

Abstract
A series of arylnaphthalene lignan lactones based on the structure of the phyllanthusmins, a class of potent natural products possessing diphyllin as the aglycone, has been synthesized and screened for activity against multiple cancer cell lines. SAR exploration was performed on both the carbohydrate and lactone moieties of this structural class. These studies have revealed the importance of functionalization of the carbohydrate hydroxy groups with both acetylated and methylated analogues showing increased potency relative to those with unsubstituted sugar moieties. In addition, the requirement for the presence and position of the C-ring lactone has been demonstrated through reduction and selective re-oxidation of the lactone ring. The most potent compound in this study displayed an IC50 value of 18 nM in an HT-29 assay with several others ranging from 50 to 200 nM. In an effort to elucidate their potential mechanism(s) of action, the DNA topoisomerase IIa inhibitory activity of the most potent compounds was examined based on previous reports of structurally similar compounds, but does not appear to contribute significantly to their antiproliferative effects.
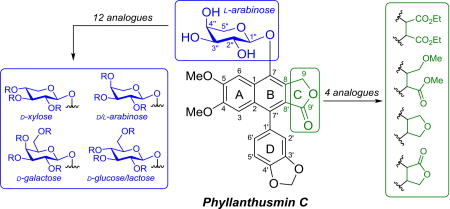
Graphical Abstract
1. Introduction
The genus Phyllanthus has historically been a rich source of natural products possessing diverse chemical structures and biological activities.1 Building in part upon this diversity, a recent effort to identify novel compounds with cytotoxic activity from Phyllanthus poilanei by Kinghorn and coworkers led to the isolation of two new natural products, phyllanthusmins D and E (Fig. 1),2 along with the previously reported phyllanthusmins A–C.3 The isolated phyllanthusmins displayed promising potent antiproliferative activity against various cancer cell lines, with phyllanthusmin D displaying the most potent activity with an IC50 value of 0.17 µM against HT-29 colon carcinoma cells and a semisynthetic analogue, 2”-acetyl-phyllanthusmin D, also exhibiting similarly potent activity in the same cell line (IC50 = 0.11 µM).2
Figure 1.

Phyllanthusmins A–E along with structurally related arylnaphthalene lignan lactones (A) and aryletralin lignan lactones (B).
From a structural perspective, the phyllanthusmins represent a subset of the arylnaphthalene lignan lactone class of natural products (Fig. 1A). Phyllanthusmins B–E are diphyllin glycosides possessing substituted arabinose units linked via a glycosidic bond to the C7 phenol of diphyllin, the aglycone portion of the molecule and a natural product itself.4 Likewise, phyllanthusmin A is also built upon a diphyllin-like core, but possesses a hydroxy group at the C4 position rather than a methoxy substituent and, more importantly, does not contain the glycosidic linkage seen in other members of this group. When considering the promising activity of these compounds, it is interesting to note that diphyllin has also been shown to display cytotoxic,5,6 antimicrobial,7 and antiviral8 activities. Other members of the arylnaphthalene lignan class and the closely related aryltetralin lignan class of natural products have also garnered significant interest due to the range of biological activities possessed by their constituents, including cytotoxic,9–13 antioxidant,14,15 antiviral,16–18 anti-inflammatory,19 cardioprotective,20,21 insecticidal,22 and neuroprotective23 properties. The relationship between the arylnaphthalene and aryltetralin classes of compounds is of interest due to their deceptive structural similarities as seen in Figure 1. The two classes differ, however, in the oxidation state of the B ring, imparting clear conformational differences that potentially lead to different modes of action. Despite this fact, both the aryltetralins, including etoposide and teniposide (Fig. 1B), and some arylnapthalenes like daurinol have been shown to possess inhibitory activity against the same biological target, DNA topoisomerase II, albeit via different interactions with the enzyme as either topoisomerase poisons or catalytic inhibitors, respectively.24,25 In addition to mechanistic considerations, the development of etoposide and teniposide, two nearly identical clinically approved aryltetralin drugs, also points to the importance of optimization of the glycosidic moiety in these compounds as these drugs display unique pharmacological properties26 and an entirely different mechanism of action than podophyllotoxin, the aglycone from which they are derived.27
Previous studies of several other structurally related arylnaphthalene compounds have also indicated the critical importance of the glycosidic sugar moiety in mediating general antiproliferative activities within the series of arylnaphthalene lignans.12,28,29 In addition to phyllanthusmins B–E,2 other diphyllin glycosides, including tuberculatin5 and D11,29 have also demonstrated more potent in vitro activity than diphyllin, their aglycone, in a variety of cancer cell lines. The impact of the glycosidic moiety can also be observed in the relative, although highly varied, antiproliferative activities of related natural products containing an array of substituted carbohydrate groups attached to diphyllin. The earliest examples of these are cleistanthins A30 and B31 (as well as cleistanthin A methyl ether)11 which have been thoroughly investigated for their antiproliferative properties as well as their inherent toxicities in rats.32–37 Acutissimalignan A, which possesses a functionalized arabinose moiety analogous to the isolated phyllanthusmins, was also recently found to possess highly potent activity against HT-29 cells following its isolation by the Kinghorn group with an IC50 value of 19 nM.38,39 Other diphyllin glycosides that have also been found to be cytotoxic towards various cancer cell lines include patentiflorin A,9 procumbenoside A,5 and cleistanthoside A tetraacetate.11,40–43
Considering the potential influence of the carbohydrate group for antiproliferative activity, this moiety was identified as a useful starting point for the exploration of the structure-activity relationships in the phyllanthusmin class of natural products. Expanding on the structure activity relationship studies reported by Shi et al and Zhao et al for this class of compounds,12,28 in this study we describe the synthesis of analogues containing various mono- and disaccharide units and the modification of the C-ring lactone in the aglycone portion of the molecules. The library of arylnaphthalene lignan glycosides generated during these studies has provided important structure-activity relationship (SAR) data across a series of cell lines previously not investigated with this class of compounds, including HT-29 (colon), MDA-MB 435/231 (breast), and OVCAR3 (ovarian cancer), as well as insight into the role of DNA topoisomerase IIα as a target for their antiproliferative activity.
2. Results and Discussion
Based on the relatively straightforward retrosynthetic disconnection of diphyllin from the carbohydrate moiety via the glycosidic linkage, the synthesis of analogues of the phyllanthusmin class of natural products, like other diphyllin glycosides, was predicated on the ability to efficiently produce diphyllin in sufficient quantities for derivatization. Although numerous elegant recent approaches to the synthesis of similar arylnaphthalene core ring systems have been reported, they are either not specifically amenable to the synthesis of diphyllin or are potentially limited by scale-up cost.44–51 The route of Charlton and coworkers,52 however, has previously been employed12,28,53–55 for the synthesis of diphyllin and related analogues based on the ease of access to the requisite starting materials and the overall efficiency of the route. Employing only minor modifications to the reported procedure, gram scale quantities of diphyllin (5) have been produced (Scheme 1). The most significant modification to the reported procedure was made based on initial difficulties observed during the isolation and purification of diphyllin following the final reduction of diester 4 using the reported sodium borohydride reduction conditions. Despite early work that indicated the potential for over-reduction of the desired lactone product in the presence of lithium aluminum hydride (LAH),56 there was literature precedent indicating that LAH could be utilized under careful dropwise inverse addition44,57 or portion-wise addition55 conditions to affect this type of transformation. In the present study, portion-wise addition of 4 equivalents of LAH cleanly and efficiently resulted in the regioselective reduction of diester 4 within five minutes as observed by thin layer chromatography. Upon workup, trituration of the crude product with methanol cleanly provided diphyllin. The overall yield of the five step sequence to prepare diphyllin ranged from 30–41% and required minimal chromatographic purification over the course of the synthesis. This method, therefore, facilitated the preparation of the aglycone in sufficient quantities for subsequent SAR studies.
Scheme 1.

Synthesis of diphyllin glycoside analogues 6a–c possessing an arabinose ring.
With diphyllin in hand, a series of simple glycosyl bromides were prepared from the corresponding carbohydrates for glycosylation of the free phenol of the aglycone. The brominated substrates were immediately subjected to phase transfer glycosylation with diphyllin, following the same procedure implemented by Zhao and coworkers.28 Utilizing this approach, the stereochemistry of the glycosidic linkage was established through neighboring group participation of the adjacent acetyl group, necessitating the presence of a C2” equatorial alcohol in all starting materials to ultimately achieve the desired stereochemical control in the glycosylation reaction. For this reason, l-arabinose, d-xylose, d-glucose, d-galactose, d-arabinose, and lactose could be effectively utilized as starting materials and were subjected to this sequence of reactions to provide phyllanthusmin (PHY) analogues 6a, 7a, 8a, 9a, 10, and 11, respectively (Scheme 1 and Fig. 2). Once introduced onto diphyllin, the carbohydrate substitution was then further manipulated. This was accomplished via hydrolysis of the acetylated compounds 6a–9a to unmask the free hydroxy groups and generate compounds 6b–9b as illustrated in Scheme 1 for compound 6. Subsequently, the newly revealed alcohols in 6b and 7b were also methylated in the presence of base to provide analogues 6c and 7c.
Figure 2.

Chemical structures of compounds 7–11.
During the methylation of compounds 6b and 7b to prepare compounds 6c and 7c (the natural product cleistanthin A methyl ether)11, however, the formation of an additional minor byproduct was also observed in each case. The minor “impurity,” although formed in only very small quantities (<2% yield), was isolated in the xylose series and was determined to be the product of ring opening of the C-ring lactone, compound 12 (Scheme 2A). Presumably, this product is generated in the presence of trace amounts of methanol (or water) through nucleophilic attack of the carbonyl and alkylation of the resulting primary alcohol.
Scheme 2.

Preparation of C-ring lactone analogues 12, 13, 17, and 18.
This result, which indicates the potential instability of the lactone ring from a chemical and/or metabolic perspective, prompted further derivatization of the C-ring to probe its significance in the antiproliferative activity of these compounds. To this end, the ample supply of the precursor to diphyllin 4 facilitated generation of diester 13, an analogue lacking the lactone ring, via phase-transfer glycosylation (Scheme 2B). Additionally, complete removal of the lactone carbonyl oxygen of 6a, leaving the tetrahydrofuran ring intact, was also explored. In order to efficiently access the desired ether functionality, the C7 phenol of diphyllin (5) was first protected as the silyl ether (Scheme 2C) and the lactone functionality was subsequently reduced utilizing a method developed by Sakai and coworkers58 that proceeds via an InBr3/Et3SiH radical mediated pathway. With the cyclic ether 14 in hand, synthesis of the desired analogue 17 was contingent on the deprotection of the silyl ether and the glycosylation of the resulting phenol to introduce the peracetylated arabinose moiety. In practice, this deprotection was readily accomplished using TBAF in THF to generate the free phenol 15, which was then glycosylated to give 17. Interestingly, however, when the deprotection reaction was run using cesium carbonate in DMF/H2O and left open to the air, in addition to the cleavage of the silyl ether, an unexpected oxidation also regiospecifically took place at the C9 position, resulting in transposition of the lactone carbonyl and generation of the C-ring type-I lactone 16 observed in other arylnaphthalene lignan lactone natural products including justicidin C.59 The position of the lactone carbonyl in 16 was confirmed using a 1D Selective Gradient NOESY NMR experiment (Supplementary Information). Similar transformations resulting in the regioselective oxidation of arylnaphthalene systems have recently also been reported by Mondal et al.60 and Yamamoto et al.,51 with the type-I lactone being produced preferentially in both cases. Glycosylation of the free phenol of 16, led to formation of compound 18, containing the transposed carbonyl, but otherwise analogous to compound 6a.
