

Abstract
A large number of SNF2 family, DNA and ATP-dependent motor proteins are needed during transcription, DNA replication, and DNA repair to manipulate protein-DNA interactions and change DNA structure. SMARCAL1, ZRANB3, and HLTF are three related members of this family with specialized functions that maintain genome stability during DNA replication. These proteins are recruited to replication forks through protein-protein interactions and bind DNA using both their motor and substrate recognition domains (SRD). The SRD provides specificity to DNA structures like forks and junctions and confer DNA remodeling activity to the motor domains. Remodeling reactions include fork reversal and branch migration to promote fork stabilization, template switching, and repair. Regulation ensures these powerful activities remain controlled and restricted to damaged replication forks. Inherited mutations in SMARCAL1 cause a severe developmental disorder and mutations in ZRANB3 and HLTF are linked to cancer illustrating the importance of these enzymes in ensuring complete and accurate DNA replication. In this review, we examine how these proteins function, concentrating on their common and unique attributes and regulatory mechanisms.
Keywords: fork reversal, checkpoint, replication stress, DNA repair, RPA, PCNA
Introduction
DNA replication is challenged by many sources of replication stress (Zeman and Cimprich, 2014). These include defects in the DNA template such as base damage and backbone breaks, collisions between the replication and transcription machineries, and inherently difficult to replicate sequences that are prone to forming polymerase stalling structures. Avoiding mutations or chromosome aberrations requires a replication stress response that involves hundreds of proteins acting in multiple pathways. One subset of this response utilizes enzymes in the SNF2 family of DNA translocases to remodel stalled DNA replication forks.
SNF2 proteins are ATP-dependent motors that act in a multiple aspects of nucleic acid metabolism. Highly studied members of this family include the catalytic subunits of the SWI/SNF, ISWI, and INO80 chromatin remodeling complexes that act during transcription and DNA repair to alter nucleosome position or composition. These proteins share a bi-lobed helicase-like domain related in structure to bacterial RecA, and they have been classified by sequence differences into multiple subfamilies (Flaus and Owen-Hughes, 2011). SMARCAL1 and ZRANB3 are part of a distant subgroup while HLTF is a member of the RAD5/16-like group named for the yeast RAD5 protein that acts in a post-replicative repair pathway (Unk et al., 2010).
SMARCAL1, ZRANB3, and HLTF associate with replication forks and their inactivation in human cells yields phenotypes consistent with critical functions in responding to replication stress (Bansbach et al., 2009, Ciccia et al., 2009, Yuan et al., 2009, Yusufzai et al., 2009, Betous et al., 2012, Ciccia et al., 2012, Yuan et al., 2012, Kile et al., 2015). For example, loss of SMARCAL1 increases the frequency of double-strand breaks in replicating cells (Yuan et al., 2009, Bansbach et al., 2010, Dungrawala et al., 2017). Loss of either SMARCAL1 or ZRANB3 reduces the ability of stalled forks to recover from replication stress challenges (Bansbach et al., 2009, Ciccia et al., 2009, Ciccia et al., 2012, Weston et al., 2012, Yuan et al., 2012). HLTF deficiency alters replication kinetics in cells experiencing replication stress and also yields increased UV-induced mutagenesis (Lin et al., 2011, Kile et al., 2015). SMARCAL1 and ZRANB3-deficient cells are hypersensitive to a variety of replication stress inducing agents (Bansbach et al., 2009, Ciccia et al., 2009, Ciccia et al., 2012, Weston et al., 2012, Yuan et al., 2012).
Atomic resolution structures of the ATPase domains of several SNF2 family members with and without DNA reveal conserved features needed for translocation on DNA (Ryan and Owen-Hughes, 2011). The ATPase domains also have unique features in loops or protrusions, but much of the differences in function of these enzymes is expected to be derived from accessory domains that provide protein-protein interaction surfaces, allow the enzymes to assemble into larger complexes, or change DNA binding properties. Unlike the chromatin remodeling complexes that contain multiple polypeptide subunits in addition to the catalytic SNF2 motor, the replication stress-responsive enzymes SMARCAL1, ZRANB3, and HLTF appear to function largely as monomers and there is no evidence that they act to remodel nucleosomes.
This review will focus on the structure, function, and regulation of these three enzymes with the goal of both summarizing the published literature, providing perspective, and pointing out important gaps in our knowledge.
SMARCAL1
SMARCAL1 is an annealing helicase
SMARCAL1 is a 954-amino acid protein containing a replication protein A (RPA) binding domain at the N-terminus followed by two HARP domains (both discussed in later sections). The two lobes of the separated ATPase domain are found in the C-terminal half of the protein (Figure 1). SMARCAL1 functions as a DNA-dependent ATPase that translocates on DNA (Coleman et al., 2000, Yusufzai and Kadonaga, 2008). Defects in this activity have severe consequences for genome integrity and human health. Bi-allelic loss-of-function mutations in SMARCAL1 result in the disease Schimke immuno-osseous dysplasia (SIOD) (Boerkoel et al., 2002).
Figure 1.

Domain structures of SMARCAL1, ZRANB3, and HLTF. The ATPase, substrate recognition, protein interaction, and other enzymatic domains are depicted. RBD, RPA binding domain; SRD, substrate recognition domain.
SIOD manifests with diverse phenotypes including renal dysfunction, immune deficiencies, microcephaly, and growth defects (Boerkoel et al., 2002). A few hundred patients have been described and most die during childhood due to infections although a few have had cancer suggesting a mild cancer predisposition (Carroll et al., 2013). However, inactivation of SMARCAL1 in mice actually decreases lymphomagenesis induced by ionizing radiation because of reduced proliferative capacity (Puccetti et al., 2016). SMARCAL1 mutations linked to SIOD typically truncate the protein or are found in critical functional domains including the ATPase and HARP domains (Boerkoel et al., 2002). Patients with missense mutations often have milder SIOD phenotypes, although a clear correlation between mutation and disease severity is lacking (Clewing et al., 2007). Only approximately half of SIOD patients were found to have SMARCAL1 mutations in coding sequences, which suggested there could be other genes involved, although none have been identified. Furthermore, more extensive sequence analyses identified mutations in non-coding regions that disrupt expression that were not identified in initial clinical testing (Carroll et al., 2015).
SMARCAL1 is also known as HepA-related protein (HARP). HepA is a bacterial DNA-dependent ATPase that binds RNA polymerase and promotes transcription (Muzzin et al., 1998, Sukhodolets et al., 2001). This transcription stimulation is achieved by recycling the RNA polymerase perhaps when it would otherwise be trapped on DNA (Sukhodolets et al., 2001). HepA also has ties to the DNA damage response. Bacteria lacking functional HepA show increased sensitivity to ultraviolet (UV) exposure (Muzzin et al., 1998). Despite the sequence similarity observed with HepA, SMARCAL1 does not perform similar functions in human cells. For example, SMARCAL1-deficient cells are not hyper-sensitive to UV exposure (Ciccia et al., 2009, Huang et al., 2010). Further, RNA polymerases have not been identified as SMARCAL1 interacting partners indicating a function in RNA polymerase recycling is not likely. Clear sequence homologs of SMARCAL1 are identifiable in vertebrates, insects, and worms, but no clear homolog is found in yeast (Coleman et al., 2000). Intriguingly, as discussed more below, structure and functional similarities suggest that the T4 bacteriophage UvsW enzyme is a SMARCAL1 homolog (Manosas et al., 2012, Mason et al., 2014).
The first biochemical activity found for SMARCAL1 other than ATP hydrolysiss was the reannealing of complementary DNA sequences (Yusufzai and Kadonaga, 2008). In a plasmid-based assay where complementary single-stranded DNA (ssDNA) is bound by the ssDNA binding protein RPA, SMARCAL1 is able to reanneal the complementary strands together, evicting RPA, to generate an entirely double-stranded DNA (dsDNA) plasmid (Figure 2) (Yusufzai and Kadonaga, 2008). DNA binding and ATP hydrolysis are required for this “annealing helicase” function of SMARCAL1 in vitro (Yusufzai and Kadonaga, 2008, Betous et al., 2012).
Figure 2.

Annealing helicase activity assay. At high concentrations, RPA will induce and stabilize a single-stranded DNA bubble in supercoiled plasmid DNA. SMARCAL1 uses the energy of ATP hydrolysis to re-anneal the complementary DNA strands and displace RPA.
SMARCAL1 annealing activity has also been observed using alternative methods, such as single-molecule magnetic tweezer experiments (Figure 3A). In this assay, a DNA hairpin is attached on one end to a glass coverslip and the other to a magnetic bead. Upon addition of a magnetic field, the hairpin becomes partially unwound to generate a larger distance between the coverslip and the magnetic bead (Manosas et al., 2012). Addition of SMARCAL1 to the reaction results in a decrease of the distance between the glass coverslip, indicating the reannealing of the complementary strands of the DNA hairpin (Betous et al., 2013a).
Figure 3.

Single molecule magnetic tweezer experiment to monitor SMARCAL1 annealing and fork reversal activities. (A) A 1.2kb DNA hairpin substrate is attached to a glass slide on one end and a magnetic bead on the other. Application of a magnetic field will stretch the DNA and unwind the duplex except for the last 20-30 base pairs because of their high GC content. MARCAL1 catalyzes re-annealing of the duplex DNA against the applied force, which is measured as a change in the distance of the bead from the glass slide. This experimental setup revealed that a single molecule of SMARCAL1 catalyzes bursts of repetitive annealing. (B) Addition of oligonucleotides to the stretched DNA allows the creation of substrates that mimic replication forks with ssDNA gaps on either the leading strand (depicted) or lagging template strands. RPA can be added to bind the ssDNA. This experimental set up revealed that RPA increases the distance SMARCAL1 moves per annealing event when it is bound to the leading template strand.
The reannealing of the DNA occurs in repetitive bursts where SMARCAL1 anneals a short stretch of DNA bases prior to pausing. These repetitive bursts of annealing are not the result of SMARCAL1 dissociation and rebinding to the DNA but rather interspersed gaps of pausing while still bound to the substrate (Betous et al., 2013a). The purpose of this pause and go action is currently unknown, but it could be a mechanism to prevent excessive reannealing of complementary sequences by SMARCAL1 during fork reversal (see below). The T4 phage enzyme UvsW, a protein with structural similarities to SMARCAL1, displays similar annealing and pausing patterns indicating this may be a conserved mechanism to regulate DNA annealing rates (Manosas et al., 2012).
SMARCAL1 fork remodeling
Stalled forks need to be stabilized to prevent nuclease cleavage or degradation to maintain genome integrity (Cortez, 2015). In situations when there is no converging replication fork, the stalled fork must also restart to complete DNA synthesis. SMARCAL1 catalyzes fork reversal of replication forks in response to replication stress that causes fork stalling (Figure 4) (Betous et al., 2012, Betous et al., 2013a, Bhat et al., 2015, Kolinjivadi et al., 2017).
Figure 4.

