

Abstract
Oligomers of N-substituted glycine, or peptoids, are versatile tools to probe biological processes and hold promise as therapeutic agents. An underlying theme in the majority of recent peptoid research is the connection between peptoid function and peptoid structure. For certain applications, well-folded peptoids are essential for activity, while unstructured peptoids appear to suffice, or even are superior, for other applications. Currently, these structure-function connections are largely made after the design, synthesis, and characterization process. However, as guidelines for peptoid folding are elucidated and the known biological activities are expanded, we anticipate these connections will provide a pathway toward the de novo design of functional peptoids. In this perspective, we review several of the peptoid structure-function relationships that have been delineated over the past five years.
TOC image
Peptoids have emerged as a valuable class of foldamers for the study of biomolecular interactions and hold promise as therapeutic agents. This perspective analyzes the importance of peptoid structure in the discovery of biologically active peptoids over the past five years.
1 Introduction
Oligomers of N-substituted glycine, or peptoids, were developed by Bartlett and co-workers in the early 1990’s.1 They were initially proposed as an accessible class of molecules from which lead compounds could be identified for drug discovery. Previous to this work, Farmer and Ariëns had introduced the term peptoid as a peptidomimetic that could mimic the biological function of peptides, but did not resemble them structurally.2,3 Bartlett and co-workers, however, defined peptoids more specifically as oligomeric N-substituted glycines and laid the foundation for research in this field. Peptoids can be described as mimics of α-peptides in which the side chain is attached to the backbone amide nitrogen instead of the α-carbon (Fig. 1). These oligomers are an attractive scaffold for biological applications because they can be generated using a straightforward, modular synthesis that allows the incorporation of a wide variety of functionality.4 This route makes peptoids highly amenable to library synthesis and high-throughput screening.5–7 Peptoids have been evaluated as tools to study biomolecular interactions,8–10 and also hold significant promise for therapeutic applications due to their enhanced proteolytic stabilities11 and increased cellular permeabilities12,13 relative to α-peptides.
Fig. 1.
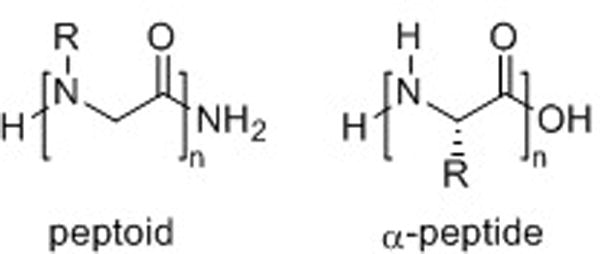
Generic structures of a peptoid and an α-peptide.
Early peptoid research focused largely on the generation of large combinatorial libraries of peptoids using split-pool methods, and the evaluation of these libraries in high-throughput screens for novel functions.5,6 Indeed, this strategy continues to be applied to identify biologically relevant peptoids.7 Biologically active peptoids have also been discovered by rational design (i.e., using molecular modeling), and were synthesized either individually or in parallel focused libraries.8,14,15 Furthermore, peptoids have been explored as drug and gene delivery agents, aided by the facile conjugation of bioactive molecules to peptoid oligomers.12,16 In all of these pursuits, displaying the proper amide side chain functionality is critical for peptoid function. For some applications, a well-defined structure is also necessary for peptoid function—to display the functionality in a particular orientation, or to adopt a conformation that promotes interaction with other molecules. However, in other biological applications, peptoids lacking defined structures appear to possess superior activities over structured peptoids.
This perspective will focus primarily on the relationship between peptoid structure and function. A comprehensive review of peptoids in drug discovery, detailing peptoid synthesis, biological applications, and structural studies, was published by Barron, Kirshenbaum, Zuckermann, and co-workers in 2004.17 Since that time, significant advances have been made in these areas, and new applications for peptoids have emerged. In addition, new peptoid secondary structural motifs have been reported, as well as strategies to stabilize those structures. For these reasons, this perspective will focus on peptoid research reported over the past five years (2004–2008). We will only discuss reports that include structural characterization or structure-activity relationships, and for brevity we must unfortunately omit several excellent accounts of peptoids being applied to important biological problems. At the outset of this perspective, a brief introduction to peptoid synthesis and structure is presented to aid the reader. Thereafter, we focus on two major areas of peptoid research: α-peptide mimics and molecular recognition. Throughout this discussion, structural features that convey biological activity are noted, making it worthwhile to also examine the structure-activity relationships elucidated thus far for cellular uptake and delivery. Lastly, the emergence of peptoid tertiary structures and progress toward peptoid-based nanostructures with biological function is discussed. We conclude by offering our perspective on future peptoid research developments.
2 Peptoid Synthesis
The relative ease of peptoid synthesis has enabled their study for a broad range of applications. Peptoids are routinely synthesized on Rink amide linker-derivatized solid supports using the submonomer synthesis method developed by Zuckermann et al. (Scheme 1).4 The peptoid monomer is constructed from C- to N-terminus using N,N-diisopropylcarbodiimide (DIC) mediated acylation with bromoacetic acid, followed by amination with a primary amine. This two-step sequence is repeated iteratively to obtain the desired oligomer. Thereafter, the Rink amide linker is cleaved using trifluoroacetic acid (TFA), yielding a primary amide at the C-terminus of the peptoid. The availability of numerous primary amines facilitates the preparation of chemically and structurally divergent peptoids. (Secondary amines can also be incorporated into peptoids, but at the N-terminal position only, as their incorporation prohibits further acylation.) In addition, the submonomer method makes the construction of peptoids relatively inexpensive compared to the preparation of α-peptides.11 Reports from Kodadek and co-workers,18 Burgess and co-workers,19 and our laboratory20 have described the use of microwave heating to accelerate the acylation and amination reactions of the peptoid submonomer synthesis, and thereby increase the efficiency of coupling sterically hindered and electronically deactivated amines. Peptoids can also be constructed by coupling N-substituted glycines using standard α-peptide synthesis methods, but this requires the synthesis of individual monomers.1 Similarly, Bradley and co-workers described microwave-assisted reaction conditions for the efficient coupling of N-substituted glycine monomers.21
Scheme 1.
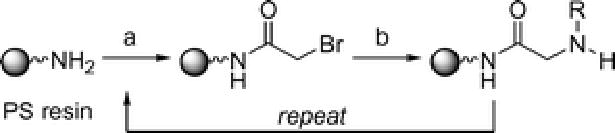
Peptoid submonomer synthesis method developed by Zuckermann and co-workers.4 Reagents and conditions: PS resin = Rink amide linker-derivatized polystyrene. (a) bromoacetic acid, N,N-diisopropylcarbodiimide (DIC), DMF. (b) amine building block NH2R, DMF. Oligomers are cleaved from the resin with 95% trifluoroacetic acid/H2O.
2.1 Peptoid Nomenclature
Peptoid nomenclature utilizes abbreviations for the amines and monomers similar to the three-letter codes used for amino acids. For example, the amine building block (S)-(1-phenylethyl) amine is abbreviated spe, and the peptoid monomer N-(S)-(1-phenylethyl)glycine is abbreviated Nspe. In most cases, the three-letter codes refer to α-chiral side chains, and the first letter designates stereochemistry (r or s for R or S). In turn, two-letter codes have been widely used for achiral side chains (e.g., Nme for N-(2-methoxyethyl)glycine). When mimicking natural amino acids, however, there is a disparity in the nomenclature—some monomers have been given peptoidic names (e.g., Npm for N-phenylmethylglycine, which is N-substituted phenylalanine) while others have retained their amino acid abbreviations (e.g., NLys for N-(4-aminobutyl)glycine, which is N-substituted lysine). Therefore, clear definitions of the abbreviations used in each peptoid study are essential for clarity.
3 Peptoid Structure
Peptoid oligomers lack conformational rigidity in comparison to α-peptides. The placement of the monomer side chains on the amide nitrogen (as opposed to the α-carbon as in α-peptides, Fig. 1) renders the peptoid backbone achiral. Likewise, amide bonds are tertiary and can isomerize between trans and cis conformations far more readily than the secondary amides in α-peptides (Fig. 2). Further, without the presence of amide protons, secondary structure cannot be stabilized by backbone hydrogen bonding in the same manner as in peptides. These characteristics make peptoid oligomers highly flexible and complicate the de novo design of well-defined secondary structures in peptoids. However, researchers have developed methods to stabilize helical, loop, and turn motifs in peptoids by incorporating amide side chains that restrict backbone conformation. Through this work, evidence has been presented that implicates several types of noncovalent interactions in peptoid folding. For example, installation of branching and bulky substituents in peptoid amide side chains engenders steric repulsion between side chains,22 and aromatic and/or negatively charged side chains cause charge-charge repulsion with backbone carbonyls.23,24 In addition, hydrophobic interactions25 and n→π* interactions26 have also been predicted to play a role in peptoid folding. The elucidation of how these and other noncovalent interactions in peptoids dictate their conformations is required to develop a fundamental understanding of the peptoid folding process.27,28
Fig. 2.
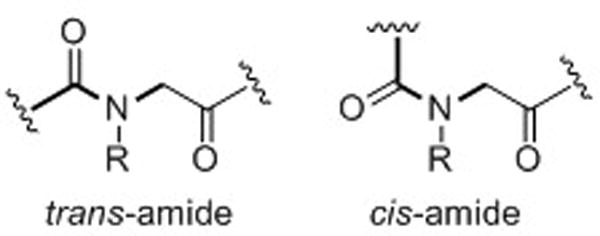
Amides in the peptoid backbone can readily access both trans and cis conformations.
The secondary structure of peptoids is typically evaluated by circular dichroism (CD) spectroscopy, as this tool allows rapid analysis relative to characterization by NMR. Furthermore, the crystallization of peptoid oligomers has been highly challenging, due in part to their relatively flexible structure. Though CD analysis is highly qualitative, the correlation of CD data to the few peptoid structures determined by NMR and X-ray (i.e., helix and loop) has proven valuable for analysis of new peptoids. In a large portion of the work discussed herein, changes in CD spectra correlated with changes in biological activity in a predictable manner, demonstrating the utility of CD analyses in evaluating structure. We note, however, that caution must be used when comparing CD data for peptoids with substantially different side chain compositions, most notably aromatic side chains, as these can affect CD spectral shape.
3.1 Peptoid Helix and Loop Structures
The peptoid helix and threaded loop represent the best characterized peptoid secondary structural motifs, and are adopted by peptoids comprised mainly of α-chiral monomers, such as N-(S)-(1-phenylethyl)glycine (Nspe), N-(S)-(sec-butyl)glycine (Nssb), and N-(S)-(1-cyclohexyl-ethyl)glycine (Nsch) (Fig. 3). The peptoid helix is a three-residue per turn helix with all cis amide bonds, and its structure has been analyzed by a range of techniques, including CD spectroscopy,29 molecular modeling studies,22 NMR spectroscopy,30 and X-ray crystallography23 (Fig. 4a). The helix can be recognized by a CD spectrum with well-defined peaks at 192, 202 and 218 nm, and this pattern serves as a useful diagnostic for helical structure in peptoids. In the early 2000’s, Barron and co-workers developed a set of predictive rules for helix formation in peptoids. First, helical structure is stabilized by the incorporation of at least 50% α-chiral monomer units into an oligomer, or if the helix contains one or more aromatic faces running parallel to the helix axis (i.e., aromatic side chains at i, i+3 positions).31 Second, peptoid helices are stabilized when the C-terminal residue is α-chiral. Third, helical structure is further stabilized at longer peptoid oligomer lengths (e.g., decamers and beyond).
Fig. 3.
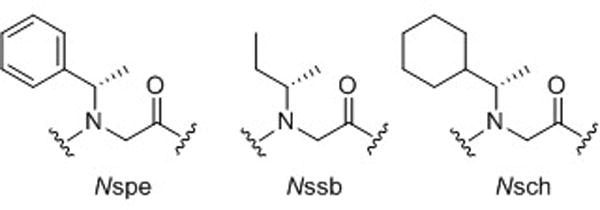
Common α-chiral monomers used to enforce structural stability in peptoids.
Fig. 4.