The antiproliferative activities of the synthesized analogues were first evaluated in colon carcinoma (HT-29), breast (MDA-MB-435 and MDA-MB-231; see Supplementary Info.), and ovarian (OVCAR3) cancer cell lines (Table 1). Structure-activity relationships were relatively consistent within both the HT-29 and MDA-MB-435 cell lines. By comparison, however, the MDA-MB-231 cell line was less susceptible to this series of compounds. The OVCAR3 cell line showed similar SAR trends to both HT-29 and MDA-MB-435, albeit with several potentially interesting outliers. In particular, analogues 7c and 8a, which display exceptional potency in HT-29 cells (0.018 and 0.040 µM), show a significant decrease in potency (>400 fold) against the OVCAR3 cells (8.0 and >10 µM). The observed discrepancies between cell lines, in combination with the higher potency observed in OVCAR3 versus HT-29 for compounds 7b and 12, suggest selectivity for specific cell lines based on the expression levels of potential cellular targets. Alternatively, this data could also point to the potential existence of multiple cellular targets within the tested cell lines. With the target(s) unknown at this time, the complete data set obtained for the test compounds against the HT-29 cell line has been utilized as the primary point of comparison and has been useful for drawing conclusions about structure-activity relationships within this series.
Table 1.
Phyllanthusmin analogues and corresponding antiproliferative data in HT-29 and OVCAR3 cell lines expressed as IC50 values in µM.
| Cmpd | Carbohydrate | HT-29 | OVCAR3 |
|---|---|---|---|
| 6a | l-arabinose (−OAc) | 0.14 | 0.29 |
| 6b | l-arabinose (−OH) | 3.2 | 4.8 |
| 6c | l-arabinose (−OMe) | 0.13 | 0.81 |
| 7a | d-xylose (−OAc) | 0.16 | 0.57 |
| 7b | d-xylose (−OH) | 1.4 | 0.69 |
| 7c | d-xylose (−OMe) | 0.018 | 7.6 |
| 8a | d-glucose (−OAc) | 0.043 | >10 |
| 8b | d-glucose (−OH) | 4.8 | >10 |
| 9a | d-galactose (−OAc) | 0.044 | 0.36 |
| 9b | d-galactose (−OH) | >10 | >10 |
| 10 | d-arabinose (−OAc) | 0.046 | 0.37 |
| 11 | Lactose (−OAc) | 2.0 | >10 |
| 12 | d-xylose (−OMe) | 1.7 | 0.45 |
| 13 | l-arabinose (−OAc) | >10 | >10 |
| 17 | l-arabinose (−OAc) | 1.4 | >10 |
| 18 | l-arabinose (−OAc) | 0.47 | >10 |
| Taxol | 0.011 | 0.011 | |
| Etoposide | 15 | 2.9 |
Against the HT-29 cells, the IC50 values for the acetylated diphyllin glycoside analogues 6a–9a, 10 and 11 ranged in potency from 0.05 – 2.0 µM. Within this series, compounds containing a C5-hydroxymethyl group were found to have increased potency over those lacking a substituent at this position (e.g. glucosylpyranoside 8a and galactosylpyranoside 9a vs xylosylpyranoside 7a and arabonsylpyranoside 6a). It is interesting to note that compound 10, possessing the d-arabinose group, displayed a slightly higher potency against the HT-29 cells than 6a, the compound with the l-arabinose group found in the natural products. Additionally, the disaccharide lactose analogue 11 resulted in the most significant loss in activity among the acetylated analogues. These observations indicate that, while a variety of different monosaccharide-containing diphyllin glycosides are well tolerated, certain disaccharide moieties may be too large to effectively elicit a biological response.
This series of synthesized analogues also allowed the relative substitution of the pendant hydroxy groups to be assessed. Compared to the acetylated compounds 6a–9a, the corresponding free alcohol containing compounds 6b–9b were much less active against the HT-29 cells. In fact, the glucose and galactose analogues 8b and 9b were found to be only weakly active or even inactive (defined in this study as having an IC50 of greater than 10 µM) in both the HT-29 and OVCAR3 cell lines. These results highlight the importance of the substitution of the hydroxy groups on the sugar moieties in this series for increased antiproliferative activity. In addition, the present findings parallel previous biological results obtained from the series of isolated phyllanthusmins2 in which increased potency correlated with a greater relative degree of substitution on the arabinose moiety. To support this argument, subsequent permethylation of these hydroxy groups resulted in restoration or even improvement of the antiproliferative activities. Analogue 7c, the permethylated xylose derivative, was not only the most potent analogue synthesized in this study (0.018 µM against HT-29), but upon comparison to 6c (0.13 µM) revealed a potential preference for equatorial (vs axial) stereochemistry at C4”. A similar correlation has also been found with HCT116 cells in a study by Zhao et al.28
Investigation into the SAR of the C-ring lactone of the aglycone has also determined the importance of this moiety as a part of the pharmacophore. The ring opened compounds 12 and 13 both showed a dramatic loss in potency in the HT-29 cells in comparison to their lactone counterparts (7c and 6a). Similarly, removal of the carbonyl moiety (compound 17) also reduced potency (in comparison to 6a). Transposition of the carbonyl in compound 18 resulted in a compound with better potency than cyclic ether 17, but still showed a 3-fold loss as compared to 6a. These results indicate that the lactone carbonyl is an important structural motif present in the diphyllin core of the phyllanthusmins required for more potent biological activity potentially due to the electronic contribution, the induced geometry for putative binding to the target site, or a combination of the two effects.
Based on the structural similarity of the synthesized compounds to the aryltetralin etoposide and the arynaphthalene daurinol, four of the most potent analogues in the tested cell lines (6a, 6c, 7c, and 8a) were selected for subsequent investigation of inhibitory activity against DNA topoisomerase IIα and for antiproliferative activity in parental human leukemia K562 cells compared to an etoposide-resistant clonal cell line, K/VP.5, with reduced levels of DNA topoisomerase II.61 These compounds comprise a useful cross-section of the analogues, sampling a variety of glycone scaffolds and substitution, as represented by both peracetylated and permethylated derivatives of the arabinose 6, xylose 7, and glucose 8 analogues.
These compounds were first examined in an in vitro assay designed to look at the effects of purified DNA topoisomerase IIα on cleavage of supercoiled pBR322 DNA. As reported previously for the natural phyllanthusmins,2 the synthetic analogues did not induce DNA topoisomerase II-mediated DNA cleavage (data not shown). However, using an assay to assess DNA topoisomerase II catalytic activity, inhibition of DNA topoisomerase IIα mediated pBR322 DNA relaxation could be observed (Figure 3) for compounds 8a, 6a, and 7c at 50 and 100 µM. This activity was more pronounced than that of diphyllin itself, although the compounds were not as potent as etoposide (100 µM). Compound 6c does not appear to show any activity in the DNA relaxation assay and, therefore, suggests some structural specificity required for inhibition of DNA topoisomerase IIα catalytic activity. Based on their observed inhibitory activity against isolated enzyme, the antiproliferative activity of these compounds was evaluated in etoposide sensitive K562 cells and in acquired etoposide resistant K/VP.5 cells, which contain ~1/5 the level of DNA topoisomerase IIα compared to parental K562 cells and which have been shown to be 30-fold resistant to etoposide in direct growth inhibition assays.61,62 The antiproliferative activities observed for the four compounds in both cell lines were nearly identical (Table 2), strongly suggesting that direct DNA topoisomerase IIα inhibition is not likely the primary mechanism of action through which these compounds mediate their effects on cancer cells.
Figure 3.

DNA topoisomerase IIα mediated pBR322 DNA relaxation in the absence or presence of etoposide, diphyllin, or PHY analogue.
Table 2.
Antiproliferative data (IC50, µM) for PHY analogues in etoposide sensitive K562 cells and isogenic etoposide resistant K/VP.5 cells, where “n” is the number of replicate experiments performed on separate days.
| Compound | K562 | K/VP.5 | Relative Resistancea |
n |
|---|---|---|---|---|
| etoposide | 1.22 | 35.36 | 28.98 | 1 |
| 8a | 0.72 ± 0.10 | 0.69 ± 0.05 | 0.96 | 3 |
| 6a | 0.85 ± 0.09 | 0.92 ± 0.13 | 1.08 | 4 |
| 6c | 1.26 ± 0.2 | 1.28 ± 0.44 | 1.02 | 4 |
| 7c | 0.47 ± 0.06 | 0.43 ± 0.09 | 0.91 | 6 |
| diphyllin | 2.38 ± 0.37 | 2.31 ± 0.35 | 0.97 | 3 |
Relative resistance is calculated by the ratio of IC50 values in K/VP.5 compared to K562 cells.
3. Conclusions
Members of the phyllanthusmin class of natural products represent promising lead compounds based on their potent cytotoxic properties. This study has demonstrated that variation of the carbohydrate portion of these compounds is well tolerated with regard to observed antiproliferative activities and establishes a foundation for future studies within this class of compounds. The mechanism(s) of action of this class remains to be determined, although evidence presented here and elsewhere suggests that DNA topoisomerase IIα activity is not a significant contributor.2 The selectivity observed for some of the compounds across the cell lines tested suggests that specific analogues may affect multiple targets within the various cancer subtypes or are dramatically affected by their relative expression levels within cells. The potential flexibility afforded by the structural variation of the carbohydrate moiety may ultimately prove to be useful for the development of novel agents with optimal physicochemical properties and could potentially be exploited to promote target selectivity in future generations of analogues.
4. Experimentals
4.1. Chemistry
All reactions were performed at room temperature, under an argon atmosphere, with reagent grade solvents unless otherwise stated. Commercially available chemicals were used without further purification. When applicable, dry solvents (THF, DCM, and DMF) were obtained from an Innovative Technology PureSolv system and oven-dried syringes were used to transfer air and moisture sensitive liquids. Reactions were monitored by thin layer chromatography (TLC) using aluminum backed pre-coated silica gel plates (w/UV254, 200 µm) from Sorbtech, using UV light as the visualizing agent and ceric ammonium molybdate (CAM) and heat as a developing agent. Flash column chromatography was carried out using Sorbtech 40–63 µm silica gel with eluent ratios expressed in v/v. All 1H- and 13C-NMR spectra were recorded at 300 K on a Bruker AV300, AVIII400, or DRX400 MHz NMR with tetramethylsilane as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm) with multiplicities when applicable (s = singlet, d = doublet, t = triplet dd = doublet of doublets, m = multiplet, etc.), and are calibrated using the residual undeuterated solvent peak (CDCl3: δ 7.26 ppm 1H NMR, 77.16 ppm 13C NMR; acetone-d6: δ 2.05 ppm 1H NMR, 29.84 ppm 13C NMR; DMSO-d6: δ 2.50 ppm 1H NMR, 39.52 ppm 13C NMR; CD3OD: δ 3.31 ppm 1H NMR, 49.00 ppm 13C NMR). As previously described by Charlton et al.,51 the hindered rotation of arylnaphthalene lignans about the C1’–C7’ bond results in observation of “additional” peaks in several 1H and 13C NMR spectra, an effect that appears to be highly solvent dependent. High resolution electrospray ionization mass spectra (HRMS-ESI) were recorded on a Thermo LTQ Orbitrap mass spectrometer. Melting points were recorded using a Thomas Hoover Melting Point Capillary Apparatus. Infrared (IR) absorption spectra were recorded on Thermo-Nicolet 6700 FTIR. Prior to biological testing, purity was determined using an ACE Excel 3 C18-PFP (150 × 4.6 mm) column on a Shimadzu HPLC system (flow rate: 1 mL/min).
