

Abstract
Currently, drug discovery approaches focus on the design of therapies that alleviate an index symptom by reengineering the underlying biological mechanism in agonistic or antagonistic fashion. For example, medicines are routinely developed to target an essential gene that drives the disease mechanism. Therapeutic overloading where patients get multiple medications to reduce the primary and secondary side effect burden is standard practice. This single-symptom based approach may not be scalable, as we understand that diseases are more connected than random and molecular interactions drive disease comorbidities. In this work, we present a proof-of-concept drug discovery strategy by combining network biology, disease comorbidity estimates, and computational drug repositioning, by targeting the risk factors and comorbidities of peripheral artery disease, a vascular disease associated with high morbidity and mortality. Individualized risk estimation and recommending disease sequelae based therapies may help to lower the mortality and morbidity of peripheral artery disease.
Introduction
Emerging role of network pharmacology in precision medicine
Many challenges in drug discovery can be mitigated by computational and predictive methods to improve success rates. Due to the compound nature of known and unknown factors at play, precise prediction of the success of drug discovery project is difficult, and this leads to the loss of both human productivity and economic loss in the setting of the pharmaceutical industry. Drug discovery budget allocations typically do not yield a successful therapy over decades of research. Further, in the real-world setting, patients take multiple drugs for sets of related clinical symptoms, and these drugs often interact and lead to new and unforeseen side effects. Network medicine approaches enable network-wide, integrated investigation of multiple biological and clinical data-types to understand key biological pathways and functional modules behind complex diseases. Such approaches could be beneficial to target and potentially intervene underlying risk factors and population scale comorbidities and hence may evolve as a potential drug discovery strategy for complex diseases with varying degree of clinical heterogeneity. The relationships between diseases and associated molecular pathways underlying them has been examined in detail by several studies in the post-genome era due to the emerging understanding on the network properties of genes and proteins (1-3). The availability of large-scale, datasets of protein-nucleic acid, protein-protein, protein-metabolic and protein-small molecule interaction helped to perform and validate such studies (4, 5). Previous studies have shown that disease comorbidities share a high-degree of functional cohesiveness and several common functional modules; protein-protein interactions and sub-units of protein complexes were found to be the underlying the molecular basis of comorbid conditions. Goh et al. have showed that several Mendelian diseases, which are comorbid, share interactions at the level of gene products using a network of Mendelian disease-gene networks (6). Lage et al. built a protein-complex centric network to illustrate a human phenome-interactome network of protein complexes implicated in genetic disorders (7). Network properties of genes that harbor disease variants or single nucleotide polymorphisms (SNPs) have also shown to have similar likelihood and phenotypic effects (8, 9). Ideker and Sharan also suggested that protein networks can be used as tools to investigate molecular basis of complex diseases (10). Additional evidence to support the pleiotropic nature of genes to influence multiple disease pathways was also revealed from network-based disease analyses. Park et al. showed that pair of comorbid conditions correlate with specific sub-cellular location of the gene products associated with the diseases and comorbidities have shown to share (11). Such integrated network based approaches have been used to find comorbidities associated with several diseases as well as specific aspects of diseases like patterns of cancer metastasis (12). Chen et al. used a personalized multi-omic study that performed longitudinal profiling of a generally healthy individual using wholegenome sequencing (WGS) together with transcriptome, proteomic, metabolomic, and autoantibody profiles showed that pathway analysis and GO term enrichment could help to understand common functional modules mediated by group of genes associated with clinically relevant phenotypes (13). Cardiovascular diseases like atherosclerosis, metabolic disorders (14-16), pulmonary diseases like chronic obstructive pulmonary disorder (COPD) (17) and asthma (18) have also been studied using network-based approaches. Recently, we have shown that exploring shared genetic architecture driving disease pairs could help in discovering disease trajectories (19, 20). In another study, we have shown that exploring shared genetic architecture in conjunction with pair-wise comorbidity would help to orthogonally validate drug repositioning success (21-23). Based upon this collective previous evidence, in this manuscript, we employed a network-based approach to delineate the molecular basis of peripheral arterial disease (PAD) comorbidities and used the common molecular sub-network driving risk factors and comorbidities as signatures to find potential drugs.
No disease is an island - clinical needs and drug discovery challenges in targeting risk trajectories and comorbidities
A measure to assess the implications of comorbidity between multiple diseases was initially used (the Charlson Index) to predict mortality rate of patients with 22 known conditions by Charlson et al. (24). Eleven years later, another index (the Elixhauser Index) that utilized hospital administrative data was proposed. This method was the first to utilize the International Classification of Disease (ICD) codes to define comorbidities (25). To perform large-scale comorbidity analyses, multiple groups have since used Medicare data encoded as ICD-9 codes. These studies that utilized Medicare databases have primarily used two metrics, (1) φ correlation and (2) relative risk (RR), to define the degree of co-occurrence between two diseases in the same patient within a defined time-period. For example, a large-scale study of diverse human disease phenotypes in a network framework using ICD-9 codes derived from raw Medicare data showed that several complex diseases share a high-degree of comorbidities (26). Another study that utilized Medicare data also showed that significant correlations exist between the underlying cellular networks and disease comorbidity patterns in the human population (27). Based on the observed correlations between several complex diseases, investigators have also proposed molecular bases for the correlations using functional modules, sub-unit of large protein-complexes, protein-protein interactions or pathways mediated by genes associated with a pair of diseases. Collective approaches to study disease mechanisms mediated by different macro and micro molecules inside the cell were considered as the basis for system-wide (systems medicine) or network-based (network medicine) approaches.