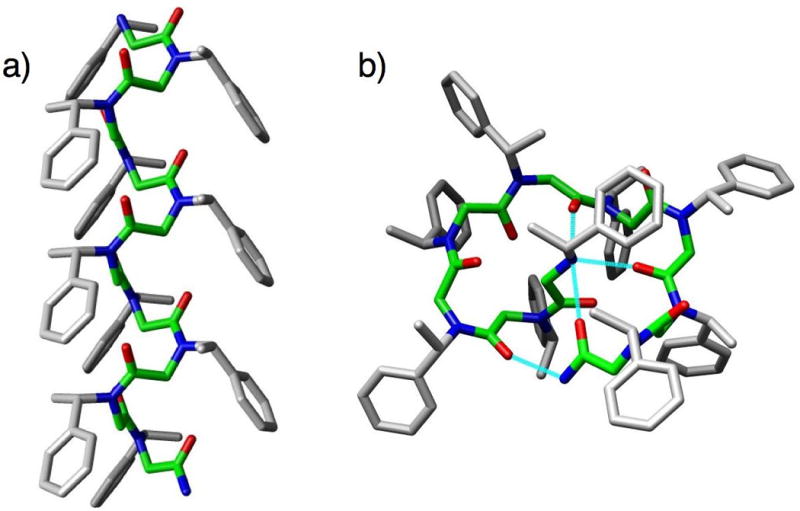
(a) The peptoid helix, shown here as the structure of (Nspe)10. Structure generated by molecular mechanics from the calculated structure of (Nspe)8; peptoid backbone highlighted in green.22 (b) The peptoid threaded loop, shown here as the structure of (Nspe)9. Structure generated by solution-phase 2D NMR analyses.32 Peptoid backbone highlighted in green and intramolecular hydrogen bonds shown in cyan. 3D-images for helix and loop generated using Chimera (v. 1.2199).36
Through careful analysis of the peptoid helix, Barron and co-workers went on to discover a second well-defined peptoid structure, the threaded loop (Fig. 4b). This structure is unique to peptoid nonamers with α-chiral side chains, and was first identified in a homononamer of Nspe (Fig. 3).32 The threaded loop structure of (Nspe)9 (as the TFA salt) was determined by solution-phase NMR spectroscopy in acetonitrile-d6, and was found to be stabilized by three intramolecular hydrogen bonds from backbone carbonyls (residues 5, 7, and 9) to the N-terminal secondary ammonium, and one intramolecular hydrogen bond from a backbone carbonyl (residue 2) to the C-terminal primary amide (Fig. 4b). The peptoid threaded loop contains four cis and four trans amide bonds, and exhibits a CD spectrum highly distinct from the peptoid helix, namely a single broad peak of significant intensity at 203 nm.29 Interestingly, the threaded loop can be converted into a peptoid helix by the addition of a solvent capable of disrupting its set of intramolecular hydrogen bonds (e.g., 50% methanol in acetonitrile). The strength of these hydrogen bonds is therefore a key factor in stabilizing the loop conformation in acetonitrile. Strategies to stabilize the helical conformation over the threaded loop have been reported. For example, both Kirshenbaum and co-workers and Vaz and Brunsveld covalently linked peptoid side chains at the i, i+3 positions to enforce helicity.33,34 In addition, our laboratory incorporated a strongly electron-withdrawing monomer (i.e., N-(S)-(1-pentafluorophenylethyl)glycine) at key positions in a peptoid nonamer to weaken or strengthen hydrogen bonding and stabilize helix or loop structures, respectively.35
3.2 Peptoid Turn Structures
More recently, turn motifs have been reported in peptoids, achieved either by macrocyclization37 or by incorporation of a heterocyclic turn-inducing unit.25 Kirshenbaum and co-workers performed head-to-tail macrocyclization of achiral peptoid oligomers and discovered that macrocyclic hexamer and octamer peptoids resembled peptide β-turns.37 These peptoids had an alternating pattern of benzyl and methoxyethyl side chains, and their crystal structures revealed that the aromatic and alkyl side chains were segregated to opposite faces of the macrocycle (hexamer shown in Fig. 5a). In each macrocycle, the two backbone amides in the turn region were cis and the remaining amides were trans, such that the pattern of cis and trans amides was (cis-cis-trans)2 for the hexamer and (cis-cis-trans-trans)2 for the octamer. Superposition of the hexamer and octamer structures showed a high degree of similarity in their turn regions. Furthermore, the peptoids were overlaid with native peptide β-turns and found to closely resemble both type I and type III β-turns (hexamer overlay shown in Fig. 5b). Elaboration of these macrocyclic peptoids via the introduction of functionalized aromatic and alkyl side chains could provide useful β-turn mimics, with the choice of a smaller hexamer or larger octamer scaffold.
Fig. 5.
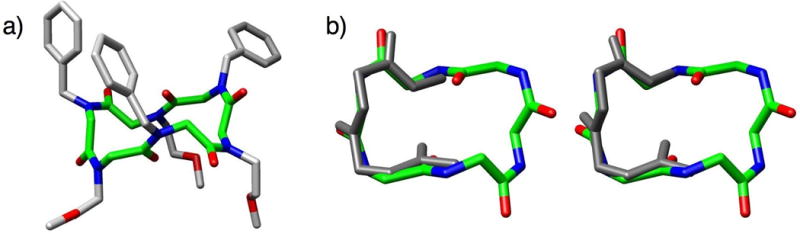
(a) X-ray crystal structure of Kirshenbaum and co-workers’ cyclic peptoid hexamer; peptoid backbone highlighted in green.37 (b) Overlay of the cyclic hexamer backbone with a type I (left) and a type III (right) β-turn. 3D-images for X-ray structure and overlays generated using Chimera (v. 1.2199).36
In 2007, Appella and co-workers designed a triazole monomer to function as a turn mimic and incorporated this unit into peptoid oligomers.25 The triazole moiety introduces a constraint in the peptoid backbone similar to that of a cis double bond, resulting in a tight turn in the peptoid structure. The triazole monomer was flanked by bulky α-chiral, aromatic monomers to further rigidify the turn motif, and structural stability increased when two hydrophobic residues were incorporated to encourage hydrophobic collapse (Fig. 6). Characterization by NMR in aqueous solution revealed a highly structured turn region and flexible termini. In the CD spectrum, the peptoid displayed a single minimum at 200 nm. This work represents the first hairpin-like structure of a linear peptoid in aqueous solution. We anticipate that this strategy for turn motif stabilization will prove fruitful in the future design of biomimetic peptoids.
Fig. 6.
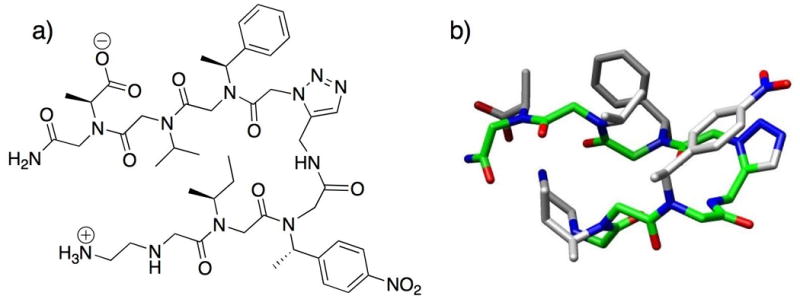
(a) Structure of Appella and co-workers’ peptoid β-hairpin mimic containing the triazole turn unit. (b) 3-D structure of the peptoid β-hairpin mimic determined by NMR analyses; backbone highlighted in green.25 3D-image for turn structure generated using Chimera (v. 1.2199).36
4 Peptoids that Mimic Biologically Active Peptides
Peptides and proteins carry out a multitude of important biological functions, ranging from gene transcription to apoptosis, with exquisite control. However, peptides largely have not been developed for clinical use, because peptide therapeutics are costly and generally have poor oral bioavailability, short half-life in the body, and/or elicit an immune response.17 These problems create a great opportunity to develop peptoid therapeutics, as peptoids have enhanced proteolytic stability relative to α-peptides and can be synthesized on large scale more cost effectively than peptides.11 In addition, peptoids can adopt folded conformations analogous to α-peptides—as outlined above, there exist robust strategies for the stabilization of peptoid helices31 and recent reports describe methods for the stabilization of loops32,35 and turns25,37 in peptoids. Along with the development of peptoid mimics, researchers have improved the properties of certain naturally occurring peptides by incorporating one or more peptoid residues to generate peptide-peptoid hybrids (or “peptomers”). This section will discuss the design and characterization of several recently reported peptoid and peptomer mimics of biologically active peptides.
4.1 Peptide to Peptoid Transformation
As the monomer-to-monomer transformation of peptide residues to peptoid residues can drastically change the folded conformation of the oligomer, constructing a peptoid mimic with the same sequence of side chains as the original peptide is most often unlikely to yield a compound with analogous activity as the peptide. Screening large, split-pool combinatorial peptoid libraries is one method to identify bioactive peptoids; however, this process can be time consuming and can waste resources. On the other hand, designing a small set of peptide mimics yields a limited set of biased compounds and may or may not prove successful in identifying active peptoids. To address this challenge, Hoffman et al. recently described a systematic method to transform a biologically active peptide into a peptoid analog with virtually equivalent biological activity.38 The systematic nature of this method can be likened to comprehensive library synthesis, but many fewer compounds are constructed than in a random or split-pool library strategy. Starting from the peptide VVSHFND, a known binder of the anti-transforming growth factor α (TGFα) monoclonal antibody (mab) Tab2, each amino acid was sequentially replaced with a peptoid monomer using a combinatorial approach. A spatially addressed array of 30 peptomers was prepared by SPOT-synthesis techniques on planar polymeric support,39 incorporating 30 different peptoid monomers at a single position in the peptide. This library was evaluated in a binding assay with mab Tab2, and the compound with the highest binding affinity was selected to design the subsequent library. Eight libraries were prepared iteratively, testing all 30 peptoid monomers at each position in the sequence. The best peptoid (comprised of the seven optimized peptoid monomer units) in the final library had a dissociation constant (KD) = 200 nM and half maximal inhibitory concentration (IC50) = 20 μM for mab Tab2, compared to KD = 90 nM and IC50 = 0.4 μM for the native peptide VVSHFND. Moreover, this peptoid had 10-fold better affinity for mab Tab2 than peptoids identified in an 8000-member randomly generated library,40 and resulted from screening just one quarter the number of compounds. This work, highly analogous to an approach for the transformation of L- into D-peptides developed by Kramer et al.,41 represents a new strategy for the discovery of biologically active peptoids and should be a useful model for future research.
4.2 Antimicrobial Peptoids
Antimicrobial peptides (AMPs) are found in myriad of organisms and are highly effective against bacterial infections.42 The mechanism of action for most AMPs is permeabilization of the bacterial cytoplasmic membrane, which is facilitated by their amphipathic structure. AMPs are short (10–50 amino acids) α-helical, β-hairpin, extended, or loop structured peptides (e.g., magainins, protegrins, indolicidin, and bactenecin, respectively) and generally have a hydrophobic and a cationic region.43 The cationic region of AMPs confers a degree of selectivity for the membranes of bacterial cells over mammalian cells, which have negatively charged and neutral membranes, respectively. The hydrophobic portions of AMPs are believed to mediate insertion into the bacterial cell membrane, lysing the cell. Although AMPs possess many positive attributes, they have not been developed as drugs due to the poor pharmacokinetics of α-peptides (see above). This problem creates an opportunity to develop peptoid mimics of AMPs as antibiotics and has sparked considerable research in this area.44
An early report of antimicrobial peptoids by Goodson et al. disclosed peptoid dimers and trimers effective against both Gram-negative and Gram-positive bacteria (minimum inhibitory concentration (MIC) = 5–40 μM), yet these compounds also displayed modest hemolytic activity (i.e., they lysed human red blood cells) at relatively low concentrations (~10% hemolysis at 50 μM).45 Toxicity problems likely arose from the incorporation of dehydroabietylamine, which was shown to be hemolytic on its own. The research discussed below details some of the recent design strategies that increase the selectivity of antimicrobial peptoids for bacterial cells.
4.2.1 Helical AMP Mimics
In 2003, Barron and co-workers reported peptoid mimics of the helical antimicrobial peptide magainin-2 that had low micromolar activity against Escherichia coli (MIC = 5–20 μM) and Bacillus subtilis (MIC = 1–5 μM).46 These peptoids were composed of α-chiral side chains known to enforce helical structure (Nspe, Nssb and Nsch; Fig. 3), and NLys was incorporated at every third position to create a cationic face on the helix. The researchers noted that peptoids with stronger helical CD signatures exhibited higher antibacterial activity, and also that more hydrophobic peptoids had greater hemolytic activity. They then conducted a more comprehensive investigation to elucidate the structure-function relationships for peptoid magainin-2 mimics, and in 2008 reported the antibacterial activity of these compounds along with their selectivity for bacterial cells over mammalian cells.14 In this more recent study, Barron and co-workers discovered that peptoid helix stability was not important for antimicrobial activity—indeed, peptoids displaying both weak and strong helical CD signatures killed bacteria (e.g., in E. coli, MIC = 3 μM for a strongly helical peptoid (1), MIC = 7 μM for a weakly helical peptoid (2); Table 1). Instead, the structural features that conferred good antibiotic activity were overall cationic charge (at least +3) and moderate hydrophobicity, which were the same requirements determined for AMPs. Importantly, this study uncovered the key structural features of peptoids that confer selectivity for bacterial cells over mammalian cells. Peptoids with strongly helical CD signatures had greater hemolytic activity. Hemolysis was also correlated with increased hydrophobicity; for example, peptoid 1 is more hydrophobic and more hemolytic than peptoid 3 (Table 1). These design strategies will aid the development of selective antimicrobial peptoids for clinical use in the future. Furthermore, this work represents an example where defined structure (at least in the absence of cells) was not required for the desired biological activity.
Table 1.
Structures of pexiganan and selected antimicrobial peptoids reported by Barron and co-workers14
| Oligomer | Sequence (N- to C-terminus) | E. coli MIC, μM | Selectivity ratiob |
|---|---|---|---|
| pexiganana | GIGKFLKKAKKFGKAFVKILKK | 3.1 | 24 |
| 1 | (NLys-Nspe-Nspe)4 | 3.5 | 6 |
| 2 | (NLys-Nsmb-Nspe)4c | 7.4 | >16 |
| 3 | (NLys-Nspe-Nspe-NLys-Nspe-NHis)2 | 3.5 | >31 |
Pexiganan is a selective AMP analog of magainin-2.
Selectivity ratio = 10% hemolytic dose/E. coli MIC.
Nsmb is N-(S)-(1-methylbutyl)glycine.