4.1.1. Preparation of diphyllin (5)
Diphyllin was synthesized according to the method of Charlton and coworkers,52 albeit with a slight modification to the last step. For the final reduction step, the diester 4 (5.86 g, 12.5 mmol) was dissolved in dry THF (250 mL) and lithium aluminum hydride (1.90 g, 50 mmol) was added portion-wise at 0 °C. The reaction mixture was allowed to warm to room temperature over a 5 minute period prior to being cooled back to 0 °C and quenched via the dropwise addition of deionized water. Once the evolution of gas was no longer observed upon further addition of water, the pH of the mixture was brought to ~2 with the slow addition of 2M HCl. The resulting aqueous mixture was then extracted three times with EtOAc. The combined organic layers were washed with brine, dried with sodium sulfate, and concentrated under reduced pressure. Trituration of the resulting crude solid with methanol provided diphyllin (3.17 g, 67%) as a pale yellow solid. The 1H and 13C spectral characteristics were identical to those previously published.52 1H NMR (400 MHz, Acetone-d6) δ 9.24 (br s, 1H), 7.69 (s, 1H), 7.09 (s, 1H), 6.96 (d, J = 7.9 Hz, 1H), 6.85 (d, J = 1.6 Hz, 1H), 6.80 (dd, J = 7.9, 1.7 Hz, 1H), 6.09 (d, J = 1.0 Hz, 1H), 6.08 (d, J = 1.0 Hz, 1H), 5.37 (s, 2H), 4.00 (s, 3H), 3.73 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 170.27, 152.21, 151.35, 148.31, 148.05, 145.69, 131.70, 131.19, 130.24, 124.80, 124.53, 122.83, 119.99, 112.01, 108.59, 106.87, 102.09, 101.34, 66.99, 56.09, 55.74; HRMS-ESI calcd for C21H16O7 (M+Na)+ 403.07882, found 403.07855.
4.1.2. Synthesis of per-acetylated diphyllin glycosides
4.1.2.1. General procedure for bromination/glycosylation
7-O-(2”,3”,4”,6”-Tetra-O-acetyl-β-d-glucopyranosyl) diphyllin (8a)
To 1,2,3,4,6-penta-O-acetyl-d-glucopyranose (1.678 g, 4.30 mmol) dissolved in DCM (10.75 mL) was added hydrogen bromide (33% in acetic acid, 2.97 mL) dropwise at 0 °C. The reaction mixture was then allowed to warm to room temperature and stir for an additional 4 h. The reaction was quenched with water and the aqueous layer was extracted with DCM. The organic layers were combined and washed with water, saturated aqueous NaHCO3, brine, and then dried with sodium sulfate and concentrated under reduced pressure to afford a clear viscous oil that was immediately added to a biphasic mixture of diphyllin (1.090 g, 2.87 mmol) and TBAB (0.933 g, 2.89 mmol) in CHCl3 (100 mL) and aqueous NaOH (0.1 M, 100 mL) at 40 °C. The mixture was maintained at 40 °C overnight. After cooling to room temperature, the layers were separated and the aqueous layer was extracted three times with CHCl3. The combined organic layers were washed with brine, dried with sodium sulfate, and concentrated under reduced pressure. Flash chromatography (silica gel, 0.3% → 1% MeOH in CHCl3) afforded compound 8a (1.928 g, 95%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.52 (s, 1H), 7.07 (s, 1H), 6.96 (d, J = 7.8 Hz, 1H), 6.82 (br s, 1H), 6.79 (br d, J = 7.9 Hz, 1H), 6.09 (br s, 1H), 6.05 (br s, 1H), 5.56 – 5.48 (m, 2H), 5.40 (br d, J = 14.8, 1H), 5.33 (t, J = 9.5 Hz, 1H), 5.23 – 5.14 (m, 2H), 4.27 (dd, J = 12.4, 6.0 Hz, 1H), 4.14 (br d, J = 12.4 Hz, 1H), 4.06 (s, 3H), 3.80 (s, 3H), 3.79 – 3.75 (m, 1H), 2.11 (s, 3H), 2.062 (s, 3H), 2.057 (s, 3H), 2.04 (br s, 3H); 13C NMR (101 MHz, CDCl3) δ 170.50, 170.27, 169.52, 169.51, 169.45, 152.14, 150.58, 147.73, 147.71, 147.69, 144.13, 144.12, 136.53, 130.87, 128.22, 127.06, 127.05, 126.30, 126.28, 123.68, 123.63, 119.32, 110.75, 110.73, 108.37, 108.34, 106.31, 101.41, 101.00, 100.99, 100.60, 100.58, 72.69, 71.75, 68.26, 66.93, 62.03, 56.32, 55.98, 20.99, 20.70, 20.68; HRMS-ESI calcd for C35H34O16 (M+Na)+ 733.17391, found 733.17317.
4.1.2.2. 7-O-(2”,3”,4”-Tri-O-acetyl-α-l-arabinopyranosyl) diphyllin (6a)
Starting from 1,2,3,4-tetra-O-acetyl-l-arabinopyranose and following the general procedure for bromination/glycosylation, flash chromatography (silica gel, 45% → 52% EtOAc in hexanes) afforded 6a (702 mg, 84%) as a white solid: (c 0.24, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.54 (s, 1H), 7.08 (s, 1H), 6.96 (d, J = 7.8 Hz, 1H), 6.84 – 6.76 (m, 2H), 6.09 (br s, 1H), 6.05 (br s, 1H), 5.70 (dd, J = 9.5, 7.0 Hz, 1H), 5.48 (d, J = 14.8 Hz, 1H), 5.41 (br d, J = 14.8 Hz, 1H), 5.39 – 5.35 (m, 1H), 5.17 (dd, J = 9.5, 3.5 Hz, 1H), 5.09 (d, J = 7.0 Hz, 1H), 4.19 (dd, J = 13.0, 3.1 Hz, 1H), 4.09 (s, 3H), 3.80 (s, 3H), 3.72 (br d, J = 12.7 Hz, 1H), 2.22 (s, 3H), 2.11 (s, 3H), 2.08 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 170.31, 170.17, 169.60, 169.55, 152.08, 150.50, 147.65, 144.28, 136.26, 130.85, 128.33, 127.31, 126.28, 123.66, 123.64, 119.41, 110.76, 108.30, 106.35, 101.48, 101.37, 100.61, 100.60, 70.26, 69.53, 67.41, 66.98, 64.09, 56.35, 55.95, 21.07, 21.04, 20.78; HRMS-ESI calcd for C32H30O14 (M+Na)+ 661.15278, found 661.15218.
4.1.2.3. 7-O-(2”,3”,4”-Tri-O-acetyl-β-d-xylopyranosyl) diphyllin (7a)
Starting from 1,2,3,4-tetra-O-acetyl-d-xylopyranose and following the general procedure for bromination/glycosylation, flash chromatography (silica gel, 45% → 55% EtOAc in hexanes) afforded 7a (511 mg, 87%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.49 (s, 1H), 7.04 (s, 1H), 6.91 (d, J = 7.7 Hz, 1H), 6.80 – 6.70 (m, 2H), 6.06 (s, 1H), 6.01 (s, 1H), 5.44 (dd, J = 9.0, 7.3 Hz, 1H), 5.43 (d, J = 15.2 Hz), 5.38 (br d, J = 15.2 Hz, 1H), 5.30 (t, J = 8.9 Hz, 1H), 5.13 (d, J = 7.0 Hz, 1H), 5.11 (dd, J = 8.6, 5.1 Hz, 1H), 4.25 (dd, J = 11.8, 5.2 Hz, 1H), 4.05 (s, 3H), 3.78 (s, 3H), 3.41 (dd, J = 11.7, 9.3 Hz, 1H), 2.11 (s, 3H), 2.08 (s, 3H), 2.05 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 170.19, 169.94, 169.51, 169.50, 152.13, 150.53, 147.70, 147.68, 147.67, 144.12, 144.11, 136.48, 130.89, 128.28, 127.89, 127.88, 126.47, 126.45, 123.69, 123.66, 119.42, 119.42, 110.77, 108.35, 108.33, 106.39, 101.59, 101.39, 100.54, 100.52, 71.80, 71.49, 68.80, 66.89, 62.97, 56.37, 55.97, 20.98, 20.82, 20.82; HRMS-ESI calcd for C32H30O14 (M+Na)+ 661.15278, found 661.15210.
4.1.2.4. 7-O-(2”,3”,4”,6”-Tetra-O-acetyl-β-d-galactopyranosyl) diphyllin (9a)
Starting from 1,2,3,4,6-penta-O-acetyl-d-galactopyranose and following the general procedure for bromination/glycosylation, flash chromatography (silica gel, 50% EtOAc in hexanes) provided compound 9a (444 mg, 68%) as an off-white solid: 1H NMR (400 MHz, CDCl3) δ 7.54 (s, 1H), 7.05 (s, 1H), 6.94 (d, J = 7.9 Hz, 1H), 6.80 (s, 1H), 6.77 (br d, J = 4.9 Hz, 1H), 6.07 (br s, 1H), 6.03 (d, J = 5.2 Hz, 1H), 5.71 (dd, J = 10.2, 8.1 Hz, 1H), 5.55 (d, J = 14.7 Hz, 1H), 5.49 (d, J = 2.7 Hz, 1H), 5.39 (br d, J = 14.7 Hz, 1H), 5.20 – 5.11 (m, 2H), 4.26 – 4.14 (m, 2H), 4.07 (s, 3H), 4.07 – 4.03 (m, 1H), 3.79 (s, 3H), 2.23 (s, 3H), 2.07 (s, 3H), 2.03 (br s, 6H); 13C NMR (101 MHz, CDCl3) δ 170.46, 170.22, 170.08, 169.68, 169.47, 169.46, 152.07, 150.55, 147.66, 144.41, 136.13, 130.78, 128.30, 125.93, 125.90, 125.65, 125.62, 123.65, 123.62, 119.35, 119.34, 110.74, 108.32, 108.30, 106.23, 101.37, 101.14, 101.13, 100.69, 100.66, 71.99, 70.78, 69.19, 67.10, 67.03, 61.95, 56.38, 55.94, 21.10, 20.77, 20.69, 20.64; HRMS-ESI calcd for C35H34O16 (M+Na)+ 733.17391, found 733.17368.
