| braf | 10,349 | 40 | 2,036 | 46 | 0.197 | 0.004 | 0.201 | 44 |
| cdk2 | 29,126 | 35 | 5,604 | 158 | 0.192 | 0.005 | 0.198 | 35 |
| csf1r | 12,720 | 43 | 2,563 | 54 | 0.201 | 0.004 | 0.206 | 47 |
| egfr | 36,274 | 43 | 6,936 | 158 | 0.191 | 0.004 | 0.196 | 44 |
| fak1 | 5,516 | 47 | 1,085 | 14 | 0.197 | 0.003 | 0.199 | 78 |
| fgfr1 | 736 | 2 | 183 | 116 | 0.249 | 0.158 | 0.406 | 2 |
| igf1r | 9,633 | 42 | 1,958 | 42 | 0.203 | 0.004 | 0.208 | 47 |
| jak2 | 6,743 | 43 | 1,343 | 32 | 0.199 | 0.005 | 0.204 | 42 |
| kit | 10,861 | 42 | 2,144 | 52 | 0.197 | 0.005 | 0.202 | 41 |
| kpcb | 9,092 | 36 | 1,732 | 42 | 0.190 | 0.005 | 0.195 | 41 |
| lck | 28,539 | 41 | 5,569 | 136 | 0.195 | 0.005 | 0.200 | 41 |
| mapk2 | 6,450 | 30 | 1,240 | 40 | 0.192 | 0.006 | 0.198 | 31 |
| met | 11,677 | 47 | 2,278 | 47 | 0.195 | 0.004 | 0.199 | 48 |
| mk01 | 4,767 | 33 | 889 | 31 | 0.186 | 0.007 | 0.193 | 29 |
| mk10 | 6,900 | 36 | 1,358 | 50 | 0.197 | 0.007 | 0.204 | 27 |
| mk14 | 37,347 | 40 | 7,265 | 184 | 0.195 | 0.005 | 0.199 | 39 |
| mp2k1 | 8,483 | 34 | 1,693 | 41 | 0.200 | 0.005 | 0.204 | 41 |
| plk1 | 7,034 | 44 | 1,402 | 32 | 0.199 | 0.005 | 0.204 | 44 |
| rock1 | 6,580 | 31 | 1,306 | 41 | 0.198 | 0.006 | 0.205 | 32 |
| src | 35,790 | 42 | 6,980 | 167 | 0.195 | 0.005 | 0.200 | 42 |
| tgfr1 | 8,958 | 31 | 1,704 | 63 | 0.190 | 0.007 | 0.197 | 27 |
| vgfr2 | 25,900 | 41 | 5,119 | 123 | 0.198 | 0.005 | 0.202 | 42 |
| wee1 | 6,371 | 46 | 1,245 | 25 | 0.195 | 0.004 | 0.199 | 50 |
2.4. Evaluation
In this study, we compare the performance of machine learning models using different feature sets and also compare the performance to the computed docking score. Docking scores are typically used for ranking compounds from most likely to least likely to bind and there is no standard that defines an exact docking score that determines a binding prediction. In order to compare binding predictions from the docking score alone to the machine learning models, the maximum Youden’s index (or J value) is calculated for each model. The best J value is calculated from the docking score receiver operator characteristic (ROC) curve and used as a cut-off to define true positive (TP), true negative (TN), false positive (FP), and false negative (FN) values for the docking results. The different feature sets are described below and we compare 6 models using the feature sets given in Table 2. All the metric presented in the Results are defined in Table 3.
Feature Set 1 (FS1): This set is selected using the entire dataset and using the active or decoy binary labels. This is to collect the most important features for making the classification in which we are interested (i.e. active vs decoy).
Feature Set 2 (FS2): This set is selected using only protein and pocket features and using the kinase as a label. We do this to ensure we have protein features to test whether or not they help identify which kinase compounds bind to and not just identify kinase inhibitors in general.
Feature Set 3 (FS3): This set is selected using the drug features with the kinase as a label. This is also used to help with kinase selectivity.
Feature Set 4 (FS4): This set contains all docking features, which includes terms for gauss1, gauss2, repulsion, hydrophobic, and hydrogen interactions for the first docked model produced using molecular docking and an average over all models (the default value of 9 models was kept when running Vina). There is also a feature for the final docking score.
