

Abstract
DNA replication in a human cell involves hundreds of proteins that copy the DNA accurately and completely each cell division cycle. In addition to the core DNA copying machine (the replisome), accessory proteins work to respond to replication stress, correct errors, and repackage the DNA into appropriate chromatin structures. New proteomic tools have been invented in the past few years to facilitate the purification, identification, and quantification of the replication, chromatin maturation, and replication stress response machineries. These tools, including iPOND (isolation of proteins on nascent DNA) and NCC (nascent chromatin capture), have yielded discoveries of new proteins involved in these processes and insights into the dynamic regulatory processes ensuring genome and chromatin integrity. In this review I will introduce these experimental approaches and examine how they have been utilized to define the replication fork proteome.
Keywords: DNA replication, iPOND, nascent chromatin capture, proteomics, SILAC, replisome, mass spectrometry, replication stress, chromatin
1. Introduction
The genome must be copied completely and accurately each cell division cycle, and the newly synthesized DNA must be re-packaged into chromatin in ways that control epigenetic regulation of gene expression and other aspects of chromosome function. Numerous challenges impede these processes including DNA damage, difficult to replicate DNA sequences, and conflicts with transcription. Large and dynamic protein machines overcome these challenges and maintain genome stability.
The replisome is the DNA copy machine that contains helicases to unwind the DNA duplex, polymerases to copy the DNA, as well as nucleases and ligases to process the discontinuous stretches of DNA on the lagging strand (Bell and Dutta, 2002). The replisome also contains accessory factors that increase efficiency and serve to couple leading and lagging strand synthesis. Proteins involved in chromatin disassembly, reassembly, and modification are tethered to the replisome ensuring a tight coordination between chromatin and DNA replication (Probst et al., 2009). Error correction mechanisms such as mismatch repair proteins, and replication stress response proteins that deal with replication challenges are also tethered via direct interactions with replisome subunits or are recruited to replication forks following changes in DNA structure (Cimprich and Cortez, 2008; Moldovan et al., 2007; Nam and Cortez, 2011).
Obtaining an inventory of all of the proteins involved in DNA and chromatin replication is a self-evident requirement to fully understand the process. Certainly most of the core essential components of the eukaryotic replisome are known. However, there continues to be a steady stream of discoveries of new accessory factors that influence the fidelity and efficiency of DNA replication. Furthermore, we have a long way to go to understand how these proteins are regulated and coordinated within these dynamic machines especially in circumstances when a replication fork encounters an obstacle.
In this review I will discuss recent advances in proteomic methods to purify, identify, and characterize replication fork-associated proteins, concentrating on approaches applicable to mammalian cells. These methods have the potential to complete the inventory of the DNA and chromatin replication proteome and will be useful in interrogating the regulation of this machinery. I will summarize the findings from these approaches, highlighting their strengths and weaknesses.
2. Replisome Purification Approaches
The most obvious way to identify proteins that replicate DNA and chromatin is to purify these protein machines and use mass spectrometry to identify them. One approach to purification is to use a known replisome subunit as the bait to fish out the interacting complex. However, this approach does not necessarily distinguish between active replisomes and complexes that are either not bound to DNA or not engaged in active synthesis. In addition, multiple protein baits would need to be utilized to try to ensure most of the machinery is purified.
A second approach is to compare chromatin-associated proteins in replicating vs. quiescent cells with the assumption that replisome subunits will only be associated with chromatin during DNA replication. The vast numbers and abundance of chromatin proteins compared to replication-specific proteins in cells makes this approach difficult, and again it does not necessarily distinguish between active and inactive complexes. Nonetheless, the robustness of replication in Xenopus egg extracts provides an opportunity to use this approach in an in vitro replication system. Chromatin mass spectrometry (CHROMASS) takes advantage of this system and was used to identify proteins that are recruited to chromatin that has been damaged with a DNA crosslinking agent (Raschle et al., 2015). Thus, CHROMASS provides an approach that is especially useful in situations where an investigator wants to interrogate chromatin composition in the context of defined DNA damage structures. However, this is an in vitro replication system, it does not directly answer whether a protein is enriched at are plication fork, and is not easily adapted to mammalian cells.
Two other purification strategies, isolation of proteins on nascent DNA (iPOND) and nascent chromatin capture (NCC) were recently developed that overcome these limitations (Alabert et al., 2014; Sirbu et al., 2011). Instead of targeting the machinery itself for purification, these methods purify the newly synthesized DNA and then interrogate the associated proteins. Proteins involved in DNA and chromatin replication are identified by their enrichment with nascent DNA compared to bulk chromatin.