4.1.2.5. 7-O-(2”,3”,4”-Tri-O-acetyl-α-d-arabinopyranosyl) diphyllin (10)
Starting from 1,2,3,4-tetra-O-acetyl-d-arabinopyranose and following the general procedure for bromination/glycosylation, flash chromatography afforded compound 10: (c 0.43, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.54 (s, 1H), 7.07 (s, 1H), 6.96 (d, J = 7.8 Hz, 1H), 6.85 – 6.76 (m, 2H), 6.10 (d, J = 1.4 Hz, 1H), 6.05 (br s, 1H), 5.70 (dd, J = 9.6, 7.0 Hz, 1H), 5.49 (d, J = 14.8 Hz, 1H), 5.42 (br d, J = 14.7 Hz, 1H), 5.39 – 5.36 (m, 1H), 5.17 (dd, J = 9.6, 3.5 Hz, 1H), 5.09 (d, J = 7.0 Hz, 1H), 4.19 (dd, J = 13.1, 3.1 Hz, 1H), 4.09 (s, 3H), 3.81 (s, 3H), 3.72 (dd, J = 13.0, 1.3 Hz, 1H), 2.22 (s, 3H), 2.11 (s, 3H), 2.08 (s, 3H); HRMS-ESI calcd for C32H30O14 (M+Na)+ 661.15278, found 661.15188.
4.1.2.6. 7-O-(Hepta-O-acetyl-β-d-galactopyranosyl-(1→4)-d-glucopyranosyl) diphyllin (11)
Starting from lactose octaacetate and following the general procedure for bromination/glycosylation, flash chromatography afforded compound 11: 1H NMR (400 MHz, CDCl3) δ 7.47 (s, 1H), 7.07 (s, 1H), 6.96 (dd, J = 7.8, 1.6 Hz, 1H), 6.83 – 6.77 (m, 2H), 6.09 (br s, 1H), 6.05 (br s, 1H), 5.50 (br d, J = 14.8, 1H), 5.46 – 5.40 (m, 2H), 5.39 – 5.36 (m, 1H), 5.34 – 5.29 (m, 1H), 5.16 – 5.09 (m, 2H), 5.01 – 4.96 (m, 1H), 4.52 (d, J = 7.9 Hz, 1H), 4.49 (br d, J = 10.3 Hz, 1H), 4.19 – 4.06 (m, 4H), 4.05 (s, 3H), 3.93 – 3.86 (m, 2H), 3.80 (s, 3H), 3.74 – 3.68 (m, 1H), 2.16 (s, 3H), 2.10 (s, 3H), 2.09 (s, 3H), 2.08 (s, 3H), 2.06 (d, J = 2.0 Hz, 3H), 2.04 (s, 3H), 1.97 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 170.50, 170.29, 170.22, 170.16, 169.80, 169.78, 169.48, 169.23, 152.15, 150.57, 147.73, 147.71, 147.70, 144.08, 136.45, 130.88, 128.26, 127.09, 126.11, 126.10, 123.70, 123.66, 119.38, 110.76, 108.38, 106.38, 101.41, 101.30, 100.83, 100.41, 100.38, 76.30, 73.63, 72.79, 72.04, 71.01, 70.99, 69.20, 66.99, 66.74, 62.12, 60.96, 56.34, 55.99, 21.04, 20.89, 20.78, 20.78, 20.77, 20.72, 20.63; HRMS-ESI calcd for C47H50O24 (M+Na)+ 1021.25842, found 1021.25795.
4.1.3. Synthesis of per-hydroxy diphyllin glycosides
4.1.3.1. General procedure for global deacetylation
7-O-α-l-arabinopyranosyl diphyllin (6b)
To a solution of 6a (324.4 mg, 0.511 mmol) in methanol (32 mL) was added K2CO3 (284 mg, 2.05 mmol) and the resulting mixture was stirred at room temperature for 30 minutes. The reaction was then quenched via the addition of aqueous HCl (2 M), filtered through filter paper, and concentrated under reduced pressure. Flash chromatography (silica gel, 2% → 10% MeOH in CHCl3) afforded compound 6b (259 mg, 99%) as an off white solid: 1H NMR (400 MHz, (CD3)2SO) δ 8.17 (s, 1H), 7.04 (d, J = 7.9 Hz, 1H), 6.98 (d, J = 2.3 Hz, 1H), 6.93 (br s, 1H), 6.80 (br d, J = 7.9, 1H), 6.13 (s, 2H), 5.81 – 5.76 (m, 1H), 5.53 (br d, J = 14.8 Hz, 1H), 5.45 (d, J = 15.0 Hz, 1H), 5.06 (d, J = 5.0 Hz, 1H), 4.79 (t, J = 7.1 Hz, 1H), 4.76 (d, J = 3.7 Hz, 1H), 3.94 (s, 3H), 3.84 (br dd, J = 12.2, 6.5 Hz, 1H), 3.77 (br d, J = 11.5 Hz, 1H), 3.70 (br s, 1H), 3.67 (s, 3H), 3.52 (br s, 1H), 3.44 (d, J = 11.5 Hz, 1H); 13C NMR (75 MHz, (CD3)2SO) δ 169.00, 151.39, 149.95, 146.90, 146.84, 144.57, 134.49, 134.44, 129.68, 128.87, 128.70, 128.24, 126.60, 123.57, 118.65, 110.84, 110.77, 107.91, 105.41, 104.82, 104.70, 101.86, 101.08, 72.24, 70.82, 67.25, 67.21, 67.03, 65.71, 65.65, 55.81, 55.19; HRMS-ESI calcd for C26H24O11 (M+Na)+ 535.12108, found 535.12058.
4.1.3.2. 7-O-β-d-Xylopyranosyl diphyllin (7b)
Starting from 7a and following the general procedure for global deacetylation, flash chromatography (2% → 10% MeOH in CHCl3) afforded compound 7b (67 mg, 88%) as a white solid: 1H NMR (400 MHz, (CD3)2SO) δ 8.12 (br s, 1H), 7.04 (d, J = 7.9 Hz, 1H), 6.99 (s, 0.5H), 6.98 (s, 0.5H), 6.93 (d, J = 1.6 Hz, 0.5H), 6.91 (d, J = 1.6 Hz, 0.5H), 6.80 (dd, J = 7.9, 1.8 Hz, 0.5H), 6.79 (dd, J = 7.9, 1.8 Hz, 0.5H), 6.12 (br s, 2H), 5.93 (dd, J = 5.4, 2.9 Hz, 1H), 5.51 (br d, J = 15.2 Hz, 1H), 5.44 (d, J = 15.1 Hz, 1H), 5.23 (d, J = 4.9 Hz, 1H), 5.07 (d, J = 4.8 Hz, 1H), 4.76 (dd, J = 7.6, 4.8 Hz, 1H), 3.94 (s, 3H), 3.81 (dd, J = 11.3, 5.2 Hz, 1H), 3.67 (s, 3H), 3.50 – 3.40 (m, 2H), 3.29 – 3.22 (m, 1H), 3.18 – 3.08 (m, 1H); 13C NMR (101 MHz, (CD3)2SO) δ 169.04, 151.47, 149.96, 146.94, 146.92, 146.89, 144.71, 144.68, 134.92, 134.89, 129.78, 129.74, 129.73, 129.65, 128.18, 126.70, 126.69, 123.59, 123.55, 118.69, 110.86, 110.75, 107.95, 107.92, 105.73, 105.68, 105.47, 101.64, 101.11, 79.16, 76.29, 73.59, 69.30, 67.00, 65.90, 55.82, 55.21; HRMS-ESI calcd for C26H24O11 (M+K)+ 551.09502, found 551.09371.
4.1.3.3. 7-O-β-d-Glucopyranosyl diphyllin (8b)
Starting from 8a and following the general procedure for global deacetylation, flash chromatography (2% → 10% MeOH in CHCl3) afforded compound 8b (49.9 mg, 98%) as a white solid: 1H NMR (400 MHz, (CD3)2SO) δ 8.19 (s, 0.5H), 8.18 (s, 0.5H), 7.04 (d, J = 7.9 Hz, 1H), 6.982 (s, 0.5H), 6.976 (s, 0.5H), 6.96 (d, J = 1.4 Hz, 0.5H), 6.92 (d, J = 1.6 Hz, 0.5H), 6.81 (dd, J = 8.1, 1.7 Hz, 0.5H), 6.79 (dd, J = 8.0, 1.7 Hz, 0.5H), 6.13 (br s, 2H), 5.99 (dd, J = 5.3, 2.9 Hz, 1H), 5.77 (d, J = 15.4 Hz, 1H), 5.48 (br d, J = 15.5, 1H), 5.25 (d, J = 5.0 Hz, 1H), 5.09 (d, J = 5.2 Hz, 1H), 4.77 – 4.72 (m, 1H), 4.70 (dd, J = 10.8, 5.4 Hz, 1H), 3.95 (s, 3H), 3.77 (br dd, J = 11.2, 4.7 Hz, 1H), 3.67 (s, 3H), 3.50 – 3.42 (m, 2H), 3.32 – 3.20 (m, 2H), 3.18 – 3.11 (m, 1H); 13C NMR (101 MHz, (CD3)2SO) δ 169.22, 151.46, 149.95, 146.96, 146.95, 146.90, 144.86, 144.83, 134.94, 134.88, 130.02, 129.88, 129.68, 128.29, 126.78, 126.74, 123.63, 123.57, 118.85, 110.94, 110.79, 107.97, 105.39, 105.12, 105.07, 101.75, 101.15, 77.28, 76.35, 73.79, 70.06, 67.42, 61.30, 55.86, 55.22; HRMS-ESI calcd for C27H26O12 (M+Na)+ 565.13165, found 565.13081.
4.1.3.4. 7-O-β-d-Galactopyranosyl diphyllin (9b)
Starting from 9a and following the general procedure for global deacetylation, flash chromatography afforded 9b as a white solid: 1H NMR (400 MHz, DMSO) δ 8.203 (s, 0.5H), 8.196 (s, 0.5H), 7.04 (d, J = 7.9 Hz, 1H), 6.983 (s, 0.5H), 6.979 (s, 0.5H), 6.95 (d, J = 1.4 Hz, 0.5H), 6.91 (d, J = 1.4 Hz, 0.5H), 6.83 – 6.77 (m, 1H), 6.13 (br s, 2H), 5.77 (dd, J = 5.3, 2.8 Hz, 1H), 5.70 (d, J = 15.3 Hz, 1H), 5.49 (br d, J = 15.2 Hz, 1H), 4.97 (d, J = 5.7 Hz, 1H), 4.71 (dd, J = 7.3, 6.4 Hz, 1H), 4.68 (dd, J = 9.6, 4.7 Hz, 1H), 4.61 (d, J = 4.3 Hz, 1H), 3.95 (s, 3H), 3.82 – 3.75 (m, 1H), 3.69 – 3.66 (m, 1H), 3.67 (s, 3H), 3.64 – 3.56 (m, 2H), 3.50 – 3.40 (m, 2H); 13C NMR (101 MHz, DMSO) δ 169.16, 151.40, 149.92, 146.92, 146.85, 145.00, 144.97, 134.77, 129.76, 129.67, 129.61, 128.29, 126.76, 123.60, 123.52, 118.80, 110.89, 110.73, 107.93, 105.69, 105.64, 105.40, 101.85, 101.09, 75.91, 73.09, 70.81, 68.43, 67.35, 60.97, 55.81, 55.19; HRMS-ESI calcd for C27H26O12 (M+H)+ 543.14970, found 543.14940.