Peripheral arterial disease
PAD is a chronic vascular disease caused by a variety of complex phenotypic characteristics including deposition of cholesterol and fat deposition in blood vessels outside of the heart. PAD is a chronic vascular disease due to atherosclerosis. Presence of PAD is considered to be a clinical surrogate of coronary artery and cerebrovascular atherosclerosis. PAD is an excellent indicator of myocardial infarction and cerebrovascular accident (stroke) risk, as it can severely limit mobility, and often ultimately leads to limb amputation. However, despite affecting eight million people in the United States and its association with significant mortality and morbidity rates, PAD is often under diagnosed. PAD is influenced by multiple genetic, proteomic, transcriptomic, metabolomic and epigenomic signals (28-31). Effective delineation of how PAD progress to other cardiovascular complications may help to develop better drug targets and may further help to manage the disease and reduce the burden for patients. In this study, we propose a disease-gene network based translational bioinformatics approach to find functional modules underlying different comorbidities associated with PAD. Further we used the molecular core that are common to two comorbidities to search for drugs capable of perturbing the biological and functional pathways.
Methods
The analysis was divided into four parts i) Identification of risk factors and comorbidities associated with PAD ii) Generation of canonical disease-gene networks iii) analyses and functional interpretation of the networks iv) Computational drug repositioning to find compounds capable of perturbing the molecular core sub-network. A workflow diagram is provided in Figure 1.
Figure 1:
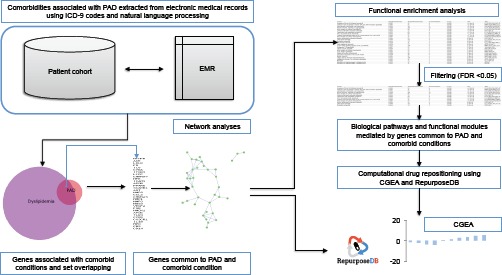
Translational bioinformatics pipeline used to identify functional modules, biological pathways and drugs targeting molecular core of PAD and it’s comorbidities
Risk factors and Comorbidities associated with PAD
We identified risk factors and comorbidities associated with PAD from a database of patients who underwent outpatient, non-invasive lower extremity arterial evaluation at the Mayo Clinic, Rochester, Minnesota, from January 1998 through December 2007, with a mean follow-up of 5.8 ± 3.1 years. Risk factors and comorbid conditions associated with PAD were ascertained on the basis of the presence of relevant ICD-9-CM diagnosis code (###.##) and procedure codes for up to six months following the date of arterial evaluation. In this study, we used a subset of PAD patients (n=10,451) and age- and sex-matched controls (n=15,779) from the vascular database to derive comorbid conditions. We defined PAD as an ankle brachial index (ABI) ≤ 0.9 and controls had an ABI > 1.0. Detailed methodologies used for the extraction of ICD-9 codes and natural language processing (NLP) algorithm to find various disease phenotypes from electronic medical records (EMR) are explained elsewhere (32, 33).
Mapping disease genes
We derived the disease-gene network from the Gene Prospector (34) database available through the HUman Genome Epidemiology (HUGE) Navigator (35, 36). We selected Gene Prospector as the resource to derive disease-gene networks as it contains comprehensive data about genes in relation to human diseases, risk factors and other clinically relevant phenotypes. For each query, Gene Prospector uses a heuristic scoring function-based gene list with numbers of publications in different categories (total, genetic association, genome-wide association, metaanalysis/pooled analysis and genetic testing) provided as the gene-disease annotation. Core genes associated with a pair of diseases were defined as follows: If a query of “peripheral arterial disease” in Gene Prospector database retrieved X genes and a query using comorbid condition “chronic kidney disorder” retrieved Y genes, we defined a subset of genes associated with a risk factor or PAD or a comorbid condition and PAD as X ⋂ Y. We calculate significance of shared genetic architecture across two diseases as explained in our previous work (20, 21).
Functional modules and pathways underlying comorbidities
We hypothesize that the functional modules and pathways enriched among the genes in the intersection of two gene lists could provide insights to functional cues specific to comorbidities. We used a Cytoscape software plug-in, ReactomeFI (http://apps.cytoscape.org/apps/reactomefiplugin) to perform cross-database pathway enrichment analyses and GO annotation enrichment analysis using the genes common to comorbid conditions. ReactomeFI enables the discovery of statistically significant relationship between a set of genes using annotations derived from multiple pathway databases (BioCarta; www.biocarta.com), Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/pathway.html), Protein ANalysis Through Evolutionary Relationship (PANTHER: http://www.pantherdb.org/pathway/), Reactome (http://reactome.org/) and GO annotations (biological process, cellular component and molecular function categories; http://www.geneontology.org/). In our post-enrichment filtering step, we used an FDR threshold of 0.05. Following the set computation and biological function enrichment, we compiled molecular core modules associated with risk factors and comorbidities. Biological plausibility of the computationally inferred networks was validated using human proteome-wide enrichment analyses using data from STRING database (https://string-db.org/).