2a. Isolation of Proteins on Nascent DNA (iPOND)
iPOND takes advantage of the nucleoside analog 5-ethynyl-2′-deoxyuridine (EdU). When added to cell culture, EdU is quickly imported into the cell, phosphorylated, and incorporated into newly synthesized DNA by the replisome polymerases(Salic and Mitchison, 2008). EdU contains an alkyne group that can be covalently linked to an azide-containing molecule using click chemistry. Thus, biotin can be conjugated to the EdU to facilitate a single-step affinity purification of the newly synthesized DNA and bound proteins using streptavidin (Figure 1A). A sonication procedure combined with copper-catalyzed DNA hydrolysis during the click chemistry step generates protein-bound DNA fragments ranging from 100-200 base pairs. Thus, the method can provide high spatial resolution largely determined by the rate of DNA polymerization and EdU labeling time (Dungrawala and Cortez, 2015; Sirbu et al., 2012; Sirbu et al., 2011). The eukaryotic replication fork elongates at rates between 0.5-2 kb/min with considerable variation in speed depending on factors such as early or late S-phase, DNA sequence, and chromatin composition. Typically, 5-10 minute incorporation times are utilizedin iPOND experiments although labeling times of as short as 2.5 minutes have been reported (Sirbu et al., 2011). Removing the EdU from the growth media and incubating in the absence of EdU can generate “chase” samples that provide spatial information of where proteins are with respect to the fork. This chase sample is essential to distinguish the proteins that participate in DNA and chromatin replication versus those that are components of bulk chromatin (Figure 2).
Figure 1.
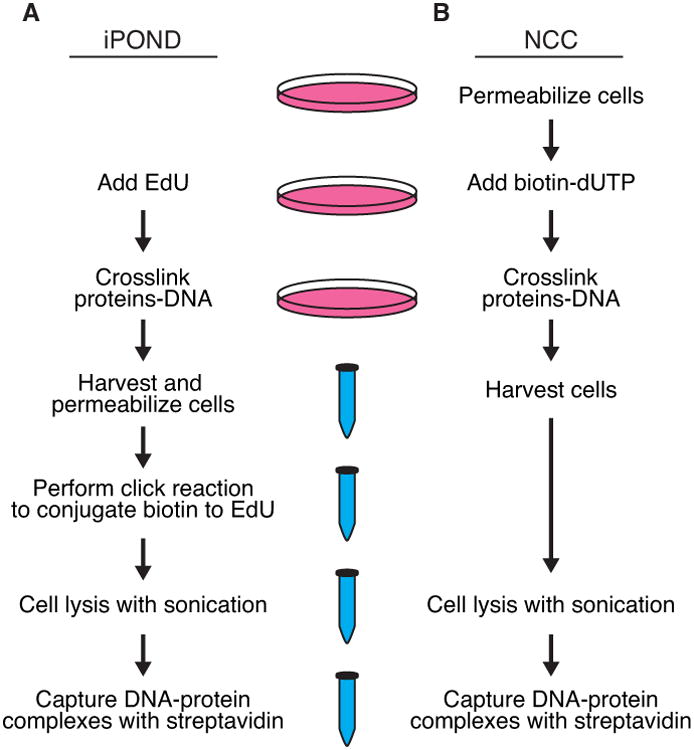
Schematic of the (A) iPOND and (B) NCC approaches to purifying proteins associated with nascent DNA.
Figure 2.

A pulse-chase protocol is essential to identify proteins that are specifically enriched near the replication fork versus proteins that are associated more generally with chromatin. This methodology also facilitates analysis of chromatin maturation.
Five studies utilized iPOND combined with different types of mass spectrometry to identify the replication fork proteome (Table 1). All compared an EdU-labeled sample with a chase sample to find proteins enriched on nascent DNA compared to mature chromatin. The primary differences in the protocols were in how the quantitative mass spectrometry was performed. Below I summarize the results from each of these studies emphasizing their strengths and weaknesses.
Table 1.
Summary of manuscripts using iPOND or NCC to identify proteins associated with replication forks.
| Study | Method | Mass Spectrometry Method | Number of replicates | Number of proteins |
|---|---|---|---|---|
| Lopez-Contreras, Cell Reports 2013 | iPOND | Label-free | 6 | 48 |
| Sirbu, J Biol Chem 2013 | iPOND | Label-free | 5 | 84 |
| Lossaint, Mol Cell 2013 | iPOND | Label-free | 3/2* | n/a** |
| Aranda, Nuc Acids Res 2014 | iPOND | Label-free | 4 | 207 |
| Alabert, Nat Cell Bio 2014 | NCC | SILAC | 3 | 462 |
| Dungrawala, Mol Cell 2015 | iPOND | SILAC | 3 | 218 |
| Lecona, Nat Struct Mol Biol 2016 | iPOND | iTRAQ | 1 | n/a** |
3 “pulse” samples and 2 “chase” samples;
These investigators did not attempt to designate which proteins should be considered significantly enriched at forks.
iPOND-Label Free-MS
Four groups utilized label-free mass spectrometry quantitation procedures combined with iPOND (Aranda et al., 2014; Lopez-Contreras et al., 2013; Lossaint et al., 2013; Sirbu et al., 2013). Label-free mass spectrometry utilizes the information derived from tandem mass spectrometry such as number of peptide spectra for a given protein or the abundance of the precursor ion to assign an abundance value to that protein within the dataset. The EdU pulse and chase samples are collected and processed independently, and then the data compared after collection (Figure 3A). Label-free mass spectrometry methods are compatible with any source of proteins and are relatively inexpensive. However, label-free methods are much less quantitatively precise and can suffer from reproducibility problems.
Figure 3.