4.1.4. Synthesis of per-methylated diphyllin glycosides
4.1.4.1. General procedure for global methylation
7-O-(2”,3”,4”-Tri-O-methoxy-α-l-arabinopyranosyl) diphyllin (6c)
Sodium hydride (43 mg, 1.072 mmol) was added to a solution of 6b (137.5 mg, 0.268 mmol) in DMF (3.58 mL) at 0 °C and stirred for 30 minutes prior to the dropwise addition of methyl iodide (133 µL, 2.144 mmol). The reaction was then stirred at 80 °C overnight. Upon cooling to room temperature, the reaction was quenched with water and extracted with a mixture of EtOAc/hexanes (1:1). The combined organic layers were washed with water, brine, and then dried with sodium sulfate and concentrated under reduced pressure. Flash column chromatography (65% → 75% EtOAc in hexanes) afforded compound 6c (107 mg, 72%) as an off white solid: 1H NMR (300 MHz, CDCl3) δ 7.92 (s, 1H), 7.08 (s, 1H), 6.95 (d, J = 7.6, 1H), 6.85 – 6.77 (m, 2H), 6.09 (d, J = 1.5 Hz, 1H), 6.04 (d, J = 1.4 Hz, 1H), 5.55 (br d, J = 15.2 Hz, 1H), 5.46 (br d, J = 15.2 Hz, 1H), 4.77 (d, J = 7.3 Hz, 1H), 4.14 (br dd, J = 13.0, 2.6 Hz, 1H), 4.07 (s, 3H), 3.81 (s, 3H), 3.80 (s, 3H), 3.78 – 3.73 (m, 1H), 3.66 – 3.63 (m, 1H), 3.57 (s, 3H), 3.53 (s, 3H), 3.34 (dd, J = 9.2, 3.4 Hz, 1H), 3.22 (br d, J = 12.4 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 169.99, 152.00, 150.30, 147.63, 144.63, 136.35, 130.94, 130.87, 130.83, 128.58, 127.24, 123.80, 123.68, 119.43, 110.91, 110.81, 108.34, 108.29, 106.36, 105.06, 101.35, 101.07, 82.58, 80.61, 74.49, 67.72, 62.40, 61.50, 58.00, 57.56, 56.30, 55.96; HRMS-ESI calcd for C29H30O11 (M+Na)+ 577.16803, found 577.16735.
4.1.4.2. 7-O-(2”,3”,4”-Tri-O-methoxy-β-d-xylopyranosyl) diphyllin (7c) and methyl 1-(benzo[d][1,3]dioxol-5-yl)-6,7-dimethoxy-3-(methoxymethyl)-4-(((2S,3R,4S,5R)-3,4,5-trimethoxytetrahydro-2H-pyran-2-yl)oxy)-2-naphthoate (12)
Starting from 7b and following the general procedure for global methylation, flash chromatography (5% EtOH, 1% EtOAc in Hexanes) afforded compound 7c (Rf = 0.23 in a 20:7.5:72.5 mixture of EtOAc/EtOH/Hex, 60 mg, 56%) as an off-white solid as well as byproduct 12 (Rf = 0.26 in a 20:7.5:72.5 mixture of EtOAc/EtOH/Hex, 2 mg, 1.7%) as a white solid. 7c: 1H NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 7.09 (s, 1H), 6.96 (br d, J = 7.2, 1H), 6.85 – 6.78 (m, 2H), 6.09 (br s, 1H), 6.05 (br s, 1H), 5.51 (d, J = 15.1, 1H), 5.42 (d, J = 15.0, 1H), 4.77 (d, J = 7.6 Hz, 1H), 4.08 (s, 3H), 4.05 – 3.99 (m, 1H), 3.83 (s, 3H), 3.81 (s, 3H), 3.68 (s, 3H), 3.49 (s, 3H), 3.43 – 3.35 (m, 2H), 3.22 (t, J = 8.7 Hz, 1H), 3.05 (dd, J = 11.6, 10.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 169.91, 152.10, 150.33, 147.66, 144.47, 136.57, 131.16, 131.13, 130.99, 128.51, 127.20, 123.77, 123.72, 119.42, 110.87, 110.83, 108.33, 106.46, 105.30, 101.37, 100.82, 85.67, 83.64, 79.53, 77.58, 77.16, 76.74, 67.46, 63.64, 61.42, 60.89, 58.92, 56.27, 55.98; HRMS-ESI calcd for C29H30O11 (M+Na)+ 577.16803, found 577.16722. 12: (c 0.09, CHCl3); IR νmax (KBr, cm−1): 2930, 2831, 1724, 1623, 1508; 1H NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 6.91 – 6.75 (m, 4H), 6.05 (br s, 1H), 6.02 (br s, 1H), 4.95 (dd, J = 12.9, 11.6 Hz, 1H), 4.77 – 4.68 (m, 2H), 4.04 (s, 3H), 3.94 (dd, J = 11.8, 5.4 Hz, 1H), 3.83 (s, 3H), 3.772 (s, 1.5H), 3.766 (s, 1.5H) 3.68 (s, 3H), 3.56 (s, 1.5H), 3.55 (s, 1.5H), 3.48 (s, 3H), 3.44 – 3.33 (m, 3H), 3.319 (s, 1.5H), 3.317 (s, 1.5H), 3.22 – 3.16 (m, 1H), 2.97 – 2.90 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 169.97, 169.94, 150.46, 150.45, 150.09, 148.65, 148.60, 147.47, 147.39, 147.10, 147.06, 133.71, 131.91, 130.57, 129.30, 129.25, 124.14, 124.05, 124.01, 123.58, 111.08, 110.74, 108.20, 108.15, 106.00, 105.98, 105.53, 102.26, 102.24, 101.24, 86.04, 84.32, 84.31, 79.74, 67.01, 63.78, 61.44, 60.95, 58.92, 58.36, 58.35, 56.17, 55.90, 55.88, 51.85, 51.84, 29.85; HRMS-ESI calcd for C31H36O12 (M+Na)+ 623.20990, found 623.21148; mp 76–78 °C.
4.1.5. Preparation of lactone (C-ring) analogues
4.1.5.1. Diethyl 1-(benzo[d][1,3]dioxol-5-yl)-6,7-dimethoxy-4-(((2S,3R,4S,5S)-3,4,5-triacetoxytetrahydro-2H-pyran-2-yl)oxy)naphthalene-2,3-dicarboxylate (13)
Starting from 1,2,3,4-tetra-O-acetyl-l-arabinopyranose and following the general procedure for bromination/glycosylation using 4 (the diester precursor to diphyllin) as the glycosidic acceptor, flash chromatography afforded compound 13 (105 mg, 45%) as a white solid: (c 0.20, CHCl3); IR νmax (KBr, cm−1): 3070, 2981, 2938, 2904, 2833, 2777, 1747, 1684, 1621; 1H NMR (300 MHz, CDCl3) δ 7.64 (s, 1H), 7.62 (s, 1H), 6.93-6.68 (m, 5H), 6.82 – 6.76 (m, 2H), 6.73 – 6.69 (m, 1H), 6.07 (d, J = 1.4 Hz, 1H), 6.06 (d, J = 1.4 Hz, 1H), 6.02 (d, J = 1.5 Hz, 1H), 6.01 (d, J = 1.5 Hz, 1H), 5.68 (dd, J = 7.0, 1.4 Hz, 1H), 5.65 (dd, J = 6.8, 1.4 Hz, 1H), 5.32 – 5.27 (m, 2H), 5.16 (d, J = 6.5 Hz, 1H), 5.14 (d, J = 6.5 Hz, 1H), 5.13 (dd, J = 3.3, 1.5 Hz, 1H), 5.10 (dd, J = 3.5, 1.3 Hz, 1H), 4.42 – 4.30 (m, 4H), 4.05 (br s, 6H), 4.05 – 3.98 (m, 4H), 3.78 (s, 3H), 3.77 (s, 3H), 3.552 (d, J = 13.0 Hz, H), 3.546 (d, J = 12.9 Hz, 1H), 2.16 (br s, 6H), 2.14 (s, 3H), 2.13 (s, 3H), 2.08 (br s, 6H), 1.40 (t, J = 7.2 Hz, 3H), 1.395 (t, J = 7.2 Hz, 3zH), 1.03 (t, J = 7.2 Hz, 3H), 1.027 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 170.34, 170.23, 169.71, 169.65, 168.13, 168.10, 166.44, 166.37, 151.22, 150.73, 150.70, 148.47, 148.43, 147.61, 147.48, 147.34, 147.28, 134.29, 134.27, 131.51, 131.42, 130.54, 130.52, 128.65, 128.51, 125.08, 124.96, 123.85, 123.33, 120.71, 120.44, 110.91, 110.52, 108.32, 108.23, 105.79, 102.80, 102.73, 102.67, 101.30, 70.31, 69.56, 69.52, 67.63, 63.69, 63.65, 61.76, 61.33, 56.11, 55.93, 55.90, 21.04, 21.00, 20.85, 14.17, 13.88; HRMS-ESI calcd for C36H38O16 (M+Na)+ 749.20521, found 749.20282; mp 117–119 °C.
4.1.5.2. ((9-(Benzo[d][1,3]dioxol-5-yl)-6,7-dimethoxy-1,3-dihydronaphtho[2,3-c]furan-4-yl)oxy)(tertbutyl) dimethylsilane (14)
A solution of diphyllin 5 (0.987 g, 2.595 mmol), TBSCl (1.173 g, 7.785 mmol), and imidazole (0.618 g, 9.083 mmol) in DMF (26 mL) was reacted at room temperature overnight. The reaction was quenched with water and extracted with EtOAc. The combined organic layers were washed with cold water, brine, and then dried over magnesium sulfate before being concentrated under reduced pressure. Flash column chromatography (2:1:17 EtOAc/DCM/Hex → 3:1:16 EtOAc/DCM/Hex) afforded TBS-protected diphyllin (828 mg, 65%) as a white crystalline solid: IR νmax (KBr, cm−1): 3074, 3010, 2955, 2932, 2887, 2859, 2831, 2776, 2255, 1766, 1613, 1599, 1507; 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1H), 7.08 (s, 1H), 6.96 (d, J = 7.8 Hz, 1H), 6.85 (d, J = 1.4 Hz, 1H), 6.81 (dd, J = 7.9, 1.7 Hz, 1H), 6.09 (d, J = 1.4 Hz, 1H), 6.05 (d, J = 1.4 Hz, 1H), 5.33 (s, 2H), 4.04 (s, 3H), 3.81 (s, 3H), 1.15 (s, 9H), 0.29 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 170.10, 162.47, 151.24, 150.17, 147.62, 147.52, 143.68, 133.56, 130.96, 128.74, 127.04, 126.54, 123.88, 119.49, 111.02, 108.27, 106.47, 101.31, 67.04, 56.18, 55.94, 25.97, 18.73, −3.05; HRMS-ESI calcd for C27H30O7Si (M+Na)+ 517.16530, found 517.16563; mp 190–191.5 °C. Following the procedure reported by Sakai and coworkers for the reduction of lactones,54 a reaction vial, charged with TBS-protected diphyllin (0.128 g, 0.259 mmol), was flushed with argon several times using a schlenck line. The starting material was then dissolved with freshly distilled CHCl3 (0.26 mL) prior to the successive addition of InBr3 (0.005 g, 0.013 mmol) and Et3SiH (0.165 mL, 1.036 mmol). The vial was immediately capped, shaken, and then stirred at 60 °C for 2 hrs. The reaction was quenched with water and then extracted with EtOAc. The combined organic layers were dried over magnesium sulfate and concentrated under reduced pressure. Flash column chromatography (1:2:17 EtOAc/CHCl3/Hex) afforded compound 14 (118 mg, 95%) as a white crystalline solid: IR νmax (KBr, cm−1): 3070, 3003, 2955, 2931, 2897, 2858, 2830, 2360, 2341, 1619, 1594, 1509; 1H NMR (400 MHz, CDCl3) δ 7.44 (s, 1H), 6.98 (s, 1H), 6.92 (d, J = 7.8 Hz, 1H), 6.82 (d, J = 1.3 Hz, 1H), 6.79 (dd, J = 7.9, 1.6 Hz, 1H), 6.07 (d, J = 1.3 Hz, 1H), 6.04 (d, J = 1.2 Hz, 1H), 5.24 (s, 2H), 4.97 (s, 2H), 4.00 (s, 3H), 3.80 (s, 3H), 1.13 (s, 9H), 0.26 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 149.46, 148.68, 147.94, 146.94, 143.28, 136.76, 132.52, 129.88, 124.85, 123.44, 123.33, 123.18, 110.25, 108.67, 104.89, 101.95, 101.27, 73.73, 72.89, 56.00, 55.86, 26.06, 18.73, −3.18; HRMS-ESI calcd for C27H32O6Si (M+Na)+ 503.18604, found 503.18721; mp 162–164 °C.