Computational drug repositioning using molecular cores of risk factors and comorbidities
We used the canonical gene list corresponding to molecular core module to find drugs capable of agonistic and antagonistic effects for perturbations. Detailed account of the method to match gene signature to corresponding drugs is explained elsewhere (37-39). Briefly, the gene list to drug matching was performed using Chemo-Genomic Enrichment Analyses approach (Manuscript in preparation) and compounds were annotated in conjunction with RepurposeDB (http://repurposedb.dudleylab.org) (21, 23, 40).
Briefly, to leverage CGEA method, we first define a biological state of interest using genes for PAD, PAD and risk factors or PAD and comorbidities, (which may reflect differential gene expression from an affected tissue in a disease of interest) in the form of “upregulated” and “downregulated” gene identifiers. The input query of gene lists is matched to compounds, and the “connectivity” between the gene signature and compound is scored after various filtering steps against the available drug-induced signatures compiled from reference databases. References resources like RepurposeDB (http://repurposedb.dudleylab.org/), Connectivity Map (CMap: https://portals.broadinstitute.org/cmap/), Genomics of Drug Sensitivity in Cancer (GDSC: http://www.cancerrxgene.org/) or Cancer Cell Line Encyclopedia (CCLE: https://portals.broadinstitute.org/ccle) are used to identify compounds that concordantly modulate the query signature in a direction “towards” or “away” from the query state. A Gaussian mixture model is to derive the “connectivity score” and assign statistical significance, and false discovery rate is estimated using Kolmogorov Smirnov test. As an output of the analyses, a ranked list of candidate compounds that may potentially modulate a biological state of interest is retrieved. Depending on the query signature and reference databases, often many candidate compounds will be extracted – such lists can be trimmed and prioritized for most likely candidates using annotations from reference databases (for example RepurposeDB, KEGG drugs, DrugBank, etc.) and also use specific characteristics including mechanism of action, side effects, chemical properties or biological targets.
Results
Seven comorbid conditions (coronary heart disease, congestive heart failure, chronic kidney disease, chronic obstructive pulmonary disease,) and four risk factors (diabetes mellitus, dyslipidemia, hypertension and smoking) were identified to be associated with PAD (Table 1) from the vascular database.
Table 1:
Comorbidities and risk-factors+ associated with PAD. 1Hypertension was considered present if there were 2 blood pressure readings of ≥140/90 mm Hg within 3 months of the date of arterial evaluation, or a prior diagnosis of hypertension and current treatment with antihypertensive medication. 2 Clinical phenotyping of these conditions was performed using presence of relevant ICD-9 codes for up to 6 months following the date of arterial evaluation. 3 Diabetes was ascertained on the basis of fasting plasma glucose ≥126 mg/dl, or random glucose >200 mg/dl, or hemoglobin A1c of >6.5%, or a prior diagnosis and use of oral hypoglycemic agent(s) or insulin
| Risk factors and Comorbidities | ICD-9 codes | PAD (n, %) | Normal (n, %) | All (n) |
|---|---|---|---|---|
| Risk factors | ||||
| Hypertension1 | 414.01 | 7517, 71.93% | 2734, 51.31% | 10251 |
| Dyslipidemia2 | 428.0 | 8016, 76.70% | 3416, 64.11% | 11432 |
| Diabetes Mellitus3 | 585, 403 | 2951, 28.24% | 889, 16.69% | 3840 |
| Smoking2 | 490–492, 494–496 | 7819, 84.17% | 3133, 67.86% | 10952 |
| Comorbidities | ||||
| Chronic kidney disease2 | 443.9 | 741, 7.08% | 201, 3.76% | 942 |
| Chronic obstructive pulmonary disease2 | 401 | 1837, 17.54% | 425, 7.96% | 2262 |
| Coronary heart disease2 | 250 | 5593, 53.52% | 1698, 31.87% | 7291 |
| Congestive heart failure2 | 272 | 1407, 13.46% | 343, 6.44% | 1750 |
| Cerebrovascular disease2 | 430-438 | 3302, 31.60% | 733, 13.76% | 4035 |
| Total (n) | -- | 10451 | 5328 | 15779 |
Disease-gene annotations
We queried the Gene Prospector database using “peripheral artery disease” and seven comorbid conditions to retrieve disease-gene lists (Supplementary File: F1). Overlap between genes associated with PAD (n=69) and proportional Venn-diagrams of PAD with various risk factors and comorbidities are provided in Figure 2 (Also See Table 2).
Shared molecular cores driving risk factors and comorbidities were identified using ReactomeFI (See Figure 2-2.). Further, we used an independent database to test whether the molecular core modules harbor higher functional interactions than expected by random. Nine molecular cores were significantly enriched for protein-protein interactions and hence potentially represent plausible drug targets that have important functional roles. In an earlier study, we have built similar functional network from public molecular databases and perturbed using experimental methods for gene prioritization and functional studies in the setting of ovarian and pancreatic cancers (41, 42).