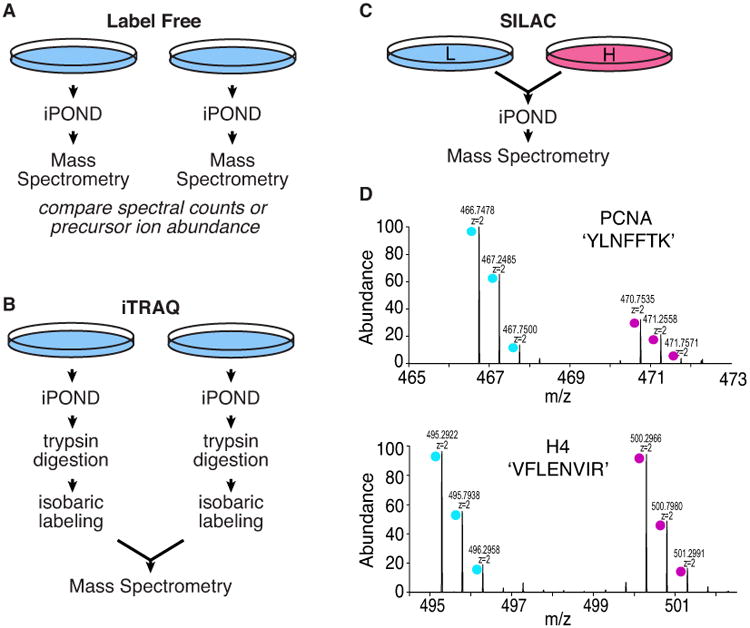
Comparison of mass spectrometry approaches that have been combined with iPOND. (A) Label free methods depend on comparing datasets that are generated independently. (B) iTRAQ improves the quantitation precision but does not eliminate variability associated with purification. (C) SILAC improves precision and reduces variation since heavy and light samples are combined prior to purification. (D) Example of data obtained from a SILAC experiment in which the light sample was labeled with EdU and the heavy sample was the chase.
Three different label-free methods have been combined with iPOND. Two studies utilized spectral counting (Lossaint et al., 2013; Sirbu et al., 2013). By counting the number of spectra corresponding to peptides from a given protein in either the pulse or chase sample, a ratio was derived to describe the relative amounts of a protein at the replication fork versus in bulk chromatin. Spectral counting is simple; however, it is also the least accurate quantification method. One of these studies performed by our research group identified 84 proteins as enriched on nascent DNA after completing five replicate samples (Sirbu et al., 2013). The second study performed smaller number of replicates, and did not attempt to provide a cutoff for what should be considered significant or provide a measure of reproducibility in the measurements (Lossaint et al., 2013).
The 84 proteins identified in our study had highest GO enrichment values for DNA metabolic process. However, the data suffers from a high false-negative rate, which is illustrated by the failure to identify many subunits of the replicative helicase (Sirbu et al., 2013). Examining the 100 proteins with the largest pulse/chase ratios in the Lossaint et al., dataset reveals that these proteins are most highly enriched in carboxylic acid metabolic processes (37 proteins) and DNA replication is only the seventh most enriched biological process (16 proteins)(Lossaint et al., 2013). Thus, the limitations of spectral counting as a quantitative measurement can generate high false-positive and false-negative discovery rates.
A second label-free mass spectrometry method employed in conjunction with iPOND is a variation of spectral counting which utilizes an algorithm that assigns a probability based score (Mascot score) to each protein identified in a sample. Mascot scores were initially designed to indicate a probability of correct protein identification so it is unclear how effectively they quantitate protein abundance (Perkins et al., 1999). Nonetheless, the investigators using this method identified 207 proteins enriched on nascent DNA based on the ratio of Mascot scores in pulse vs. chase samples (Aranda et al., 2014). The list is highly enriched in DNA metabolism and replication proteins, but it does not contain a quantitative measurement of reproducibility. Furthermore, for many proteins, quantitative ratios were not generated. Despite these limitations the data set generated seems superior to either of the spectral counting datasets based on the number of known replication proteins identified and their enrichment values in gene ontology analyses.
The third label-free method coupled to iPOND quantitated the precursor ion abundance in the MS-MS data (Lopez-Contreras et al., 2013). This method is more accurate than spectral counting but still suffers from relatively high variability. The lack of precision is illustrated by the large variations in the reported abundance of the six subunits of the MCM2-7 complex in this dataset. For example, among the biological replicates the log2 of the abundance ratio of MCM2 varied from -5.75 to +11.46 (where a positive value indicates enrichment at the fork, zero indicates no enrichment, and a negative value indicates exclusion from the fork). To try to compensate for the lack of precision, six biological replicates were performed and only proteins that were observed to be enriched at least 8-fold in at least all experiments but one in which they were observed were reported to be enriched on the nascent DNA. This list included 48 proteins and the authors acknowledge the method suffers from a high false-negative rate. Only 3 of the 6 MCM2-7 subunits met the criteria for a fork-associated protein. Despite the stringent criteria applied to the dataset, it also seems likely that it contains false-positives since predominantly cytoplasmic proteins like Tubulin and a ribosomal protein were scored as hits.