Diagram depicting fork reversal and fork restoration. Fork reversal anneals the parental (black) and nascent (silver) DNA strands in a concerted reaction generating a chicken foot structure. Migrating the four-way junction in the opposite direction yields fork restoration. A ssDNA gap is diagramed on the leading strand template in this example.
Fork reversal (also called fork regression) involves a concerted reannealing of the parental template strands to reverse the direction of the replication fork and generate a four-way junction, also termed the “chicken foot” (Figure 4) (Neelsen and Lopes, 2015). Fork reversal is thought to stabilize the fork, promote repair of the parental DNA template, or serve as a template switching mechanism to bypass the source of replication stress. Once the source of stress has been addressed, SMARCAL1 can also catalyze the reverse reaction (called fork restoration), to reset the replication fork to a three-way junction from the reversed chicken foot structure (Figure 4) (Betous et al., 2013a, Bhat et al., 2015). Many other proteins including ZRANB3 and HLTF catalyze fork reversal and the RAD51 recombinase is also needed for this process (Blastyak et al., 2010, Achar et al., 2011, Ciccia et al., 2012, Zellweger et al., 2015, Kolinjivadi et al., 2017). There are many questions about fork reversal that remain to be answered such as how the ATPase motor proteins cooperate with RAD51 to promote reversal, where does the replication machinery go during this process, and how dynamic is fork reversal. We point the reader to a recent review on fork reversal for further discussion of this process (Neelsen and Lopes, 2015).
SMARCAL1 mutant proteins found in patients with SIOD are unable to catalyze fork reversal illustrating the importance of these fork remodeling functions of SMARCAL1 (Betous et al., 2012, Carroll et al., 2013). It is also clear, however, that these functions must be tightly regulated to prevent excessive fork reversal or failure to restore the replication fork. For example, overexpression of catalytically active SMARCAL1 interferes with DNA replication and causes fork breakage (Bansbach et al., 2009). Checkpoint kinases negatively regulate SMARCAL1 to prevent excessive fork reversal and fork breakage (see below, (Couch et al., 2013)). Similarly, the checkpoint inhibits fork reversal in S. cerevisiae (Sogo et al., 2002). Furthermore, failure to regulate fork reversal in cells deficient for the recently described RADX protein also yields double-strand breaks (Dungrawala et al., 2017), and fork reversal can facilitate nascent strand degradation (Kolinjivadi et al., 2017). Thus, inappropriate, excessive, or persistent fork reversal can be deleterious.
The anneal-and-pause activity observed for SMARCAL1 in the magnetic tweezer experiments might be one mechanism preventing excessive fork reversal. Short bursts of annealing followed by a pausing event could provide a window of opportunity for downstream processing of the replication stress to occur. Additionally, a pause after short bursts of annealing would prevent fork reversal extending uninhibited for long distances and, therefore, prevent delays in replication. This idea is supported by magnetic tweezer experiments with the T4 UvsW enzyme in which fork reversal and template switching to bypass a DNA lesion were reconstituted (Manosas et al., 2012).
SMARCAL1 Domain Structure and Function
SMARCAL1 does not bind DNA substrates composed entirely of ssDNA or dsDNA; however, it displays a strong preference for binding DNA substrates with a ssDNA/dsDNA junction (Muthuswami et al., 2000, Yusufzai and Kadonaga, 2008, Ghosal et al., 2011, Betous et al., 2012, Mason et al., 2014). ATPase activity closely correlates with DNA binding affinity (Betous et al., 2012). SMARCAL1 displays equal affinity for DNA substrates with a 3′ and 5′ ssDNA overhangs (Betous et al., 2012). In addition to the simple junction DNA structures, SMARCAL1 also binds a variety of other DNA substrates commonly found during DNA replication and repair including 3-way DNA junctions, 4-way Holliday junctions, a splayed arm, and dsDNA structures with internal ssDNA gaps (Betous et al., 2012).
DNA binding is likely mediated through the combined functions of the ATPase and HARP domains. Structures of related proteins indicate that the ATPase domain may bind dsDNA. However, it has not been possible to purify the ATPase domain by itself so a formal demonstration that it binds DNA has not been possible. Although a stable ATPase domain has yet to be isolated, the core catalytic domain of SMARCAL1 has been identified from limited proteolysis and small angle X-ray scattering (SAXS) data. The minimal stable protein contains the HARP2 and ATPase domains (Betous et al., 2012). This catalytic core has been purified and displays levels of DNA binding and annealing activity similar to wild-type protein on naked DNA substrates. Structural studies with this core catalytic unit of SMARCAL1 in the presence of DNA substrates could be informative for characterizing the contribution of the ATPase domain to DNA binding and to clarify the mechanism of translocation.
Part of the DNA binding activity of SMARCAL1 resides in its HARP domains located in the N-terminal half of the protein. The importance of these domains is highlighted by the identification of mutations in the HARP domains found in patients with SIOD (Boerkoel et al., 2002). SMARCAL1 protein lacking the function of the first HARP domain (HARP1) displays much milder defects in DNA binding, ATP hydrolysis, and fork remodeling compared to protein lacking the function of the second HARP domain (HARP2) (Betous et al., 2012). Although the phenotypes associated with HARP2 deficiencies are more severe in vitro, both domains are important biologically as point mutations in the HARP1 domain also result in the severe phenotypes associated with SIOD.
The HARP domains function as substrate recognition domains (SRDs) that confer DNA binding preference for junction DNA (Figure 5 and (Betous et al., 2012, Mason et al., 2014)). Chimeric protein containing the ATPase domain of related SNF2 enzymes fused to the HARP domains of SMARCAL1 possess annealing helicase activity (Ghosal et al., 2011). Furthermore, some defects associated with SMARCAL1 deficiency may be rescued by overexpression of these chimeric proteins indicating they are able to recognize, bind, and act on endogenous DNA substrates addressed by SMARCAL1 in cells (Ghosal et al., 2011).
Figure 5.
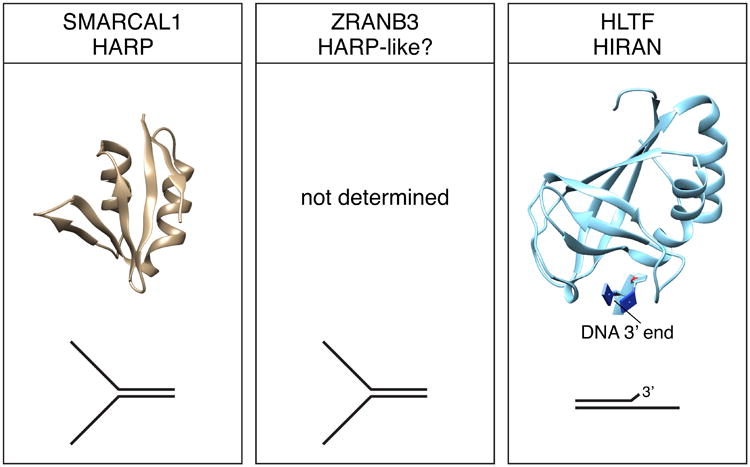
Substrate recognition domains of SMARCAL1, ZRANB3, and HLTF. Structures of the SMARCAL1 HARP and HLTF HIRAN domains were derived from PDB 4S0N and 4XZF respectively. The DNA binding specificity of the domains is illustrated. The structure of the ZRANB3 SRD domain has not been determined.
An atomic resolution structure of the HARP1 domain of SMARCAL1 showed that it forms a compact domain composed of a four-stranded anti-parallel beta sheet decorated with two alpha helices (Figure 5) (Mason et al., 2014). Importantly, this domain fold is shared in other proteins including XPB and UvsW. XPB is a helicase that acts during transcription and nucleotide excision repair. UvsW, like SMARCAL1, catalyzes fork reversal during DNA replication. The HARP-like domain of UvsW is located N-terminal to the ATPase domains similar to the HARP domains of SMARCAL1 indicating a structure conservation for these fork reversal enzymes. The SMARCAL1 HARP1 domain can substitute for the HARP-like domain in UvsW to yield a functional UvsW enzyme (Mason et al., 2014).
The number of HARP domains in SMARCAL1 decreases from two in higher eukaryotes to one in lower eukaryotes such as Drosophila melanogaster (Ghosal et al., 2011). This decrease in the number of SRDs could reflect the differences in biochemical activities observed between the human and fly SMARCAL1 proteins. Although SMARCAL1 from Drosophila functions as an annealing helicase similar to the human protein, it cannot catalyze branch migration of replication fork structures like human SMARCAL1 (Kassavetis and Kadonaga, 2014). However, only a single HARP domain in human SMARCAL1 and UvsW is required to catalyze fork reversal in vitro suggesting that the second HARP domain in vertebrate SMARCAL1 may have another function in cells.
SMARCAL1 Regulation
Knockdown of SMARCAL1 in human cells results in double-strand break formation during S-phase even without the addition of any external genotoxic agents. However, tight regulation of SMARCAL1 is essential since overexpression also results in double-strand breaks (Bansbach et al., 2009). This overexpression induced DNA damage is dependent on both its ATPase and RPA binding domains suggesting that excessive localization of an active protein to undamaged forks causes inappropriate fork remodeling and breakage. Therefore, SMARCAL1 is regulated at multiple levels including transcription, post-translational modifications, and protein-protein interactions.
Regulation by RPA
RPA is an essential protein complex composed of three subunits – RPA70, RPA32, and RPA14. The heterotrimer binds ssDNA with high affinity and coordinates the recruitment and exchange of proteins at sites of DNA damage (Oakley and Patrick, 2010). RPA also functions in normal DNA replication during elongation and Okazaki fragment maturation on the lagging strand. To achieve these functions, RPA interacts with multiple proteins using protein interaction domains at the N-terminus of the RPA70 subunit (70N) and the C-terminus of the RPA32 (RPA32C).
SMARCAL1 interacts with the RPA32 subunit using a conserved motif (Bansbach et al., 2009, Ciccia et al., 2009, Postow et al., 2009, Yuan et al., 2009, Yusufzai et al., 2009). This RPA binding domain (RBD) shares sequence similarity to many other proteins that interact with RPA32, including DNA damage response proteins like ETAA1 and nucleotide excision repair protein XPA (Matsuzaki et al., 2015, Bass et al., 2016, Haahr et al., 2016). As many proteins bind the same surface on RPA, it is important to understand how these interactions are coordinated to permit rapid repair of DNA damage. The interaction between SMARCAL1 and RPA is required to localize SMARCAL1 to sites of replication stress (Bansbach et al., 2009, Ciccia et al., 2009, Postow et al., 2009, Yuan et al., 2009, Yusufzai et al., 2009). Furthermore, the RPA interaction is required for SMARCAL1 to prevent fork damage (Bansbach et al., 2009, Ciccia et al., 2009, Yuan et al., 2009, Bansbach et al., 2010).