Table 2:
Evaluation Models
| Model | Feature Set | Model | Feature Set | Model | Feature Set |
|---|---|---|---|---|---|
| 1 | FS1 | 3 | FS1 + FS3 | 5 | FS1 + FS2 + FS3 + FS4 |
| 2 | FS4 | 4 | FS1 + FS3 + FS4 | 6 | all features |
Table 3:
Metrics used in this study
| Name | Definition | Formula |
|---|---|---|
| Youden’s index | Performance of dichotomous test. The value 1 indicates a perfect test and -1 indicates a useless test. | |
| F1 | Harmonic mean of precision and recall | |
| Precision | Positive predictive value | |
| Recall | True positive rate |
3. Results
3.1. Youden’s Index for Docking Scores
The maximum Youden’s index (or J value) is calculated and used to define TP, FP, TN, and FN values using docking scores. The best J values and docking score cut-off for each kinase and on the dataset overall all are given in Table 4.
Table 4:
Youden’s Index.
| Kinase | Youdens Index | Best docking score | Kinase | Youdens Index | Best docking score |
|---|---|---|---|---|---|
| abl1 | 0.35 | -9.1 | lck | 0.29 | -8.9 |
| akt1 | 0.02 | -8 | mapk2 | 0.45 | -8 |
| akt2 | 0.22 | -8.5 | met | 0.51 | -8.9 |
| braf | 0.53 | -9.6 | mk01 | 0.6 | -9.1 |
| cdk2 | 0.33 | -8.2 | mk10 | 0.4 | -8.7 |
| csf1r | 0.23 | -8.9 | mk14 | 0.28 | -8.5 |
| egfr | 0.15 | -8.7 | mp2k1 | 0.13 | -7.8 |
| fak1 | 0.52 | -8.4 | plk1 | 0.2 | -8.6 |
| fgfr1 | 0.01 | -8 | rock1 | 0.4 | -7.7 |
| igf1r | 0.46 | -8.4 | src | 0.17 | -8.1 |
| jak2 | 0.37 | -9.3 | tgfr1 | 0.56 | -9.6 |
| kit | 0.24 | -8.5 | vgfr2 | 0.36 | -9 |
| kpcb | 0.33 | -8.5 | wee1 | 0.76 | -10 |
| Overall | 0.23 | -8.6 | |||
3.2. PCA of Feature Sets
We performed a PCA of each Feature Set (FS) described in the Methods section. Figures 1 and 2 plot the first two components for FS1-FS4. FS1 contains 776 features all which are drug features. FS2 contains three protein features that are most important in determining the kinase. These are [G3.1.1.1.19], [G4.1.23.3], [G4.3.17.2] and come from the PROFEAT Webserver [10]. These correspond to an autocorrelation descriptors based on the distribution of amino acid types along the protein sequence, a protein-ligand binding site propensity descriptor, and a protein-DNA interface propensity descriptor, respectively. FS3 gives us 191 features which has an overlap of 134 features with FS1, so this set has 57 new features in it. FS4 has 11 features.
Figure 1:
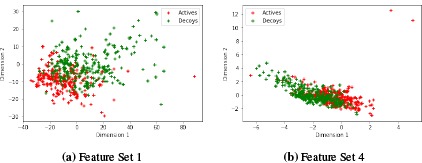
PCA of FS1 and FS4
Figure 2:
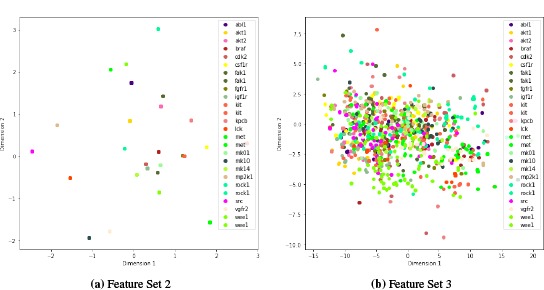
PCA of FS2 and FS3
3.3. Evaluation of Models Containing Different Feature Sets
Metrics (which are defined in Table 3) for each model are given in Table 5 and a comparison to the docking metrics obtained using the test set is also given. All models do very well at identifying the decoy compounds or negative class. However, given the ratio of decoys to actives, a model could always predict decoy and give good results. Therefore, our focus is on the positive class metrics. Interestingly Model 2 which is all docking features and would hopefully be highly predictive, has the worst performance in all metrics. Model 2 still performs better than docking alone by F1-score. Model 5 gives the best F1-score which is a key metric as it is a balance between the precision and recall. It also gives the best recall of 0.92. Additionally, this is the model that includes protein features and we are interested in the added benefit of including them.