Overall, the value of combining iPOND with the label-free mass spectrometry methods is primarily limited to the generation of a starting list for a candidate approach to finding new replication machinery proteins. The high false-positive and false-negative rates preclude a description of the replication fork proteome from these datasets. Two more quantitative mass spectrometry methods, iTRAQ and SILAC provide superior data to overcome these limitations.
iPOND-iTRAQ-MS
iTRAQ (isobaric tags for relative and absolute quantification) involves labeling peptides from different experimental conditions with different mass tags, combining the labeled peptides, and then performing mass spectrometry (Figure 3B). iTRAQ yields highly quantitative results and has some other advantages. Since it involves labeling peptides after cell lysis, it can be used in systems like samples from intact tissues where other methods such as SILAC (stable isotope labeling with amino acids in cell culture) are difficult or impossible. Second, iTRAQ can be used to compare four or more samples in a single experiment. Unfortunately, the method tends to minimize abundance differences—compressing the relative abundance ratios (Rauniyar and Yates, 2014). It also is less precise and reproducible than SILAC due to differences in peptide labeling efficiencies and the need to process individual samples through the purification procedure separately prior to combining the labeled peptides for mass spectrometry.
The dataset reported by Lecona et al., illustrates the abundance ratio compression problem (Lecona et al., 2016). Only 31 proteins were found to be at least two-fold enriched on the nascent DNA with the highest log2 abundance ratio being 2.1 for PCNA. Most known replication fork proteins including all the subunits of the replicative helicase were enriched at ratios that would be difficult to know are significant. For example, the average log2 ratio for the MCM2-7 complex subunits was only 0.26 and 570 proteins had higher or equal enrichment than the lowest MCM subunit (MCM4, log2=0.15). This dataset also did not include biological replicates. Perhaps for these reasons the authors did not attempt to define which proteins were actually enriched on the nascent DNA and instead used the dataset to identify candidates for further analyses.
iPOND-SILAC-MS
Our group combined iPOND with SILAC mass spectrometry (Dungrawala et al., 2015). SILAC relies on the incorporation of isotopically distinct amino acids during protein synthesis in cells (Ong and Mann, 2005). Two cell populations are prepared by growing in “light” or “heavy” media for several generations to isotopically label nearly 100% of the proteome. These cell populations are equivalent to one another with the exception of the small mass differences in each protein. Thus, EdU-pulse and chase samples can be compared directly by examining the abundance of the heavy vs. light version of a peptide. SILAC minimizes experimental variability since the samples to be compared are mixed prior to performing the iPOND procedure (Figure 3C and D). It can also provide highly quantitative results with measured variability of less than 20% in many cases (Dungrawala et al., 2015). Disadvantages include the relatively high cost and limitation to systems that are amenable to metabolic labeling like cell culture. Generally only two samples can be compared although this can be stretched to additional comparison by utilizing high precision mass spectrometers.
The advantages of iPOND-SILAC-MS are readily apparent from the datasets that are generated. In the studies from our lab, three iPOND biological replicates generated a list of 218 proteins with average enrichment ratios at replication forks compared to mature chromatin of greater than two (Dungrawala et al., 2015). These 218 proteins included all of the known replication elongation proteins except GINS2, POLD4, POLE3, DNA2, and RPA3 (Figure 4A). Four of these proteins are small with few tryptic peptides likely explaining why their abundance was difficult to quantitate. The fourth, DNA2, was observed but did not quite meet the stringent cutoff for significance. Thus, the method has a lower false-negative rate than any of the other methods. It also likely has a low false-positive rate (see below).
Figure 4.
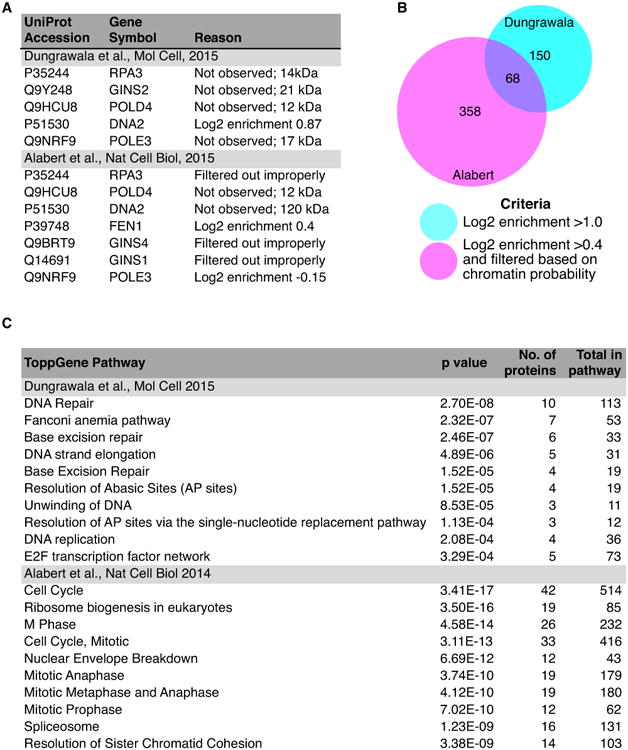
Comparison of the iPOND-SILAC and NCC-SILAC datasets. (A) Known replication fork proteins that were not identified or filtered out by the bioinformatics approach used in the NCC study. (B) Comparison of the total numbers of proteins identified in each dataset and the criteria utilized. (C) Gene ontology analysis of the 150 and 358 proteins uniquely identified in the Dungrawala et al., or Alabert et al., datasets.