A recent publication by Shin and co-workers described the incorporation of peptoid residues into the antimicrobial peptide melittin and the identification of cell-selective analogs.47 Melittin is known to adopt an amphipathic helical structure and contains a leucine zipper motif. This zipper motif is believed to promote the self-association of melittin, leading to the formation of a transmembrane pore in cell membranes and effecting cell lysis. The researchers designed melittin analogs with peptoid residue replacements (NAla, NLeu, NPhe, and NLys) of key residues in the leucine zipper (Leu-6, Leu-13, Ile-20; Fig. 7). The analogs containing NLeu, NPhe, and NLys showed strong antimicrobial activity against three Gram-negative bacterial strains (MIC = 4–16 μM; melittin MIC = 2 μM), three Gram-positive bacterial strains (MIC = 1–4 μM; melittin MIC = 0.5–1 μM), and four antibiotic-resistant bacterial strains (MIC = 2–8 μM; melittin MIC = 1–8 μM). Structures of the active peptoids and selected data are shown in Table 2. Most gratifyingly, the peptoid analogs did not cause hemolysis of human erythrocytes at concentrations as high as 100 μM (note, melittin was hemolytic at 1 μM). Further experiments demonstrated that the peptoid analogs were unable to permeate mammalian cell membranes and were not cytotoxic to mammalian cells. Structural characterization by CD spectroscopy revealed that the peptoid analogs had a random structure in aqueous solution, while melittin displayed a helical CD signature. Similar to the conclusions derived by Barron and co-workers for magainin-2 mimics,14 this study showed that destabilizing helical structure in peptomers, at least in the absence of cell membranes, appears to reduce toxicity in mammalian cells.
Fig. 7.
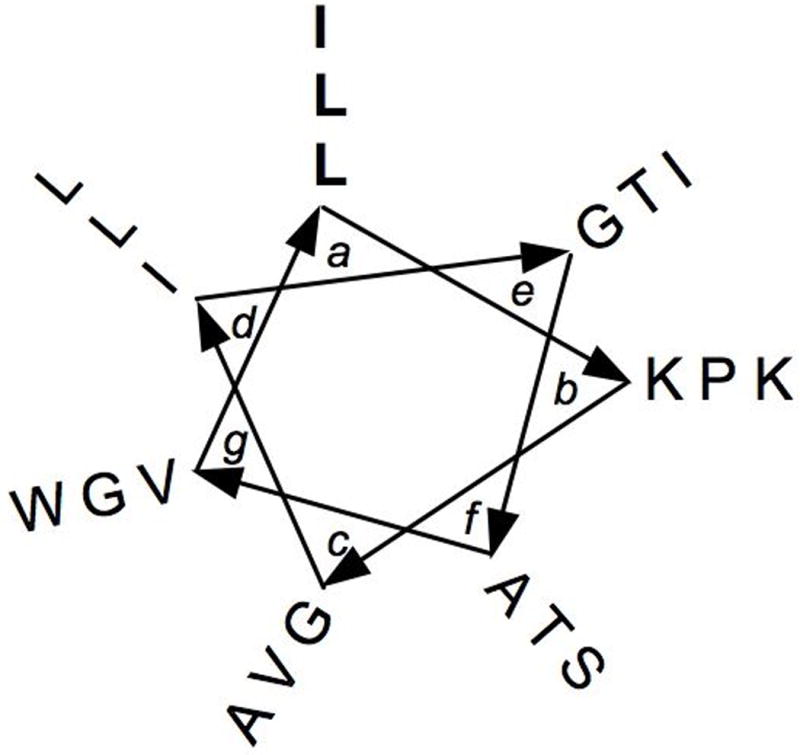
Helical wheel diagram of the peptide melittin. The residues in bold were replaced by peptoid monomers in the melittin mimics developed by Shin and co-workers.47
Table 2.
Structures of melittin and the antimicrobial peptomers reported by Shin and co-workers47
| Oligomer | Sequence (N- to C-terminus) | E. coli MIC, μM | MRSA MIC, μMa |
|---|---|---|---|
| Melittin | GIGAVLKVLTTGLPALISWIKRKRQQb | 2 | 1 |
| 4 | GIGAV-NAla-KVLTTG-NAla-PALISW-NAla-KRKRQQ | 16 | 32 |
| 5 | GIGAV-NLeu-KVLTTG-NLeu-PALISW-NLeu-KRKRQQ | 4 | 8 |
| 6 | GIGAV-NPhe-KVLTTG-NPhe-PALISW-NPhe-KRKRQQ | 4 | 4 |
| 7 | GIGAV-NLys-KVLTTG-NLys-PALISW-NLys-KRKRQQ | 4 | 2 |
Methicillin-resistant S. aureus strain CCARM 3543.
Residues shown in bold were replaced by peptoid monomers.
The structural characteristics of peptoids and peptomers that confer antimicrobial and hemolytic activity have also been studied computationally by Nandel and Saini.48 These researchers computed the energy-minimized structures for all-cis and all-trans amide bond conformations of trimer to octamer heteropeptoids comprised of NIle, NLys, and NPhe residues (with acetylated N-termini and methylamide C-termini). These peptoids were designed based on the antimicrobial peptide magainin, and were similar to the peptoid magainin-2 mimics studied earlier by Patch and Barron.46 In addition, the ability of the energy minimized peptoid structures to interact with and insert into a membrane was modeled. First, Nandel and Saini analyzed peptoids containing only NIle and NLys residues. The energy minimized structures of Ac-NIle(NLysNIleNIle)n-NHMe (n = 1, 2) lacked a regular repeating structure and the bulky NIle side chains masked the NLys side chains, preventing a charged interaction with the cell membrane. They concluded that this designed peptoid would have negligible antimicrobial activity. A similar peptoid studied by Patch and Barron, NIleNIle(NLysNIleNIle)5, had a weak CD spectrum suggesting a random structure and had no antimicrobial activity.46 Second, the authors analyzed peptoids containing only NPhe and NLys residues: Ac-NPhe(NLysNPheNPhe)n-NHMe (n = 1, 2). The modeled heptamer peptoid had a defined helical structure and was shown to be capable of cell membrane disruption. The authors noted that the plane of the NPhe aromatic ring was perpendicular to the plane of the membrane in the computational model, which was predicted to confer both good antimicrobial activity and higher hemolytic activity. Similarly, Patch and Barron had previously discovered that both (NLysNspeNspe)4 and NspeNspe(NLysNspeNspe)5 had good antimicrobial activity (E. coli MIC = 5–8 μM, B. subtilis MIC = 1 μM) and high hemolytic activity; moreover, the latter peptoid, which contained more aromatic residues and was thus more hydrophobic, had greater hemolytic activity than the former.46 Third, Nandel and Saini analyzed peptoids containing NIle, NLys, and NPhe residues: Ac-(NLysNIleNPhe)2-NHMe and Ac-NIleNPhe(NLysNIleNPhe)2-NHMe. These peptoids were found to adopt ordered, yet non-helical structures that suggested good antimicrobial activity. In the model of a cell membrane, the plane of the NPhe aromatic rings in these peptoids was at an angle of less than 90° to the plane of the membrane, which the authors suggested would reduce hemolytic activity. These computational results were consistent with the experimental results of Patch and Barron, which showed that peptoids (NLysNIleNspe)5 and NIleNspe(NLysNIleNspe)5 had good antimicrobial activity (E. coli MIC = 10–20 μM, B. subtilis MIC = 1–5 μM) and low hemolytic activity (0% hemolysis up to 200 μg/mL). These computationally derived design principles should prove valuable in the further refinement of antimicrobial peptoid activity. Further, these methods now provide a more exacting prediction of peptoid hemolytic activity a priori.
4.2.2 Cyclic Peptomers for Antimicrobial Applications
A peptomer scaffold was used by Robinson and co-workers to mimic protegrin I, a cationic β-hairpin antibiotic.49 In protegrin I, the β-hairpin motif is enforced by two disulfide bridges; however, the authors introduced a hairpin-stabilizing, α-peptide template (D-Pro-L-Pro) into a macrocyclic structure to build their peptomer mimic of protegrin-I (Fig. 8). They synthesized both the α-peptide (8) and peptomer (9, single peptoid residue substitution of Arg-6 with NLys; Fig. 8a) versions of the mimic, characterized their secondary structures, and evaluated their antimicrobial and hemolytic activities. NMR studies of peptide 8 showed that it had an unordered structure in water, but adopted a largely β-hairpin conformation in the presence of micelles.50 In contrast, peptomer 9 adopted a relatively stable β-hairpin conformation in water alone, which resembled a type-II´ β-turn at the hairpin tip (Fig. 8b). The antibacterial activities of peptide 8 and peptomer 9 were lower than that of protegrin I, but 8 and 9 still displayed good activity against Gram-negative bacteria (MIC = 4–16 μg/mL). Indeed, peptomer 9 displayed slightly enhanced activity over peptide 8 in both Gram-negative and Gram-positive strains. Moreover, the hemolytic activity of 8 and 9 (1.4% and 0.5%, respectively, at 100 μg/mL) was much lower than that of protegrin I (37% at 100 μg/mL). Thus, this work indicates that the incorporation of just one peptoid residue into a protegrin I mimic led to enhancement in structural stability, antibacterial activity, and selectivity. Furthermore, in conjunction with the peptomer mimics of melittin discussed above, this work suggests that the incorporation of peptoid residues in biologically active peptides can lead to improved selectivity for bacterial cells over mammalian cells.
Fig. 8.
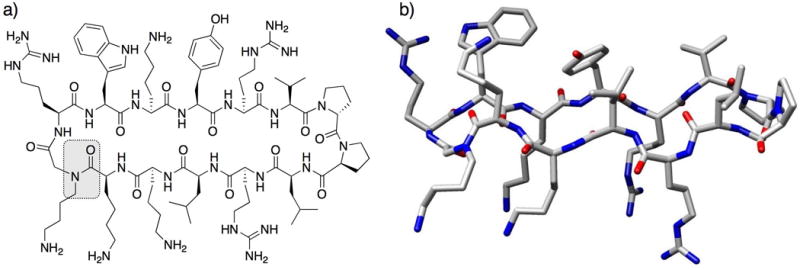
(a) Structure of Robinson and co-workers’ macrocyclic peptomer 9, a mimic of protegrin-I. The NLys replacement at Arg-6 is highlighted. (b) 3D-image of peptomer 9. The macrocycle adopts a stable β-hairpin conformation in aqueous solution, as determined by NMR analyses.49 Image generated using Chimera (v. 1.2199).36
Recent studies in our laboratory are focused on an emerging approach to antimicrobial therapy: the inhibition of bacterial virulence through the modulation of quorum sensing (QS). QS is a method of bacterial cell-cell communication, in which bacteria use small molecules (Gram-negative) or peptides (Gram-positive) to sense their population density and regulate group behaviors.51,52 We reasoned that peptoids were well-suited to mimic the QS signaling molecules of Staphylococcus aureus, termed autoinducing peptides (AIPs)53 and designed a mimic of one of these signals, AIP-I.15 AIP-I is a macrocyclic thiolactone, and acyclic versions of this peptide are inactive.54 Notably, removal of the linear tail portion of AIP-I yields an inhibitor of the AIP receptor proteins essential for QS. Our scaffold design was a “tail-free”, macrocyclic peptomer and contained two peptoid units to probe whether N-substitution of key residues impacted activity (peptomer 10; Fig. 9a). Using molecular modeling, we observed that peptomer 10 overlaid moderately well on the macrocyclic portion of AIP-I (Fig. 9b). We synthesized a small (11-member) library of peptomer variants of 10 containing different peptoid side chains. From this small set of compounds, we identified one peptomer capable of promoting biofilm formation in S. aureus, a phenotype linked to inhibition of the AIP-I receptor protein. Examination of the solution-phase structure of these AIP-I mimics will be an important next step in improving biological activity. As peptoid-based scaffolds have been shown to cyclize more readily than α-peptides (see Kirshenbaum work above),37 peptoids should prove valuable in the development of other non-native QS modulators for S. aureus and other related Gram-positive bacteria.
Fig. 9.
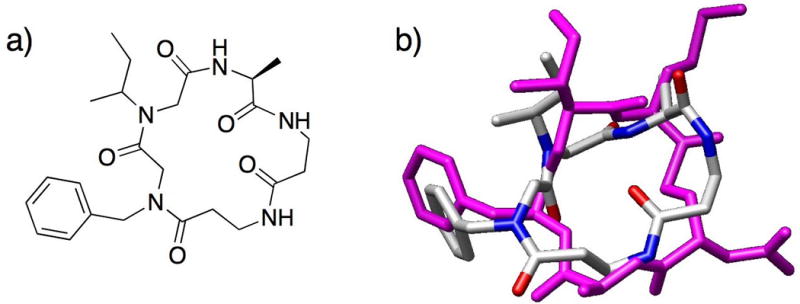
(a) Structure of macrocyclic peptomer 10, a mimic of AIP-I, reported by Fowler et al. (b) Overlaid computed models of the macrocyclic portion of AIP-I (magenta) and peptomer 10 (colored by atom type). Molecular mechanics performed in MOE (v. 2006.08).55
4.3 Lung Surfactant Mimics
Lung surfactant (LS) is a naturally-occurring material that is essential for proper respiration in humans, and is composed of lipids and proteins that reduce and regulate surface tension at the air-liquid interface in the lungs.56 A deficiency in functional LS leads to respiratory distress syndrome, a leading cause of mortality in premature infants and of respiratory impairment or failure in children and adults. Treatment with current synthetic surfactant formulations is less effective than natural LS, and animal-extracts of natural LS have several disadvantages, such as causing an immune response. Barron and co-workers have reported peptoid mimics of the two helical, hydrophobic surfactant proteins (SP) found in natural LS, termed SP-B and SP-C. The peptoid SP mimics could serve as LS replacements in a peptoid-lipid mixture formulation. Baron’s team designed mimics of the N-terminal segment of SP-B, SP-B1–25, which has a hydrophobic insertion region (residues 1–9) that is hypothesized to insert into the lipid layer of the air-liquid interface in the lung, and an amphipathic helix containing Arg and Lys that interacts with the charged lipid head groups. The peptoid SP-B mimics contained α-chiral aromatic (Nspe) or α-chiral aliphatic (Nssb) monomers (Fig. 3) as hydrophobic residues and to promote helicity, and NArg and/or NLys as cationic residues (Table 3).57 The authors also examined the effect of including a helical insertion region (i.e., residues 1–8 in peptoids B2, B4, and B5) and whether NArg could be replaced with NLys (i.e., peptoid B4 versus B5) in these peptoids.