4.1.5.3. 9-(Benzo[d][1,3]dioxol-5-yl)-6,7-dimethoxy-1,3-dihydronaphtho[2,3-c]furan-4-ol (15)
A solution of 14 (0.075 g, 0.156 mmol) and TBAF (1 M in THF, 0.234 mL) in DCM (1.7 mL) was stirred at room temperature for 30 minutes. The reaction mixture was quenched with water and several drops of HCl (aq., 2M) before being extracted with DCM. The combined organic layers were washed with water, brine, and then dried over sodium sulfate and concentrated under reduced pressure. Flash column chromatography (2:1:17 EtOAc/EtOH/Hex) afforded compound 15 (53 mg, 93%) as an orange solid: IR νmax (KBr, cm−1): 3271, 3091, 3003, 2939, 2902, 2860, 2833, 2780, 1620, 1597, 1509; 1H NMR (400 MHz, Acetone-d6) δ 7.62 (s, 1H), 7.02 (s, 1H), 6.98 (d, J = 7.9 Hz, 1H), 6.87 (d, J = 1.7 Hz, 1H), 6.83 (dd, J = 7.9, 1.7 Hz, 1H), 6.09 (s, 1H), 6.07 (s, 1H), 5.18 (s, 2H), 4.92 (d, J = 12.4 Hz, 1H), 4.88 (d, J = 12.3 Hz, 1H), 3.93 (s, 3H), 3.71 (s, 3H); 13C NMR (75 MHz, Acetone-d6) δ 150.74, 149.83, 148.83, 147.68, 145.16, 137.37, 133.52, 130.36, 123.99, 123.54, 121.29, 119.22, 110.89, 109.23, 105.65, 102.14, 101.99, 73.76, 72.26, 55.89, 55.68; HRMS-ESI calcd for C21H18O6 (M+Na)+ 389.09956, found 389.09906.
4.1.5.4. 4-(Benzo[d][1,3]dioxol-5-yl)-9-hydroxy-6,7-dimethoxynaphtho[2,3-c]furan-1(3H)-one (16)
A mixture of 14 (0.100 g, 0.208 mmol) and Cs2CO3 (0.068 g, 0.208 mmol) in DMF/H2O (10:1, v/v, 0.229 mL) was stirred overnight at room temperature open to the air. The reaction was quenched with HCl (aq., 2M) and extracted with EtOAc. The combined organic layers were washed with HCl (aq., 2M), water, brine, and then dried over sodium sulfate. Flash column chromatography (25 → 30% EtOAc in Hex) then afforded compound 16 (15 mg, 19%) as a pale yellow solid: 1H NMR (400 MHz, Acetone-d6) δ 9.02 (s, 1H), 7.67 (s, 1H), 7.14 (s, 1H), 7.03 (d, J = 7.9 Hz, 1H), 6.98 (d, J = 1.5 Hz, 1H), 6.94 (dd, J = 7.9, 1.7 Hz, 1H), 6.12 (s, 1H), 6.10 (s, 1H), 5.31 (d, J = 14.7 Hz, 1H), 5.26 (d, J = 14.7 Hz, 1H) 4.00 (s, 3H), 3.79 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 173.22, 153.59, 153.17, 150.52, 149.20, 148.28, 138.24, 134.53, 130.90, 124.17, 119.96, 110.82, 109.60, 105.59, 104.22, 102.52, 102.36, 70.73, 56.11, 55.87; HRMS-ESI calcd for C21H16O7 (M+Na)+ 403.07882, found 403.07804.
4.1.5.5. (2S,3R,4S,5S)-2-((9-(Benzo[d][1,3]dioxol-5-yl)-6,7-dimethoxy-1,3-dihydronaphtho[2,3-c]furan-4-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (17)
Starting from 1,2,3,4-tetra-O-acetyl-l-arabinopyranose and following the general procedure for bromination/glycosylation replacing diphyllin with compound 15, flash chromatography (3:6:11 EtOAc/CHCl3/Hex → 4:6:10 EtOAc/CHCl3/Hex) afforded compound 17 (63 mg, 33%) as a white solid: (c 0.12, CHCl3); IR νmax (KBr, cm−1): 3072, 3002, 2958, 2905, 2859, 2834, 2780, 2255, 1748, 1621, 1592, 1508; 1H NMR (400 MHz, CDCl3) δ 7.53 (s, 1H), 6.95 (s, 1H), 6.93 (d, J = 7.8 Hz, 1H), 6.79 (d, J = 1.9 Hz, 1H), 6.77 (dd, J = 7.7, 1.9 Hz, 1H), 6.07 (br s, 1H), 6.04 (br s, 1H), 5.68 (dd, J = 9.6, 7.2 Hz, 1H), 5.38 (br d, J = 12.5 Hz, 1H), 5.35 (br s, 1H), 5.29 (br d, J = 12.4 Hz, 1H), 5.14 (dd, J = 9.7, 3.5 Hz, 1H), 5.01 (d, J = 7.1 Hz, 1H), 4.97 (br d, J = 13.9 Hz, 1H), 4.93 (br d, J = 12.8 Hz, 1H), 4.16 (br d, J = 13.1, 1H), 4.04 (br s, 3H), 3.80 (br s, 3H), 3.69 (br d, J = 13.1 Hz, 1H), 2.21 (br s, 3H), 2.07 (br s, 6H); 13C NMR (101 MHz, CDCl3) δ 170.45, 170.30, 169.75, 149.84, 149.36, 147.99, 147.12, 144.63, 136.73, 131.99, 129.73, 127.28, 123.29, 123.04, 122.93, 122.70, 110.03, 109.95, 108.78, 108.76, 104.59, 101.50, 101.36, 73.29, 72.41, 70.46, 69.56, 67.62, 64.10, 56.16, 55.86, 21.12, 20.85; HRMS-ESI calcd for C32H32O13 (M+Na)+ 647.17351, found 647.17458; mp 117–119.5 °C.
4.1.5.6. (2S,3R,4S,5S)-2-((9-(Benzo[d][1,3]dioxol-5-yl)-6,7-dimethoxy-3-oxo-1,3-dihydronaphtho[2,3-c]furan-4-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (18)
Starting from 1,2,3,4-tetra-O-acetyl-l-arabinopyranose and following the general procedure for bromination/glycosylation replacing diphyllin with compound 16, flash chromatography (0.75% MeOH in CHCl3) afforded compound 18 (20 mg, 80%) as a pale-orange solid: (c 0.09, CHCl3); IR νmax (KBr, cm−1): 3073, 3005, 2958, 2936, 2835, 2780, 2256, 1749, 1619, 1598, 1507; 1H NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 6.96 (dd, J = 8.0, 2.2 Hz, 1H), 6.93 (s, 1H), 6.80 – 6.75 (m, 2H), 6.22 (d, J = 7.3 Hz, 1H), 6.09 (br s, 1H), 6.06 (br s, 1H) 5.70 (dd, J = 9.8, 7.3 Hz, 1H), 5.36 (br s, 1H), 5.27 (dd, J = 9.9, 3.6 Hz, 1H), 5.15 (br d, J = 15.2 Hz, 1H), 5.10 (br d, J = 15.0 Hz, 1H), 4.10 (s, 3H), 4.09 – 4.01 (m, 2H), 3.86 (br s, 1H), 3.83 (s, 3H), 2.16 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 170.39, 170.35, 170.12, 169.54, 152.61, 149.90, 149.46, 148.41, 148.39, 147.69, 138.60, 133.55, 129.61, 127.16, 123.84, 123.03, 109.83, 109.18, 109.15, 108.52, 103.77, 101.56, 101.33, 70.23, 69.92, 69.01, 67.99, 64.46, 56.32, 56.03, 21.10, 21.09, 20.83; HRMS-ESI calcd for C32H30O14 (M+Na)+ 661.15278, found 661.15392; mp 112–114.5 °C.
4.2. Biology
4.2.1. HT-29 cell proliferation assay
The cytotoxicity of compounds 6a–c, 7a–c, 12, and 17–18 was screened against HT-29 cell lines using the previously reported protocol.63
4.2.2. HT-29 and OVCAR3 cell proliferation assays
The cytotoxicity of the tested compounds was screened against HT-29 (compounds 8a–b, 9a–b, 10–11, and 13) and OVCAR3 cell lines using the following protocol:
4.2.2.1 Cell Culture
Ovarian cancer cell line, OVCAR3, and colon cancer cell line, HT-29, were purchased from the American Type Culture Collection. OVCAR3 cells were grown in RPMI 1640 medium supplemented with 20% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S), and 10 µg/mL insulin. HT-29 cells were grown in RPMI 1640 with 10% FBS and 1% P/S. Cultured cells were maintained in a humidified incubator at 37°C in 5% CO2. Cells were passaged a maximum of 20 times after resuscitation from frozen stocks. Cell lines were validated by STR in 2015 and 2017 and tested mycoplasma free in 2017.
4.2.2.2 Cell Viability Assay
Cells were seeded in 96-well, clear, flat-bottomed plates at 2,500 to 5,000 cells per well, depending on the cell line, and allowed to attach overnight. Compounds suspended in DMSO were diluted to final concentrations as noted in figures in the appropriate media and added to the cells. The final vehicle concentration was 0.25% to achieve the widest dose range possible. Cells were incubated for 24, 48, or 72 hours. The amount of cellular protein content attached to the plate bottom after fixation at the end of the treatment period was dyed and measured as previously described with sulforhodamine B (SRB) assay as a measure of cell survival.64 Treatment measurements were normalized to vehicle, and dose response curves with corresponding IC50 values were generated using Graphpad Prism Software.