Table 5:
Comparison of performance for both classes on the testing set.
| Model | Class | Precision | Recall | F1-Score | Class | Precision | Recall | F1-Score |
|---|---|---|---|---|---|---|---|---|
| 1 | 0 | 1.00 | 1.00 | 1.00 | 1 | 0.83 | 0.92 | 0.87 |
| 2 | 0 | 0.98 | 0.98 | 0.98 | 1 | 0.26 | 0.30 | 0.28 |
| 3 | 0 | 1.00 | 1.00 | 1.00 | 1 | 0.83 | 0.92 | 0.87 |
| 4 | 0 | 1.00 | 1.00 | 1.00 | 1 | 0.83 | 0.91 | 0.87 |
| 5 | 0 | 1.00 | 1.00 | 1.00 | 1 | 0.84 | 0.92 | 0.88 |
| 6 | 0 | 1.00 | 1.00 | 1.00 | 1 | 0.85 | 0.89 | 0.87 |
| Docking | 0 | 0.99 | 0.58 | 0.73 | 1 | 0.04 | 0.67 | 0.07 |
In Table 6 we further analyze the per kinase performance. All metrics here are using Model 5 and again the metrics based on docking scores alone are also given. The “Per kinase” columns are metrics on each individual kinase from the analysis given in Table 5. The “Leave-one-out” columns are additional models using the same feature sets as Model 5, but in which one kinase is left out for testing while all the other kinases are in the training set.
Table 6:
Evaluation metrics per kinase for the positive class
| Per Kinase | Leave-one-out | Docking | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Kinase | Prec. | Recall | F1 | Prec. | Recall | F1 | Prec. | Recall | F1 |
| abl1 | 0.92 | 0.95 | 0.93 | 0.86 | 0.95 | 0.9 | 0.06 | 0.72 | 0.11 |
| akt1 | 0.86 | 0.96 | 0.91 | 0.84 | 0.81 | 0.82 | 0.03 | 0.29 | 0.05 |
| akt2 | 0.94 | 0.94 | 0.94 | 0.83 | 0.8 | 0.81 | 0.04 | 0.59 | 0.07 |
| braf | 0.85 | 0.98 | 0.91 | 0.76 | 0.85 | 0.8 | 0.06 | 0.80 | 0.11 |
| cdk2 | 0.85 | 0.77 | 0.81 | 0.6 | 0.31 | 0.41 | 0.04 | 0.78 | 0.08 |
| csf1r | 0.82 | 0.83 | 0.83 | 0.62 | 0.59 | 0.6 | 0.03 | 0.63 | 0.06 |
| egfr | 0.85 | 0.91 | 0.88 | 0.76 | 0.75 | 0.75 | 0.03 | 0.81 | 0.05 |
| fak1 | 0.75 | 0.86 | 0.8 | 0.8 | 0.77 | 0.78 | 0.03 | 0.86 | 0.06 |
| igf1r | 0.93 | 1.00 | 0.97 | 0.9 | 0.92 | 0.91 | 0.05 | 0.86 | 0.09 |
| jak2 | 0.97 | 0.94 | 0.95 | 0.87 | 0.77 | 0.82 | 0.06 | 0.5 | 0.10 |
| kit | 0.83 | 0.92 | 0.87 | 0.74 | 0.88 | 0.81 | 0.05 | 0.58 | 0.08 |
| kpcb | 0.70 | 0.93 | 0.80 | 0.45 | 0.32 | 0.37 | 0.04 | 0.86 | 0.08 |
| lck | 0.94 | 0.93 | 0.94 | 0.8 | 0.83 | 0.82 | 0.05 | 0.49 | 0.09 |
| mapk2 | 0.93 | 0.97 | 0.95 | 0.72 | 0.37 | 0.49 | 0.06 | 0.65 | 0.10 |
| met | 0.87 | 0.96 | 0.91 | 0.75 | 0.77 | 0.76 | 0.05 | 0.81 | 0.09 |
| mk01 | 0.91 | 0.97 | 0.94 | 0.78 | 0.69 | 0.73 | 0.16 | 0.71 | 0.26 |
| mk10 | 0.88 | 0.86 | 0.87 | 0.67 | 0.51 | 0.58 | 0.06 | 0.82 | 0.11 |
| mk14 | 0.90 | 0.85 | 0.88 | 0.76 | 0.5 | 0.6 | 0.05 | 0.56 | 0.09 |
| mp2k1 | 0.80 | 0.95 | 0.87 | 0.59 | 0.48 | 0.53 | 0.03 | 0.93 | 0.05 |
| plk1 | 0.82 | 0.84 | 0.83 | 0.73 | 0.5 | 0.59 | 0.03 | 0.91 | 0.06 |
| rock1 | 0.88 | 0.85 | 0.86 | 0.63 | 0.32 | 0.42 | 0.05 | 0.88 | 0.10 |
| src | 0.93 | 0.97 | 0.95 | 0.86 | 0.87 | 0.86 | 0.03 | 0.88 | 0.06 |
| tgfr1 | 0.94 | 0.98 | 0.96 | 0.89 | 0.83 | 0.86 | 0.09 | 0.84 | 0.17 |
| vgfr2 | 0.93 | 0.90 | 0.92 | 0.8 | 0.79 | 0.8 | 0.05 | 0.65 | 0.10 |