The robustness of the method is evident from the highly similar quantitative data obtained for the subunits of the MCM2-7 complex, which differed in mean enrichment values by less than 10% (Figure 5). In fact, the quantitation is so robust that it can predict protein complexes (Dungrawala et al., 2015). Proteins that function as part of a stoichiometric protein complex should be captured equally in any given iPOND experiment. Unsupervised hierarchical clustering of the approximately 1500 proteins quantitated in multiple experiments was capable of predicting both known and new protein complexes based on similarities in their subunit abundance ratios across different experiments. This property of the iPOND-SILAC-MS data was used to identify ZNF644 as a new subunit of the G9a/GLP methyltransferase complex that travels just behind the replication fork to modify newly deposited histones and illustrates the accuracy and reproducibility of iPOND-SILAC-MS (Dungrawala et al., 2015).
Figure 5.

Comparison of the enrichment of selected replisome proteins at replication forks calculated in five proteomic datasets. A log2 transformation of the mean enrichment comparing fork/chromatin (pulse/chase) is depicted. Larger positive values indicate increased enrichment at forks compared to bulk chromatin. Error bars were calculated as SEM where possible. The data illustrates the reproducibility and precision of SILAC quantitation compared to other methods.
Variations on iPOND
Variations to the iPOND procedure that may be useful in specific circumstances have been reported. A similar methodology was developed independently and named Dm-ChP for DNA-mediated chromatin pull-down (Kliszczak et al., 2011). This method was combined with mass spectrometry, but the lack of a chase sample precludes analysis of which proteins are specifically enriched at replication forks versus simply being chromatin associated. In another iPOND variation we omitted the formaldehyde crosslinking step to examine histone modifications in a procedure we termed native iPOND corresponding to native chromatin immunoprecipitation methods (Sirbu et al., 2012). A similar procedure was also reported by another group and called aniPOND (Leung et al., 2013). An advantage of native iPOND is that the formaldehyde crosslinking step in regular iPOND can interfere with detection of proteins by western blotting due to destruction of epitopes or incomplete crosslink reversal yielding aberrant migration on gels. This issue is especially problematic for large proteins, but does not interfere with detection by mass spectrometry.
2b. Nascent chromatin capture (NCC)
NCC is similar to iPOND except that biotin-dUTP is utilized instead of EdU (Figure 1B and (Alabert et al., 2014)). Thus, no biotin-conjugation step is required in NCC. However, unlike EdU, biotin-dUTP cannot be imported into intact cells. Thus, a cell permeabilization step such as incubating cells in hypotonic conditions must be utilized to allow the biotin-dUTP to cross the cell membrane. Similar to iPOND, a chase step after the biotin-dUTP labeling allows the identification of proteins that are enriched near the fork versus those that are simply bound to chromatin.
Comparison of iPOND and NCC
Both iPOND and NCC depend on the polymerase to be capable of incorporating and extending from the modified base. Furthermore, both depend on the cell being unable to recognize the modified base as DNA damage and assume the modification does not interfere with the binding of proteins to the DNA. In short-term assays these assumptions appear to be justified; however, there is a long literature indicating that these types of base modifications are not completely neutral. For example, incorporation of halogenated nucleoside analogs like BrdU can activate the DNA damage response (Masterson and O'Dea, 2007). Less is known about EdU and biotin-dUTP. Assays of DNA replication did not observe any consequences of EdU in short-term assays, although long-term assays indicated decreased cell proliferation and increased DNA damage signaling (Kohlmeier et al., 2013). It is likely that the much larger biotin group on biotin-dUTP could cause similar or worse problems.
iPOND has the advantage over NCC in that it is quite simple to add or remove the EdU from the culture media to label for defined times. Thus, it is possible to incorporate the EdU at stressed forks such as those challenged by hydroxyurea or DNA damaging agents, which may incorporate very slowly and require labeling periods of several hours. Since the biotin-nucleotide used in NCC is not cell permeable, long-term labeling is not possible.
An additional concern with NCC is the need to alter the cell growth conditions to permeabilize cells to the biotin-dUTP. The osmotic stress used to achieve permeabilization can affect multiple cellular processes. For example, osmotic stress activates the ATM DNA damage response kinase which can modify replisome proteins and alter DNA replication kinetics (Bakkenist and Kastan, 2003). Thus, permeabilization conditions need to be optimized for each cell type to minimize unwanted effects.
The Groth lab has combined NCC with SILAC-MS and reported the identification of 426 proteins enriched on nascent DNA (Alabert et al., 2014). This list was selected by a combination of enrichment criteria from the mass spectrometry data (with a log2 enrichment cutoff of >0.4 that yielded a list of 1296 proteins) and filtering by bioinformatics comparison to a previous dataset of chromatin associated proteins. This list of 426 proteins has relatively few false-negatives with only seven of the well-established replication fork elongation proteins missing the cutoff because of filtering, lack of sufficient enrichment, or non-detection (Figure 4A). As might be expected, the use of SILAC provides highly quantitative data. Examining the reproducibility of the MCM2-7 complex subunit quantifications illustrates its quantitation accuracy is second only to the iPOND-SILAC-MS dataset and both are far more reproducible than label-free mass spectrometry (Figure 5).