Table 3.
Sequences of lung surfactant proteins SP-B and SP-C and peptoid mimics thereof reported by Barron and co-workers57,58
| Oligomer | Sequence (N- to C-terminus) |
|---|---|
| Peptide SP-B1–25 | FPIPLPYAWLARALIKRIQAMIPKG |
| Peptoid B1 | Nspe2-(NLys-Nspe-Nspe)5 |
| Peptoid B2 | Nspe8-NLys-Nspe2-NLys-NLys-Nspe4-NLys-Nspe2 |
| Peptoid B3 | Nssb2-(NLys-Nssb-Nssb)5 |
| Peptoid B4 | Nssb8-NArg-Nssb2-NLys-NArg-Nssb4-NLys-Nssb2 |
| Peptoid B5 | Nssb8-NLys-Nssb2-NLys-NLys-Nssb4-NLys-Nssb2 |
|
| |
| Peptide SP-CLff | FGIPFFPVHLKRLLILLLLLLLILLLILGALLMGL |
| Peptoid C1, n = 8 | NPhe-NPhe-Pro-NVal-NPhe-NLeu-NLys-NArg-(Nspe)n |
| Peptoid C2, n = 11 | |
| Peptoid C3, n = 14 | |
| Peptoid C4, n = 8 | NPhe-NPhe-Pro-NVal-NPhe-NLeu-NLys-NArg-(Nssb)n |
| Peptoid C5, n = 11 | |
| Peptoid C6, n = 14 | |
Peptide sequences are represented by their one letter amino acid codes. NLys, NArg, NPhe, NVal and NLeu are the peptoid monomers of the corresponding amino acids.
The peptoid SP-B mimics were evaluated by CD spectroscopy, and all were shown to be helical in methanol. The helices formed by aromatic peptoids (B1 and B2; Table 3) were more stable (based on CD intensity) than the helices formed by aliphatic peptoids (B3–B5). Next, the surface-active behavior of the peptoids in a model lipid mixture was evaluated. The aliphatic peptoids (B3–B5) were found to have better surface activity (e.g., increased lift-off areas and more rapid adsorption to the air-liquid interface) than the aromatic peptoids (B1 and B2), and the authors postulated that the more flexible helices of the aliphatic peptoids might interact with lipids more easily. In addition, the peptoids containing an N-terminal helical insertion region (B2, B4, and B5) were able to maintain lower surface tensions than the shorter peptoids (B1 and B3). The presence of this hydrophobic insertion region was also hypothesized to enhance surface activity by improving interactions with the lipids. Finally, the similar surface activities of B4 and B5 suggested that NArg residues were not required for function and could be replaced with NLys residues. Collectively, these data for peptoid mimics indicate that although mimicking the helicity of SP-B is important, peptoid conformational flexibility is critical to the activity of peptoid-lipid mixtures as LS replacements.
Barron and co-workers also designed peptoid mimics of the LS protein SP-C.58 Like SP-B, SP-C is a helical hydrophobic protein and an essential component of functional LS. In addition to disorders caused by a deficiency of LS, accumulation of misfolded SP-C can lead to lung disease. SP-C has two positively charged residues, Lys and Arg, at positions 11 and 12 that interact with phospholipid head groups, and a helical C-terminal region that interacts with the lipid acyl chains. The high hydrophobicity of SP-C makes it difficult with which to work, and for this reason, the modified peptide SP-CLff was used as a control in this study (Table 3). The designed peptoids, C1–C6, contained a C-terminal helix that was either all α-chiral aromatic (Nspe; C1–C3) or all α-chiral aliphatic (Nssb; C4–C6) and was eight, 11, or 14 residues in length. The N-terminal portion of the peptoids mimicked the residues in positions 5–12 of SP-C. All six peptoids (C1–C6) were determined to be helical in methanol by CD spectroscopy. Aromatic peptoids C1–C3 displayed the double minimum CD signature characteristic of α-chiral aromatic peptoids and were stable helices regardless of chain length. Likewise, the aliphatic peptoids C4–C6 displayed the CD signature characteristic of helices for this class of α-chiral peptoids, but with an increase in helix stability at longer chain lengths. Previous work had shown that both Nspe and Nssb peptoid helices are three residue per turn right-handed helices, but that Nspe helices are tighter, more rigid structures (helical pitch ~6.0 Å)30 than Nssb helices (helical pitch ~6.7 Å).23 As the helicity of SP-C is essential for its surface activity, Barron and co-workers sought to discover which type of peptoid helix would best mimic SP-C.
The surface-active behaviors of the peptoids in a model lipid mixture were evaluated and compared to SP-CLff. In general, the aromatic peptoids (C1–C3, Table 3) were found to have better surface activity (e.g., increased lift-off areas, superior adsorption, and less compression to reach low surface tension) than the aliphatic peptoids (C4–C6). Of the three aromatic peptoids, the longest (C3), exhibited superior lift-off area and adsorbed to lower surface tension lipid compositions. A study of the film morphology of SP-CLff, C3, and C6 showed that the morphology of the aromatic peptoid C3 closely resembled that of SP-CLff. The authors reasoned that the α-chiral, aromatic peptoids were the best mimics of SP-C in this series because (1) the more rigid helix better modulated surface film, and (2) the increased hydrophobicity better mimicked SP-C and may have facilitated insertion into the lipid film. In addition, all of the peptoid surfactants in this study were less prone to aggregation and had more stable secondary structures than natural SP-C, making them attractive for the development of new LS replacement therapies.
4.4 Amylin Mimics
Liskamp and co-workers recently reported peptoid mimics of the peptide amylin, which is implicated in the onset of type II diabetes.59 Amylin readily aggregates and forms amyloid fibrils in the insulin producing islet β-cells60 via a cross β-sheet topology.61 Liskamp and co-workers designed and synthesized peptoid 11 and retropeptoid 12 (residues in reverse order, see Table 4) as analogs of the amylin core region, amylin(20-29), and studied the structures of these peptoids and their ability to disrupt amylin aggregation. The secondary structures of amylin(20-29) and peptoids 11 and 12 were evaluated by CD spectroscopy: amylin(20-29) displayed a CD signature characteristic of a β-sheet, while 11 and 12 had weak, random CD spectra indicating no defined secondary structure. The presence of a cross β-sheet topology was identified in amylin(20-29) by a diagnostic peak at 1630 cm−1 (type I amide absorption) in a Fourier transform infrared (FTIR) spectrum. The FTIR spectrum of 11 lacked an absorption at 1630 cm−1, further evidence that it was unable to form a β-sheet conformation. Transmission electron microscopy (TEM) was used to assess the ability of these peptoids to form amyloid fibrils. A solution of amylin(20-29) in 0.1% TFA/H2O rapidly formed an opalescent gel (in less than 10 min), and TEM analysis revealed amyloid fibril formation. However, solutions of 11 and 12 remained clear (up to three weeks) under the same conditions. No amyloid fibrils or other aggregates of 11 were visible by TEM. In contrast, TEM analysis revealed that retropeptoid 12 formed supramolecular assemblies like ribbons and tapes.
Table 4.
Structures of amylin(20-29) and peptoid 11 and retropeptoid 12 reported by Liskamp and co-workers59
| Oligomer | Sequence (N- to C-terminus) |
|---|---|
| amylin(20-29) | SNNFGAILSS |
| peptoid 11 | NSer-NAsn-NAsn-NPhe-Gly-NAla-NIle-NLeu-NSer-NSer |
| retropeptoid 12 | NSer-NSer-NLeu-NIle-NAla-Gly-NPhe-NAsn-NAsn-NSer |
Next, Liskamp and co-workers evaluated the ability of the peptoids to inhibit β-sheet and amyloid fibril formation in amylin(20-29). A 1:1 (w/w) mixture of amylin(20-29) and 11 did not display a CD spectrum indicative of a β-sheet, evidence that 11 was able to inhibit the β-sheet formation of amylin(20-29). In addition, this mixture of amylin(20-29) and 11 was ~20% as turbid as amylin(20-29) alone, demonstrating the ability of 11 to inhibit aggregation (i.e., amyloid formation). The retropeptoid 12, however, was only a moderate inhibitor of aggregation, as the turbidity of a 1:1 (w/w) mixture of amylin(20-29) and 12 was at ~50% the level of amylin(20-29) alone. The supramolecular assemblies of 12 (observed by TEM) potentially prevent the peptoid from being an effective inhibitor of amyloid formation. The authors suggested that, in general, peptoids may disrupt β-sheets by terminating the hydrogen bond networks (i.e., peptoids lack hydrogen bond donors, preventing further assembly). Since this characteristic was common to both 11 and 12, it was perhaps the lack of supramolecular structure in 11 that allowed it to interact effectively with amylin(20-29). This work showed that lack of both secondary and higher order structures in peptoids was necessary to disrupt β-sheet formation in amylin(20-29), and represents an example of lack of structure being essential for peptoid function. Further development of peptoids such as those described here could lead to new treatments for diseases caused by fibril and plaque formation.
5 Molecular Recognition
The ability to design synthetic molecules that can bind to specific protein targets in the body and effect a biological response (i.e., pharmaceutical drugs) has revolutionized the treatment of human disease and improved our quality of life. As researchers strive to inhibit more complex interactions at the biomolecular level, nondrug-like molecules are being incorporated into the chemist’s toolbox to gain specificity and potency. Peptoids are currently being studied for their potential to serve as such pharmaceutical agents and as chemical tools to study complex biomolecular interactions. Peptoid-protein interactions were first demonstrated in a 1994 report by Zuckermann and co-workers, where the authors examined the high-affinity binding of peptoid dimers and trimers to G-protein-coupled receptors.5 Shortly thereafter, Lim and co-workers designed peptomers that could bind selectively to a Src homology 3 (SH3) domain and potentially could be utilized to inhibit protein-protein interactions involving SH3 domains.62,63 These groundbreaking studies have led to the identification of several peptoids with moderate to good affinity and, more importantly, excellent selectivity for protein targets that are implicated in a range of human diseases. In the following sections, we provide several recent examples of such molecular recognition events modulated by peptoids.
5.1 Peptoid-Protein Interactions
Synthetic molecules capable of activating the expression of specific genes would be valuable for the study of biological phenomena and could be therapeutically useful. From a library of ~100,000 peptoid hexamers, Kodadek and co-workers recently identified three peptoids (13–15) with low micromolar binding affinities for the coactivator CREB-binding protein (CBP) in vitro (Fig. 10).7,64 This coactivator protein is involved in the transcription of a large number of mammalian genes, and served as a target for the isolation of peptoid activation domain mimics. Of the three peptoids, only 13 was selective for CBP, while peptoids 14 and 15 showed higher affinities for bovine serum albumin. The authors concluded that the promiscuous binding of 14 and 15 could be attributed to their relatively “sticky” natures (i.e., aromatic, hydrophobic amide side chains). Although only peptoid 13 was selective for CBP, the researchers next assessed the cell permeability of all three peptoids. Peptoid 13 and 14 had good and moderate cell permeability, respectively, while 15 displayed poor permeability. Peptoid 13 is relatively hydrophilic and the most cell permeable, a correlation observed in other studies (see below). Interestingly, 14 is the most hydrophobic of the three and still quite permeable, while 15 has intermediate hydrophobicity but displays poor cell permeability. Such connections between primary structure and cell permeability are highly relevant in the further development of peptoids for in vivo applications.
Fig. 10.
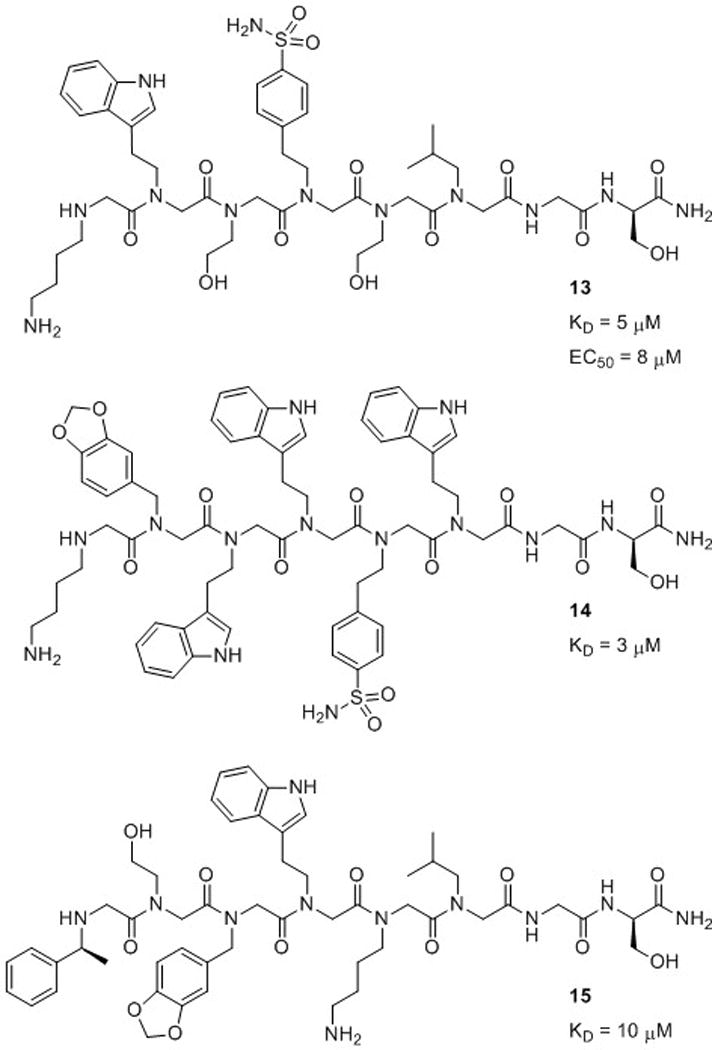
Peptoid hexamers 13, 14, and 15 reported by Kodadek and co-workers and their dissociation constants (KD) for coactivator CBP.7,64 Peptoid 13 was able to function as a transcriptional activation domain mimic (EC50 = 8 μM).