Biological pathways mediated by genes shared by genes associated with PAD, its risk factors and comorbidities
Individual lists of PAD genes common to risk factors and comorbid conditions were used to perform enrichment analysis using ReactomeFI. Functional interactions were obtained for all lists (Supplemental Data) and shared pathways were inferred using KEGG and Reactome annotations (Figure 2-3 and 2-4). Four independent enrichment analyses were performed using different gene lists using ReactomeFI to identify specific biological pathways and functional modules (protein domain annotations, biological process, cellular compartment and molecular function; Supplemental Data). Biological process annotated across the risk factors and comorbidities include the digestion of dietary lipids, sterol uptake, the formation and turnover of lipoproteins (chylomicrons, VLDL, LDL, and HDL), and the mobilization of fatty acids through the action of hormone-sensitive lipases. Pathways driving key process across risk factors and comorbidities suggests that Cell surface interactions at the vascular wall, formation of fibrin clot, formation of platelet plug and integrin cell surface and metabolism of lipids and lipoproteins are common across both risk factors.
Computational Drug Repositioning using Molecular Core Module suggests new therapeutic interventions
We tested the molecular core shared across two diseases to individual risk factors and comorbidities across 1309 compounds, 743 of which have some approval status in the global pharmaceutical market as indicated in DrugBank. In this analysis, we focused on the already approved subset of 743 compounds to explore the feasibility of our approach. Briefly the gene set-drug matching data was compiled and annotated using data from the Connectivity Map, Anatomical Therapeutic Chemical (ATC) Classification System, PubChem, SIDER, Offsides and Drug Bank. Results compiled from CGEA consist of compound information including chemoinformatics signatures, drug target information, indications, mechanism of action and side effects. The rank-scored compound list that can perturb the molecular core sub network are provided in the Supplemental Data and the summary of top ranked compounds are provided in Table 4. Several of the compound associations that we discovered are new; a compound fluspirilene is of particular interest. Fluspirilene is diphenylbutylpiperidine typical antipsychotic drug and it targets dopamine receptor, 5HT2A receptor and voltage-dependent calcium channel gamma-1 subunit. Optimal calcium ion flux is critical in regulating cardiac function. Fluspirilene has shown experimental evidence in the in vitro studies for various cardiovascular diseases including cardiomyopathy (43).
Discussion
Precision medicine approaches are now leveraging molecular profiling data to recommend medications based an individualized risk. These approaches are now emerging in oncology and potentially expanding to other therapeutic domains. However, it should be noted that suggesting index disease based approach may lead to a “precision therapeutics-deluge”; for example, multiple rare genetic variants driving complex diseases like cancer and cardiovascular disorders. Hence there is an urgent need for a novel drug discovery approach. We envisage that a drug discovery strategy that combines epidemiology, network biology and computational approaches may yield better drug repositioning and discovery productivity (Figure 3). In this study, we have shown a method and initial results by applying the method to PAD. Compared to traditional drug repositioning and discovery strategies, network pharmacology and systems medicine approaches address both depth and breadth of biological knowledge to infer molecular connections to discover therapies. To the best of our knowledge, no previous studies have integrated comorbidities from a patient cohort database with follow-up functional module discovery study for PAD. While multiple novel therapeutic themes are now emerging in cardiovascular therapeutic space, including PCSK9 inhibitors and loss-of-function based therapies, precise modulation of classical pathways and systematic control of lipid homeostasis may also improve optimal outcomes in these patients. Concordant with emerging findings, we also found NOS3, a key regulator of vascular disease and common gene across the risk factors and comorbidities (44, 45). A previous report on drug repositioning for PAD suggests that anti-inflammatory molecules may serve as potential candidates for the systemic control of inflammation and associated pathways in the setting of PAD (46). Similarly we also found several existing cardiovascular therapies as top candidate drugs in our approach: this includes doxazosin, midodrine, and nadolol. Our findings are concordant to previous therapeutic indications for PAD as we found anti-inflammatory medications including felbinac in the ranked list of repurposed drugs that modulate the molecular core driving risk factors and PAD comorbidities. Interestingly, we also observe antivirals (aciclovir), antibacterials (ceftazidime, vancomycin) and antifungals (natamycin) as potential agents that could target the risk trajectories and comorbidities. This discovery opens up several possibilities including the need for the precise control of infection, inflammation and immune responses in the early stages of the PAD to potentially save patients from long-term detrimental effects. We also noted that a subnetwork motif composed of APOB, APOE and LDLR as a consensus theme across various risk factors and comorbidities. Developing novel molecules to monitor and target these genes may also yield novel cardiovascular therapies including CRISPR/Cas9 based therapies (47).
Limitations of the current study
In this proof of concept bioinformatics investigation, we leveraged the molecular core shared across risk factors or comorbidities associated with an index disease as a complement of disease signatures. Patient derived gene-expression data would be a more ideal resource than the canonical gene sets used in the current investigation. Data elements like protein-protein interactions are also generated using reference database; compiling this from patients would be ideal. While our approach has been able to recommend drugs, extensive experimental validation and clinical trials are required before recommending the risk factor, comorbidity and disease sequelae based preventive therapies in cardiology (21). Also, in this study, we focus on a specific sub-module that is shared across a risk factor or comorbidity to find potential drug repositioning opportunities. Other modules including the particular set of genes that are unique to index diseases, risk factors or comorbidities or the entire set of genes may also lead to potential therapeutic opportunities. However, we did not evaluate these additional modules as part of this study. Also, gene expression profiling studies are usually reported for an index disease, not from patients with a specific risk factors or comorbidity. In this context, we compiled the gene set as a canonical gene list and did not consider the directionality of the query genes in the querying step. Also, the matching algorithm is sensitive to directionality, and the limited number of shared genes in the query, hence same drugs have been shown to modulate a given modulate in a different direction across different modules. Various studies suggests that polypharmacy leads to increase in prescription drug could that in turn increase primary side effects and secondary side effects due to drug-drug interactions. It is impossible to capture the entire spectrum of medications (over the counter, inpatient, ambulatory, surgery, ICU, emergency, etc.) to perform personalized medication reconciliation for all patients. Hence we hypothesize that using comorbidity targeting we may reduce the number of medications needed. However, we have only used pair-wise comorbidity in this analyses, expanding the number of shared diseases and finding the common module, if any, across all related diseases of an index condition could lead to better sub-typing and identification of optimal therapies instead of “pill-burden” and extensive side effect management using additional pills.