Comparing the 426 proteins from the NCC-SILAC approach to the 218 in the iPOND-SILAC dataset indicates that the NCC methodology likely yielded a much higher false-positive detection rate. It is possible to decrease the false-positive rate by requiring a larger enrichment value such as the two-fold enrichment criteria in the iPOND-SILAC-MS dataset, but that would also increase the false-negative frequency. There are 68 proteins in common between the two datasets (Figure 4B and Table 2). These are mostly core replisome proteins, proteins tethered to the replisome to facilitate chromatin deposition and remodeling, or replication stress response proteins. Subtracting the common proteins from the iPOND data still yields a list of 150 proteins highly enriched for processes related to DNA replication including DNA repair (Figure 4C). On the other hand, subtracting these proteins from the NCC dataset leaves a list of 358 proteins enriched in nuclear functions, but not necessarily DNA metabolism (Figure 4C).
Table 2.
Replication fork proteins identified in both Dungrawala et al., Mol Cell 2015 and Alabert et al., Nat Cell Biol 2014.
| Uniprot | Symbol | Gene ID |
|---|---|---|
| O75419 | CDC45 | 8318 |
| Q92674 | CENPI | 2491 |
| Q13111 | CHAF1A | 10036 |
| Q13112 | CHAF1B | 8208 |
| Q8WVB6 | CHTF18 | 63922 |
| Q9HAW4 | CLSPN | 63967 |
| Q6PJP8 | DCLRE1A | 9937 |
| P26358 | DNMT1 | 1786 |
| Q9BVC3 | DSCC1 | 79075 |
| Q9NZJ0 | DTL | 51514 |
| Q56NI9 | ESCO2 | 157570 |
| Q9UQ84 | EXO1 | 9156 |
| Q9BXW9 | FANCD2 | 2177 |
| Q9NVI1 | FANCI | 55215 |
| P16383 | GCFC2 | 6936 |
| Q9BRX5 | GINS3 | 64785 |
| Q15004 | KIAA0101 | 9768 |
| P18858 | LIG1 | 3978 |
| Q7L590 | MCM10 | 55388 |
| P49736 | MCM2 | 4171 |
| P25205 | MCM3 | 4172 |
| P33991 | MCM4 | 4173 |
| P33992 | MCM5 | 4174 |
| Q14566 | MCM6 | 4175 |
| P33993 | MCM7 | 4176 |
| Q6ZRQ5 | MMS22L | 253714 |
| P49959 | MRE11A | 4361 |
| P43246 | MSH2 | 4436 |
| P20585 | MSH3 | 4437 |
| P52701 | MSH6 | 2956 |
| O60934 | NBN | 4683 |
| Q86W56 | PARG | 8505 |
| P12004 | PCNA | 5111 |
| Q14181 | POLA2 | 23649 |
| P28340 | POLD1 | 5424 |
| P49005 | POLD2 | 5425 |
| Q15054 | POLD3 | 10714 |
| Q07864 | POLE | 5426 |
| P56282 | POLE2 | 5427 |
| P49642 | PRIM1 | 5557 |
| P49643 | PRIM2 | 5558 |
| Q9Y606 | PUS1 | 80324 |
| Q9NS91 | RAD18 | 56852 |
| Q92878 | RAD50 | 10111 |
| Q9Y4B4 | RAD54L2 | 23132 |
| Q99638 | RAD9A | 5883 |
| O94762 | RECQL5 | 9400 |
| P35251 | RFC1 | 5981 |
| P35250 | RFC2 | 5982 |
| P40938 | RFC3 | 5983 |
| P35249 | RFC4 | 5984 |
| P40937 | RFC5 | 5985 |
| Q5TBB1 | RNASEH2B | 79621 |
| P27694 | RPA1 | 6117 |
| P15927 | RPA2 | 6118 |
| Q9BQI6 | SLF1 | 84250 |
| Q8IX21 | SLF2 | 55719 |
| Q9NZC9 | SMARCAL1 | 50485 |
| Q08945 | SSRP1 | 6749 |
| Q96FV9 | THOC1 | 9984 |
| Q9UNS1 | TIMELESS | 8914 |
| Q9BVW5 | TIPIN | 54962 |
| Q92547 | TOPBP1 | 11073 |
| O94842 | TOX4 | 9878 |
| Q9BSV6 | TSEN34 | 79042 |
| O94782 | USP1 | 7398 |
| O75717 | WDHD1 | 11169 |
| Q6PJT7 | ZC3H14 | 79882 |
3. Additional Applications
Purifying replication stress response proteins
In addition to identifying proteins at unperturbed DNA replication forks, iPOND has been utilized to interrogate the replication fork proteome after a challenge that perturbs replication (Table 3 and (Dungrawala et al., 2015; Lossaint et al., 2013; Olcina et al., 2016; Ribeyre et al., 2016; Sirbu et al., 2013)). For example, combining the EdU label with drugs like hydroxyurea or aphidicolin allowed the identification of proteins at stalled replication forks. Combining EdU with camptothecin or alternatively with hydroxyurea and a selective inhibitor of the ATR checkpoint kinase identifies proteins recruited to collapsed replication forks (Dungrawala et al., 2015; Ribeyre et al., 2016; Sirbu et al., 2013). iPOND is particularly good for these types of experiments since the EdU labeling time can be varied easily. Thus, the amount of DNA labeling can be optimized to ensure equal amounts of capture. Often these experiments are done with a short pre-incubation with EdU followed by addition of the replication stress agent. Since even high doses of hydroxyurea do not completely stop replication fork movement (Dungrawala et al., 2015), it is important to maintain EdU in the growth media during long HU time courses to ensure the DNA adjacent to the fork continues to be labeled. Alternatively, the drug can be added prior to the EdU, but it is important to normalize the amount of EdU incorporation to facilitate sample comparisons.