Evaluation of peptoids 13–15 as activation domain surrogates in mammalian cells revealed that, as expected, no detectable transcription was induced by 15, since it could not appreciably accumulate inside the cell. Peptoid 14 also failed to induce transcription, indicating that while it could enter cells and was capable of binding to CBP, it did not function as an activation domain. The authors suggested that this was likely due to the ability of peptoid 14 to bind to other proteins. In contrast, peptoid 13 was capable of serving as an activation domain, with a half maximal effective concentration (EC50) = 8 μM. This study elucidated important structural considerations for designing peptoids that are capable of selective protein binding in cells. The more hydrophilic peptoid (13) displayed the best cell permeability. In addition, the peptoids in this study with numerous aromatic, hydrophobic moieties showed increased nonselective protein binding.
5.2 Inhibiting Protein-Protein Interactions
Protein-protein interactions control many cellular processes and represent an emerging target for the development of a range of therapeutics. Proteins commonly interact by making intermolecular contacts over a large binding domain; this domain is often a nonpolar cleft. Designing synthetic molecules to bind to these clefts with specificity is a challenging endeavor. The protein clefts are large and the topologies of different proteins share similar features, which make these domains difficult to target with small molecules. Recently, Appella and co-workers reported the design of peptoids that inhibit the human double minute 2 protein (HDM2)-p53 interaction.8 Functioning as a transcription factor, active p53 leads to cell cycle arrest and apoptosis. However, p53 is inactivated when bound to HDM2, allowing for cell proliferation. In many cancerous cells, p53 is mutated (preventing DNA binding) or HDM2 is overexpressed (inactivating p53), resulting in uncontrolled cell growth and the development of tumors. Inhibitors of the HDM2-p53 interaction therefore hold promise for cancer therapy.
The HDM2 binding domain of p53 (residues 18–26) is an amphipathic α-helix and binds tightly to HDM2 by making three hydrophobic interactions through a set of Phe, Trp and Leu side chains that are proximal on a helix face. Appella and co-workers designed α-chiral peptoid helices that displayed these three amino acid side chains in a similar arrangement, and used molecular modeling to show good alignment of these side chains in the native p53 peptide and peptoid helices.8 They evaluated the helicity of the peptoids by CD spectroscopy in aqueous solution and the ability of the peptoids to bind to HDM2 in a competition assay with a fluorescein-labeled p53 fragment. Interestingly, the authors found that the helical peptoids had very weak binding affinities for HDM2 (IC50 ~ 200 μM), while the nonhelical peptoids had good binding affinities for HDM2 (IC50 ~ 16 μM; for comparison, the p53 peptide binds HDM2 at IC50 = 3 μM). Following this discovery, Appella and co-workers synthesized a series of peptoids to optimize cell permeability and binding affinity. They gained significant binding affinity by replacing the indole side chain (Trp mimic) with 6-chloroindole (IC50 = 10 μM). However, an analog of the chloroindole peptoid with all achiral side chains had even better affinity for HDM2 (16, IC50 = 7 μM; Fig. 11). This peptoid could not be evaluated by CD spectroscopy, but was presumed to be nonhelical. The authors did note, however, that a helical conformation may be induced in 16 upon binding of the peptoid to HDM2. Regardless, this study suggests again that a defined secondary structure was not required for the peptoid to possess biological activity. In addition, this research demonstrates that the ability of peptoids to display key residues for protein binding is a feature that could be exploited for the further development of peptoids targeting protein-protein interactions.
Fig. 11.
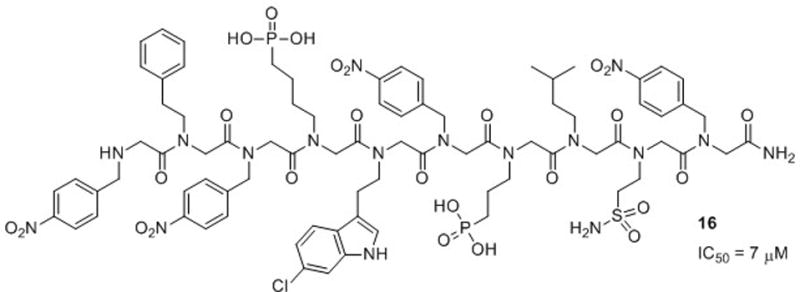
Structure of the achiral peptoid 16 reported by Appella and co-workers that inhibits the HDM2-p53 interaction.8
Inhibitors of proteasome function that can intercept proteins targeted for degradation would be valuable as both research tools and therapeutic agents. The 26S proteasome is responsible for most non-lysosomal protein degradation in eukaryotic cells. The 19S regulatory particle (RP), a portion of the 26S proteasome, binds polyubiquitinated proteins, unfolds them, and guides them into the proteasome interior for proteolytic degradation. In 2007, Kodadek and co-workers identified the first chemical modulator of the 19S RP, a purine-capped peptoid heptamer (17, Fig. 12a).65 In an in vitro assay, peptoid 17 inhibited the protein unfolding activity of the 19S RP with an IC50 = 3 μM. In addition, peptoid 17 inhibited 26S-mediated proteolysis in cells, although less effectively (IC50 ~ 30–50 μM). Next, the researchers identified the minimal pharmacophore of this 19S RP inhibitor by performing a glycine scan of 17 (analogous to alanine scanning in α-peptides).66 This experiment showed that only the core tetrapeptoid was required for activity (18, Fig. 12b). In the same in vitro assay used to evaluate heptamer 17, tetrapeptoid 18 inhibited protein unfolding with an IC50 ~ 5–10 μM, which was only a 2- to 3-fold decrease relative to 17. However, in the cell-based assay, tetramer 18 inhibited 26S-mediated proteolysis with an IC50 ~ 10–15 μM, a 3- to 5-fold increase in activity relative to 17. The increased activity of 18 in cellulo was likely due to increased cellular uptake, as 18 is approximately half the size of 17 and does not contain charged residues. Based on these data, the authors concluded that 18 was indeed the minimal pharmacophore of a peptoid inhibitor of the 19S RP. We anticipate that the structural features uncovered here for in cellulo activity may be applicable to the development of peptoids for other biological applications.
Fig. 12.
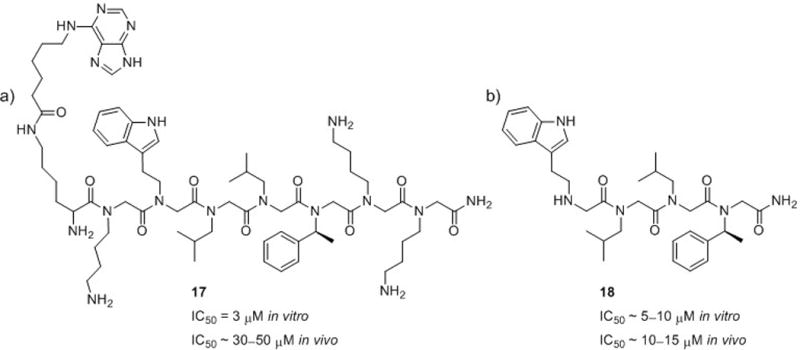
Peptoids reported by Kodadek and co-workers that inhibit the interaction of 19S RP with polyubiquitinated proteins, preventing their degradation. (a) Purine-capped peptoid heptamer 17.65 (b) Peptoid tetramer 18.66
Another study by Kodadek and co-workers in 2008 uncovered a peptoid that antagonizes the vascular endothelial growth factor receptor 2 (VEGFR2).10 The binding of VEGF to VEGFR2 is a critical event in angiogenesis, and blocking this hormone-receptor interaction represents a possible strategy for the treatment of certain cancers (especially those involving solid tumors) and macular degeneration. To date, monoclonal antibodies, protein-based molecules, and peptides have been shown to inhibit the VEGF pathway, but these agents have limited tumor penetration and are often immunogenic. Small molecule-based treatments, on the other hand, often have side effects due to interaction with other receptors similar to VEGFR2 in the body. By screening a library of 300,000 peptoid nonamers, Kodadek and co-workers identified five peptoids that bound selectively to VEGFR2 in vitro with low micromolar affinity. Each of these peptoids contained four or five NLys residues, two NLeu residues, and one or two aromatic residues, and two peptoids were selected for further study (19 and 20; Fig. 13a). However, these two peptoids were only weak antagonists of VEGFR2 autophosphorylation in a whole cell assay (autophosphorylation of the kinase domain of VEGFR2 is an early step in angiogenesis and occurs upon VEGF binding). Building on the knowledge that VEGFR2 functions as a homodimer, the authors designed homodimers of the peptoid ligands, and found that the dimer of 19 containing a relatively long linker had a binding affinity of 30 nM for VEGFR2 (Fig. 13b). This 19-dimer was a low micromolar antagonist (IC50 ~ 1 μM) in the VEGFR2 autophosphorylation whole cell assay. Moreover, this peptoid was active in vivo and inhibited tumor growth in a mouse model. At the end of a 21-day study, mice that had received a continuous treatment of 19-dimer had tumors that were five times smaller than saline-treated control mice. Interestingly, further study showed that the peptoids did not bind VEGFR2 competitively with VEGF.67 Indeed, Kodadek and co-workers were able to effect simultaneous binding of a peptoid and VEGF to VEGFR2. However, the effect of this peptoid binding to VEGFR2 produced the same phenotype (tumor growth inhibition) as was expected from inhibition of VEGF-VEGFR2 binding. Overall, this work successfully utilized a peptoid-dimerization strategy to target a protein that functions as a homodimer, and is relevant to the modulation of protein-protein interactions in other dimeric protein systems.
Fig. 13.
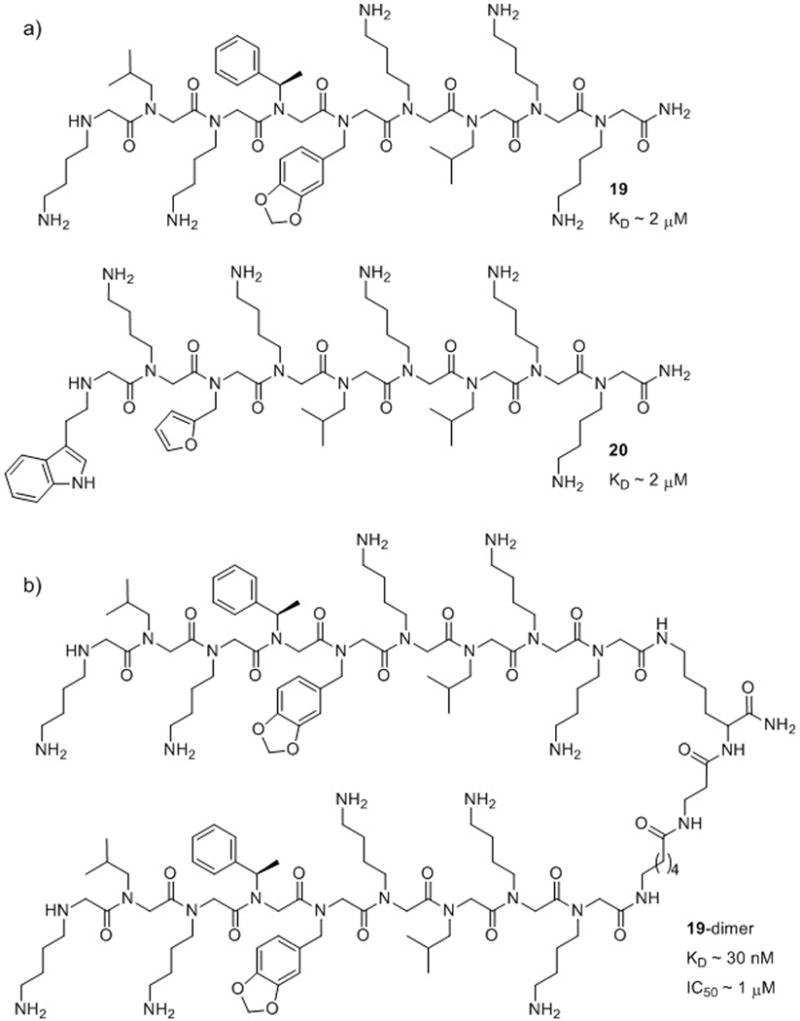
(a) Two of the peptoids (19 and 20) found to bind to VEGFR2 by Kodadek and co-workers.10 (b) Dimerization of 19 via a flexible linker (to yield 19-dimer) resulted in an inhibitor of VEGFR2 and suppressed tumor growth in a mouse model.10,67
Pérez-Payá and co-workers recently identified a peptoid inhibitor of the apoptotic protease-activating factor Apaf-1.68 Apaf-1 is a central protein component of the apoptosome, and the formation of this multiprotein complex is a key event in apoptosis activation. Disruption of protein-protein interactions involving Apaf-1 represents a promising strategy for the treatment of several disorders, including immune and neurodegenerative diseases. Although Pérez-Payá and co-workers found that tripeptoid 21 (Fig. 14) was potent in vitro, it displayed only modest inhibition of apoptosis in cells. Improved in cellulo efficacy was achieved through two strategies: conjugation of the peptoid to poly-L-glutamic acid (to generate a pro-drug, PGA-21) and peptoid backbone cyclization (22; Fig. 14).69 While the bioconjugate PGA-21 displayed higher in cellulo efficacy (up to 100% inhibition of apoptosis) relative to the cyclic peptoid 22 (up to 60% inhibition), it required much higher concentrations of compound to achieve such activity (50 μM PGA-21 versus 1 μM 22). This is a common effect among PGA pro-drugs, which usually display activity at concentrations 10-fold higher than the parent molecule. The reduced flexibility in the cyclic peptoid 22 (while counterintuitive) may explain its lower efficacy, as it may be restricted to a sub-optimal conformation for binding to Apaf-1. But, the increased potency of 22 relative to 21 suggests it is a viable lead compound for future development.