| wee1 | 0.93 | 1.00 | 0.96 | 0.75 | 0.58 | 0.65 | 0.14 | 0.8 | 0.24 |
4. Discussion
We can see from the PCA (Figures 1 and 2) that FS1 (drug features selected based on active or decoy classification) has a fairly good separation of the two classes and alone gives good predictions with an F1-score of 0.87. FS4 (docking features) has some separation between the classes but also a sizable overlap and only an F1-score of 0.28 (Model 2) for classifying the compounds. FS3 (drug features selected based on kinase classification) do not do a great job at kinase classification and do not improve the model over using just FS1 (Model 3 vs Model 1). Only three protein features were selected for their ability to classify the kinase (FS2) and this model, Model 5, gives the best F1 score.
Even though the drug features are by far the most informative features in these models, they cannot account for kinase selectivity. When including features that are informative at classifying the kinase, we had a slight increase in F1 score. Even though kinase inhibitors are promiscuous and kinases have a high sequence and structural similarity, this provides hope that protein features can be informative in universal (multi-protein) models for drug binding.
We can see that the per kinase performamce is much better when each kinase is represented in the training set by comparing the “Per kinase” and “Leave-one-out” columns in Table 6. However, many kinases still perform very well in the “leave-one-out” analysis. We believe that as we add more kinases to the model the “leave-one-out” analysis will improve. Recent results using a diverse set of proteins also show promise to improve models.
To exemplify the usefulness of our method, we have identified a compound that would never be identified in a docking virtual screen as an active compound (receiving a score of +95.19, when the most negative scores predict binding), that has been saved using this machine learning model. An example of such a compound is CHEMBL448926, an ackt1 active compound. This compound is an actual ackt1 inhibitor patented by Merck and Co Inc. (Patent ID US7544677) and directed to chemotherapeutic compositions. The reason this compound may be lost during docking is it a potent allosteric inhibitor [20].
Docking is often used as a tool to enrich a subset of data and therefore early enrichment is a common important metric. For example, if a large virtual drug set has 2% unknown active compounds in it then hopefully the top subset of scored drugs by docking will have maybe 10% active compounds in it. This would allow researchers to select a smaller set of drugs for experimental testing and have a greater success rate than randomly selecting a subset for testing. We can see here that the precision is always low for docking, therefore to recover the same amount of active compounds using docking you would always have a much higher false positive rate making the number of compounds needed for experimental validation to be much higher for the same success rate.