4.2.3. K562 and K/VP.5 cell proliferation assays
A cell proliferation assay65 was performed by using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS), the CellTiter 96® AQueous One Solution Reagent (Promega, Madison, WI) which measures the ability of viable cells to enzymatically reduce MTS. DMSO used as control or various concentrations of phyllanthusmin analogs dissolved in DMSO were added to one ml cell suspensions (7.5 × 104 cell/ml) of K562 and K/VP.5 cells (final DMSO concentration 0.5%) followed by addition of triplicate 0.1 ml aliquots from these cell suspensions to 96 well plates. After 72 hr incubation at 37°C, 6 µL of MTS reagent was added to each well followed by incubation for 1 hr at 37°C. Reduced MTS was measured by absorbance at 490 nm using a SynergyH1 Hybrid Reader (Biotek, Winooski, Vermont). The IC50 values for growth inhibition were determined using a logistic four parameter fit of concentration versus absorbance curves (Sigma Plot, Systat Software Inc. San Jose, CA, USA).
4.2.4. Topoisomerase IIα-catalyzed plasmid DNA relaxation
Reaction mixtures (20 µL) contained 125 ng of topoisomerase IIα, isolated as previously described,66 150 ng of negatively supercoiled pBR322 DNA, 1 mM ATP in assay buffer [10 mM Tris-HCl, 50 mM KCl, 50 mM NaCl, 0.1 mM Na2EDTA, 5 mM MgCl2, 2.5% (v/v) glycerol, pH 8.0, and 1 µL of DMSO control, etoposide, phyllanthusmins, or diphyllin (all drugs in DMSO solvent). Components of the assay mixture were assembled and mixed on ice prior to addition of drugs/DMSO. Reactions were initiated by addition of enzyme and experimental mixtures were then incubated at 37°C for 15 min. DNA relaxation was quenched by addition of 2 µL of a stop solution (0.77% SDS, 77 mM Na2EDTA). 4 µL of 10× DNA loading solution (Invitrogen) at 45°C was added to the mixture and incubated for 2 min. DNA bands were separated by electrophoresis (50 V for 10 min then overnight at 15 V) on a TBE agarose gel [1% (w/v)]. The agarose gel was then stained with ethidium bromide (2 µg/ml) for 30 min. DNA bands were visualized under UV light on a Molecular Imager (ChemiDoc™ XRS, Bio-Rad). Conversion from supercoiled pBR322 DNA to relaxed topoisomer bands was monitored as a readout of enzyme activity or its inhibition by tested agents.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health [grant 2P01 CA125066 (to ADK)] and predoctoral fellowships from the NIH/OSU Chemistry-Biology Interface Training Program [GM08512 (to JLW)], the American Foundation for Pharmaceutical Education (to JLW), and an OSU University Fellowship (to ACH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Qi W, Hua L, Gao K. Chemical constituents of the plants from the genus phyllanthus. Chem Biodivers. 2014;11:364–395. doi: 10.1002/cbdv.201200244. [DOI] [PubMed] [Google Scholar]
- 2.Ren Y, Lantvit DD, Deng Y, Kanagasabai R, Gallucci JC, Ninh TN, Chai H-B, Soejarto DD, Fuchs JR, Yalowich JC, Yu J, Swanson SM, Kinghorn AD. Potent cytotoxic arylnaphthalene lignan lactones from Phyllanthus poilanei. J Nat Prod. 2014;77:1494–1504. doi: 10.1021/np5002785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu S-J, Wu T-S. Cytotoxic arylnaphthalene lignans from Phyllanthus oligospermus. Chem Pharm Bull (Tokyo) 2006;54:1223–1225. doi: 10.1248/cpb.54.1223. [DOI] [PubMed] [Google Scholar]
- 4.Murakami T, Matsushima A. Studies on the constituents of Japanese Podophyllaceae plants. I. On the constituent of the root of Diphylleia grayi. J Pharm Soc Jpn. 1961;81:1596. [PubMed] [Google Scholar]
- 5.Day S-H, Lin Y-C, Tsai M-L, Tsao L-T, Ko H-H, Chung M-I, Lee J-C, Wang J-P, Won S-J, Lin C-N. Potent cytotoxic lignans from Justicia procumbens and their effects on nitric oxide and tumor necrosis factor-α production in mouse macrophages. J Nat Prod. 2002;65:379–381. doi: 10.1021/np0101651. [DOI] [PubMed] [Google Scholar]
- 6.Fukamiya N, Lee K-H. Antitumor agents, 81. Justicidin-A and diphyllin, two cytotoxic principles from Justicia procumbens. J Nat Prod. 1986;49:348–350. doi: 10.1021/np50044a030. [DOI] [PubMed] [Google Scholar]
- 7.Di Giorgio C, Delmas F, Akhmedjanova V, Ollivier E, Bessonova I, Riad E, Timon-David P. In vitro antileishmanial activity of diphyllin isolated from Haplophyllum bucharicum. Planta Med. 2005;71:366–369. doi: 10.1055/s-2005-864106. [DOI] [PubMed] [Google Scholar]
- 8.Asano J, Chiba K, Tada M, Yoshii T. Antiviral activity of lignans and their glycosides from Justicia procumbens. Phytochemistry. 1996;42:713–717. doi: 10.1016/0031-9422(96)00024-6. [DOI] [PubMed] [Google Scholar]
- 9.Susplugas S, Hung NV, Bignon J, Thoison O, Kruczynski A, Sévenet T, Guéritte F. Cytotoxic arylnaphthalene lignans from a Vietnamese acanthaceae Justicia patentiflora. J Nat Prod. 2005;68:734–738. doi: 10.1021/np050028u. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y-J, Litaudon M, Bousserouel H, Martin M-T, Thoison O, Léonce S, Dumontet V, Sévenet T, Guéritte F. Sesquiterpenoids and cytotoxic lignans from the bark of Libocedrus chevalieri. J Nat Prod. 2007;70:1368–1370. doi: 10.1021/np070124q. [DOI] [PubMed] [Google Scholar]
- 11.Tuchinda P, Kumkao A, Pohmakotr M, Sophasan S, Santisuk T, Reutrakul V. Cytotoxic arylnaphthalide lignan glycosides from the aerial parts of Phyllanthus taxodiifolius. Planta Med. 2006;72:60–62. doi: 10.1055/s-2005-873141. [DOI] [PubMed] [Google Scholar]
- 12.Shi D-K, Zhang W, Ding N, Li M, Li Y-X. Design, Synthesis and biological evaluation of novel glycosylated diphyllin derivatives as topoisomerase II inhibitors. Eur J Med Chem. 2012;47:424–431. doi: 10.1016/j.ejmech.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda R, Nagao T, Okabe H, Nakano Y, Matsunaga H, Katano M, Mori M. Antiproliferative constituents in umbelliferae plants. IV. Constituents in the fruits of Anthriscus sylvestris HOFFM. Chem Pharm Bull. 1998;46:875–878. doi: 10.1248/cpb.46.875. [DOI] [PubMed] [Google Scholar]
- 14.Yang X-W, He H-P, Du Z-Z, Liu H-Y, Di Y-T, Ma Y-L, Wang F, Lin H, Zuo Y-Q, Li L, Hao X-J. Tarennanosides A–H, eight new lignan glucosides from Tarenna attenuata and their protective effect on H2O2-induced impairment in PC12 cells. Chem Biodivers. 2009;6:540–550. doi: 10.1002/cbdv.200800022. [DOI] [PubMed] [Google Scholar]
- 15.Zheng C-J, Huang B-K, Han T, Zhang Q-Y, Zhang H, Rahman K, Qin L-P. Nitric oxide scavenging lignans from Vitex negundo seeds. J Nat Prod. 2009;72:1627–1630. doi: 10.1021/np900320e. [DOI] [PubMed] [Google Scholar]
- 16.Charlton JL. Antiviral activity of lignans. J Nat Prod. 1998;61:1447–1451. doi: 10.1021/np980136z. [DOI] [PubMed] [Google Scholar]
- 17.Hara H, Fujihashi T, Sakata T, Kaji A, Kaji H. Tetrahydronaphthalene lignan compounds as potent anti-HIV type 1 agents. AIDS Res Hum Retroviruses. 1997;13:695–705. doi: 10.1089/aid.1997.13.695. [DOI] [PubMed] [Google Scholar]
- 18.Janmanchi D, Tseng YP, Wang K-C, Huang RL, Lin CH, Yeh SF. Synthesis and the biological evaluation of arylnaphthalene lignans as anti-hepatitis B virus agents. Bioorg Med Chem. 2010;18:1213–1226. doi: 10.1016/j.bmc.2009.12.038. [DOI] [PubMed] [Google Scholar]
- 19.Prieto JM, Recio MC, Giner RM, Máñez S, Massmanian A, Waterman PG, Ríos JL. Topical anti-inflammatory lignans from Haplophyllum hispanicum. Z Für Naturforschung C. 1996;51:618–622. doi: 10.1515/znc-1996-9-1002. [DOI] [PubMed] [Google Scholar]
- 20.Chen C-C, Hsin W-C, Ko F-N, Huang Y-L, Ou J-C, Teng C-M. Antiplatelet arylnaphthalide lignans from Justicia procumbens. J Nat Prod. 1996;59:1149–1150. doi: 10.1021/np960443+. [DOI] [PubMed] [Google Scholar]
- 21.Leung Y-M, Tsou Y-H, Kuo C-S, Lin S-Y, Wu P-Y, Hour M-J, Kuo Y-H. Arylnaphthalene lignans from Taiwania cryptomerioides as novel blockers of voltage-gated K+ channels. Phytomedicine. 2010;18:46–51. doi: 10.1016/j.phymed.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Xu H, Zhang X, Tian X, Lu M, Wang Y. Synthesis and insecticidal activity of novel 4β-Halogenated benzoylamino podophyllotoxins against Pieris rapae LINNAEUS. Chem Pharm Bull (Tokyo) 2002;50:399–402. doi: 10.1248/cpb.50.399. [DOI] [PubMed] [Google Scholar]
- 23.Loers G, Yashunsky DV, Nifantiev NE, Schachner M. Neural cell activation by phenolic compounds from the siberian larch (Larix sibirica) J Nat Prod. 2014;77:1554–1561. doi: 10.1021/np4009738. [DOI] [PubMed] [Google Scholar]
- 24.Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang K, Oh SH, Yun JH, Jho EH, Kang J-H, Batsuren D, Tunsag J, Park KH, Kim M, Nho CW. A novel topoisomerase inhibitor, daurinol, suppresses growth of HCT116 cells with low hematological toxicity compared to etoposide. Neoplasia N Y N. 2011;13:1043–1057. doi: 10.1593/neo.11972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark PI, Slevin ML. The clinical pharmacology of etoposide and teniposide. Clin Pharmacokinet. 1987;12(4):223–252. doi: 10.2165/00003088-198712040-00001. [DOI] [PubMed] [Google Scholar]
- 27.Phillippe M, Dechaux E, Monneret C, Bertounesque E. Etoposide: discovery and medicinal chemistry. Curr Med Chem. 2004;11:2443–2466. doi: 10.2174/0929867043364531. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y, Ni C, Zhang Y, Zhu L. Synthesis and bioevaluation of diphyllin glycosides as novel anticancer agents. Arch Pharm (Weinheim) 2012;345:622–628. doi: 10.1002/ardp.201200035. [DOI] [PubMed] [Google Scholar]