Fig. 14.
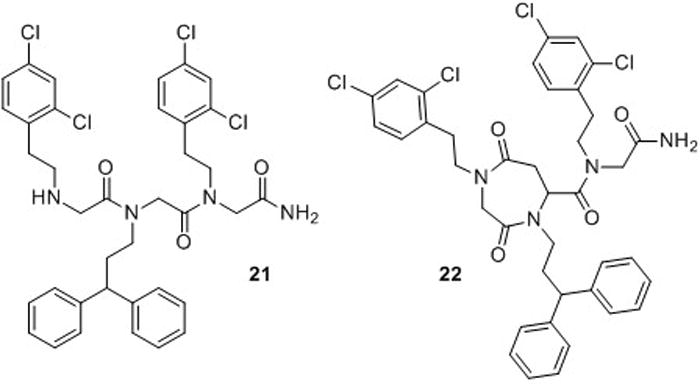
Peptoid inhibitors of Apaf-1 (21 and 22) developed by Pérez-Payá and co-workers.68,69
5.3 Multivalent Peptoid Ligands
The coupling of two or more noncompetitive ligands has been shown to be an effective strategy to create multivalent protein binding agents.70 Such a multivalent ligand can possess protein affinity and specificity greater than the sum of its parts. Oligomeric molecules have been beneficial for displaying an array of recognition elements for binding. Peptoid-based multivalent oligomers are advantageous due to their ease of synthesis relative to other multivalent ligands (e.g., oligosaccharides), and their precise display of recognition elements as opposed to ligands generated by block copolymerization or dendrimer synthesis.
In 2004, Kodadek and co-workers reported a procedure for identifying bivalent ligands with high affinity and specificity for their target protein.71 A chalcone known to bind to the p53 binding domain of murine double minute 2 protein (MDM2, the mouse homolog of HDM2) with low affinity (KD = 220 μM) was selected for conjugation to peptoid decamers. A split-pool library of chalcone-capped peptoids (~78,000 compounds) was screened for MDM2 binding in the presence of a 10,000-fold excess of other proteins. This strategy was employed to eliminate nonspecific binding. Three peptoid-chalcone hybrids were identified as high-affinity binders for MDM2, and the structures were determined to be NLys-rich sequences. Further analysis of one of the hits revealed that peptoid-chalcone hybrid 23 had a KD = 1.3 μM for MDM2 (Fig. 15a), a significant improvement over the chalcone alone. For comparison, the peptoid was synthesized without the chalcone-cap, and its KD was 378 μM. This work highlights the improvement in activity that can be gained through multivalent display. In addition, the conjugation of small organic molecules to peptoid oligomers represents an important new avenue for the discovery of lead compounds.
Fig. 15.
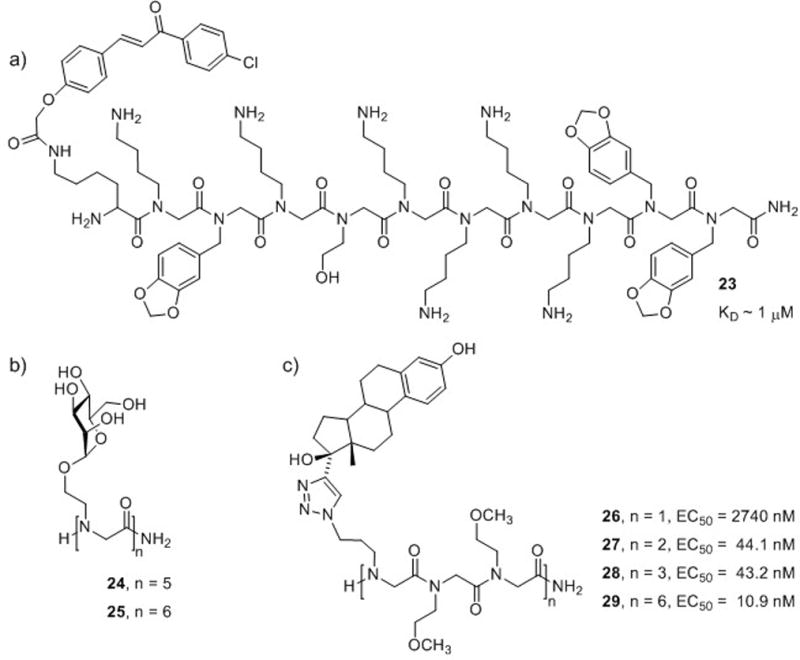
Multivalent peptoid ligands for various protein targets. (a) A chalcone-peptoid hybrid 23 that binds selectively to MDM2.71 (b) Pentamer (24) and hexamer (25) mannosylpeptoids that bind to ConA.72 (c) Four estradiol-containing peptoids (26–29) that bind to the estrogen receptor.73
A 2007 report by Yuasa and co-workers described oligomannosylpeptoids as potential substitutes for glycopeptides or oligosaccharides.72 The major advantage of these peptoid mimics was their ease of synthesis relative to that of native carbohydrate structures. The researchers synthesized peptoid oligomers displaying one to six mannoses (monomer to hexamer; Fig. 15b), and observed that only the pentamer (24) and hexamer (25) were capable of appreciable binding to concanavalin A (ConA, a lectin protein that specifically binds mannosyl and glucosyl residues). Hence, multivalent display of the mannosyl residue (i.e., a minimum of five mannoses) in the peptoid structures was determined to be critical for binding. This work indicates a role for peptoids in the development of non-native carbohydrate derivatives, and also underscores the critical role of oligomer length in the design of peptoid mimics.
Kirshenbaum and co-workers have utilized Cu-catalyzed azide-alkyne [3+2] cycloadditions (“click chemistry”) to conjugate biologically relevant molecules (e.g., nucleobases and fluorophores) to peptoid oligomers.74 This technique allows the conjugation of one molecule at desired sites in the oligomer by performing click chemistry after peptoid synthesis, or the conjugation of a variety of molecules by sequential click reactions during peptoid chain elongation.75 Thus, multivalent peptoid conjugates may be constructed to display several copies of one functionality or a variety of functionalities. The latter strategy could potentially have application in the covalent linkage of functionalities identified through fragment-based drug discovery, for example. Kirshenbaum and co-workers recently demonstrated the utility of this methodology in the construction of multivalent estradiol-peptoid conjugates.73 Peptoids of methoxyethyl side chains (to enhance water solubility) were constructed with azidopropane side chains at every third position. Following oligomer synthesis, 17α-ethynylestradiol was coupled to the azides to yield the desired estradiol-peptoids (26–29; Fig. 15c). Using a competitive binding assay, EC50 values for binding of these peptoid-estradiol conjugates to the estrogen receptor were determined by displacement of tritium-labeled 17β-estradiol, the native ligand for the estrogen receptor. The conjugates showed increasing activities with increasing multivalency, and hexavalent peptoid 29 was the most active in the series. These “click” techniques represent a highly straightforward approach to construct multivalent peptoid ligands displaying one or several types of recognition elements, and could find wide application.
6 Cellular Uptake and Delivery
Zuckermann and co-workers first demonstrated the ability of peptoids to serve as cellular delivery agents in 1998.76 Cationic peptoids containing lysine-like residues were able to form complexes with DNA and mediate cell transfection with efficiencies similar to that of traditional cationic lipids. However, peptoids had the advantage of retaining high levels of activity in the presence of serum. The authors went on to construct peptoid-lipid hybrids, and found that these compounds displayed transfection efficiencies up to 20 times higher than that of lipids alone.77 In both studies, the researchers observed that peptoids with a repeating trimeric side chain motif (cationic, neutral, neutral) displayed the highest transfection activities. Subsequent work by Wender et al. in 2000 demonstrated rapid cellular uptake of guanidinylated peptoids relative to L- and D-Arg polypeptides.12 This collective work has prompted further investigations into the ability of peptoids to serve as drug and gene delivery agents, and several recent examples are highlighted below.
6.1 Cellular Uptake
In 2005, Kodadek and co-workers developed a high-throughput assay for assessing the mammalian cell permeability of synthetic molecules.78 Using this method, the researchers evaluated the cell permeability of peptoids relative to their corresponding α-peptide analogs.13 In the evaluation of six sequences, the peptoids displayed 3- to 26-fold increases in cell permeability over the corresponding α-peptides, and relative permeability was inversely proportional to oligomer length. The authors suggested that the lack of amide hydrogens in the peptoid backbone, which increases the overall lipophilicity of the molecule, could enhance movement across the cell membrane. Kodadek and co-workers then performed a more comprehensive study of the criteria for cell permeability in peptoids.79 Using split-pool techniques, libraries of peptoid and α-peptide tetramers were prepared, and the relative cell permeabilities of 350 peptoids and α-peptides were analyzed to generate structure-activity relationships. The average cell permeability of the peptoids was twice that of the α-peptides. Collectively, these peptoids were slightly less lipophilic than their α-peptide counterparts; however, the topological polar surface area (i.e., the solvent-accessible surface areas of heteroatoms) was lower in the peptoids. This was attributed to the lower polarity of peptoid backbone tertiary amides relative to peptide secondary amides. Furthermore, the authors noted that the fewer hydrogen bond donors and acceptors in a peptoid, the greater its cell permeability. Lastly, peptoids and peptides with the greatest permeabilities had highly similar side chain composition, namely one-third hydrophobic residues and two-thirds hydrophilic residues. This result suggests that direct translation of peptide sequences into peptoids could result in improved cellular uptake.
6.2 Delivery
Recently, Bräse and co-workers reported the synthesis of (NLys)6, (NArg)5, and (NArg)6 homopeptoids conjugated to fluorophores and evaluated their cellular uptake.80 The researchers observed that uptake was more rapid in guanidinium functionalized peptoids (NArg) compared to amino functionalized peptoids (NLys). Furthermore, the location of transport differed depending on amide side chain functionality—amino peptoids were transported to the cytosol, while guanidinium peptoids had greater, but not exclusive, accumulation in the nucleus. Thus, in this study, peptoid primary structure had a significant effect on the rate of cellular uptake and the location of delivery.
In 2008, Bradley and co-workers described peptoid dendrimers that functioned as efficient gene transfection agents.81 First, second, and third generation dendrimers were synthesized containing lysine-like amide side chains, which displayed four, eight, or 16 amines, respectively. The researchers observed efficient uptake of DNA by the third generation peptoid dendrimer (~3-fold higher transfection relative to a polyamidoamine (PAMAM) dendrimer). The researchers also demonstrated that the active peptoid dendrimer had no cytotoxicity in human cells, while the PAMAM dendrimer showed a slight reduction in cell viability. This work suggests that efficient transfection is dependent on sufficient cationic charge in peptoid dendrimers, as only the dendrimer displaying 16 amines effected transfection.
7 Toward the Design of Peptoid-Derived Artificial Proteins
The ability to design peptoids with defined tertiary structures that can perform biological functions represents a pathway to the development of artificial proteins. Such foldamers would have far-ranging applications and could prove useful as therapeutics for diseases ranging from genetic disorders to cancer. However, considering the difficulties associated with stabilizing well-defined secondary structures in peptoids (see above), mimicking the complex folded states of natural proteins constitutes a huge challenge. Working toward this goal, Zuckermann, Dill, and co-workers have designed peptoids that adopt defined tertiary structures in aqueous solution82 and, more recently, reported the incorporation of a zinc-binding function into these structures.83 This work represents an important first step toward peptoid-based, biomimetic structures with enzyme-like function.
In 2005, Dill and co-workers reported the design of a stable, multi-helical tertiary structure in peptoids.82 Previous to this work, Zuckermann and co-workers had discovered that amphiphilic peptoid helices associated in aqueous solution to form helical multimers.84 One of these amphiphilic peptoid 15-mers, which self-assembled into a trimer, was chosen by Dill and co-workers as a basis for the design of a peptoid helix bundle. This peptoid contained α-chiral side chains to stabilize helical secondary structure, and hydrophobic side chains at every third residue to create a hydrophobic face on the helix (30; Fig. 16). For comparison, α-chiral peptoids with hydrophobic side chains at every other residue, α-chiral peptoids lacking hydrophobic side chains, and achiral peptoids were synthesized as controls. The peptoid 15-mers were covalently linked (via flexible disulfide and oxime linkers) to generate 30-mer, 45-mer and 60-mer peptoids, which could potentially form two-, three-, and four-helix bundles, respectively.