With the test case presented here evaluated with Model 5, which includes protein features and gives the best F1 score, 97% of the test data is classified as non-binding. Therefore, 97% of the data can immediately be discarded and you would lose less than 10% of the binding compounds at this prediction stage. Experimentally testing the predicted active compounds would give an 83% success rate at identifying active compounds. Testing the same number of compounds based on docking score alone would have less than a 27% success rate. Part of the problem here is that docking scores are not a good indicator of binding when looking at multiple proteins. The range of docking scores varies per protein. This demonstrates a huge advantage to our machine learning approach for a multi-protein model.
4.1. Limitations
While our results highlight the potential of our approach, there are several limitations of our evaluation that warrant further investigation. The main limitation is that we do not know whether a compound that is active for one kinase is not active for another. There is some overlap between the 26 different active sets but it is not much. Since selectively inhibiting a kinase is difficult it should be experimentally validated before marking a compound that is active for one kinase as not for another one. It is also difficult to tell if a given active for one kinase is in the decoy set for another kinase because the active and decoy compounds in DUD-e come from different databases, CHEMBL [21] and ZINC [22], respectively. Having an all-to-all set of connections where we know whether every drug in our dataset binds or does not bind to every protein in our dataset may uncover important features for this selectivity. Also, due to concerns with the fgfr1 dataset (i.e. the proportion of actives to decoys does not match what is expected from DUD-e), we have excluded fgfr1 compounds from our test set.
4.2. Future Work
Some potential future directions include (1) evaluating different ways of using the pocket features that may correlate better with predictions, (2) incorporating information on multiple possible binding sites in the model, and (3) incorporating diverse proteins in the dataset.
5. Conclusion
We successfully created a model of several kinases that makes good binding predictions. We found that the features we collected greatly increased binding predictions when used in a machine learning model over docking scores alone. A model using features selected based on which kinase they belong to gave the best F1 score which balances precision and recall. We calculate a nearly 60% increase in success rate for discovering active compounds over docking.
6 Acknowledgements
This research was supported by the Cancer Research Informatics Shared Resource Facility of the University of Kentucky Markey Cancer Center (P30CA177558). This research used computational resources at the University of Kentuckys Center for Computational Sciences and the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. This work was supported by the National Institutes of Health(NIH) National Center for Advancing Translational Science grant KL2TR000116 and1KL2TR001996-01.This work was supported by the Director, Office of Science, Office of Advanced Scientific Computing Research, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We would like to acknowledge the assistance of Radoslav Krivak and David Hoksza in using the p2rank software.
References
- 1.Joanne Bowes, Andrew J Brown, Jacques Hamon, Wolfgang Jarolimek, Arun Sridhar, Gareth Waldron, Steven Whitebread. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nature reviews. Drug discovery. 2012;11(12):909. doi: 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- 2.Ian Roberts, Irene Kwan, Phillip Evans, Steven Haig. Does animal experimentation inform human healthcare? observations from a systematic review of international animal experiments on fluid resuscitation. BMJ: British Medical Journal. 2002;324(7335):474. doi: 10.1136/bmj.324.7335.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kun-Yi Hsin, Samik Ghosh, Hiroaki Kitano. Combining machine learning systems and multiple docking simulation packages to improve docking prediction reliability for network pharmacology. PloS one. 2013;8(12):e83922. doi: 10.1371/journal.pone.0083922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarah L Kinnings, Nina Liu, Peter J Tonge, Richard M Jackson, Lei Xie, Philip E Bourne. A machine learning-based method to improve docking scoring functions and its application to drug repurposing. Journal of chemical information and modeling. 