In addition to localization, RPA also confers substrate specificity to SMARCAL1 reactions by inhibiting it in some cases while activating it in others. When RPA binds a replication fork with a ssDNA gap on the lagging strand – a substrate that mimics normal replication intermediates – it acts as a block to SMARCAL1-catalyzed fork reversal thereby allowing DNA replication to proceed (Figure 6A) (Betous et al., 2013b, Bhat et al., 2015). Conversely, RPA stimulates SMARCAL1 fork reversal on DNA substrates when it is bound to a ssDNA gap on the leading template strand – a substrate mirroring a replication fork that has stalled (Betous et al., 2013a, Bhat et al., 2015). This relationship is reversed when considering the fork restoration functions of SMARCAL1. RPA inhibits SMARCAL1 fork restoration activity on a DNA substrate whose restoration would yield a gap on the leading strand while it stimulates restoration on DNA structures that result in a gap on the lagging strand (Betous et al., 2013a, Bhat et al., 2015).
Figure 6.

The orientation of RPA binding to the replication fork substrate differentially regulates SMARCAL1. (A) Fork reversal is stimulated when RPA is bound to the leading strand template and is inhibited when it is bound to the lagging strand template. Fork restoration is activated when RPA is bound to a longer nascent leading strand and inhibited when it is bound to the nascent lagging strand. (B) RPA binds asymmetrically to ssDNA using four DNA binding domains with varying affinities. RPA stimulates SMARCAL1 when the two highest affinity DNA binding domains are located next to the fork junction and inhibits SMARCAL1 when the low affinity binding domains are located next to the junction.
An important question is how RPA achieves this dual role – inhibiting SMARCAL1 when bound to some substrates, and activating it when bound to others. The mechanism of SMARCAL1 inhibition is most likely by acting as a block that must be removed from the DNA substrate to generate the product. However, the activation of SMARCAL1 on damaged replication fork structures is more complex. DNA binding affinity measurements and DNA footprinting experiments indicate RPA doesn't alter how SMARCAL1 binds DNA. Furthermore, RPA binding doesn't alter ATPase activity (Betous et al., 2013a). RPA binding by SMARCAL1 is essential for stimulation even though the exact mode of binding is not. In fact, a SMARCAL1 protein that has been engineered to bind RPA70 instead of RPA32 still is stimulated when RPA is bound on the leading strand template of a model replication fork (Bhat et al., 2015).
Single molecule, magnetic tweezer experiments revealed at least a partial answer to how RPA activates SMARCAL1 (Figure 3B). Measuring enzyme processivity and rates of fork reversal in the presence and absence of RPA revealed that when RPA is on the DNA substrate, SMARCAL1 is able to catalyze annealing of longer stretches of DNA prior to pausing. (Betous et al., 2013a). Importantly, just as in the solution biochemistry experiments, this stimulation of processive power by RPA is specific to DNA structures with a gap on the leading template strand. Conversely, RPA decreases the distance traveled by SMARCAL1 when it is bound to a lagging strand gap substrate. This substrate specificity is exactly what would be needed to avoid reversing replication forks that are actively elongating since RPA is normally found on the lagging strand during DNA synthesis. Therefore, it provides selectivity so SMARCAL1 only works on stalled forks with RPA bound to the leading strand template (Betous et al., 2013a).
The ability of RPA to enhance or inhibit SMARCAL1 fork remodeling on different substrates may be due to the asymmetry of RPA binding to DNA substrates. RPA is composed of four DNA binding domains of varying affinity (Oakley and Patrick, 2010). The polarity of these DNA binding domains with respect to the junction of the replication fork differs depending on which substrate RPA is bound (Figure 6B). The two high affinity DNA binding domains of RPA are found close to the replication fork junction in substrates where SMARCAL1 function is stimulated, and, paradoxically, reducing their affinity for DNA by mutating these domains reduces the stimulation of SMARCAL1 (Bhat et al., 2015). Multiple models were proposed to explain this observation. First, the proximity of the high affinity DNA binding domains to the replication fork junction could facilitate the RPA-mediated melting of complementary DNA strands to allow for the appropriate fork remodeling reactions. In cases of fork reversal, RPA could destabilize the complementary lagging parental and nascent DNA duplex to promote reannealing of the parental template. Conversely, the fork restoration reaction could be stimulated upon destabilization of the four-way junction by placing the high affinity DNA binding domains of RPA next to the junction. Alternatively, stimulation of SMARCAL1 activity could involve a SMARCAL1 protein conformation change that is specific to RPA DNA binding orientation.
Currently there is no data to differentiate these models. Switching the polarity of how RPA binds to ssDNA would be one approach to testing these ideas, but it is unlikely to be feasible. Structural studies with SMARCAL1, RPA, and DNA in a complex would provide information to support one model over the other. Although the exact mechanism remains unresolved, the ability of RPA to direct SMARCAL1 activity to specific substrates is an important differentiating factor that is not shared by ZRANB3 (Betous et al., 2013a). Whether HLTF is regulated by RPA remains untested.
Regulation by phosphorylation
Phosphorylation is a common form of regulation for proteins involved in the DNA damage response. The DNA damage activated phosphoinositol 3-kinase-related (PIKK) family of enzymes regulates many aspects of the DNA damage response including fork reversal by SMARCAL1 (Lovejoy and Cortez, 2009, Blackford and Jackson, 2017). This kinase family includes ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related (ATR), and DNA-dependent protein kinase (DNA-PK). ATR is activated by ssDNA while ATM and DNA-PK are activated by dsDNA breaks (Lovejoy and Cortez, 2009).
All three PIKK family kinases phosphorylate SMARCAL1 following DNA damage (Bansbach et al., 2009, Postow et al., 2009). SMARCAL1 phosphorylation by ATR on the conserved serine 652 (S652), which is found in the linker between the lobes of the ATPase domains, decreases its ATPase activity and reduces its fork remodeling activities (Figure 7) (Couch et al., 2013). Importantly, this modification only occurs after SMARCAL1 binds to DNA. Localization to a stalled fork through its interaction with RPA is insufficient for phosphorylation. Since S652 is in the linker between the ATPase lobes, it is likely that the site is inaccessible until DNA binding induces a conformational change that allows S652 to be recognized and phosphorylated. Since S652 phosphorylation decreases its activity, its phosphorylation after localization and DNA binding may allow it to perform its fork remodeling functions prior to phosphorylation-dependent inhibition. ATR phosphorylation of S652 is one mechanism by which ATR prevents replication fork collapse (Couch et al., 2013).
Figure 7.

ATR inhibits SMARCAL1. ATRIP binding to the 70N domain of RPA recruits ATR to replication forks. The SMARCAL1 RBD binds RPA32C to bring SMARCAL1 to replication forks. Once SMARCAL1 binds to the forked DNA via its ATPase and HARP domains, it becomes a substrate for ATR phosphorylation. Phosphorylation within the linker between the ATPase domains inhibits SMARCAL1 activity.
Another SMARCAL1 phosphorylation happens near the C-terminus on serine 889 (S889). A phospho-mimetic mutation results in a hyperactive protein suggesting this post-translational modification alleviates auto-inhibition (Carroll et al., 2014). Consistent with this idea, deletion of the entire C-terminal region after the ATPase domains also renders the protein hyperactive in vitro. It is unclear which kinase is responsible for this modification as the phosphorylation site does not match the conserved sequence motif recognized by the PIKK kinases (Kim et al., 1999, Carroll et al., 2014). Additional studies are also needed to determine when this modification occurs since it is not induced by DNA damage.
Several additional SMARCAL1 phosphorylation sites have been reported. Following treatment with hydroxyurea, SMARCAL1 is phosphorylated on S173 and S919, two residues found within non-conserved regions of the protein (Couch et al., 2013). Mutations in these residues had no effect on SMARCAL1 ATPase activity so their functional importance remains unknown. Other phosphorylation sites include S112, S123, S129, S172, and S198 (Carroll et al., 2014). All of these sites fall within a region of SMARCAL1 predicted to be disordered. How phosphorylation at these other sites affects SMARCAL1 function and which kinases are responsible remain to be determined.
Several additional forms of SMARCAL1 regulation have also been proposed. One report suggests SMARCAL1 localization is cell cycle regulated where it is predominately nuclear during S phase through mitosis but is exported to the cytoplasm during the first growth phase of the cell cycle (G1) (Haokip et al., 2016). This study also indicated SMARCAL1 transcript expression is DNA damage-dependent (Haokip et al., 2016). However, these observations require further validation in additional systems. Additionally, some evidence in zebrafish indicate SMARCAL1 expression is regulated by the cell cycle (Huang et al., 2010), but whether this is true in other organisms remains unknown. In any case, the regulation of SMARCAL1 function is essential to prevent excessive activity while also permitting SMARCAL1 to perform its required functions in preserving genome stability.
Where is SMARCAL1 needed?
SMARCAL1-deficient cells are sensitive to a diverse set of DNA damaging agents including hydroxyurea (HU), aphidicolin, mitomycin C, ionizing radiation, and camptothecin (Bansbach et al., 2009, Ciccia et al., 2009, Yuan et al., 2009). However, SMARCAL1 is also required to address endogenous sources of replication stress. Cells without SMARCAL1 display high levels of DNA damage in the absence of any added drugs (Bansbach et al., 2009, Ciccia et al., 2009, Yuan et al., 2009, Bansbach et al., 2010, Poole et al., 2015) and some SMARCAL1 protein is observed at elongating replication forks in untreated cells (Betous et al., 2012, Dungrawala et al., 2017). Thus, it is important to determine what endogenous sources of replication stress require SMARCAL1 for resolution.
S phase functions of SMARCAL1
Our laboratory reported the first endogenous source of replication stress that requires the function of SMARCAL1 for genome integrity to be telomere replication (Poole et al., 2015, Poole and Cortez, 2016). Telomeres are highly repetitive DNA sequences that cap the ends of linear chromosomes. SMARCAL1 deficient cells exhibit signs of telomere damage. Specifically, the DNA damage markers 53BP1 and RPA are found at elevated levels at telomeres in SMARCAL1-deficient cells in the absence of any genotoxic agents (Poole et al., 2015).
Telomeric DNA damage suggests that SMARCAL1 may be required to ensure replication forks successfully traverse telomeric sequences. Consistent with this interpretation, SMARCAL1-deficient human and mouse cells accumulate extrachromosomal telomere circles (C-circles) (Poole et al., 2015). These increased extra-chromosomal telomere circles are specifically observed in mouse and human cells that have long telomeres and their appearance is dependent on DNA synthesis. A likely explanation is forks moving through telomeric sequences stochastically stall, and when SMARCAL1 is not present, they are broken and processed into the C-circles. Longer telomere sequences increase the chance of stalling and increase the need for SMARCAL1.
Prior to this report, C-circles had been exclusively associated with the alternative lengthening of telomeres (ALT) telomere elongation mechanism that occurs in about 15% of cancers (Henson et al., 2009). ALT cells are characterized by increased frequencies of telomere recombination and ALT-associated promyelocytic leukemia (PML) bodies (APBs), changes in telomere length, and accumulation of C-circles (Henson et al., 2009, Cesare and Reddel, 2010). However, the increased C-circles caused by SMARCAL1-deficiency happened in telomerase-positive cells and no other phenotypes associated with ALT were found in these cells (Poole et al., 2015).