Fig. 16.
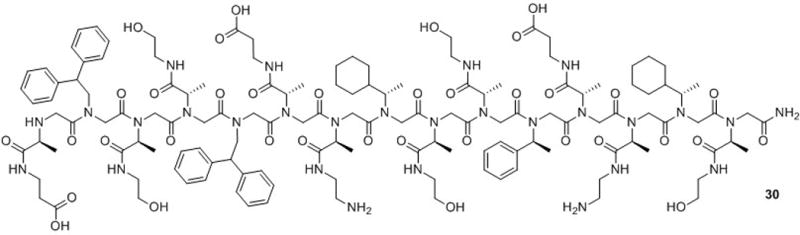
Structure of a peptoid 15-mer (30) that can self-assemble into a helical trimer in aqueous solution. Dill and co-workers constructed peptoid helix bundles by covalent linkage of two, three, or four such peptoid 15-mers.82
In the first phase of Dill and co-workers’ study, they examined the cooperative unfolding of the peptoid 30-mer structures in aqueous solution. The 30-mer peptoids contained a fluorescence donor at the N-terminus and a fluorescence quencher at the C-terminus, and fluorescence was quenched in the folded state. The Förster resonance energy transfer (FRET) efficiencies were measured for each peptoid in increasing concentrations of acetonitrile in water, and sizable changes in FRET were observed. Notably, the addition of a denaturing solvent did not disrupt secondary structure in the 30-mers (as confirmed by CD spectroscopy); thus, the authors deduced that the FRET differences resulted from the unfolding of tertiary structure. Cooperative unfolding occurred in both α-chiral and achiral 30-mer peptoids with hydrophobic side chains at every third residue. The achiral peptoids displayed greater folding cooperativity relative to the α-chiral peptoids, suggesting more hydrophobic burial in the folded state. The authors postulated that the increased flexibility in the achiral peptoids may facilitate tighter packing of the hydrophobic groups. In contrast, cooperative unfolding was not observed in peptoids lacking three-residue periodicity of hydrophobic side chains or in those lacking hydrophobic side chains. Based on these results, Dill and co-workers inferred that the formation of a hydrophobic core by the packing of hydrophobic helix faces was the driving force for self-assembly in water. In the second part of this study, the authors compared the stabilities of the tertiary structures in the 30-mer, 45-mer, and 60-mer α-chiral peptoids. The cooperative unfolding of these structures revealed that the three-helix bundle 45-mer was a more stable structure than either the 30-mer or 60-mer helix bundles. This was attributed to greater burial of hydrophobic side chains in the folded state of the 45-mer three-helix bundle.
In 2008, Zuckermann and co-workers described the incorporation of a zinc-binding motif into peptoid two-helix bundles.83 The zinc-binding motif was included because zinc can stabilize native protein structures and act as a cofactor in enzyme catalysis.85,86 The authors designed peptoids that would only be capable of binding zinc if properly folded into the helix bundle structure in aqueous solution. The peptoids had a general sequence of Nspe(Nsce)2Nspe(Nae)2, which was two-thirds α-chiral to enforce helicity and had hydrophobic Nspe residues at every third position to form an amphipathic helix (Fig. 17a). For zinc binding, thiol (NCys) and imidazole (NHis) substitutions were made at different sites in the primary sequence. Two such 15-mer peptoids were covalently linked via flexible, glycine-rich chains (Gly-Pro-Gly-Gly or (Gly)12) to form the two-helix bundles (Fig. 17b). The researchers analyzed the peptoids for their ability to bind zinc and for alteration of structure in the presence of zinc. First, cooperative unfolding of the peptoids by addition of organic solvent was observed in the absence of zinc, confirming that the peptoids adopted a tertiary structure in aqueous solution. Second, there was no indication of unfolding by organic solvent in the presence of zinc, demonstrating that zinc stabilized the two-helix bundle. This was analogous to the ability of zinc to stabilize tertiary structure in native proteins. Furthermore, the peptoids displayed nearly identical CD spectra in the presence or absence of zinc, indicating that zinc had no effect on their helical secondary structures.
Fig. 17.
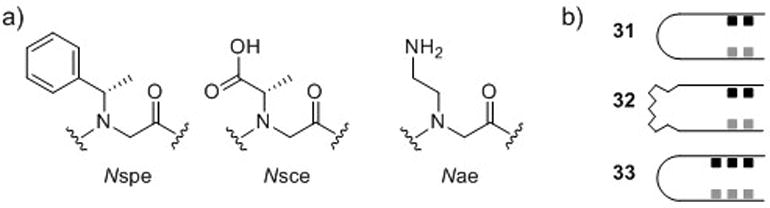
(a) Zuckermann and co-workers’ peptoid two-helix bundles were constructed from the monomers Nspe (see Fig. 3), Nsce, and Nae. (b) Schematic of two-helix bundles with two thiols, two imidazoles, and short linker (31), two thiols, two imidazoles, and long linker (32), and three thiols, three imidazoles, and short linker (33).83
Next, Zuckermann and co-workers investigated the effects of linker length (31 v. 32, Fig. 17b) and the quantity of NCys and NHis residues (31 v. 33, Fig. 17b) on zinc binding affinity. Apparent KD values were calculated based on FRET efficiencies for peptoids derivatized with fluorescence donors and quenchers. Peptoid 31 containing two thiols, two imidazoles, and the short linker had low micromolar zinc binding affinity (apparent KD = 1.2 μM). However, high-affinity zinc binding was observed with the longer linker in peptoid 32 (apparent KD ~ 0.4 nM). When three thiols and three imidazoles were incorporated into the peptoid (33), high-affinity zinc binding was also observed (apparent KD ~ 0.3 nM). The authors proposed that overlapping multivalency in 33 was responsible for tight binding, while the longer linker in 32 could accommodate optimal zinc-coordination geometry (Fig. 18). In addition, the authors demonstrated that the thiol and imidazole binding motif was selective for zinc, as several other divalent metal ions had peptoid binding affinities that were an order of magnitude higher than zinc. Such selectivity for a particular metal ion has also been observed in native proteins.87 Overall, this work represents substantial progress toward the goal of engineering protein-like structure and function into a non-natural polymer.
Fig. 18.
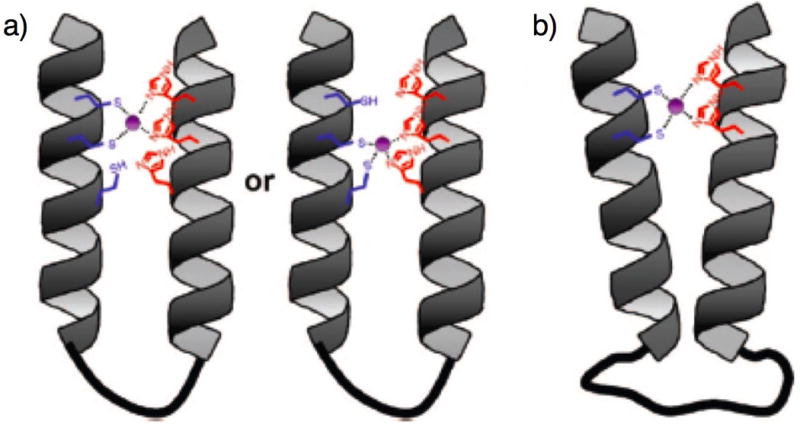
Schematic of proposed models for peptoid two-helix bundles with high-affinity zinc binding sites.83 (a) Trivalent peptoid 33. (b) Peptoid 32 with long flexible linker. Reprinted with permission from J. Am. Chem. Soc., 2008, 130, 8847-8855. Copyright 2008 American Chemical Society.
8 Summary
This perspective has analyzed the importance of peptoid structure in the discovery of biologically active peptoids over the past five years. In some cases, defined secondary structure was required for activity (e.g., lung surfactant mimics), and in other cases, the lack of defined secondary structure (at least in the absence of a cell membrane or target protein) resulted in the most active compound (e.g., HDM2-binding peptoids). There were also instances where both structurally well-defined and structurally undefined peptoids possessed equally good activity, as observed in antimicrobial peptoids, but the peptoids that adopted stable helical structures had poorer selectivities for bacterial cells over mammalian cells. For the inhibition of protein-protein interactions, the extent of hydrophobicity affected selectivity for a specific protein domain, as highly hydrophobic peptoids were found to bind to many different protein clefts. Analysis of peptoid primary structure showed that molecular weight and polarity can significantly influence cell permeability, an important consideration for in vivo activity. Lastly, in protein mimetics, properly folded peptoid tertiary structure relied on the formation of a hydrophobic core and, as dictated by the design, was required for function (zinc-binding).
Biomimicry by peptoids has not been straightforward thus far—the conformational flexibility of peptoids has made the design of well-folded peptoids a formidable challenge. However, defined structure does not appear to be a stringent requirement for biological function, and peptoids, whether structured or not, have proven to be valuable tools for biological and medicinal research. Furthermore, as additional strategies are developed for the design of well-folded peptoid structures a priori, researchers will be able to expand the array and complexity of structures that can be mimicked by peptoids. In the short term, new biological applications for peptoids are expected to evolve – for example, from the recent reports describing β-turn-like peptoid structures. A larger goal for future peptoid research is the creation of novel peptoid tertiary structures incorporating helix, loop, and turn regions. In addition, the relative ease of peptoid synthesis and the high cell permeability of peptoids will continue to aid the design and application of peptoid mimics of bioactive molecules. In these ways, and certainly many more that are currently being pursued, we anticipate that peptoids will remain an important and versatile approach to the study of biological phenomena.