Table 3.
Summary of studies that examined replication stress proteomes.
| Study | Perturbation | Method | Number of replicates | Number of proteins |
|---|---|---|---|---|
| Sirbu, J Biol Chem 2013 | Hydroxyurea | iPOND Label-free | 5 | 139* |
| Sirbu, J Biol Chem 2013 | Hydroxyurea + ATR inhibitor | iPOND Label-free | 5 | 137* |
| Lossaint, Mol Cell 2013 | Hydroxyurea | iPOND Label-free | 3 | n/a |
| Dungrawala, Mol Cell 2015 | Hydroxyurea | iPOND-SILAC | 18 | 192 |
| Dungrawala, Mol Cell 2015 | Aphidicollin | iPOND-SILAC | 2 | n/a |
| Dungrawala, Mol Cell 2015 | Hydroxyurea + ATR inhibitor | iPOND-SILAC | 11 | 151 |
| Dungrawala, Mol Cell 2015 | Aphidicolin + ATR inhibitor | iPOND-SILAC | 2 | n/a |
| Ribeyre, Cell Reports 2016 | Camptothecin | iPOND-Label-free | 6 | 21 |
| Olcina, Tumor Microenvironment 2016 | Hypoxia | iPOND | n/a | n/a |
| Raschle, Science 2015 | Psoralen | Chromass-Label-free | 42** | 198*** |
These numbers included proteins that are at undamaged forks since the comparison was to a chase sample.
Additional samples were analyzed containing other inhibitors in addition to psoralen.
112 of these proteins were enriched in a replication-dependent manner.
Another consideration in using iPOND with genotoxic drugs or genetic perturbations is that some conditions could alter replication initiation. For example, comparison of the stalled replication fork proteome after hydroxyurea treatment with the proteome after combining hydroxyurea with an ATR inhibitor initially suggested that most replisome proteins actually became more abundant in the ATR-inhibited sample (Dungrawala et al., 2015). However, the abundance differences are entirely explained by increased replication initiation in the ATR-inhibited samples due to the inhibition of the checkpoint response. Thus, comparisons like these need to consider whether equal numbers of forks are being purified in addition to considerations of whether equal amounts of nascent DNA are captured.
As with experiments examining the unperturbed replisome, the mass spectrometry methodology makes a substantial difference in the results. The most complete datasets completed with iPOND-SILAC mass spectrometry have identified 192 proteins recruited to stalled forks and 151 recruited to collapsed forks (Dungrawala et al., 2015). Among these proteins are the known DNA damage response proteins, but a significant number of potential new replication stress response proteins are also identified. For example, a new ATR activating protein, ETAA1, was identified from iPOND-derived stalled replication fork proteomes (Bass et al., 2016).
Analysis of Chromatin deposition and maturation
Both iPOND and NCC are particularly useful for studying the process of chromatin deposition and maturation following DNA replication. By examining multiple chase time points, it is possible to follow the assembly of the histones on the nascent DNA and changes in their post-translational modifications. For example, the timing of histone H1 deposition in relation to DNA replication, the changes in histone acetylation and their genetic dependencies, and H2AX phosphorylation spreading from a stalled fork were followed by iPOND (Nagarajan et al., 2013; Sirbu et al., 2011). The Groth lab combined NCC with pulsed SILAC-MS to monitor changes in the modifications of new histones deposited on the nascent DNA as a function of time after DNA replication. This procedure allowed them to determine how rapidly histone marks are copied to the newly synthesized histones after DNA replication (Alabert et al., 2015).
Analysis of viral replication and other opportunities
Hundreds of studies have utilized iPOND and NCC since their development primarily to study DNA replication, chromatin deposition/maturation, and replication stress responses. There are also opportunities beyond studying nuclear genome replication. In principle, any process that involves the synthesis of new DNA could be analyzed. For example, iPOND was recently utilized to identify proteins that function in Herpes Simplex Virus replication, genome processing, and packaging (Dembowski and DeLuca, 2015). Other opportunities include the study mitochondrial DNA metabolism, DNA repair, chromatin re-establishment after DNA repair synthesis, and break-induced replication. Additionally, the DNA that is captured in the iPOND protocol can be analyzed instead of the proteins. This served as the basis for the analysis of the nucleosome landscape following DNA replication in a methodology called MINCE-Seq (Ramachandran and Henikoff, 2016).
4. Summary and Conclusions
The development of procedures to purify newly synthesized DNA and associated proteins has provided an opportunity to define the replication fork proteome and identify proteins that contribute to DNA and chromatin replication. The 68 proteins in common between the iPOND-SILAC-MS and NCC-SILAC-MS nascent DNA datasets is the highest confidence list of replication fork associated proteins in mammalian cells (Table 2). Most of these proteins are well known parts of the DNA copying, replication stress response, or chromatin modification machinery. However, approximately 10% of these proteins have never been experimentally linked to DNA replication previously. Furthermore, the proteins unique to each of the iPOND-SILAC-MS and NCC-SILAC-MS datasets provide high probability candidates for further study. Since each of these datasets were derived from a single cell line with only three biological replicates, further interrogation of the fork proteomes using these methods promises to be a high value approach. Furthermore, combining these techniques with drug and genetic perturbations, methods to label new and recycled histones, and mass spectrometry approaches to study post-translational modifications provide opportunities to better understand the regulation of DNA and chromatin replication and responses to replication challenges.