A second group also identified SMARCAL1 as a protein required for telomere stability. However, they concluded that the function of SMARCAL1 was limited to ALT-positive cells (Cox et al., 2016). This group used two telomerase-positive cell lines with average telomere length possibly explaining why they did not observe a need for SMARCAL1 during telomere replication in telomerase-positive cells. In addition to C-circles, they also found that SMARCAL1 knockdown in ALT cells caused increased frequencies of chromosome fusions and APBs. However, these effects were not observed in another ALT-positive cell type (U2OS), suggesting there may be cell type-specific differences (Poole et al., 2015).
The identification of telomeres as regions of the genome requiring SMARCAL1 function was an important step in understanding the requirement for this protein during DNA replication. However, the exact source of replication stress was not identified. The telomeric repeats are thought to pose as obstacles to replication because they can form complex DNA structures such as G-quadruplexes. In addition, the chromosome end is usually hidden in a telomere loop (t-loop), and the repeats are coated with telomere-binding protein complexes (Gilson and Geli, 2007). In principle, any of these properties could be the source of replication stress counteracted by SMARCAL1.
Interestingly, RPA may be dispensable for the function of SMARCAL1 at telomeres (Poole et al., 2015). A mutant of SMARCAL1 lacking the RPA binding domain was able to rescue the telomere instability phenotypes to the same extent as wild-type protein. Thus, this mutant may provide a separation of function between SMARCAL1 function at telomeres and its functions elsewhere in the genome (Bansbach et al., 2009). This result raises the important questions of how SMARCAL1 is recruited and regulated at telomeres. Another important question is whether telomere replication defects contribute to any of the SIOD phenotypes observed in patients.
As yet, the other genomic contexts requiring SMARCAL1 function remain unidentified. Future studies to characterize other sources of endogenous stress will be necessary to understand the requirement for SMARCAL1 during DNA replication and the etiology of SIOD.
SMARCAL1 functions outside replication forks
Although most of the studies of SMARCAL1 have concentrated on its functions during DNA replication, there is also evidence for other functions. SMARCAL1 was reported to be required for non-homologous end-joining (NHEJ) repair of dsDNA breaks during G1 (Keka et al., 2015). This report proposed that SMARCAL1 anneals unwound DNA at breaks to facilitate the retention of canonical NHEJ factors, like the Ku complex and DNA-PK, at the DNA end. This function of SMARCAL1 in NHEJ requires an interaction with RPA (Keka et al., 2015). Perhaps SMARCAL1 counteracts end resection activities at breaks; however, further validation of its role in NHEJ is needed.
SMARCAL1 also has been reported to have a regulatory role in the transcription of a subset of genes (Baradaran-Heravi et al., 2012b, Sharma et al., 2015, Haokip et al., 2016, Patne et al., 2017). Specifically, SMARCAL1 was reported to directly modify the chromatin near promoter sequences to regulate gene expression especially in cells experiencing replication stress (Sharma et al., 2015, Haokip et al., 2016, Patne et al., 2017). However, SMARCAL1 binds DNA in a sequence independent manner and has not been found in complexes with any transcription factors or components of ATP-dependent chromatin remodeling complexes (Betous et al., 2012). Thus, how it would be recruited to specific genes is unclear. SMARCAL1 could resolve transcription replication conflicts, which could manifest as a change in gene expression. SMARCAL1 is capable of resolving RNA-DNA structures lending support to this idea (Kassavetis and Kadonaga, 2014). Another proposal is that SMARCAL maintains DNA topology to promote transcription (Baradaran-Heravi et al., 2012a). In support of a function in regulating gene expression, there may be differences in gene expression when SMARCAL1 is knocked out in mice and flies (Baradaran-Heravi et al., 2012a). However, unpublished RNA sequencing experiments from our lab using SMARCAL1 knockout human cells do not support a major function in gene-specific transcription regulation. Further studies are needed to reconcile these observations.
ZRANB3
ZRANB3 is an annealing helicase, fork remodeler, and structure specific nuclease
ZRANB3 (zinc finger, RAN-binding domain containing 3) is a 1079-amino acid protein and, based on sequence homology, is the most closely related SNF2 protein to SMARCAL1. The separated lobes of the ZRANB3 ATPase domain are located at the N-terminal portion of the protein followed by several supplementary domains that will be discussed in later sections (Figure 1). Defects in ZRANB3 function cause genome instability. For example, ZRANB3-deficient cells display higher rates of replication fork stalling and increases in sister chromatid exchanges (SCEs), a marker of hyper-recombination (Ciccia et al., 2012). ZRANB3 deficiency also causes hyper-sensitivity to diverse DNA damaging agents (Ciccia et al., 2012, Weston et al., 2012, Yuan et al., 2012). Despite these striking phenotypes, ZRANB3 deficiency has not been directly associated with any human diseases; however, reports of ZRANB3 mutations in endometrial cancers suggest it may function as a tumor suppressor (Lawrence et al., 2014).
Current evidence suggests that like SMARCAL1, ZRANB3 functions primarily in replication stress responses and not in chromatin remodeling or transcription regulation. Cells lacking ZRANB3 display higher levels of replication fork stalling and subsequent cell death following treatment with the replication stress-inducing agent HU (Ciccia et al., 2012, Yuan et al., 2012). Like SMARCAL1, ZRANB3 lacks helicase activity but can act to anneal complementary DNA using the plasmid assay containing an RPA-induced bubble in the DNA duplex (Yusufzai and Kadonaga, 2010). Furthermore, like SMARCAL1, ZRANB3 catalyzes both fork reversal and restoration (Ciccia et al., 2012, Betous et al., 2013a). Although it is able to catalyze these same reactions in vitro, ZRANB3 displays different substrate preferences compared to SMARCAL1 (Betous et al., 2013a). These proteins are not functioning redundantly since depleting both further sensitizes cells to replication stress compared to individual depletions (Ciccia et al., 2012). Unlike SMARCAL1, ZRANB3 deficiency does not result in fork breakage in the absence of replication stress, and ZRANB3 has not been linked to telomere replication (Poole et al., 2015, Dungrawala et al., 2017).
In addition to catalyzing fork remodeling, ZRANB3 also remodels displacement loop (D-loop) structures in vitro (Ciccia et al., 2012). The formation of a D-loop is a critical step in homologous recombination (HR) DNA repair and during a template switching pathway used for replication fork restart (Ulrich and Walden, 2010, Jasin and Rothstein, 2013). This structure is formed when a DNA end invades the duplex of a homologous template, a reaction catalyzed by the RAD51 recombinase. Failure to resolve D-loops in cells may result in DNA crossovers that can have deleterious effects on the integrity of the genome. Interestingly, ZRANB3 is able to dissolve pre-formed D-loops and prevent their formation in vitro. Thus, ZRANB3 could dissolve D-loops after DNA repair or damage bypass and prevent excessive strand invasion to maintain the appropriate levels of recombination intermediates during DNA replication. Failure to perform these functions results in the increased frequency of SCEs observed in ZRANB3-deficient cells (Ciccia et al., 2012). SMARCAL1 can also catalyze D-loop dissolution suggesting a common mechanism with ZRANB3 (Ciccia et al., 2012). However, unlike ZRANB3, SMARCAL1 is not able to prevent D-loop formation by RAD51. The source and functional significance of this difference is unclear.
In contrast to SMARCAL1 and other SNF2 family members, ZRANB3 possesses endonuclease activity in addition to its other enzymatic functions (Weston et al., 2012, Badu-Nkansah et al., 2016, Sebesta et al., 2017). The endonuclease activity depends on ATP hydrolysis by the intact motor domain as well as a C-terminal nuclease domain. DNA cleavage happens on one of the two strands of a DNA duplex, but it requires adjacent ssDNA. The preferred DNA substrate is a splayed arm with a minimum of 20 nucleotides of ssDNA (Weston et al., 2012). ZRANB3 will also cleave replication fork structures as long as there is ssDNA present at the fork junction (Weston et al., 2012). On these substrates, ZRANB3 generates a nick two nucleotides into the DNA duplex on the leading strand template.
The coupling of ATP hydrolysis and fork remodeling with an endonuclease activity is unusual. Exactly how these activities work together at stalled forks is unknown. One proposed model is that ZRANB3 nicks the leading strand duplex and catalyzes fork reversal to prevent the formation of a double-strand break (Weston et al., 2012). The nicking activity leaves a 3′OH group that could be extended by a polymerase. If the polymerase displaces the damaged DNA, then flap cleavage by FEN1 could yield a religatable nick and successful repair of the lesion (Fig 8).
Figure 8.

Model for how ZRANB3 nuclease and fork remodeling activities could be coordinated to repair a leading strand template lesion (red star). ZRANB3 endonuclease cuts two nucleotides into the parental duplex. Fork reversal could then stabilize the fork and allow for strand displacement DNA synthesis. A flap endonuclease could then remove the damaged DNA and permit fork restoration to restart the fork. This model is adapted from Weston et al., Genes and Development (Weston et al., 2012).
Some cancer associated mutations in ZRANB3 inactivate its nuclease activity without affecting its ATPase activity (Sebesta et al., 2017), and the nuclease domain contributes to ZRANB3 localization to damaged forks (Weston et al., 2012). Thus, nuclease activity may be important for its genome protection functions, but further studies will be needed to determine if nuclease inactivation actually drives tumorigenesis.
ZRANB3 Domain Structures and Function
ZRANB3 displays similar DNA binding activities as SMARCAL1 with a preference for ssDNA/dsDNA junctions over substrates composed entirely of ssDNA or dsDNA (Yuan et al., 2012). ZRANB3 also binds 3-way junctions and a splayed arm substrate; however, extensive studies similar to those done for SMARCAL1 to fully characterize DNA binding preferences and generate footprinting information have not yet been completed. Given its similar biochemical activities to SMARCAL1, the expectation is that ZRANB3 will bind DNA using similar domains. Thus, two groups have looked for a SRD that would confer DNA binding specificity (Yuan et al., 2012, Badu-Nkansah et al., 2016). Initially, a region between amino acids 712-818 was suggested to be a HARP-like SRD based on limited sequence similarity and functional analyses (Yuan et al., 2012). This HARP-like domain was described as dispensable for DNA binding and ATPase activity but required for annealing helicase activity in vitro. This conclusion was based on biochemical studies of a purified protein lacking the putative HARP-like domain and is unexpected since the HARP domains of SMARCAL1 are essential for DNA binding and ATPase activity (Betous et al., 2012). Our laboratory revisited this conclusion and found that the same ZRANB3 mutant lacking amino acids 712-818 did not bind DNA or hydrolyze ATP. The lack of an ATPase-deficient negative control protein in the original study suggests that a contaminating DNA-dependent ATPase could have been responsible for the different result.