Supplementary Material
{kind=link}
Acknowledgments
Peptoid research in our laboratory is supported by the NSF (CHE-0449959), ONR (N000140710255), Burroughs Wellcome Fund, Research Corporation, Johnson & Johnson, and 3M. H.E.B is an Alfred P. Sloan Foundation fellow. S.A.F. acknowledges the ACS Division of Medicinal Chemistry for a pre-doctoral fellowship (sponsored by Sanofi-Aventis) and Pfizer Global R&D for a Diversity in Organic Chemistry Fellowship. We thank the following individuals for the coordinates for peptoid images used in this perspective: Prof. Annelise Barron (Fig. 4), Prof. Kent Kirshenbaum (Fig. 5), Prof. Daniel Appella (Fig. 6), and Prof. John Robinson (Fig. 8). These images were produced using the UCSF Chimera software package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
References
- 1.Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, Spellmeyer DC, Tan R, Frankel AD, Santi DV, Cohen FE, Bartlett PA. Proc Natl Acad Sci U S A. 1992;89:9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farmer PS, Ariëns EJ. Trends Pharmacol Sci. 1982;3:362–365. [Google Scholar]
- 3.This concept was applied to the development of novel cholecystokinin (CCK) analogs by Horwell et al. Horwell DC, Beeby A, Clark CR, Hughes J, editors. J Med Chem. 1987;30:729. doi: 10.1021/jm00387a027. [DOI] [PubMed] [Google Scholar]
- 4.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 5.Zuckermann RN, Martin EJ, Spellmeyer DC, Stauber GB, Shoemaker KR, Kerr JM, Figliozzi GM, Goff DA, Siani MA, Simon R, Banville SC, Brown EG, Wang L, Richter LS, Moos WH. J Med Chem. 1994;37:2678–2685. doi: 10.1021/jm00043a007. [DOI] [PubMed] [Google Scholar]
- 6.Ng S, Goodson B, Ehrhardt A, Moos WH, Siani M, Winter J. Bioorg Med Chem. 1999;7:1781–1785. doi: 10.1016/s0968-0896(99)00132-7. [DOI] [PubMed] [Google Scholar]
- 7.Liu B, Alluri PG, Yu P, Kodadek T. J Am Chem Soc. 2005;127:8254–8255. doi: 10.1021/ja0515295. [DOI] [PubMed] [Google Scholar]
- 8.Hara T, Durell SR, Myers MC, Appella DH. J Am Chem Soc. 2006;128:1995–2004. doi: 10.1021/ja056344c. [DOI] [PubMed] [Google Scholar]
- 9.Xiao XS, Yu P, Lim HS, Sikder D, Kodadek T. J Comb Chem. 2007;9:592–600. doi: 10.1021/cc070023a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. J Am Chem Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 11.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Bioorg Med Chem Lett. 1994;4:2657–2662. [Google Scholar]
- 12.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwon YU, Kodadek T. J Am Chem Soc. 2007;129:1508–1509. doi: 10.1021/ja0668623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Proc Natl Acad Sci U S A. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fowler SA, Stacy DM, Blackwell HE. Org Lett. 2008;10:2329–2332. doi: 10.1021/ol800908h. [DOI] [PubMed] [Google Scholar]
- 16.Schröder T, Schmitz K, Niemeier N, Balaban TS, Krug HF, Schepers U, Bräse S. Bioconjugate Chem. 2007;18:342–354. doi: 10.1021/bc0602073. [DOI] [PubMed] [Google Scholar]
- 17.Patch JA, Kirshenbaum K, Seurynck SL, Zuckermann RN, Barron AE. Pseudopeptides in Drug Development, ed P E Nielsen, Wiley-VCH, Weinheim, Germany. 2004:1–31. [Google Scholar]
- 18.Olivos HJ, Alluri PG, Reddy MM, Salony D, Kodadek T. Org Lett. 2002;4:4057–4059. doi: 10.1021/ol0267578. [DOI] [PubMed] [Google Scholar]
- 19.Nnanabu E, Burgess K. Org Lett. 2006;8:1259–1262. doi: 10.1021/ol052867d. [DOI] [PubMed] [Google Scholar]
- 20.Gorske BC, Jewell SA, Guerard EJ, Blackwell HE. Org Lett. 2005;7:1521–1524. doi: 10.1021/ol0502984. [DOI] [PubMed] [Google Scholar]
- 21.Fara MA, Díaz-Mochón JJ, Bradley M. Tetrahedron Lett. 2006;47:1011–1014. [Google Scholar]
- 22.Armand P, Kirshenbaum K, Falicov A, Dunbrack RL, Jr, Dill KA, Zuckermann RN, Cohen FE. Fold Des. 1997;2:369–375. doi: 10.1016/S1359-0278(97)00051-5. [DOI] [PubMed] [Google Scholar]
- 23.Wu CW, Kirshenbaum K, Sanborn TJ, Patch JA, Huang K, Dill KA, Zuckermann RN, Barron AE. J Am Chem Soc. 2003;125:13525–13530. doi: 10.1021/ja037540r. [DOI] [PubMed] [Google Scholar]
- 24.Shin SBY, Kirshenbaum K. Org Lett. 2007;9:5003–5006. doi: 10.1021/ol702207n. [DOI] [PubMed] [Google Scholar]
- 25.Pokorski JK, Miller Jenkins LM, Feng H, Durell SR, Bai Y, Appella DH. Org Lett. 2007;9:2381–2383. doi: 10.1021/ol070817y. [DOI] [PubMed] [Google Scholar]
- 26.Gorske BC, Bastian BL, Geske GD, Blackwell HE. J Am Chem Soc. 2007;129:8928–8929. doi: 10.1021/ja071310l. [DOI] [PubMed] [Google Scholar]
- 27.Gellman SH. Acc Chem Res. 1998;31:173–180. [Google Scholar]
- 28.Hill DJ, Mio MJ, Prince RB, Hughes TS, Moore JS. Chem Rev. 2001;101:3893–4012. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- 29.Wu CW, Sanborn TJ, Zuckermann RN, Barron AE. J Am Chem Soc. 2001;123:2958–2963. doi: 10.1021/ja003153v. [DOI] [PubMed] [Google Scholar]
- 30.Armand P, Kirshenbaum K, Goldsmith RA, Farr-Jones S, Barron AE, Truong KTV, Dill KA, Mierke DF, Cohen FE, Zuckermann RN, Bradley EK. Proc Natl Acad Sci U S A. 1998;95:4309–4314. doi: 10.1073/pnas.95.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu CW, Sanborn TJ, Huang K, Zuckermann RN, Barron AE. J Am Chem Soc. 2001;123:6778–6784. doi: 10.1021/ja003154n. [DOI] [PubMed] [Google Scholar]
- 32.Huang K, Wu CW, Sanborn TJ, Patch JA, Kirshenbaum K, Zuckermann RN, Barron AE, Radhakrishnan I. J Am Chem Soc. 2006;128:1733–1738. doi: 10.1021/ja0574318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holub JM, Jang H, Kirshenbaum K. Org Lett. 2007;9:3275–3278. doi: 10.1021/ol071169l. [DOI] [PubMed] [Google Scholar]
- 34.Vaz B, Brunsveld L. Org Biomol Chem. 2008;6:2988–2994. doi: 10.1039/b806847j. [DOI] [PubMed] [Google Scholar]
- 35.Gorske BC, Blackwell HE. J Am Chem Soc. 2006;128:14378–14387. doi: 10.1021/ja065248o. [DOI] [PubMed] [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Shin SBY, Yoo B, Todaro LJ, Kirshenbaum K. J Am Chem Soc. 2007;129:3218–3225. doi: 10.1021/ja066960o. [DOI] [PubMed] [Google Scholar]
- 38.Hoffmann B, Ast T, Polakowski T, Reineke U, Volkmer R. Protein Peptide Lett. 2006;13:829–833. doi: 10.2174/092986606777841299. [DOI] [PubMed] [Google Scholar]
- 39.Frank R. Tetrahedron. 1992;48:9217–9232. [Google Scholar]
- 40.Heine N, Ast T, Schneider-Mergener J, Reineke U, Germeroth L, Wenschuh H. Tetrahedron. 2003;59:9919–9930. [Google Scholar]
- 41.Kramer A, Stigler RD, Knaute T, Hoffmann B, Schneider-Mergener J. Protein Eng. 1998;11:941–948. doi: 10.1093/protein/11.10.941. [DOI] [PubMed] [Google Scholar]
- 42.Peschel A, Sahl HG. Nat Rev Microbiol. 2006;4:529–536. doi: 10.1038/nrmicro1441. [DOI] [PubMed] [Google Scholar]
- 43.Hancock REW, Sahl HG. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 44.For a review of antimicrobial peptoids, see:; Masip I, Pérez-Payá E, Messeguer A. Comb Chem High Throughput Screen. 2005;8:235. doi: 10.2174/1386207053764567. [DOI] [PubMed] [Google Scholar]
- 45.Goodson B, Ehrhardt A, Ng S, Nuss J, Johnson K, Giedlin M, Yamamoto R, Moos WH, Krebber A, Ladner M, Giacona MB, Vitt C, Winter J. Antimicrob Agents Chemother. 1999;43:1429–1434. doi: 10.1128/aac.43.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patch JA, Barron AE. J Am Chem Soc. 2003;125:12092–12093. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]
- 47.Zhu WL, Song YM, Park Y, Park KH, Yang ST, Kim JI, Park IS, Hahm KS, Shin SY. Biochim Biophys Acta. 2007;1768:1506–1517. doi: 10.1016/j.bbamem.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 48.Nandel FS, Saini A. Macromol Theory Simul. 2007;16:295–303. [Google Scholar]
- 49.Shankaramma SC, Moehle K, James S, Vrijbloed JW, Obrecht D, Robinson JA. Chem Commun. 2003:1842–1843. doi: 10.1039/b304310j. [DOI] [PubMed] [Google Scholar]
- 50.Shankaramma SC, Athanassiou Z, Zerbe O, Moehle K, Mouton C, Bernardini F, Vrijbloed JW, Obrecht D, Robinson JA. ChemBioChem. 2002;3:1126–1133. doi: 10.1002/1439-7633(20021104)3:11<1126::AID-CBIC1126>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 51.Waters CM, Bassler BL. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 52.Lyon GJ, Muir TW. Chem Biol. 2003;10:1007–1021. doi: 10.1016/j.chembiol.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 53.Gorske BC, Blackwell HE. Org Biomol Chem. 2006;4:1441–1445. doi: 10.1039/b517681f. [DOI] [PubMed] [Google Scholar]
- 54.Chan WC, Coyle BJ, Williams P. J Med Chem. 2004;47:4633–4641. doi: 10.1021/jm0400754. [DOI] [PubMed] [Google Scholar]
- 55.For information on MOE software. see: http://www.chemcomp.com/index.htm.
- 56.Notter RH. Lung Surfactants: Basic Science and Clinical Applications. Marcel Dekker; New York: 2000. [Google Scholar]
- 57.Seurynck-Servoss SL, Dohm MT, Barron AE. Biochemistry. 2006;45:11809–11818. doi: 10.1021/bi060617e. [DOI] [PubMed] [Google Scholar]
- 58.Brown NJ, Wu CW, Seurynck-Servoss SL, Barron AE. Biochemistry. 2008;47:1808–1818. doi: 10.1021/bi7021975. [DOI] [PubMed] [Google Scholar]
- 59.Elgersma RC, Mulder GE, Kruijtzer JAW, Posthuma G, Rijkers DTS, Liskamp RMJ. Bioorg Med Chem Lett. 2007;17:1837–1842. doi: 10.1016/j.bmcl.2007.01.042. [DOI] [PubMed] [Google Scholar]
- 60.Höppener JWM, Ahrén B, Lips CJM. N Engl J Med. 2000;343:411–419. doi: 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- 61.Westermark P, Engström U, Johnson KH, Westermark GT, Betsholtz C. Proc Natl Acad Sci U S A. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nguyen JT, Turck CW, Cohen FE, Zuckermann RN, Lim WA. Science. 1998;282:2088–2092. doi: 10.1126/science.282.5396.2088. [DOI] [PubMed] [Google Scholar]
- 63.Nguyen JT, Porter M, Amoui M, Miller WT, Zuckermann RN, Lim WA. Chem Biol. 2000;7:463–473. doi: 10.1016/s1074-5521(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 64.Alluri P, Liu B, Yu P, Xiao X, Kodadek T. Mol BioSyst. 2006;2:568–579. doi: 10.1039/b608924k. [DOI] [PubMed] [Google Scholar]
- 65.Lim HS, Archer CT, Kodadek T. J Am Chem Soc. 2007;129:7750–7751. doi: 10.1021/ja072027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lim HS, Archer CT, Kim YC, Hutchens T, Kodadek T. Chem Commun. 2008:1064–1066. doi: 10.1039/b717861a. [DOI] [PubMed] [Google Scholar]
- 67.Udugamasooriya DG, Ritchie C, Brekken RA, Kodadek T. Bioorg Med Chem. 2008;16:6338–6343. doi: 10.1016/j.bmc.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Malet G, Martin AG, Orzáez M, Vicent MJ, Masip I, Sanclimens G, Ferrer-Montiel A, Mingarro I, Messeguer A, Fearnhead HO, Pérez-Payá E. Cell Death Differ. 2006;13:1523–1532. doi: 10.1038/sj.cdd.4401828. [DOI] [PubMed] [Google Scholar]
- 69.Mondragón L, Orzáez M, Sanclimens G, Moure A, Armiñán A, Sepúlveda P, Messeguer A, Vicent MJ, Pérez-Payá E. J Med Chem. 2008;51:521–529. doi: 10.1021/jm701195j. [DOI] [PubMed] [Google Scholar]
- 70.For pertinent reviews of multivalency, see:; (a) Mammen M, Choi S, Whitesides GM. Angew Chem Int Ed. 1998;37:2754. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; (b) Kiessling LL, Gestwicki JE, Strong LE. Angew Chem Int Ed. 2006;45:2348. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reddy MM, Bachhawat-Sikder K, Kodadek T. Chem Biol. 2004;11:1127–1137. doi: 10.1016/j.chembiol.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 72.Yuasa H, Honma H, Hashimoto H, Tsunooka M, Kojima-Aikawa K. Bioorg Med Chem Lett. 2008;17:5274–5278. doi: 10.1016/j.bmcl.2006.12.075. [DOI] [PubMed] [Google Scholar]
- 73.Holub JM, Garabedian MJ, Kirshenbaum K. QSAR Comb Sci. 2007;26:1175–1180. [Google Scholar]
- 74.Jang HJ, Fafarman A, Holub JM, Kirshenbaum K. Org Lett. 2005;7:1951–1954. doi: 10.1021/ol050371q. [DOI] [PubMed] [Google Scholar]
- 75.Holub JM, Jang H, Kirshenbaum K. Org Biomol Chem. 2006;4:1497–1502. doi: 10.1039/b518247f. [DOI] [PubMed] [Google Scholar]
- 76.Murphy JE, Uno T, Hamer JD, Cohen FE, Dwarki V, Zuckermann RN. Proc Natl Acad Sci U S A. 1998;95:1517–1522. doi: 10.1073/pnas.95.4.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang CY, Uno T, Murphy JE, Lee S, Hamer JD, Escobedo JA, Cohen FE, Radhakrishnan R, Dwarki V, Zuckermann RN. Chem Biol. 1998;5:345–354. doi: 10.1016/s1074-5521(98)90173-9. [DOI] [PubMed] [Google Scholar]
- 78.Yu P, Liu B, Kodadek T. Nat Biotechnol. 2005;23:746–751. doi: 10.1038/nbt1099. [DOI] [PubMed] [Google Scholar]
- 79.Tan NC, Yu P, Kwon YU, Kodadek T. Bioorg Med Chem. 2008;16:5853–5861. doi: 10.1016/j.bmc.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schröder T, Niemeier N, Afonin S, Ulrich AS, Krug HF, Bräse S. J Med Chem. 2008;51:376–379. doi: 10.1021/jm070603m. [DOI] [PubMed] [Google Scholar]
- 81.Diaz-Mochon JJ, Fara MA, Sanchez-Martin RM, Bradley M. Tetrahedron Lett. 2008;49:923–926. [Google Scholar]
- 82.Lee BC, Zuckermann RN, Dill KA. J Am Chem Soc. 2005;127:10999–11009. doi: 10.1021/ja0514904. [DOI] [PubMed] [Google Scholar]
- 83.Lee BC, Chu TK, Dill KA, Zuckermann RN. J Am Chem Soc. 2008;130:8847–8855. doi: 10.1021/ja802125x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Burkoth TS, Beausoleil E, Kaur S, Tang D, Cohen FE, Zuckermann RN. Chem Biol. 2002;9:647–654. doi: 10.1016/s1074-5521(02)00140-0. [DOI] [PubMed] [Google Scholar]
- 85.Coleman JE. Annu Rev Biochem. 1992;61:897–946. doi: 10.1146/annurev.bi.61.070192.004341. [DOI] [PubMed] [Google Scholar]
- 86.Berg JM, Godwin HA. Annu Rev Biophys Biomol Struct. 1997;26:357–371. doi: 10.1146/annurev.biophys.26.1.357. [DOI] [PubMed] [Google Scholar]
- 87.Ghering AB, Shokes JE, Scott RA, Omichinski JG, Godwin HA. Biochemistry. 2004;43:8346–8355. doi: 10.1021/bi035673j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.