Conclusion
In this study, we present a drug discovery workflow that demonstrates how epidemiological information from a vascular disease registry can be effectively used for downstream translational studies to identify molecular bases underlying disease comorbidities. Biologically relevant pathways and functional categories obtained from the subset of genes common to a pair of conditions can be used to prioritize potential primary or secondary drug targets. This analysis also utilized multiple open access bioinformatics resources for finding protein-protein interaction or pathway sub-graphs that are common to various diseases, thus encouraging open access and reproducibility. We also plan to extend the study to systematically search for molecular core modules driving pair-wise diseases compiled from a phenome-wide library of 1988 disease conditions and 37, 282 disease pairs. Further, such a resource will be an indispensable resource for personalized, data-driven drug discovery.
Figure 2:
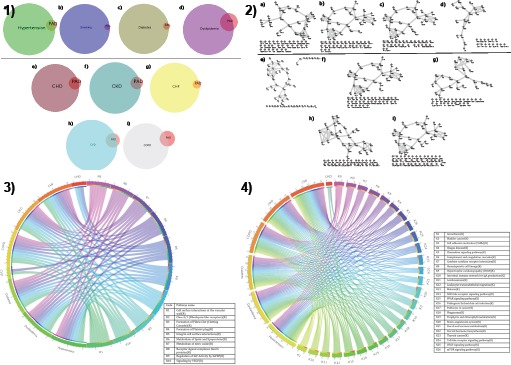
1) Proportional Venn-diagrams of genes associated with risk factors (a, b, c and d) and comorbid conditions (e: coronary heart disease; f: chronic kidney disease; g: congestive heart failure; h: cerebrovascular disease; i: chronic obstructive pulmonary disease) 2) Functional interactions mediated by gene products common to risk factors (dyslipidemia, diabetes mellitus, hypertension and smoking) and comorbidities associated with PAD (coronary heart disease, congestive heart failure, chronic kidney disease, cerebrovascular disease, chronic obstructive pulmonary disease) visualized using Cytoscape. Nodes are genes and an edge indicates a common pathway (derived from BioCarta, KEGG, NCI-Pathways database, PANTHER or Reactome) or functional category term shared by two genes. 3) Reactome molecular events mediated by genes common to PAD and seven comorbidities 4) KEGG pathways associated with genes common to PAD and comorbidities
Figure 3:
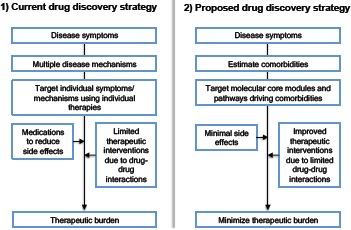
Comparison of current and proposed drug discovery approach
Table 2:
Summary of disease-gene annotations, functional interactions and orthogonal validation of molecular core using protein-protein interaction using STRING database for PAD and its comorbidities Disease annotations were compiled using HuGE Navigator ^Genes common across PAD and comorbid conditions ^^Collective term to define relationship between two genes. Relationship could be association with a biological pathway or common functional roles (biological processes or molecular function) or cellular localizations.