Using sequence conservation and secondary structure predictions as guides, we identified a human ZRANB3 SRD consisting of amino acids 721-869 (Badu-Nkansah et al., 2016). As predicted from the SMARCAL1 studies, this SRD is essential for DNA binding, ATPase function, and annealing helicase activity. Deletions or amino acid substitution mutations in the SRD are sufficient to abolish all ZRANB3 enzymatic activity including its nuclease activity (Badu-Nkansah et al., 2016). Furthermore, the ZRANB3 SRD by itself is able to bind splayed arm DNA but not double or single-stranded DNA indicating that it provides specificity to fork junctions. The binding affinity of the SRD is significantly less than the full-length protein as would be expected if other domains including the ATPase motor contact DNA. Fusing the ZRANB3 SRD to the ATPase domain is sufficient to reconstitute a minimal ZRANB3 enzymatic unit that can catalyze fork reversal (Badu-Nkansah et al., 2016). Despite the functional similarities of the SMARCAL1 HARP domains and ZRANB3 SRD, it is not yet clear if they adopt a similar three-dimensional structure.
In addition to the ATPase and SRD domains, the ZRANB3 nuclease domain also binds DNA. Nuclease activity requires a C-terminal HNH domain, named for the conserved amino acids that compose this domain, that is characteristic of numerous bacterial and fungal nucleases (Yusufzai and Kadonaga, 2010). HNH nucleases can act as sequence-specific homing endonucleases and RNA guided nucleases like Cas9 (Jinek et al., 2012). The purified ZRANB3 HNH domain can bind 4-way Holliday junctions and structures with 5′ DNA flaps (Weston et al., 2012). Interestingly, ZRANB3 is the only protein present in vertebrates described to contain this domain. Further, structure comparisons of the HNH of ZRANB3 with other HNH domains from lower organisms indicate that ZRANB3 contains an evolutionarily conserved unique sequence insert in the HNH domain that is absent in most other organisms (Sebesta et al., 2017). Deletions in this ZRANB3-specific region abolish nuclease activity. This structure could be important for recognition of ZRANB3-specific DNA substrates for cleavage, but future studies are needed to confirm this hypothesis. Importantly, the nuclease activity conferred by the ZRANB3 HNH domain is dependent on an intact motor domain, ATP hydrolysis, and an intact SRD domain (Weston et al., 2012, Badu-Nkansah et al., 2016, Sebesta et al., 2017). How this linkage is achieved is not known.
ZRANB3 Regulation
Regulation by PCNA
Unlike SMARCAL1 that utilizes RPA to localize to sites of replication stress, ZRANB3 does not interact with RPA (Yusufzai and Kadonaga, 2010). Instead, ZRANB3 utilizes multiple motifs to bind to proliferating cell nuclear antigen (PCNA) (Figure 1). ZRANB3 contains a conserved canonical PCNA-interacting protein (PIP) box that is similar to several other PCNA-binding proteins (Ciccia et al., 2012, Weston et al., 2012, Yuan et al., 2012). ZRANB3 also contains an APIM (AlkB homology 2 PCNA interaction motif) that contributes to its interactions with PCNA. Both of these motifs are important for facilitating ZRANB3 localization to sites of replication stress (Ciccia et al., 2012, Weston et al., 2012, Yuan et al., 2012). Mutations in either domain impair ZRANB3 co-localization with PCNA after replication stress, but it is not until both domains are mutated that ZRANB3 localization is abolished.
A crystal structure of the PIP box of ZRANB3 with PCNA shows this motif associates with the same hydrophobic region on the surface of PCNA as other PIP boxes (Sebesta et al., 2017). This study also reported that the ZRANB3 APIM interaction with PCNA is on the same surface that is contacted with the PIP box, indicating these two motifs could compete with each other for PCNA binding. Since PCNA exists as a homotrimer in cells, both the PIP and APIM motifs could bind to different subunits of the same trimeric ring. Alternatively, both motifs could be binding the same surface on a single subunit but in a temporally regulated manner. Once one motif dissociates from PCNA, the other would bind and allow the interaction between PCNA and ZRANB3 to persist.
Over 200 proteins contain a PIP box and perhaps another 200 contain an APIM (Mailand et al., 2013). Thus, ZRANB3 must outcompete hundreds of proteins for binding to the PCNA homotrimer and respond to replication stress in a timely manner. Further, this binding would likely need to be regulated such that ZRANB3 is only localized to sites of replication stress so as not to outcompete PCNA interacting partners that are required for normal elongation during DNA replication. To solve this problem, ZRANB3 also contains an NZF (NPL4 zinc-finger) domain that binds poly-ubiquitin (Ciccia et al., 2012). When the replisome encounters an obstacle to replication, PCNA is polyubiquitinated on lysine 164 (K164) which acts as a signal for fork restart through error-free methods such as template switching (Mailand et al., 2013). PCNA polyubiquitination may be mediated by two other members of the SNF2 family, HLTF and SHPRH, discussed more below. Mutations in the NZF domain abolish ZRANB3 binding to polyubiquitinated PCNA both in cells and in vitro (Ciccia et al., 2012, Weston et al., 2012). ZRANB3 is the first human protein shown to read this poly-ubiquitin PCNA signal.
PCNA also regulates ZRANB3 function beyond controlling localization. PCNA stimulates ZRANB3-mediated cleavage of a splayed arm DNA substrate in vitro (Sebesta et al., 2017). Mutations in either the PIP box or the APIM completely abolished the stimulatory effect of PCNA on ZRANB3 nuclease activity. Interestingly, the double mutant that lacks both a functional PIP box and APIM does not display an additive effect. One model to explain this observation is that although either domain is sufficient to anchor ZRANB3 to PCNA, ZRANB3 may need to directly contact PCNA with both domains concurrently to stimulate nuclease activity. As yet no studies have investigated the effect of PCNA on the fork remodeling functions of ZRANB3.
Regulation by RPA
Although ZRANB3 does not contain an RPA binding domain, RPA affects the fork remodeling functions of ZRANB3 in vitro. In the absence of RPA, ZRANB3 is able to reverse replication forks with gaps on the leading and lagging strands with equal efficiency (Betous et al., 2013a). When RPA is added, a situation mirroring what is happening in cells during DNA replication, fork reversal on DNA substrates containing a lagging strand gap remain unchanged; whereas the presence of RPA on replication forks with a leading strand gap results in a strong inhibition of fork reversal (Betous et al., 2013a). These results contrast with the stimulation of SMARCAL1 when RPA is bound to the leading strand. Unlike SMARCAL1, RPA also inhibits ZRANB3-mediated restoration of a replication fork with a gap on the lagging strand.
How RPA regulates the fork remodeling function of ZRANB3 has yet to be determined. RPA could simply act as a steric block to ZRANB3 that prevents fork remodeling when bound to DNA. Since ZRANB3 does not contain a way to interact with RPA, the block could persist and prevent fork remodeling. However, the presence of RPA does not affect fork regression of DNA substrates with lagging strand gaps. One explanation is that the block may depend on which DNA strand ZRANB3 is tracking. While SNF2 enzymes act as double-strand DNA translocases, they are still thought to travel along one of the two duplexed strands. DNA footprinting studies suggest that SMARCAL1 binds to the leading strand template and may track on that strand (Betous et al., 2013a). If ZRANB3 also tracks on the leading strand, perhaps this would explain why a protein bound to that strand blocks its activity. Of course, this explanation cannot account for why SMARCAL1 is stimulated by RPA on the same substrate. Additional analyses of how these proteins actually accomplish DNA translocation are needed.
Regulation by phosphorylation
Initial microscopy experiments indicate ZRANB3 co-localization with PCNA is enhanced after DNA damage when ATR or ATM are inhibited (Ciccia et al., 2012). Whether this effect is mediated by direct phosphorylation of ZRANB3 is unknown. ATM and ATR inhibition does not alter the initial ZRANB3 recruitment to sites of damage; instead, it affects ZRANB3 retention. It is unclear whether this regulation is functionally important, but it is reminiscent of the negative regulation of SMARCAL1 by ATR (Couch et al., 2013). Thus, it may point to another mechanism by which fork remodeling reactions are restrained by the checkpoint kinases. Mapping of the relevant phosphorylation sites will be needed to better understand this regulation.
HLTF
HLTF is an annealing helicase, fork remodeler, and E3 ubiquitin ligase
Like SMARCAL1 and ZRANB3, HLTF functions primarily in the replication stress response. Although named as a helicase-like transcription factor, it is unlikely to directly regulate gene expression. Defects in HLTF function without exogenous stress do not result in strong genome instability phenotypes in cell culture (Blastyak et al., 2010). However, HLTF is commonly silenced in colorectal cancers indicating it may function as a tumor suppressor like ZRANB3 (Moinova et al., 2002, Sandhu et al., 2012, Lawrence et al., 2014). Like SMARCAL1 and ZRANB3, HLTF is an ATP-dependent dsDNA translocase (Blastyak et al., 2010). The function of HLTF in the replication stress response is multi-faceted and dependent on both its motor domain and an associated ubiquitin ligase activity. Failure to perform any of these functions results in increases in replication fork collapse and decreased cell viability after treatment with UV radiation or methyl methanesulfonate (MMS) (Motegi et al., 2008, Unk et al., 2008, Blastyak et al., 2010).
HLTF shares a high degree of sequence conservation with the yeast protein Rad5, a ubiquitin ligase that promotes post-replication repair of DNA damage through an error-free pathway (Unk et al., 2008). When a replication fork stalls in yeast, PCNA is monoubiquitinated through the concerted efforts of Rad6, a ubiquitin conjugating enzyme, and Rad18, a ubiquitin ligase that modifies PCNA on K164 (Figure 9) (Unk et al., 2010). Rad5 then transfers K63 polyubiquitin chains to PCNA that were assembled by the E2 ubiquitin conjugating enzyme complex Mms2/Ubc13 (Hoege et al., 2002). Yeast cells lacking Rad5 are hyper-sensitive to UV radiation, similar to HLTF-deficiency in human cells. Initial studies suggested human HLTF could compensate for Rad5 in S. cerevisiae; however, this result has not been reproduced by other groups (Motegi et al., 2008, Unk et al., 2008, MacKay et al., 2009). Nonetheless, HLTF is able to polyubiquitinate PCNA indicating it may share some functions with Rad5 (Motegi et al., 2008, Unk et al., 2008, Lin et al., 2011, Masuda et al., 2012). This ubiquitin ligase function of HLTF is required for genome stability as mutations in the responsible RING domain result in increases in replication fork collapse after treatment DNA damaging agents (Blastyak et al., 2010).
Figure 9.

Model for cooperativity between HLTF and ZRANB3. PCNA is monoubiquitinated by the RAD6/RAD18 complex after the replication fork stalls. Polyubiquitin chains are added by MMS2/UBC13 with the ubiquitin ligase HLTF. ZRANB3 then binds polyubiquitinated PCNA and remodels or cleaves the stalled replication fork. Ub, ubiquitin.