- 29.Gui M, Shi D-K, Huang M, Zhao Y, Sun Q-M, Zhang J, Chen Q, Feng J-M, Liu C-H, Li M, Li Y-X, Geng M, Ding J. D11, a novel glycosylated diphyllin derivative, exhibits potent anticancer activity by targeting topoisomerase IIα. Invest New Drugs. 2011;29:800–810. doi: 10.1007/s10637-010-9425-3. [DOI] [PubMed] [Google Scholar]
- 30.Govindachari TR, Sathe SS, Viswanathan N, Pai BR, Srinivasan M. Chemical constituents of Cleistanthus collinus. Tetrahedron. 1969;25:2815–2821. [Google Scholar]
- 31.Lakshmi TG, Srimannarayana G, Rao NVS. A new glucoside from Cleistanthus collinus. Curr Sci. 1970;39:395–396. [Google Scholar]
- 32.Meenakshi J, Shanmugam G. Cleistanthin A, a diphyllin glycoside from Cleistanthus collinus is cytotoxic to PHA-stimulated (proliferating) human lymphocytes. Drug Dev Res. 2000;51:187–190. [Google Scholar]
- 33.Parasuraman S, Raveendran R, Rajesh NG, Nandhakumar S. Sub-chronic toxicological evaluation of cleistanthin A and cleistanthin B from the leaves of Cleistanthus collinus (Roxb.) Toxicol Rep. 2014;1:596–611. doi: 10.1016/j.toxrep.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pradheepkumar CP, Panneerselvam N, Shanmugam G. Cleistanthin A causes DNA strand breaks and induces apoptosis in cultured cells. Mutat Res. 2000;464:185–193. doi: 10.1016/s1383-5718(99)00179-5. [DOI] [PubMed] [Google Scholar]
- 35.Kumar CPP, Panneerselvam N, Rajesh S, Shanmugam G. Cytotoxic and genotoxic effects of cleistanthin B in normal and tumour cells. Mutagenesis. 1996;11:553–557. doi: 10.1093/mutage/11.6.553. [DOI] [PubMed] [Google Scholar]
- 36.Kumar CP, Pande G, Shanmugam G. Cleistanthin B causes G1 arrest and induces apoptosis in mammalian cells. Apoptosis. 1998;3:413–419. doi: 10.1023/a:1009658518998. [DOI] [PubMed] [Google Scholar]
- 37.Thummar VR, Parasuraman S, Basu D, Raveendran R. Evaluation of in vivo antitumor activity of cleistanthin B in swiss albino mice. J Tradit Complement Med. 2016;6:383–388. doi: 10.1016/j.jtcme.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuchinda P, Kornsakulkarn J, Pohmakotr M, Kongsaeree P, Prabpai S, Yoosook C, Kasisit J, Napaswad C, Sophasan S, Reutrakul V. Dichapetalin-type triterpenoids and lignans from the aerial parts of Phyllanthus acutissima. J Nat Prod. 2008;71:655–663. doi: 10.1021/np7007347. [DOI] [PubMed] [Google Scholar]
- 39.Ren Y, Yuan C, Deng Y, Kanagasabai R, Ninh TN, Tu VT, Chai H-B, Soejarto DD, Fuchs JR, Yalowich JC, Yu J, Kinghorn AD. Cytotoxic and natural killer cell stimulatory constituents of Phyllanthus songboiensis. Phytochemistry. 2015;111:132–140. doi: 10.1016/j.phytochem.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sastry K, Rao E. Isolation and structure of cleistanthoside A. Planta Med. 1983;47:227–229. doi: 10.1055/s-2007-969993. [DOI] [PubMed] [Google Scholar]
- 41.Himakoun L, Tuchinda P, Puchadapirom P, Tammasakchai R, Leardkamolkarn V. Evaluation of genotoxic and anti-mutagenic properties of cleistanthin A and cleistanthoside A tetraacetate. Asian Pac J Cancer Prev. 2011;12:3271–3275. [PubMed] [Google Scholar]
- 42.Wanitchakool P, Jariyawat S, Suksen K, Soorukram D, Tuchinda P, Piyachaturawat P. Cleistanthoside A tetraacetate-induced DNA damage leading to cell cycle arrest and apoptosis with the involvement of P53 in lung cancer cells. Eur J Pharmacol. 2012;696:35–42. doi: 10.1016/j.ejphar.2012.09.029. [DOI] [PubMed] [Google Scholar]
- 43.Puchadapirom P, Himakhun W, Tuchinda P, Himakoun L, Koomsang T, Suwannalert P. The effects of cleistanthoside A tetraacetate synthesis on acute toxicity and bone marrow micronucleus in ICR mice. Walailak J Sci Technol WJST. 2015;12:605–611. [Google Scholar]
- 44.Naresh G, Kant R, Narender T. Silver(I)-catalyzed regioselective construction of highly substituted α-naphthols and its application toward expeditious synthesis of lignan natural products. Org Lett. 2015;17:3446–3449. doi: 10.1021/acs.orglett.5b01477. [DOI] [PubMed] [Google Scholar]
- 45.Kim HY, Oh K. A facile access to 4-substituted-2-naphthols via a tandem friedel–crafts reaction: a β-chlorovinyl ketone pathway. Org Lett. 2014;16:5934–5936. doi: 10.1021/ol502951v. [DOI] [PubMed] [Google Scholar]
- 46.Kocsis LS, Brummond KM. Intramolecular dehydro-Diels-Alder reaction affords selective entry to arylnaphthalene or aryldihydronaphthalene lignans. Org Lett. 2014;16:4158–4161. doi: 10.1021/ol501853y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao P, Liu J, Wei Y. Hypervalent iodine(III)-mediated benzannulation of enamines with alkynes for the synthesis of polysubstituted naphthalene derivatives. Org Lett. 2013;15:2872–2875. doi: 10.1021/ol401206g. [DOI] [PubMed] [Google Scholar]
- 48.Jiang H, Cheng Y, Zhang Y, Yu S. De novo synthesis of polysubstituted naphthols and furans using photoredox neutral coupling of alkynes with 2-bromo-1,3-dicarbonyl compounds. Org Lett. 2013;15:4884–4887. doi: 10.1021/ol402325z. [DOI] [PubMed] [Google Scholar]
- 49.Patel RM, Argade NP. Palladium-promoted [2 + 2 + 2] cocyclization of arynes and unsymmetrical conjugated dienes: synthesis of justicidin B and retrojusticidin B. Org Lett. 2013;15:14–17. doi: 10.1021/ol3028658. [DOI] [PubMed] [Google Scholar]
- 50.Peng S, Wang L, Wang J. Direct access to highly substituted 1-naphthols through palladium-catalyzed oxidative annulation of benzoylacetates and internal alkynes. Chem – Eur J. 2013;19:13322–13327. doi: 10.1002/chem.201302740. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto Y, Mori S, Shibuya M. A combined transition-metal-catalyzed and photopromoted process: synthesis of 2,3-fused 4-phenylnaphthalen-1-yl carboxylates from 1,7-diaryl-1,6-diynes. Chem - Eur J. 2015;21:9093–9100. doi: 10.1002/chem.201500978. [DOI] [PubMed] [Google Scholar]
- 52.Charlton JL, Oleschuk CJ, Chee G-L. Hindered rotation in arylnaphthalene lignans. J Org Chem. 1996;61:3452–3457. [Google Scholar]
- 53.Hui J, Zhao Y, Zhu L. Synthesis and in vitro anticancer activities of novel aryl-naphthalene lignans. Med Chem Res. 2012;21:3994–4001. [Google Scholar]
- 54.Wang Y, Xia C, Zhang W, Zhao Y. Synthesis and biological evaluation of novel lignan glycosides as anticancer agents. Chem Biol Drug Des. 2016;88:562–567. doi: 10.1111/cbdd.12785. [DOI] [PubMed] [Google Scholar]
- 55.Liu L, Hu Y, Liu H, Liu D-Y, Xia J-H, Sun J-S. First Total Synthesis of the bioactive arylnaphthyl lignan 4-O-glycosides phyllanthusmin D and 4”-O-acetylmananthoside B. Eur. J. Org. Chem. 2017:3674–3680. [Google Scholar]
- 56.Arnold BJ, Mellows SM, Sammes PG. Photochemical Reactions. Part I. A New Route to Tetradehydropodophyllotoxin, Taiwanin E, Related Compounds. J Chem Soc [Perkin 1] 1973 [Google Scholar]
- 57.Singh O, Tapadiya S, Deshmukh R. Process for the Synthesis of Cleistanthin. 2010089778 (A2) WO. 2010 Aug 12;
- 58.Sakai N, Moriya T, Konakahara T. An Efficient One-Pot Synthesis of Unsymmetrical Ethers: A Directly Reductive Deoxygenation of Esters Using an InBr3/Et3SiH Catalytic System. J Org Chem. 2007;72:5920–5922. doi: 10.1021/jo070814z. [DOI] [PubMed] [Google Scholar]
- 59.Ohta K, Munakata K. Justicidin C and D, the 1-methoxy-2,3-naphthalide lignans, isolated from Justicia procumbens L. Tetrahedron Lett. 1970;11:923–925. [Google Scholar]
- 60.Mondal S, Maji M, Basak A. A Garratt-Braverman route to aryl naphthalene lignans. Tetrahedron Lett. 2011;52:1183–1186. [Google Scholar]
- 61.Ritke MK, Yalowich JC. Altered gene expression in human leukemia K562 cells selected for resistance to etoposide. Biochem Pharmacol. 1993;46:2007–2020. doi: 10.1016/0006-2952(93)90643-b. [DOI] [PubMed] [Google Scholar]
- 62.Ritke MK, Roberts D, Allan WP, Raymond J, Bergoltz VV, Yalowich JC. Altered stability of etoposide-induced topoisomerase II-DNA complexes in resistant human leukemia K562 cells. Br J Cancer. 1994;69(4):687–697. doi: 10.1038/bjc.1994.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ren Y, Matthew S, Lantvit DD, Ninh TN, Chai H, Fuchs JR, Soejarto DD, de Blanco EJC, Swanson SM, Kinghorn AD. Cytotoxic and NF-κB inhibitory constituents of the stems of Cratoxylum cochinchinense and their semisynthetic analogues. J Nat Prod. 2011;74:1117–1125. doi: 10.1021/np200051j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 65.Hasinoff BB, Wu X, Patel D, Kanagasabai R, Karmahapatra S, Yalowich JC. Mechanisms of action and reduced cardiotoxicity of pixantrone; a topoisomerase II targeting agent with cellular selectivity for the topoisomerase II α isoform. J Pharmacol Exp Ther. 2016;356:397–409. doi: 10.1124/jpet.115.228650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hasinoff BB, Wu X, Krokhin OV, Ens W, Standing KG, Nitiss JL, Sivaram T, Giorgianni A, Yang S, Jiang Y, Yalowich JC. Biochemical and proteomics approaches to characterize topoisomerase IIα cysteines and DNA as targets responsible for cisplatin-induced inhibition of topoisomerase Iiα. Mol Pharmacol. 2005;67:937–947. doi: 10.1124/mol.104.004416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.