| Risk factors and comorbidities | Disease-gene annotations | Overlap^ | Functional interactions^^ | PPI enrichment P |
|---|---|---|---|---|
| Index disease | ||||
| Peripheral artery disease | 69 | 69 | 38 | <0.001 |
| Risk factors | ||||
| Hypertension | 1459 | 57 | 35 | <0.001 |
| Dyslipidemia | 567 | 48 | 29 | <0.001 |
| Diabetes Mellitus | 3675 | 61 | 36 | <0.001 |
| Smoking | 4949 | 67 | 38 | <0.001 |
| Comorbidities | ||||
| Chronic kidney disease | 1198 | 56 | 32 | <0.001 |
| Chronic obstructive pulmonary disease | 614 | 32 | 24 | <0.001 |
| Coronary heart disease | 1019 | 57 | 34 | <0.001 |
| Congestive heart failure | 2491 | 58 | 36 | <0.001 |
| Cerebrovascular disease | 900 | 57 | 33 | <0.001 |
Table 3:
Reactome molecular events shared by genes implicated in risk factors and PAD comorbidities; NA = molecular event is not associated with gene set; DM= Diabetes Mellitus; DL=dyslipidemia; CHD=coronary heart disease; CHF=coronary heart failure; CKD= chronic kidney disease; COPD=chronic obstructive pulmonary diseases; CVD=cerebrovascular disease
| Reactome events | Risk factors | Comorbidities | |||||
|---|---|---|---|---|---|---|---|
| DM | DL | CHD | CHF | CKD | COPD | CVD | |
| Cell surface interactions at the vascular wall | MMP1, APOB, F2, ITGB3, SELP | ITGB3, APOB, SELP, F2 | NA | MMP1, APOB, F2, ITGB3, SELP | MMP1, APOB, F2, ITGB3, SELP | ITGB3, MMP1, APOB | APOB |
| Class A/1 (Rhodopsin-like receptors) | AGTR1, CCR5, CX3CR1, F2, AGT | AGTR1, AGT, F2, CX3CR1 | NA | AGTR1, CCR5, CX3CR1, F2, AGT | AGTR1, CCR5, CX3CR1, F2, AGT | AGT, CCR5, CX3CR1 | NA |
| Formation of Fibrin Clot (Clotting Cascade) | F13A1, F12, F7, F5, F2, FGG, FGA, FGB | F13A1, FGB, F12, F7, F5, F2 | F13A1, F12, F7, F5, F2, FGG, FGA, FGB | F13A1, F12, F7, F5, F2, FGG, FGA, FGB | F13A1, F12, F7, F5, F2, FGG, FGA, FGB | FGB, F5 | NA |
| Formation of Platelet plug | F13A1, F5, VEGFA, F2, GNB3, ITGB3, FGG, FGA, FGB, SELP | F13A1, ITGB3, FGB, SELP, F5, VEGFA, F2, GNB3 | F13A1, F5, VEGFA, F2, GNB3, ITGB3, FGG, FGA, FGB, SELP | F13A1, F5, F2, VEGFA, GNB3, ITGB3, FGG, FGA, FGB, SELP | F13A1, F5, VEGFA, F2, GNB3, ITGB3, FGG, FGA, FGB, SELP | ITGB3, FGB, F5, VEGFA | NA |
| Integrin cell surface interactions | ICAM1, ITGB3, FGG, FGA, FGB | ITGB3, FGB, ICAM1 | ICAM1, ITGB3, FGG, FGA, FGB | ICAM1, ITGB3, FGG, FGA, FGB | ICAM1, ITGB3, FGG, FGA, FGB | ITGB3, FGB, ICAM1 | NA |
| Metabolism of lipids and lipoproteins(R) | LDLR, APOB, APOE, SCARB1, MTTP, LIPC | LDLR, APOB, APOE, SCARB1, MTTP, LIPC | NA | LDLR, APOB, APOE, SCARB1, MTTP, LIPC | LDLR, APOB, APOE, SCARB1, MTTP, LIPC | LDLR, APOB, APOE, SCARB1, LIPC | APOB, LDLR, APOE |
| Metabolism of nitric oxide(R) | NOS3 | NOS3 | NA | NOS3 | NOS3 | NOS3 | NA |
| Receptor-ligand complexes bind G proteins(R) | AGTR1, CCR5, GNB3, AGT | AGTR1, AGT, GNB3 | NA | AGTR1, CCR5, GNB3, AGT | AGTR1, CCR5, GNB3, AGT | AGT, CCR5 | NA |
| Regulation of IGF Activity by IGFBP(R) | MMP1, F2 | F2 | NA | MMP1, F2 | MMP1, F2 | MMP1 | NA |
| Signaling by VEGF(R) | VEGFA | VEGFA | NA | VEGFA | VEGFA | VEGFA | NA |
Table 4:
Top compounds to perturb the molecular core of risk factors and comorbidities
| Risk factors and comorbidities | Drugs to down regulate the module | Drugs to upregulate the module |
|---|---|---|
| PAD (Index disease) | fluspirilene, metyrapone, liothyronine | nadolol, felbinac, aciclovir |
| Risk factors | ||
| Hypertension Dyslipidemia | midodrine, felbinac, vancomycin, fluspirilene, metyrapone, methacholine chloride | fluspirilene, metyrapone, colecalciferol felbinac, cloxacillin, lisuride, amantadine |
| Diabetes Mellitus | fluspirilene, metyrapone, ceftazidime | vancomycin, midodrine, lisuride |
| Smoking | aciclovir, vancomycin, nadolol | fluspirilene, metyrapone, liothyronine |
| Comorbidities | ||
| Chronic kidney disease | vancomycin, midodrine, aciclovir | clioquinol, fluspirilene, daunorubicin |
| Chronic obstructive pulmonary disease | benzathine benzylpenicillin, lisuride, iodixanol | atropine, doxazosin, fluspirilene |
| Coronary heart disease | lisuride, felbinac, vancomycin | fluspirilene, metyrapone, blebbistatin |
| Congestive heart failure | vancomycin, midodrine, felbinac | clioquinol, metyrapone, fluspirilene |
| Cerebrovascular disease | vancomycin, midodrine, felbinac | fluspirilene, natamycin, metyrapone |
Acknowledgement
This work was supported by a grant from the National Institutes of Health, National Center for Advancing Translational Sciences (NCATS), Clinical and Translational Science Awards (UL1TR001433-01). We would like to thank Harris Center for Precision Wellness (http://precisionwellness.org/) and Icahn Institute for Genomics and Multiscale Biology (http://icahn.mssm.edu/research/genomics) for infrastructural support.