Acknowledgments
Research using iPOND in the Cortez lab is supported by NIH grant R01GM116616 and the Breast Cancer Research Foundation. I thank Dr. Huzefa Dungrawala for his input.
Bibliography
- Alabert C, Barth TK, Reveron-Gomez N, Sidoli S, Schmidt A, Jensen ON, Imhof A, Groth A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 2015;29:585–590. doi: 10.1101/gad.256354.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, Groth A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol. 2014;16:281–293. doi: 10.1038/ncb2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda S, Rutishauser D, Ernfors P. Identification of a large protein network involved in epigenetic transmission in replicating DNA of embryonic stem cells. Nucleic acids research. 2014;42:6972–6986. doi: 10.1093/nar/gku374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, Cortez D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol. 2016;18:1185–1195. doi: 10.1038/ncb3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembowski JA, DeLuca NA. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS pathogens. 2015;11:e1004939. doi: 10.1371/journal.ppat.1004939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Cortez D. Purification of proteins on newly synthesized DNA using iPOND. Methods in molecular biology. 2015;1228:123–131. doi: 10.1007/978-1-4939-1680-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Molecular cell. 2015;59:998–1010. doi: 10.1016/j.molcel.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliszczak A, Rainey M, Harhen B, Boisvert F, Santaocanale C. DNA mediated chromatin pull-down for the study of chromatin replication. Scientific Reports. 2011;1:1–7. doi: 10.1038/srep00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier F, Maya-Mendoza A, Jackson DA. EdU induces DNA damage response and cell death in mESC in culture. Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology. 2013;21:87–100. doi: 10.1007/s10577-013-9340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecona E, Rodriguez-Acebes S, Specks J, Lopez-Contreras AJ, Ruppen I, Murga M, Munoz J, Mendez J, Fernandez-Capetillo O. USP7 is a SUMO deubiquitinase essential for DNA replication. Nat Struct Mol Biol. 2016;23:270–277. doi: 10.1038/nsmb.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KH, Abou El Hassan M, Bremner R. A rapid and efficient method to purify proteins at replication forks under native conditions. BioTechniques. 2013;55:204–206. doi: 10.2144/000114089. [DOI] [PubMed] [Google Scholar]
- Lopez-Contreras AJ, Ruppen I, Nieto-Soler M, Murga M, Rodriguez-Acebes S, Remeseiro S, Rodrigo-Perez S, Rojas AM, Mendez J, Munoz J, et al. A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep. 2013;3:1105–1116. doi: 10.1016/j.celrep.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, Constantinou A. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Molecular cell. 2013;51:678–690. doi: 10.1016/j.molcel.2013.07.023. [DOI] [PubMed] [Google Scholar]
- Masterson JC, O'Dea S. 5-Bromo-2-deoxyuridine activates DNA damage signalling responses and induces a senescence-like phenotype in p16-null lung cancer cells. Anti-cancer drugs. 2007;18:1053–1068. doi: 10.1097/CAD.0b013e32825209f6. [DOI] [PubMed] [Google Scholar]
- Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Nagarajan P, Ge Z, Sirbu B, Doughty C, Agudelo Garcia PA, Schlederer M, Annunziato AT, Cortez D, Kenner L, Parthun MR. Histone acetyl transferase 1 is essential for mammalian development, genome stability, and the processing of newly synthesized histones H3 and H4. PLoS Genet. 2013;9:e1003518. doi: 10.1371/journal.pgen.1003518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam EA, Cortez D. ATR signalling: more than meeting at the fork. The Biochemical journal. 2011;436:527–536. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcina MM, Giaccia AJ, Hammond EM. Isolation of Proteins on Nascent DNA in Hypoxia and Reoxygenation Conditions. Advances in experimental medicine and biology. 2016;899:27–40. doi: 10.1007/978-3-319-26666-4_3. [DOI] [PubMed] [Google Scholar]
- Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nature chemical biology. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- Ramachandran S, Henikoff S. Transcriptional Regulators Compete with Nucleosomes Post-replication. Cell. 2016;165:580–592. doi: 10.1016/j.cell.2016.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle M, Smeenk G, Hansen RK, Temu T, Oka Y, Hein MY, Nagaraj N, Long DT, Walter JC, Hofmann K, et al. DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science. 2015;348:1253671. doi: 10.1126/science.1253671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauniyar N, Yates JR., 3rd Isobaric labeling-based relative quantification in shotgun proteomics. Journal of proteome research. 2014;13:5293–5309. doi: 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeyre C, Zellweger R, Chauvin M, Bec N, Larroque C, Lopes M, Constantinou A. Nascent DNA Proteomics Reveals a Chromatin Remodeler Required for Topoisomerase I Loading at Replication Forks. Cell Rep. 2016;15:300–309. doi: 10.1016/j.celrep.2016.03.027. [DOI] [PubMed] [Google Scholar]
- Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Cortez D. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nature protocols. 2012;7:594–605. doi: 10.1038/nprot.2012.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem. 2013;288:31458–31467. doi: 10.1074/jbc.M113.511337. [DOI] [PMC free article] [PubMed] [Google Scholar]