In addition to its ubiquitin ligase functions, HLTF is also able to catalyze fork reversal like SMARCAL1 and ZRANB3 (Blastyak et al., 2010, Achar et al., 2011). HLTF reverses replication forks either lacking any ssDNA or containing a gap on the leading strand in a process dependent on the ATPase domain and ATP hydrolysis (Blastyak et al., 2010, Achar et al., 2011, Achar et al., 2015, Kile et al., 2015). However, studies with other DNA substrates, such as lagging strand gapped substrates, have not been reported. Further, it has not yet been determined if HTLF can catalyze the restoration of a replication fork like ZRANB3 and SMARCAL1.
Interestingly, replication elongation actually proceeds faster in HLTF-deficient cells exposed to replication stress induced by low concentrations of HU compared to control cells (Kile et al., 2015). The authors of this study attribute this phenotype to a lack of HLTF-mediated fork reversal in response to replication stress since inactivation of the ATPase domain yields the same phenotype. Decreasing the frequency of fork reversal could yield faster overall elongation rates although it might come at the expense of genome stability.
One report also found that HLTF catalyzes D-loop formation in vitro (Burkovics et al., 2014). Unlike the canonical pathway of D-loop formation that requires invasion of a DNA duplex by a RAD51-coated ssDNA, HLTF is able to catalyze D-loop formation in the absence of any ssDNA binding proteins. The D-loop product is able to be extended by DNA polymerases in vitro indicating the production of a functional template. This function of HLTF was proposed as an alternative to fork reversal for promoting the restart of stalled replication forks. Interestingly, this process does not require ATP. How the D-loop is formed and which domains of HLTF are required for this function have not yet been determined.
HLTF Domain Structures and Function
HLTF is a 1009-amino acid protein with a HIRAN domain located at the N-terminus that acts as a SRD (Fig 1 and 5). The HIRAN domain is highly conserved throughout evolution. In addition to being present in HTLF and Rad5, the HIRAN domain is found as a stand-alone protein in some prokaryotic organisms and linked to other functional domains in some eukaryotes (Iyer et al., 2006). The HIRAN domain is required for DNA binding and fork remodeling catalyzed by HLTF but is dispensable for the ubiquitin ligase activity (Achar et al., 2015, Kile et al., 2015). In contrast to the HARP and HARP-like domains of SMARCAL1 and ZRANB3, the HIRAN domain of HLTF is not required for efficient DNA-dependent ATP hydrolysis (Kile et al., 2015).
Similar to SMARCAL1 and ZRANB3, HLTF binds dsDNA with low affinity; however, it can bind ssDNA and it prefers to bind a replication fork-like structure (Blastyak et al., 2010, Hishiki et al., 2015, Kile et al., 2015). The HIRAN domain by itself recognizes ssDNA overhangs with a preference for 3′ overhangs (Hishiki et al., 2015, Kile et al., 2015). High-resolution structural studies of the HIRAN domain in HLTF indicate the structure of this domain is strongly conserved throughout evolution (Achar et al., 2015, Hishiki et al., 2015, Kile et al., 2015, Korzhnev et al., 2016). Studies performed in the presence of DNA show that the HIRAN domain recognizes a 3′-hydroxyl group (3′OH) (Hishiki et al., 2015, Kile et al., 2015). The affinity for DNA substrates is decreased by adding a bulky fluorophore or phosphate group to the 3′ end (Hishiki et al., 2015, Kile et al., 2015). Even replacing the 3′-OH with a hydrogen reduces the binding capabilities of HIRAN (Kile et al., 2015).
The presence of a 3′-OH stimulates HIRAN binding and also stimulates the fork reversal activity of HLTF (Kile et al., 2015). Phosphorylating the 3′ end of the nascent leading strand inhibits fork reversal by HLTF. This strong preference for 3′-OH end is not shared by SMARCAL1, which can catalyze fork regression of 3′-OH and 3′-phosphorylated substrates at equal efficiencies. Thus, this mode of regulation is not a common mechanism shared by the SNF2 annealing helicases but rather a function specific to HLTF and the HIRAN domain.
This binding preference would fit a model where a replication fork has stalled and the 3′-OH end of the leading strand is exposed near the fork junction. The HIRAN domain of HLTF could capture the 3′-OH end of the nascent DNA during fork reversal to facilitate the unwinding of the template-nascent strand duplex to create a 4-way junction and stabilize the replication fork (Kile et al., 2015). Alternatively, it is possible that HLTF works after SMARCAL1 or ZRANB3 reverses the fork sufficiently for the 3′OH group to be in proximity to the fork junction assuming the HLTF ATPase binds the parental DNA duplex.
The ubiquitin ligase activity of HLTF is conferred by a RING domain located in the linker between the two lobes of the ATPase domain (Figure 1). Like Rad5, HLTF catalyzes the polyubiquitination of PCNA (Motegi et al., 2008, Unk et al., 2008). In human cells, PCNA is monoubiquitinated on K164 by the RAD6/RAD18 complex that serves an equivalent role as its yeast homolog (Figure 9) (Mailand et al., 2013). HLTF can catalyze polyubiquitination with the E2 ubiquitin conjugating complex MMS2/UBC13 (Masuda et al., 2012). Like the ATPase domain, the function of this domain has strong implications on DNA replication as mutations in the RING domain result in increased frequencies of fork stalling and fork collapse after treatment with DNA damaging agents (Blastyak et al., 2010). Recognition of polyubiquitinated PCNA by ZRANB3 provides one explanation for the functional consequence of this modification in human cells. This recognition suggests HLTF may function upstream of ZRANB3 in a common pathway (Figure 9).
In addition to HLTF, the protein SHPRH also shares sequence homology with the yeast Rad5 protein (Unk et al., 2010). SHPRH can polyubiquitinate PCNA in cells and in vitro (Unk et al., 2006, Motegi et al., 2008). SHPRH-deficient cells display high rates of MMS-induced mutagenesis (Lin et al., 2011). Rather than functioning redundantly, HLTF and SHPRH are utilized differentially depending on the type of damage present at the replication fork. HLTF primarily functions in response to UV-induced damage while SHPRH responds to the damage from MMS (Lin et al., 2011). While HLTF and SHPRH provide two complementary pathways that result in PCNA polyubiquitination, there may be other PCNA ubiquitin ligases since some polyubiquitination persists even when both enzymes are inactivated (Lin et al., 2011).
HLTF Regulation
Regulation of fork reversal
Studies using iPOND indicate HLTF is enriched at replication forks (Dungrawala et al., 2015, Kile et al., 2015). However, the mechanism of recruitment to forks is still unknown. Unlike SMARCAL1 and ZRANB3, canonical protein interaction domains and motifs have not been identified in HLTF. One study looking to identify HLTF interacting partners in unperturbed cells identified RPA70 and the dsDNA break repair regulatory protein PTIP (Pax transactivation domain-interacting protein) (MacKay et al., 2009). Further studies to investigate the importance of these interactions have not yet been completed, but it's possible these interactions could recruit HLTF to sites of replication stress.
Alternatively, these proteins could also regulate the fork remodeling functions of HLTF. Initial biochemical studies indicate RPA has no effect on the HLTF fork reversal activity when bound to a leading strand DNA gapped substrate (Achar et al., 2011). HLTF is also able to catalyze fork reversal in the presence of PCNA, and the PCNA loading enzyme replication factor C (RFC) (Achar et al., 2011). However, careful kinetic studies to characterize the effects of RPA and PCNA with various substrates would be useful to better define if these proteins affect HLTF function. No in vitro studies have been completed with other validated HLTF interacting proteins.
Conclusions and Perspectives
Table 1 summarizes the similarities and differences between SMARCAL1, ZRANB3, and HTLF. Since all catalyze similar fork remodeling reactions in vitro an important question is why cells have three related enzymes. Certainly the addition of a nuclease domain to ZRANB3 and a ubiquitin ligase domain to HLTF provides added functionality to these proteins compared to SMARCAL1. However, if all three enzymes catalyze fork reversal using their motor domains, other differences must be important to explain why this function is retained in all three enzymes. Perhaps different fork structures are formed by various forms of replication stress that require enzymes with different specificities or modes of regulation. Diverse types of replication stress that generate stalled forks with unique characteristics result in fork reversal in human cells (Zellweger et al., 2015). The differences in the SRD domains in each of the proteins may help them recognize and work on forks with different structures.
Table 1.
Properties of SMARCAL1, ZRANB3, and HLTF.
| SMARCAL1 | ZRANB3 | HLTF | ||
|---|---|---|---|---|
| Biochemistry | Annealing helicase? | Yes | Yes | ? |
| Fork remodeling activities | Fork reversal, Fork restoration | Fork reversal, Fork restoration | Fork reversal | |
| D loop activities | D loop dissolution | D loop dissolution, D loop inhibition | D loop formation | |
| Other enzymatic activities | ATP-dependent endonuclease | Ubiquitin ligase | ||
| Protein Structure | Substrate recognition domain (SRD) | HARP | HARP-like? | HIRAN |
| SRD DNA preference | Splayed arm junction | Splayed arm junction | ssDNA with 3′-OH | |
| Replication fork recruitment mechanism | RPA | Polyubiquitinated PCNA | ? | |
| Evolutionary conservation | Metazoans, Bacteriophage | Metazoans | Metazoans to yeast | |
| In cells | Reported locations of function | Telomeres | ? | ? |
| Loss of function replication phenotypes | dsDNA breaks, fork restart deficiency | SCEs, fork restart deficiency | faster fork elongation with mild stress | |
| Drug sensitivity | HU, CPT, MMC, IR, aphidicolin | HU, CPT, MMS, MMC, IR, cisplatin | UV, MMS | |
| Human disease | Schimke Immunooseous Dysplasia | endometrial cancer? | colorectal cancer |
Regulation of their recruitment to stalled forks also differentiates these proteins. iPOND studies of replication fork proteomes indicates that ZRANB3 and HLTF localization to forks differs significantly from SMARCAL1. SMARCAL1 abundance at forks closely tracks RPA abundance (Dungrawala et al., 2015). Thus, stalling forks with HU-treatment yields a large increase in SMARCAL1 localization to replication forks. In contrast, ZRANB3 abundance tracks with PCNA and its abundance actually decreases at stalled forks as PCNA abundance decreases (Dungrawala et al., 2015). Thus, fork stalling by HU without a DNA lesion appears to favor SMARCAL1 recruitment over ZRANB3. HU treatment does not yield large increases in recruitment of the HLTF and SHPRH ubiquitin ligases or high levels of polyubiquitinated PCNA likely explaining why ZRANB3 is not more abundant at stalled forks in these circumstances. These observations point to the source of replication stress as a determinant of which enzyme is utilized. Consistent with this idea, SMARCAL1 is required for replication through telomeres; however, silencing HLTF or ZRANB3 did not yield telomere instability (Poole et al., 2015). Furthermore, only silencing SMARCAL1 in unstressed caused spontaneous double-strand breaks (Dungrawala et al., 2017).