Supplemental Data: https://figshare.com/s/a82c06922a265bb0b464
References
- 1.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411(6833):41–2. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 2.Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5(2):101–13. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 3.Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nature genetics. 2003;(33 Suppl):228–37. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 4.Yamada T, Bork P. Evolution of biomolecular networks: lessons from metabolic and protein interactions. Nat Rev Mol Cell Biol. 2009;10(11):791–803. doi: 10.1038/nrm2787. [DOI] [PubMed] [Google Scholar]
- 5.Schadt EE, Friend SH, Shaywitz DA. A network view of disease and compound screening. Nat Rev Drug Discov. 2009;8(4):286–95. doi: 10.1038/nrd2826. [DOI] [PubMed] [Google Scholar]
- 6.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104(21):8685–90. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lage K, Karlberg EO, Storling ZM, Olason PI, Pedersen AG, Rigina O, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nature biotechnology. 2007;25(3):309–16. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 8.Feldman I, Rzhetsky A, Vitkup D. Network properties of genes harboring inherited disease mutations. Proc Natl Acad Sci U S A. 2008;105(11):4323–8. doi: 10.1073/pnas.0701722105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Lee Y, Chen J, Rebman E, Li J, Lussier Y. Complex-disease networks of trait-associated single-nucleotide polymorphisms (SNPs) unveiled by information theory. Journal of the American Medical Informatics Association: JAMIA. 2012;19(2):295–305. doi: 10.1136/amiajnl-2011-000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ideker T, Sharan R. Protein networks in disease. Genome Res. 2008;18(4):644–52. doi: 10.1101/gr.071852.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park S, Yang JS, Shin YE, Park J, Jang SK, Kim S. Protein localization as a principal feature of the etiology and comorbidity of genetic diseases. Mol Syst Biol. 2011;7:494. doi: 10.1038/msb.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen LL, Blumm N, Christakis NA, Barabasi AL, Deisboeck TS. Cancer metastasis networks and the prediction of progression patterns. Br J Cancer. 2009;101(5):749–58. doi: 10.1038/sj.bjc.6605214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Miriami E, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148(6):1293–307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu S, Lusis A, Drake T. A systems-based framework for understanding complex metabolic and cardiovascular disorders. Journal of lipid research. 2009;50(Supplement):S358–63. doi: 10.1194/jlr.R800067-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lusis A, Weiss J. Cardiovascular Networks. Circulation. 2010;121(1):157–70. doi: 10.1161/CIRCULATIONAHA.108.847699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan S, White K, Loscalzo J. Deciphering the molecular basis of human cardiovascular disease through network biology. Current opinion in cardiology. 2012;27(3):202–9. doi: 10.1097/HCO.0b013e3283515b31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agusti A, Sobradillo P, Celli B. Addressing the complexity of chronic obstructive pulmonary disease: from phenotypes and biomarkers to scale-free networks, systems biology, and P4 medicine. Am J Respir Crit Care Med. 2011;183(9):1129–37. doi: 10.1164/rccm.201009-1414PP. [DOI] [PubMed] [Google Scholar]
- 18.Lu X, Jain V, Finn P, Perkins D. Hubs in biological interaction networks exhibit low changes in expression in experimental asthma. Mol Syst Biol. 2007;3(1) doi: 10.1038/msb4100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glicksberg BS, Li L, Badgeley MA, Shameer K, Kosoy R, Beckmann ND, et al. Comparative analyses of population-scale phenomic data in electronic medical records reveal race-specific disease networks. Bioinformatics. 2016;32(12):i101–i10. doi: 10.1093/bioinformatics/btw282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glicksberg BS, Li L, Cheng WY, Shameer K, Hakenberg J, Castellanos R, et al. An integrative pipeline for multi-modal discovery of disease relationships. Pac Symp Biocomput. 2015:407–18. [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson KW, Shameer K, Glicksberg BS, Readhead B, Sengupta PP, Björkegren JLM, et al. Enabling Precision Cardiology Through Multiscale Biology and Systems Medicine. JACC: Basic to Translational Science. 2017;2(3):311. doi: 10.1016/j.jacbts.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodos RA, Kidd BA, Shameer K, Readhead BP, Dudley JT. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip Rev Syst Biol Med. 2016;8(3):186–210. doi: 10.1002/wsbm.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shameer K, Readhead B, Dudley JT. Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr Top Med Chem. 2015;15(1):5–20. doi: 10.2174/1568026615666150112103510. [DOI] [PubMed] [Google Scholar]
- 24.Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373–83. doi: 10.1016/0021-9681(87)90171-8. [DOI] [PubMed] [Google Scholar]