2011;51(2):408–419. doi: 10.1021/ci100369f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ali Akbar Jamali, Reza Ferdousi, Saeed Razzaghi, Jiuyong Li, Reza Safdari, Esmaeil Ebrahimie. DrugMiner: comparative analysis of machine learning algorithms for prediction of potential druggable proteins. Drug Discov. Today. 2016 May;21(5):718–724. doi: 10.1016/j.drudis.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Michael M Mysinger, Michael Carchia, John J Irwin, Brian K Shoichet. Directory of useful decoys, enhanced (dud-e): better ligands and decoys for better benchmarking. Journal of medicinal chemistry. 2012;55(14):6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amos Bairoch, Rolf Apweiler, Cathy H Wu, Winona C Barker, Brigitte Boeckmann, Serenella Ferro, Elisabeth Gasteiger, Hongzhan Huang, Rodrigo Lopez, Michele Magrane, et al. The universal protein resource (uniprot) Nucleic acids research. 2005;33(suppl_1):D154-D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elisabeth Gasteiger, Christine Hoogland, Alexandre Gattiker, S’everine Duvaud, Marc R Wilkins, Ron D Appel, Amos Bairoch. Protein identification and analysis tools on the ExPASy server. In: John M Walker., editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005. pp. 571–607. [Google Scholar]
- 9.Claudio Mirabello, Gianluca Pollastri. Porter, PaleAle 4.0: high-accuracy prediction of protein secondary structure and relative solvent accessibility. Bioinformatics. 2013 Aug 15;29(16):2056–2058. doi: 10.1093/bioinformatics/btt344. [DOI] [PubMed] [Google Scholar]
- 10.Peng Zhang, Lin Tao, Xian Zeng, Chu Qin, Shangying Chen, Feng Zhu, Zerong Li, Yuyang Jiang, Weiping Chen, Yu-Zong Chen. A protein network descriptor server and its use in studying protein, disease, metabolic and drug targeted networks. Brief. Bioinform. 2016 Aug 19; doi: 10.1093/bib/bbw071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radoslav Krivak, David Hoksza. Improving protein-ligand binding site prediction accuracy by classification of inner pocket points using local features. Journal of cheminformatics. 2015;7(1):12. doi: 10.1186/s13321-015-0059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kode srl. Dragon (software for molecular descriptor calculation) version 7.0.6. 2016. https://chm.kode-solutions.net.
- 13.William Humphrey, Andrew Dalke, Klaus Schulten. Vmd: visual molecular dynamics. Journal of molecular graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 14.Sally R Ellingson, Jeremy C Smith, Jerome Baudry. VinaMPI: Facilitating multiple receptor high-throughput virtual docking on high-performance computers. J. Comput. Chem. 2013 Sep 30;34(25):2212–2221. doi: 10.1002/jcc.23367. [DOI] [PubMed] [Google Scholar]
- 15.Oleg Trott, Arthur J Olson. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leo Breiman. Random forests. Mach. Learn. 2001 Oct 1;45(1):5–32. [Google Scholar]
- 17.Bjoern H Menze, B Michael Kelm, Ralf Masuch, Uwe Himmelreich, Peter Bachert, Wolfgang Petrich, Fred A Hamprecht. A comparison of random forest and its gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC Bioinformatics. 2009 Jul 10;10:213. doi: 10.1186/1471-2105-10-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali Anaissi, Paul J Kennedy, Madhu Goyal, Daniel R Catchpoole. A balanced iterative random forest for gene selection from microarray data. BMC Bioinformatics. 2013 Aug 27;14:261. doi: 10.1186/1471-2105-14-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilles Louppe, Louis Wehenkel, Antonio Sutera, Pierre Geurts. Understanding variable importances in forests of randomized trees. In: C J C Burges, L Bottou, M Welling, Z Ghahramani, K Q Weinberger., editors. Advances in Neural Information Processing Systems 26. Curran Associates, Inc.; 2013. pp. 431–439. [Google Scholar]
- 20.Tony Siu, Jun Liang, Jeannie Arruda, Yiwei Li, Raymond E Jones, Deborah Defeo-Jones, Stanley F Barnett, Ronald G Robinson. Discovery of potent and cell-active allosteric dual akt 1 and 2 inhibitors. Bioorganic & medicinal chemistry letters. 2008;18(14):4186–4190. doi: 10.1016/j.bmcl.2008.05.085. [DOI] [PubMed] [Google Scholar]
- 21.Anna Gaulton, Louisa J Bellis, A Patricia Bento, Jon Chambers, Mark Davies, Anne Hersey, Yvonne Light, Shaun McGlinchey, David Michalovich, Bissan Al-Lazikani, et al. Chembl: a large-scale bioactivity database for drug discovery. Nucleic acids research. 2011;40(D1):D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.John J Irwin, Teague Sterling, Michael M Mysinger, Erin S Bolstad, Ryan G Coleman. Zinc: a free tool to discover chemistry for biology. Journal of chemical information and modeling. 2012;52(7):1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]