Besides influencing localization through protein interaction partners, the source of replication stress might also influence the DNA structures present at replication forks. This, in turn, might influence the preferential recruitment or function of HLTF, SMARCAL1, and ZRANB3. All three enzymes can catalyze fork reversal in the absence of any additional proteins. The efficiency of these reactions begins to change once DNA gaps are introduced to the substrate and supplemental DNA binding proteins are added to the reactions. Fork reversal by SMARCAL1 on a leading strand gapped DNA substrate is stimulated by the presence of RPA whereas fork reversal by ZRANB3 is inhibited and HLTF function may be unaffected. The SRD HIRAN in HLTF specifically recognizes a 3′-OH while SMARCAL1 does not display a bias for this functional group. These differences in substrate preferences, conferred by the SRDs, would diversify the pool of potential substrates and could contribute to the requirement for each of these enzymes in cells.
Another model is that these enzymes operate sequentially or cooperatively at a single stalled fork. PCNA ubiquitylation by HLTF could be part of the signal that directs ZRANB3 to act on the fork (Figure 9). Also, the DNA substrate changes during fork reversal. A leading strand gapped fork with RPA bound could be a good substrate for SMARCAL1, but as soon as the fork has reversed far enough to remove RPA and bring the 3′ end of the leading strand close to the fork junction, it would be an excellent substrate for HLTF recognition. Furthermore, nuclease actions at the fork both before and after fork reversal could change the fork structure making it better or worse for one of the enzymes. Finally, regulatory signals such as ATR phosphorylation of SMARCAL1 could signal some kind of hand-off mechanism. Thus, these enzymes could function cooperatively even in response to a single replication stress challenge. In this model, the differences in phenotypic outcomes caused by inactivation could be due to alternative functions such as the proposed function of SMARCAL1 in double-strand break repair or transcriptional regulation. Additionally, failures at any one step in a fork repair process would cause shunting of the stalled fork into alternative pathways that may differ depending on which enzyme was inactivated.
Much of the in vitro work to identify the DNA binding and substrate preferences has been done with naked DNA or in the presence of one additional protein like RPA and PCNA. The biochemical studies have not yet even incorporated proteins like RAD51 that are important for fork reversal and might be expected to alter the enzymatic activities of the DNA translocases. The fork is also a very crowded place since the replisome does not disassemble when the fork is stalled (Dungrawala et al., 2015), raising the question of how the fork remodelers gain access to the fork junction. Also, where the replisome goes during the fork reversal process itself is unknown. Specifically, the replicative helicase must be retained on the DNA since forks restart rapidly after the replication stress is removed and it cannot be reloaded once S-phase has begun. Does the helicase get pushed away from the junction and trapped in a bubble within the duplex? Also, while nucleosomes are removed from the immediate vicinity of the fork, their deposition on the newly synthesized DNA would be expected to limit the distances that replication forks can reverse and could have implications on the rates of fork restoration. Even the DNA itself is not adequately modeled biochemically since things like torsional stress are not accounted for at least in the solution biochemistry experiments. Extending these in vitro assays to reflect more complex biological conditions, such as including histones and the replication machinery, might identify different or supplemental mechanisms of regulation.
In addition to the SNF2 annealases, there are also several other proteins that have replication fork remodeling functions. RECQ helicases, the Fanconi Anemia helicase FANCM, and combining RAD54 with RAD51 can all catalyze fork reversal in vitro (Neelsen and Lopes, 2015). How all these proteins coordinate in cells to remodel replication forks is unknown. Finally, the consequences for human disease caused by their inactivation or misregulation can be profound. Describing how SMARCAL1 works at a mechanistic level is important, but does little to help SIOD patients and their families deal with the devastating outcomes of SMARCAL1 mutations. HLTF inactivation appears to be a cancer driver, but the kinds of genetic changes that happen in HLTF-deficient cells that drive tumorigenesis are unknown. Thus, future studies to describe where, when, and how SMARCAL1, ZRANB3 and HLTF act to maintain the genome will require structure, solution and single-molecule biochemistry, model organism genetics, human cell experiments, and animal models.
Acknowledgments
We thank Brandt Eichman for critically reading the manuscript.
Disclosure of Interest: Studies of SMARCAL1 and ZRANB3 in the Cortez lab is partially funded by NIH grants P01CA092584 and R01CA160432. L.P. is funded by NIH grant F31CA189375.
References
- Achar YJ, Balogh D, Haracska L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc Natl Acad Sci U S A. 2011;108:14073–8. doi: 10.1073/pnas.1101951108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achar YJ, Balogh D, Neculai D, Juhasz S, Morocz M, Gali H, Dhe-Paganon S, Venclovas C, Haracska L. Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res. 2015;43:10277–91. doi: 10.1093/nar/gkv896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badu-Nkansah A, Mason AC, Eichman BF, Cortez D. Identification of a Substrate Recognition Domain in the Replication Stress Response Protein Zinc Finger Ran-binding Domain Containing Protein 3 (ZRANB3) J Biol Chem. 2016 doi: 10.1074/jbc.M115.709733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansbach CE, Betous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–14. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansbach CE, Boerkoel CF, Cortez D. SMARCAL1 and replication stress: an explanation for SIOD? Nucleus. 2010;1:245–8. doi: 10.4161/nucl.1.3.11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baradaran-Heravi A, Cho KS, Tolhuis B, Sanyal M, Morozova O, Morimoto M, Elizondo LI, Bridgewater D, Lubieniecka J, Beirnes K, Myung C, Leung D, Fam HK, Choi K, Huang Y, Dionis KY, Zonana J, Keller K, Stenzel P, Mayfield C, Lucke T, Bokenkamp A, Marra MA, Van Lohuizen M, Lewis DB, Shaw C, Boerkoel CF. Penetrance of biallelic SMARCAL1 mutations is associated with environmental and genetic disturbances of gene expression. Hum Mol Genet. 2012a;21:2572–87. doi: 10.1093/hmg/dds083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baradaran-Heravi A, Raams A, Lubieniecka J, Cho KS, Dehaai KA, Basiratnia M, Mari PO, Xue Y, Rauth M, Olney AH, Shago M, Choi K, Weksberg RA, Nowaczyk MJ, Wang W, Jaspers NG, Boerkoel CF. SMARCAL1 deficiency predisposes to non-Hodgkin lymphoma and hypersensitivity to genotoxic agents in vivo. Am J Med Genet A. 2012b;158A:2204–13. doi: 10.1002/ajmg.a.35532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, Cortez D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol. 2016;18:1185–1195. doi: 10.1038/ncb3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betous R, Couch FB, Mason AC, Eichman BF, Manosas M, Cortez D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013a;3:1958–69. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betous R, Glick GG, Zhao R, Cortez D. Identification and characterization of SMARCAL1 protein complexes. PLoS One. 2013b;8:e63149. doi: 10.1371/journal.pone.0063149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, Cortez D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–62. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Betous R, Cortez D. High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. J Biol Chem. 2015;290:4110–7. doi: 10.1074/jbc.M114.627083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Blastyak A, Hajdu I, Unk I, Haracska L. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol. 2010;30:684–93. doi: 10.1128/MCB.00863-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, Andre JL, Bogdanovic R, Burguet A, Cockfield S, Cordeiro I, Frund S, Illies F, Joseph M, Kaitila I, Lama G, Loirat C, Mcleod DR, Milford DV, Petty EM, Rodrigo F, Saraiva JM, Schmidt B, Smith GC, Spranger J, Stein A, Thiele H, Tizard J, Weksberg R, Lupski JR, Stockton DW. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet. 2002;30:215–20. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- Burkovics P, Sebesta M, Balogh D, Haracska L, Krejci L. Strand invasion by HLTF as a mechanism for template switch in fork rescue. Nucleic Acids Res. 2014;42:1711–20. doi: 10.1093/nar/gkt1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll C, Badu-Nkansah A, Hunley T, Baradaran-Heravi A, Cortez D, Frangoul H. Schimke Immunoosseous Dysplasia associated with undifferentiated carcinoma and a novel SMARCAL1 mutation in a child. Pediatr Blood Cancer. 2013;60:E88–90. doi: 10.1002/pbc.24542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll C, Bansbach CE, Zhao R, Jung SY, Qin J, Cortez D. Phosphorylation of a C-terminal auto-inhibitory domain increases SMARCAL1 activity. Nucleic Acids Res. 2014;42:918–25. doi: 10.1093/nar/gkt929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll C, Hunley TE, Guo Y, Cortez D. A novel splice site mutation in SMARCAL1 results in aberrant exon definition in a child with Schimke immunoosseous dysplasia. Am J Med Genet A. 2015;167A:2260–4. doi: 10.1002/ajmg.a.37146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319–30. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–25. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, Livingston DM, Haracska L, Elledge SJ. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clewing JM, Fryssira H, Goodman D, Smithson SF, Sloan EA, Lou S, Huang Y, Choi K, Lucke T, Alpay H, Andre JL, Asakura Y, Biebuyck-Gouge N, Bogdanovic R, Bonneau D, Cancrini C, Cochat P, Cockfield S, Collard L, Cordeiro I, Cormier-Daire V, Cransberg K, Cutka K, Deschenes G, Ehrich JH, Frund S, Georgaki H, Guillen-Navarro E, Hinkelmann B, Kanariou M, Kasap B, Kilic SS, Lama G, Lamfers P, Loirat C, Majore S, Milford D, Morin D, Ozdemir N, Pontz BF, Proesmans W, Psoni S, Reichenbach H, Reif S, Rusu C, Saraiva JM, Sakallioglu O, Schmidt B, Shoemaker L, Sigaudy S, Smith G, Sotsiou F, Stajic N, Stein A, Stray-Pedersen A, Taha D, Taque S, Tizard J, Tsimaratos M, Wong NA, Boerkoel CF. Schimke immunoosseous dysplasia: suggestions of genetic diversity. Hum Mutat. 2007;28:273–83. doi: 10.1002/humu.20432. [DOI] [PubMed] [Google Scholar]
- Coleman MA, Eisen JA, Mohrenweiser HW. Cloning and characterization of HARP/SMARCAL1: a prokaryotic HepA-related SNF2 helicase protein from human and mouse. Genomics. 2000;65:274–82. doi: 10.1006/geno.2000.6174. [DOI] [PubMed] [Google Scholar]