- 25.Elixhauser A, Steiner C, Harris DR, Coffey RM. Comorbidity measures for use with administrative data. Med Care. 1998;36(1):8–27. doi: 10.1097/00005650-199801000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Hidalgo CA, Blumm N, Barabasi AL, Christakis NA. A dynamic network approach for the study of human phenotypes. PLoS Comput Biol. 2009;5(4):e1000353. doi: 10.1371/journal.pcbi.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park J, Lee DS, Christakis NA, Barabasi AL. The impact of cellular networks on disease comorbidity. Mol Syst Biol. 2009;5:262. doi: 10.1038/msb.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leeper NJ, Kullo IJ, Cooke JP. Genetics of peripheral artery disease. Circulation. 2012;125(25):3220–8. doi: 10.1161/CIRCULATIONAHA.111.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chi YW, Jaff MR. Peripheral artery disease and genetics: is there a cause-and-effect relationship? Postgrad Med. 2010;122(4):170–6. doi: 10.3810/pgm.2010.07.2183. [DOI] [PubMed] [Google Scholar]
- 30.Masud R, Shameer K, Dhar A, Ding K, Kullo IJ. Gene expression profiling of peripheral blood mononuclear cells in the setting of peripheral arterial disease. J Clin Bioinforma. 2012;2:6. doi: 10.1186/2043-9113-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kullo IJ, Shameer K, Jouni H, Lesnick TG, Pathak J, Chute CG, et al. The ATXN2-SH2B3 locus is associated with peripheral arterial disease: an electronic medical record-based genome-wide association study. Front Genet. 2014;5:166. doi: 10.3389/fgene.2014.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kullo IJ, Fan J, Pathak J, Savova GK, Ali Z, Chute CG. Leveraging informatics for genetic studies: use of the electronic medical record to enable a genome-wide association study of peripheral arterial disease. J Am Med Inform Assoc. 2010;17(5):568–74. doi: 10.1136/jamia.2010.004366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arain FA, Ye Z, Bailey KR, Chen Q, Liu G, Leibson CL, et al. Survival in patients with poorly compressible leg arteries. Journal of the American College of Cardiology. 2012;59(4):400–7. doi: 10.1016/j.jacc.2011.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu W, Wulf A, Liu T, Khoury MJ, Gwinn M. Gene Prospector: an evidence gateway for evaluating potential susceptibility genes and interacting risk factors for human diseases. BMC bioinformatics. 2008;9:528. doi: 10.1186/1471-2105-9-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin BK, Clyne M, Walsh M, Gomez O, Yu W, Gwinn M, et al. Tracking the epidemiology of human genes in the literature: the HuGE Published Literature database. Am J Epidemiol. 2006;164(1):1–4. doi: 10.1093/aje/kwj175. [DOI] [PubMed] [Google Scholar]
- 36.Yu W, Gwinn M, Clyne M, Yesupriya A, Khoury MJ. A navigator for human genome epidemiology. Nature Genetics. 2008;40(2):124–5. doi: 10.1038/ng0208-124. [DOI] [PubMed] [Google Scholar]
- 37.Jahchan NS, Dudley JT, Mazur PK, Flores N, Yang D, Palmerton A, et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 2013;3(12):1364–77. doi: 10.1158/2159-8290.CD-13-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sirota M, Dudley JT, Kim J, Chiang AP, Morgan AA, Sweet-Cordero A, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med. 2011;3(96):96ra77. doi: 10.1126/scitranslmed.3001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dudley JT, Deshpande T, Butte AJ. Exploiting drug-disease relationships for computational drug repositioning. Brief Bioinform. 2011;12(4):303–11. doi: 10.1093/bib/bbr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iskar M, Zeller G, Blattmann P, Campillos M, Kuhn M, Kaminska KH, et al. Characterization of drug-induced transcriptional modules: towards drug repositioning and functional understanding. Mol Syst Biol. 2013;9:662. doi: 10.1038/msb.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giri K, Shameer K, Zimmermann MT, Saha S, Chakraborty PK, Sharma A, et al. Understanding protein-nanoparticle interaction: a new gateway to disease therapeutics. Bioconjug Chem. 2014;25(6):1078–90. doi: 10.1021/bc500084f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saha S, Xiong X, Chakraborty PK, Shameer K, Arvizo RR, Kudgus RA, et al. Gold Nanoparticle Reprograms Pancreatic Tumor Microenvironment and Inhibits Tumor Growth. ACS Nano. 2016;10(12):10636–51. doi: 10.1021/acsnano.6b02231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drawnel FM, Boccardo S, Prummer M, Delobel F, Graff A, Weber M, et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 2014;9(3):810–21. doi: 10.1016/j.celrep.2014.09.055. [DOI] [PubMed] [Google Scholar]
- 44.Bleda S, De Haro J, Florez A, Varela C, Esparza L, Acin F. Long-term pleiotropic effect of statins upon nitric oxide and C-reactive protein levels in patients with peripheral arterial disease. Heart Asia. 2011;3(1):130–4. doi: 10.1136/heartasia-2011-010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barbato JE, Tzeng E. Nitric oxide and arterial disease. J Vasc Surg. 2004;40(1):187–93. doi: 10.1016/j.jvs.2004.03.043. [DOI] [PubMed] [Google Scholar]
- 46.Chu LH, Annex BH, Popel AS. Computational drug repositioning for peripheral arterial disease: prediction of antiinflammatory and pro-angiogenic therapeutics. Front Pharmacol. 2015;6:179. doi: 10.3389/fphar.2015.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang L, Hua Z, Xiao H, Cheng Y, Xu K, Gao Q, et al. CRISPR/Cas9-mediated ApoE-/- and LDLR-/- double gene knockout in pigs elevates serum LDL-C and TC levels. Oncotarget. 2017;8(23):37751–60. doi: 10.18632/oncotarget.17154. [DOI] [PMC free article] [PubMed] [Google Scholar]