- Cortez D. Preventing replication fork collapse to maintain genome integrity. DNA Repair (Amst) 2015;32:149–57. doi: 10.1016/j.dnarep.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, Cortez D. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610–23. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox KE, Marechal A, Flynn RL. SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell Rep. 2016 doi: 10.1016/j.celrep.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, Wessel SR, Sathe AA, Zhao R, Cortez D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol Cell. 2017 doi: 10.1016/j.molcel.2017.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol Cell. 2015;59:998–1010. doi: 10.1016/j.molcel.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaus A, Owen-Hughes T. Mechanisms for ATP-dependent chromatin remodelling: the means to the end. FEBS J. 2011;278:3579–95. doi: 10.1111/j.1742-4658.2011.08281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal G, Yuan J, Chen J. The HARP domain dictates the annealing helicase activity of HARP/SMARCAL1. EMBO Rep. 2011;12:574–80. doi: 10.1038/embor.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson E, Geli V. How telomeres are replicated. Nat Rev Mol Cell Biol. 2007;8:825–38. doi: 10.1038/nrm2259. [DOI] [PubMed] [Google Scholar]
- Haahr P, Hoffmann S, Tollenaere MA, Ho T, Toledo LI, Mann M, Bekker-Jensen S, Raschle M, Mailand N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol. 2016;18:1196–1207. doi: 10.1038/ncb3422. [DOI] [PubMed] [Google Scholar]
- Haokip DT, Goel I, Arya V, Sharma T, Kumari R, Priya R, Singh M, Muthuswami R. Transcriptional Regulation of Atp-Dependent Chromatin Remodeling Factors: Smarcal1 and Brg1 Mutually Co-Regulate Each Other. Sci Rep. 2016;6:20532. doi: 10.1038/srep20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson JD, Cao Y, Huschtscha LI, Chang AC, Au AY, Pickett HA, Reddel RR. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat Biotechnol. 2009;27:1181–5. doi: 10.1038/nbt.1587. [DOI] [PubMed] [Google Scholar]
- Hishiki A, Hara K, Ikegaya Y, Yokoyama H, Shimizu T, Sato M, Hashimoto H. Structure of a Novel DNA-binding Domain of Helicase-like Transcription Factor (HLTF) and Its Functional Implication in DNA Damage Tolerance. J Biol Chem. 2015;290:13215–23. doi: 10.1074/jbc.M115.643643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jensch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- Huang C, Gu S, Yu P, Yu F, Feng C, Gao N, Du J. Deficiency of smarcal1 causes cell cycle arrest and developmental abnormalities in zebrafish. Dev Biol. 2010;339:89–100. doi: 10.1016/j.ydbio.2009.12.018. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Babu MM, Aravind L. The HIRAN domain and recruitment of chromatin remodeling and repair activities to damaged DNA. Cell Cycle. 2006;5:775–82. doi: 10.4161/cc.5.7.2629. [DOI] [PubMed] [Google Scholar]
- Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol. 2013;5:a012740. doi: 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassavetis GA, Kadonaga JT. The annealing helicase and branch migration activities of Drosophila HARP. PLoS One. 2014;9:e98173. doi: 10.1371/journal.pone.0098173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keka IS, Mohiuddin, Maede Y, Rahman MM, Sakuma T, Honma M, Yamamoto T, Takeda S, Sasanuma H. Smarcal1 promotes double-strand-break repair by nonhomologous end-joining. Nucleic Acids Res. 2015;43:6359–72. doi: 10.1093/nar/gkv621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, Cimprich KA. HLTF's Ancient HIRAN Domain Binds 3′ DNA Ends to Drive Replication Fork Reversal. Mol Cell. 2015;58:1090–100. doi: 10.1016/j.molcel.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE, Kastan MB. Substrate Specificities and Identification of Putative Substrates of ATM Kinase Family Members. Journal of Biological Chemistry. 1999;274:37538–37543. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, Krejci L, Costanzo V. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell. 2017 doi: 10.1016/j.molcel.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzhnev DM, Neculai D, Dhe-Paganon S, Arrowsmith CH, Bezsonova I. Solution NMR structure of the HLTF HIRAN domain: a conserved module in SWI2/SNF2 DNA damage tolerance proteins. J Biomol NMR. 2016;66:209–219. doi: 10.1007/s10858-016-0070-9. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Zeman MK, Chen JY, Yee MC, Cimprich KA. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol Cell. 2011;42:237–49. doi: 10.1016/j.molcel.2011.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy CA, Cortez D. Common mechanisms of PIKK regulation. DNA Repair (Amst) 2009;8:1004–8. doi: 10.1016/j.dnarep.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay C, Toth R, Rouse J. Biochemical characterisation of the SWI/SNF family member HLTF. Biochem Biophys Res Commun. 2009;390:187–91. doi: 10.1016/j.bbrc.2009.08.151. [DOI] [PubMed] [Google Scholar]
- Mailand N, Gibbs-Seymour I, Bekker-Jensen S. Regulation of PCNA-protein interactions for genome stability. Nat Rev Mol Cell Biol. 2013;14:269–82. doi: 10.1038/nrm3562. [DOI] [PubMed] [Google Scholar]
- Manosas M, Perumal SK, Croquette V, Benkovic SJ. Direct observation of stalled fork restart via fork regression in the T4 replication system. Science. 2012;338:1217–20. doi: 10.1126/science.1225437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason AC, Rambo RP, Greer B, Pritchett M, Tainer JA, Cortez D, Eichman BF. A structure-specific nucleic acid-binding domain conserved among DNA repair proteins. Proc Natl Acad Sci U S A. 2014;111:7618–23. doi: 10.1073/pnas.1324143111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda Y, Suzuki M, Kawai H, Hishiki A, Hashimoto H, Masutani C, Hishida T, Suzuki F, Kamiya K. En bloc transfer of polyubiquitin chains to PCNA in vitro is mediated by two different human E2-E3 pairs. Nucleic Acids Res. 2012;40:10394–407. doi: 10.1093/nar/gks763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K, Borel V, Adelman CA, Schindler D, Boulton SJ. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev. 2015;29:2532–46. doi: 10.1101/gad.272740.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moinova HR, Chen WD, Shen L, Smiraglia D, Olechnowicz J, Ravi L, Kasturi L, Myeroff L, Plass C, Parsons R, Minna J, Willson JK, Green SB, Issa JP, Markowitz SD. HLTF gene silencing in human colon cancer. Proc Natl Acad Sci U S A. 2002;99:4562–7. doi: 10.1073/pnas.062459899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi A, Liaw HJ, Lee KY, Roest HP, Maas A, Wu X, Moinova H, Markowitz SD, Ding H, Hoeijmakers JH, Myung K. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc Natl Acad Sci U S A. 2008;105:12411–6. doi: 10.1073/pnas.0805685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswami R, Truman PA, Mesner LD, Hockensmith JW. A eukaryotic SWI2/SNF2 domain, an exquisite detector of double-stranded to single-stranded DNA transition elements. J Biol Chem. 2000;275:7648–55. doi: 10.1074/jbc.275.11.7648. [DOI] [PubMed] [Google Scholar]
- Muzzin O, Campbell EA, Xia L, Severinova E, Darst SA, Severinov K. Disruption of Escherichia coli HepA, an RNA Polymerase-associated Protein, Causes UV Sensitivity. Journal of Biological Chemistry. 1998;273:15157–15161. doi: 10.1074/jbc.273.24.15157. [DOI] [PubMed] [Google Scholar]
- Neelsen KJ, Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol. 2015;16:207–20. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- Oakley GO, Patrick SM. Replication protein A: directing traffic at the intersection of replication and repair. Frontiers in Bioscience. 2010:883–900. doi: 10.2741/3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patne K, Rakesh R, Arya V, Chanana UB, Sethy R, Swer PB, Muthuswami R. BRG1 and SMARCAL1 transcriptionally co-regulate DROSHA, DGCR8 and DICER in response to doxorubicin-induced DNA damage. Biochim Biophys Acta. 2017;1860:936–951. doi: 10.1016/j.bbagrm.2017.07.003. [DOI] [PubMed] [Google Scholar]
- Poole LA, Cortez D. SMARCAL1 and telomeres: Replicating the troublesome ends. Nucleus. 2016;7:270–4. doi: 10.1080/19491034.2016.1179413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole LA, Zhao R, Glick GG, Lovejoy CA, Eischen CM, Cortez D. SMARCAL1 maintains telomere integrity during DNA replication. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1510750112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow L, Woo EM, Chait BT, Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem. 2009;284:35951–61. doi: 10.1074/jbc.M109.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puccetti MV, Fischer MA, Arrate MP, Boyd KL, Duszynski RJ, Betous R, Cortez D, Eischen CM. Defective replication stress response inhibits lymphomagenesis and impairs lymphocyte reconstitution. Oncogene. 2016 doi: 10.1038/onc.2016.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DP, Owen-Hughes T. Snf2-family proteins: chromatin remodellers for any occasion. Curr Opin Chem Biol. 2011;15:649–56. doi: 10.1016/j.cbpa.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu S, Wu X, Nabi Z, Rastegar M, Kung S, Mai S, Ding H. Loss of HLTF function promotes intestinal carcinogenesis. Mol Cancer. 2012;11:18. doi: 10.1186/1476-4598-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebesta M, Cooper CDO, Ariza A, Carnie CJ, Ahel D. Structural insights into the function of ZRANB3 in replication stress response. Nat Commun. 2017;8:15847. doi: 10.1038/ncomms15847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma T, Bansal R, Haokip DT, Goel I, Muthuswami R. SMARCAL1 Negatively Regulates C-Myc Transcription By Altering The Conformation Of The Promoter Region. Sci Rep. 2015;5:17910. doi: 10.1038/srep17910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- Sukhodolets MV, Cabrera JE, Zhi H, Jin DJ. RapA, a bacterial homolog of SWI2/SNF2, stimulates RNA polymerase recycling in transcription. Genes Dev. 2001;15:3330–41. doi: 10.1101/gad.936701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich HD, Walden H. Ubiquitin signalling in DNA replication and repair. Nat Rev Mol Cell Biol. 2010;11:479–89. doi: 10.1038/nrm2921. [DOI] [PubMed] [Google Scholar]
- Unk I, Hajdu I, Blastyak A, Haracska L. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair (Amst) 2010;9:257–67. doi: 10.1016/j.dnarep.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Unk I, Hajdu I, Fatyol K, Hurwitz J, Yoon JH, Prakash L, Prakash S, Haracska L. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc Natl Acad Sci U S A. 2008;105:3768–73. doi: 10.1073/pnas.0800563105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk I, Hajdu I, Fatyol K, Szakal B, Blastyak A, Bermudez V, Hurwitz J, Prakash L, Prakash S, Haracska L. Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc Natl Acad Sci U S A. 2006;103:18107–12. doi: 10.1073/pnas.0608595103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston R, Peeters H, Ahel D. ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev. 2012;26:1558–72. doi: 10.1101/gad.193516.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–9. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Ghosal G, Chen J. The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol Cell. 2012;47:410–21. doi: 10.1016/j.molcel.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T, Kadonaga JT. HARP Is an ATP-Driven Annealing Helicase. Science. 2008;322:748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T, Kadonaga JT. Annealing helicase 2 (AH2), a DNA-rewinding motor with an HNH motif. Proc Natl Acad Sci U S A. 2010;107:20970–3. doi: 10.1073/pnas.1011196107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T, Kong X, Yokomori K, Kadonaga JT. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009;23:2400–4. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol. 2015;208:563–79. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]